

Abstract
Pancreatic cancer has one of the worst prognoses among all cancers due to the late manifestation of identifiable symptoms and high resistance to chemo- and radiation therapies. In recent years, a cancer development phase termed epithelial-mesenchymal transition (EMT) has gained increasing research focus. The process is implicated in tumour metastasis, and emerging evidence suggests EMT also contributes or induces chemoresistance in several cancers. Nevertheless, the applicability of therapeutic targeting of EMT faces many challenges. In this mini-review, we summarise the evidence supporting the role of EMT in pancreatic cancer progression, focusing particularly on its association with chemoresistance.
Keywords: Epithelial-mesenchymal transition, Drug resistance, Pancreatic cancer, Chemotherapy
Core tip: This mini-review examines the role of epithelial-mesenchymal transition in the development of drug resistant pancreatic cancer and a possible use of this process as a drug target.
INTRODUCTION
Epithelial-mesenchymal transition (EMT) is a stage of phenotypic alteration in cancer cells instigated by several paracrine and autocrine stimuli, leading to a morphological transformation of epithelial-like cancer cells to an elongated mesenchymal phenotype. The phenotypic change is thought to derive from a shift in the balance between epithelial (e.g., E-cadherin and Claudin-1) and mesenchymal (e.g., N-cadherin, Snail, Zeb-1 and Twist-1) factors. Once described as a key step for successful metastasis in some types of cancers[1], the role of EMT in chemotherapy resistance has attracted much interest recently. Indeed, EMT has been shown to contribute to drug resistance in pancreatic cancer. For instance, in a recent study, the patterns of sensitivity and resistance to three conventional chemotherapeutic agents with divergent mechanisms of action were investigated in several pancreatic cancer cell lines[2]. Interestingly, gene expression profiling revealed that the sensitive and resistant cells formed two distinct groups with resistant cells showing several features consistent with EMT. Additionally, an inverse correlation between E-cadherin and its transcriptional suppressor, Zeb-1, was observed in the gene expression. Moreover, silencing of Zeb-1 restored drug sensitivity in pancreatic cancer cells. The implication of this study is that effectors of EMT, in particular Zeb-1, are potential molecules to target to overcome drug resistance. Recently, a study casts doubt on the role of EMT in metastasis, while a separate study strongly corroborates the key role of EMT in drug resistance. Indeed, both Fischer et al[3] and Zheng et al[4], demonstrated that not only was EMT able to aid chemotherapy resistance in pancreatic and lung cancer cells but that EMT was not needed for metastasis, as inhibition of EMT did not prevent metastasis of pancreatic and lung cancer cells.
EMT SIGNALLING PATHWAYS IN PANCREATIC CANCER
Several signalling pathways can regulate EMT during tumorigenesis[5-7]. Major pathways which can induce EMT include those activated by growth factors such as transforming growth factor-β (TGFβ), epidermal growth factor and hepatocyte growth factor or the Wnt/beta-catenin and Notch pathway. Notch-2 activation was identified in gemcitabine-resistant (GR) pancreatic cancer cells that have acquired EMT. By using pancreatic cancer cells that have developed gemcitabine resistance through exposure to increasing concentrations of gemcitabine, Wang et al[8] were the first to delineate the EMT profile of GR cells. GR but not gemcitabine sensitive (GS) cells had an elongated and irregular shape, increased expression of Zeb-1, vimentin, fibronectin and alpha-SMA, as well as translocation of E-cadherin from the plasma membrane to the nucleus. In concordance with previous reports regarding the role of Notch signalling in EMT, Wang et al[8] further characterised the Notch activation status in GR cells. Notch-2 and its ligands, Jagged-1 and Notch-4, were upregulated in GR cells. The Notch signalling also upregulates NF-κB, a critical mediator of the EMT process. Indeed, NF-κB was upregulated in GR cells as well as its downstream target, matrix metallopeptidase 9 (MMP-9). The activity of MMP-9 is governed by urinary plasminogen activator (uPA), also upregulated in GR cells, and both proteins are known for their role in cancer invasion and metastasis. Using Notch-2 and Jagged-1 siRNA, the interrelation between Notch, EMT and NF-κB were further delineated. Notch-2 and Jagged-1 knockdown in GR cells led to mesenchymal-epithelial transition (MET) morphological changes which include upregulation of E-cadherin, and reduction of vimentin, slug, snail, Zeb-1, and NF-κB (p65 subunit). As expected, Notch-2 and Jagged-1 downregulation by siRNA also led to a reduction in GR cells invasion and migration[8]. The study characterised for the first time the Notch-driven EMT pathway in GR pancreatic cancer cells, however, the authors did not address whether silencing or inhibition of Notch could reverse GR.
