

Abstract
The glucocorticoid receptor single-nucleotide polymorphism (SNP) N363S has been reported to be associated with metabolic syndrome, type 2 diabetes, and cardiovascular disease. Our aim was to determine how the N363S SNP modifies glucocorticoid receptor signaling in a healthy population of individuals prior to the onset of disease. We examined the function of the N363S SNP in a cohort of subjects from the general population of North Carolina. Eighteen N363S heterozygous carriers and 36 noncarrier, control subjects were examined for clinical and biochemical parameters followed by a low-dose dexamethasone suppression test to evaluate glucocorticoid responsiveness. Serum insulin measurements revealed that N363S carriers have higher levels of insulin, although not statistically significant, compared with controls. Glucocorticoid receptor protein levels evaluated in peripheral blood mononuclear cells from each clinical subject showed no difference between N363S and control. However, investigation of gene expression profiles in macrophages isolated from controls and N363S carriers using microarray, quantitative RT-PCR, and NanoString analyses revealed that the N363S SNP had an altered profile compared with control. These changes in gene expression occurred in both the absence and the presence of glucocorticoids. Thus, our observed difference in gene regulation between normal N363S SNP carriers and noncarrier controls may underlie the emergence of metabolic syndrome, type 2 diabetes, and cardiovascular disease associated with the N363S polymorphism.
Keywords: metabolic syndrome, insulin resistance, type 2 diabetes, cardiovascular disease
metabolic syndrome is a group of factors that include abdominal obesity, insulin resistance, elevated fasting blood glucose levels, elevated blood pressure, and elevated cholesterol. In combination, these conditions increase an individual's risk of serious diseases such as type 2 diabetes (T2D) and cardiovascular disease (CVD) (17). These complex diseases are also influenced by many factors including environmental and genetic variability (14). One such factor is the glucocorticoid receptor gene NR3C1, which encodes a nuclear transcription factor and mediates glucocorticoid signaling through the hypothalamic-pituitary-adrenal (HPA) axis (42). Glucocorticoid receptors are expressed in most cells of the body and regulate multiple genetic pathways (8). Several polymorphisms in the glucocorticoid receptor gene have been reported, and these genetic variations may influence an individual's response to glucocorticoids (2, 6, 11, 19, 27, 34, 47). The glucocorticoid receptor single-nucleotide polymorphism (SNP) N363S (rs6195) has been associated with increased sensitivity to glucocorticoids in direct response to exogenously administered dexamethasone (DEX) (21). In addition, the N363S SNP has been associated with increased body mass index (BMI) (5, 12, 21, 31, 32), increased insulin response (21) and shown to be increased in patients with T2D (16, 44) and coronary artery disease (CAD) (33). These diagnoses are all hallmarks or potential outcomes of metabolic syndrome. Conversely, other reports have shown no association between N363S and obesity (3, 15, 18, 43). However, all of these conclusions are based on assessment of glucocorticoid receptor function in N363S carriers with overt disease which does not permit causation.
Previously, we (24) demonstrated in stable cell lines expressing either the human glucocorticoid receptor (hGR) or the N363S SNP that N363S regulated a novel set of genes compared with hGR. Furthermore, our data showed that the family of serum amyloid A, which are involved in inflammation and have been shown to be associated with obesity, were highly elevated at the mRNA level in the N363S cell line. This led us to examine whether genetic changes potentially leading to metabolic syndrome were associated with the glucocorticoid receptor N363S polymorphism in healthy subjects. These subjects, recruited from the Environmental Polymorphisms Registry (EPR), a cohort of 3,695 individuals from the general population of North Carolina and identified as noncarriers, controls (did not carry the GR9βA3669G or N363S polymorphisms), or heterozygous for the N363S SNP, were examined for differences in gene regulation in macrophages differentiated from peripheral blood mononuclear cells and treated ex vivo with DEX. Our findings report an association of the N363S polymorphism with altered gene regulation in the insulin signaling and inflammatory pathways which may underlie the reported metabolic syndrome phenotype.
MATERIALS AND METHODS
Human subjects.
A total of 54 healthy subjects (24–69 yr old) were included in this study, with 18 subjects genotyped as heterozygous N363S carriers (N363S) and 36 subjects who did not carry the glucocorticoid receptor N363S SNP or the GR9βA3669G SNP (control). Subjects were recruited from the EPR under the approved National Institute of Environmental Health Sciences (NIEHS) Institutional Review Board Protocol 10-E-0130. The EPR is a long-term project to collect and store DNA samples from a racially diverse population in the greater North Carolina Triangle Region, USA. These DNA samples were collected to be used in research studies to examine environmental risk factors for human disease (7). Each subject signed an informed consent form after a thorough explanation of the study. Inclusion criteria required participants to be healthy as defined by the International Red Cross guidelines, at least 18 yr of age, willing to fast for 12 h, and with no glucocorticoid use within 14 days of scheduled visits, and females could not be pregnant or currently breastfeeding. Exclusion criteria for the dexamethasone suppression test (DST) included no chronic inflammatory diseases, no immunosuppressive disorders or hypothyroidism, no current diagnosis of cancer or depression, and not taking hormonal contraceptives or barbiturates.
Screening procedures consisted of medical history, which included diagnosed medical conditions and medication history, followed by physical examination including blood pressure, weight, height, hip and waist circumference measurements, a pregnancy test for all women, and blood draw. Serum total cholesterol, cholesterol fractionation (HDL, LDL), triglycerides, glucose, insulin, C-reactive protein (CRP), creatinine, and cortisol were measured at the NIH Clinical Center (Bethesda, MD). Participants eligible for the modified DST (15 N363S and 32 controls) were provided one 0.25-mg tablet of DEX to take at 11 PM the night prior to their scheduled appointment (1–14 days after the first visit) and instructed to fast for 12 h prior to their appointment time. After the first clinical visit, any participants who reported taking the medications metformin, rosiglitazone, or glimepiride for any condition or were identified with T2D during physical examination were also excluded from all analysis. A small number of African Americans, Caucasians, and Hispanics identified as carrying the N363S SNP were eliminated from our study.
Genotyping.
Genotyping was performed by the NIEHS Genomics Core. Briefly, genomic DNA was prepared from human blood samples using the QIAGEN Autopure LS system. DNA samples were genotyped for the glucocorticoid receptor N363S SNP (rs6195) as well as the following glucocorticoid receptor SNPs: E22E (rs6189), R23K (rs6190), GR9βA3669G (rs6198), GRβG3134T (rs6191), and Bcl1 (rs41423247), using the Illumina customized GoldenGate Assay (San Diego, CA).
Flow cytometry analysis of glucocorticoid receptor levels in peripheral blood mononuclear cells.
Patient blood (100 μl), collected in a Vacuette K3 EDTA tube (Greiner Bio-One, Monroe, NC), was incubated with a phenotyping antibody panel for 30 min at room temperature: anti-CD3-V450