EMT AND GEMCITABINE RESISTANCE
The development of gemcitabine resistance and its association with EMT phenotype has drawn attention to a possible role of gemcitabine in inducing EMT. Güngör et al[9] examined the role of Midkine (MK) in orchestrating the interplay between Notch, EMT and gemcitabine resistance. MK is a heparin-binding growth factor overexpressed in some types of cancers[10,11]. High MK mRNA expression was detected in pancreatic cancer cell lines that developed gemcitabine resistance following repeated exposure to gemcitabine, and in PDAC tumour samples isolated from patients who underwent total pancreaticoduodenectomy. Gemcitabine treatment in GR cells led to a dose-dependent increase in MK mRNA expression and protein secretion, an effect not observed in GS cells. Knockdown of MK with siRNA restored gemcitabine sensitivity in GR cells, while the addition of recombinant human MK (rh-MK) to MK-knockdown or GS cells, restored or induced resistance, respectively. GR cells overexpressing MK displayed EMT characteristics while MK-depleted GR cells displayed MET characteristics as evidenced by downregulation of vimentin and NF-κB, and upregulation of E-cadherin. As expected, there was a reduction in migration and invasion in MK-depleted cells compared to control. As Notch-2 and EMT are associated with gemcitabine resistance, Güngör et al[9] further examined the impact of MK on Notch-2 activation. Treatment of GR cells with rh-MK resulted in enhanced cleavage of NotchICD and expression of Hes-1 (Notch-2 target). Expectedly, silencing of Notch-2 improved gemcitabine efficacy in GR cells. Güngör et al[9] were the first to pinpoint the role of MK in gemcitabine resistance and its impact on Notch-2 activation and EMT phenotype.
The role of Notch-2 activation in EMT, metastasis and chemotherapy resistance has attracted attention to target Notch-2 in pancreatic cancer. Palagani et al[12] showed the effect of the γ-secretase inhibitor (GSI IX) in preventing Notch-2 activation, EMT, and cancer cell proliferation and migration in vitro and pancreatic tumour-initiating CD44+/EpCAM+ xenograft growth and metastasis in vivo. Future studies are needed to examine the effect of GSI IX in reducing gemcitabine resistance in pancreatic cancer.
A few microRNAs are implicated in EMT and chemoresistance. Li et al[13] were the first to introduce a novel way of reducing gemcitabine resistance in pancreatic cancer (PaCa) through modulation of microRNAs. GR PaCa cells showed downregulation of miR-200 in addition to the typical EMT signature discussed earlier. Upregulation of miR-200 either through the reintroduction of miR-200 or the treatment with the natural compound, isoflavone, resulted in MET as demonstrated by decreased levels of mesenchymal markers (Zeb1, Vimentin and Slug) and induction of epithelial-associated morphological changes, and thus reducing gemcitabine resistance[13]. Using a similar methodology, Ma et al[14] recently showed a positive influence of miR-233 in EMT, invasion, migration and gemcitabine resistance in PaCa cells.
The conversion in cancer cells from epithelial to mesenchymal phenotype and its requirement for cancer invasion and migration were clearly demonstrated through in vitro studies. However, the consequence of EMT on cancer metastasis had not been investigated in vivo until recently. Using the well-established mouse model that mirrors human pancreatic cancer (KPC mice), Zheng et al[4], produced KPC mice absent in Snail or Twist protein. Although accumulating evidence suggests the requirement of EMT process for cancer migration, the authors showed the ability of pancreatic cancer to metastasize despite deleted EMT-inducing factors, Snail and Twist. Deletion of either one of these proteins did not affect local invasion, metastasis or overall survival compared to control KPC mice. It also resulted in a reduction in expression of other mesenchymal markers (e.g., Slug, Zeb1 and alpha-SMA) while enhancing the expression of the epithelial factor, E-cadherin. Although the apoptosis of cancer cells was not affected by deletion of Snail or Twist, the proliferation rate of cancer cells significantly increased, while blood dissemination remained unchanged compared to control KPC mice. Examination of EMT profile of the metastatic pancreatic cancer cells at secondary sites (liver, lung and spleen) showed positive for E-cadherin, and negative for Snail or Twist. Moreover, the ability of cancer cells, isolated from either control KPC mice or KPC with deleted Snail or Twist, to form tumour spheres were comparable. In the study, EMT program did not appear essential for primary cancer growth, local invasion, blood dissemination and metastasis. Interestingly, gemcitabine sensitivity was improved in KPC mice with deleted Snail or Twist compared to KPC control mice which could be explained by a significant upregulation of equiliberative nucleoside transporter 1 and concentrating nucleoside transporter 3 (receptors that mediate uptake of nucleosides) in cancer cells lacking Snail or Twist.
GENERAL CONSIDERATIONS ON PHARMACOLOGICAL APPROACHES TO TARGET EMT
Several strategies have been proposed for the design of EMT-based therapies as recently and extensively described and reviewed by Davis et al[1]. While major challenges and questions remain regarding the possibility of targeting EMT to counteract metastasis specifically, stronger evidence is accumulating on the use of anti-EMT agents in cancer chemoresistance settings. However, targeting a single receptor, enzyme or transporter that is associated with EMT faces many limitations since several redundant pathways are involved in this process. Strategies focused on targeting micro-RNAs regulating EMT such as miR-200, or transcription factors might represent a more effective approach since they influence the process more broadly. In addition, key components of the tumour microenvironment are also attractive targets for therapeutic intervention. Indeed, recent evidence has revealed that local tumour microenvironment represents a main driving force for EMT, chemotherapy resistance and cancer progression. Inflammatory cells such as neutrophils and macrophages are contained in the tumour microenvironment, which offers multidirectional interactions leading in some cases to increased chemotherapy resistance and metastasis[15-18]. Neutrophils have been shown to induce EMT in pancreatic cancer while macrophages induce gemcitabine resistance via promoting cytidine deaminase mediated drug inactivation[15,19,20]. Similarly, platelets were recently shown to be capable of inducing EMT in cancer[21,22]. Pancreatic cancer is associated with a high risk of venous thromboembolism (VTE) caused by tumour-derived or tumour-elicited tissue factor which can indirectly induce platelets aggregation[23,24]. Whether targeting platelets can offer an indirect way to reduce EMT and chemoresistance as well as the risk of VTE in pancreatic cancer is yet to be demonstrated.
Therefore, a systematic testing of different methods for targeting EMT in combination with existing chemotherapeutic agents is required for each model of therapy relapse. The excitement elicited by the new reinforcement of the link between EMT and chemoresistance will surely result in a surge of studies in this field, and consequently, further in-depth investigations are warranted, especially in pancreatic cancer.
CONCLUSION
In summary, resistance to treatments such as Gemcitabine in pancreatic cancer can be mediated by several EMT-dependent or independent pathways (Figure 1)[15,25-29], making the EMT process an attractive target for reducing chemotherapy resistance in pancreatic cancer. EMT can be regulated by blocking extracellular signalling molecules such as TGFβ1 (a cytokine mediator of EMT in many types of cancer) and EMT signal transduction pathways[1]. Loss of E-cadherin-mediated cell adhesion is a hallmark of EMT and subsequent invasion and metastasis. Since Snail and Zeb family of transcriptional factors mediate E-cadherin translocation, loss of function or downregulation, they can potentially be targeted to avert EMT at its initial steps. Several studies have reported disruption in TGFβ signalling in pancreatic cancer[30-32]. Therefore, the mesenchymal transcriptional factors may be better druggable targets compared to TGF-β receptors to reduce EMT-derived chemoresistance in pancreatic cancer. Despite a significant number of emerging studies examining the role of EMT in cancer, the interplay between different signalling pathways that drive EMT is more complex than we initially thought. The fact that the phenotypic alteration is transient and triggered by several dynamics encountered by tumour cells during their development or metastasis emphasises the challenge of utilising EMT as a lone druggable target. The diversity of EMT-inducing transcriptional factors may enable cancer cells to adapt and survive a single targeted molecular therapy. The association between EMT and chemotherapy resistance is well established in the literature, but it is not well understood how EMT can affect cancer cell survival pathways, drug transporters and drug metabolising enzymes. Delineation of these interactions may uncover novel approaches to inhibit chemoresistance in pancreatic cancer. Nevertheless, at present, there may be potential benefits in tempering cancer EMT in a combined immunotherapy and molecular-targeted drug strategies to treat pancreatic cancer.
Figure 1.
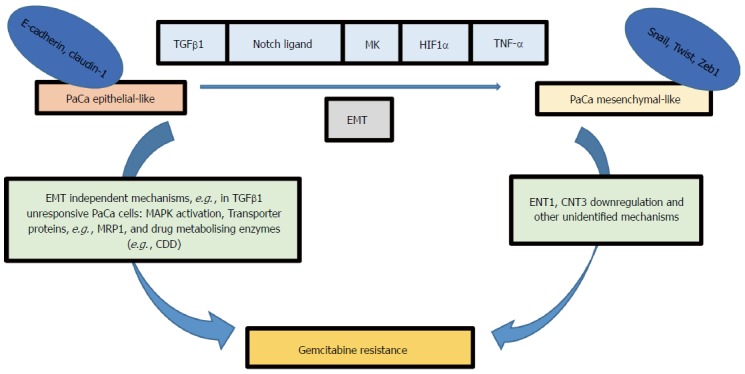
Several signalling pathways can induce epithelial-mesenchymal transition in pancreatic cancer cells for example Notch, transforming growth factor-β, Midkine, hypoxia-inducible factor-1α and tumor necrosis factor-α. EMT phenotype is associated with gemcitabine resistance; however, the signalling pathway relating EMT factors (e.g., Snail, Twist and Zeb1) to gemcitabine resistance is not clearly identified, although ENT1 and CNT3 upregulation was observed in KPC mice models with deleted Snail or Twist. EMT independent pathways can also lead to gemcitabine resistance for example MAPK activation, transporter proteins, and gemcitabine metabolising enzymes (e.g., CDD). MRP1: Multidrug resistance protein1; CDD: Cytidine deaminase; TGFβ: Transforming growth factor-β; MK: Midkine; HIF1α: Hypoxia-inducible factor-1α; TNF-α: Tumor necrosis factor-α; EMT: Epithelial-mesenchymal transition; ENT1: Equilibrative nucleoside transporter 1; CNT3: Concentrative nucleotide transporter.
ACKNOWLEDGMENTS
The authors acknowledge the infrastructure and staff support provided by CHIRI, School of Biomedical Sciences and Faculty of Health Sciences, Curtin University. Work in the Falasca lab is supported by Avner Pancreatic Cancer Foundation.
Footnotes
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Australia
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): 0
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): E
Peer-review started: September 18, 2016
First decision: October 21, 2016
Article in press: November 22, 2016
P- Reviewer: Barreto S, Fu D, Wani IA S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Davis FM, Stewart TA, Thompson EW, Monteith GR. Targeting EMT in cancer: opportunities for pharmacological intervention. Trends Pharmacol Sci. 2014;35:479–488. doi: 10.1016/j.tips.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong D, Wang Z, Sarkar SH, Li Y, Banerjee S, Saliganan A, Kim HR, Cher ML, Sarkar FH. Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26:1425–1435. doi: 10.1634/stemcells.2007-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569–581. doi: 10.1172/JCI21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Güngör C, Zander H, Effenberger KE, Vashist YK, Kalinina T, Izbicki JR, Yekebas E, Bockhorn M. Notch signaling activated by replication stress-induced expression of midkine drives epithelial-mesenchymal transition and chemoresistance in pancreatic cancer. Cancer Res. 2011;71:5009–5019. doi: 10.1158/0008-5472.CAN-11-0036. [DOI] [PubMed] [Google Scholar]
- 10.Aridome K, Tsutsui J, Takao S, Kadomatsu K, Ozawa M, Aikou T, Muramatsu T. Increased midkine gene expression in human gastrointestinal cancers. Jpn J Cancer Res. 1995;86:655–661. doi: 10.1111/j.1349-7006.1995.tb02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garver RI, Radford DM, Donis-Keller H, Wick MR, Milner PG. Midkine and pleiotrophin expression in normal and malignant breast tissue. Cancer. 1994;74:1584–1590. doi: 10.1002/1097-0142(19940901)74:5<1584::aid-cncr2820740514>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 12.Palagani V, El Khatib M, Kossatz U, Bozko P, Müller MR, Manns MP, Krech T, Malek NP, Plentz RR. Epithelial mesenchymal transition and pancreatic tumor initiating CD44+/EpCAM+ cells are inhibited by γ-secretase inhibitor IX. PLoS One. 2012;7:e46514. doi: 10.1371/journal.pone.0046514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma J, Fang B, Zeng F, Ma C, Pang H, Cheng L, Shi Y, Wang H, Yin B, Xia J, et al. Down-regulation of miR-223 reverses epithelial-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Oncotarget. 2015;6:1740–1749. doi: 10.18632/oncotarget.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, Gil Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33:3812–3819. doi: 10.1038/onc.2013.357. [DOI] [PubMed] [Google Scholar]
- 16.Gardian K, Janczewska S, Olszewski WL, Durlik M. Analysis of pancreatic cancer microenvironment: role of macrophage infiltrates and growth factors expression. J Cancer. 2012;3:285–291. doi: 10.7150/jca.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71:2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 18.Su MJ, Aldawsari H, Amiji M. Pancreatic Cancer Cell Exosome-Mediated Macrophage Reprogramming and the Role of MicroRNAs 155 and 125b2 Transfection using Nanoparticle Delivery Systems. Sci Rep. 2016;6:30110. doi: 10.1038/srep30110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaida MM, Günther F, Wagner C, Friess H, Giese NA, Schmidt J, Hänsch GM, Wente MN. Expression of the CXCR6 on polymorphonuclear neutrophils in pancreatic carcinoma and in acute, localized bacterial infections. Clin Exp Immunol. 2008;154:216–223. doi: 10.1111/j.1365-2249.2008.03745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grosse-Steffen T, Giese T, Giese N, Longerich T, Schirmacher P, Hänsch GM, Gaida MM. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: the role of neutrophils and neutrophil-derived elastase. Clin Dev Immunol. 2012;2012:720768. doi: 10.1155/2012/720768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guillem-Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A, Grande R, Alberti S, Arena V, Cirillo M, et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget. 2016;7:32462–32477. doi: 10.18632/oncotarget.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerotziafas GT, Galea V, Mbemba E, Khaterchi A, Sassi M, Baccouche H, Prengel C, van Dreden P, Hatmi M, Bernaudin JF, et al. Tissue factor over-expression by human pancreatic cancer cells BXPC3 is related to higher prothrombotic potential as compared to breast cancer cells MCF7. Thromb Res. 2012;129:779–786. doi: 10.1016/j.thromres.2011.07.049. [DOI] [PubMed] [Google Scholar]
- 24.Geddings JE, Hisada Y, Boulaftali Y, Getz TM, Whelihan M, Fuentes R, Dee R, Cooley BC, Key NS, Wolberg AS, et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J Thromb Haemost. 2016;14:153–166. doi: 10.1111/jth.13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, Adler G, Gress TM. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001;61:4222–4228. [PubMed] [Google Scholar]
- 26.Zheng C, Jiao X, Jiang Y, Sun S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J Int Med Res. 2013;41:300–306. doi: 10.1177/0300060512474128. [DOI] [PubMed] [Google Scholar]
- 27.König J, Hartel M, Nies AT, Martignoni ME, Guo J, Büchler MW, Friess H, Keppler D. Expression and localization of human multidrug resistance protein (ABCC) family members in pancreatic carcinoma. Int J Cancer. 2005;115:359–367. doi: 10.1002/ijc.20831. [DOI] [PubMed] [Google Scholar]
- 28.Nath S, Daneshvar K, Roy LD, Grover P, Kidiyoor A, Mosley L, Sahraei M, Mukherjee P. MUC1 induces drug resistance in pancreatic cancer cells via upregulation of multidrug resistance genes. Oncogenesis. 2013;2:e51. doi: 10.1038/oncsis.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skrypek N, Vasseur R, Vincent A, Duchêne B, Van Seuningen I, Jonckheere N. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget. 2015;6:10853–10867. doi: 10.18632/oncotarget.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YW, Hsiao PJ, Weng CC, Kuo KK, Kuo TL, Wu DC, Hung WC, Cheng KH. SMAD4 loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer. 2014;14:181. doi: 10.1186/1471-2407-14-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang JH, Liu C, Cheng H, Lu Y, Qin Y, Xu YF, Xu J, Long J, Liu L, Ni QX, et al. Epithelial-mesenchymal transition in pancreatic cancer: Is it a clinically significant factor? Biochim Biophys Acta. 2015;1855:43–49. doi: 10.1016/j.bbcan.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Simeone DM, Pham T, Logsdon CD. Disruption of TGFbeta signaling pathways in human pancreatic cancer cells. Ann Surg. 2000;232:73–80. doi: 10.1097/00000658-200007000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]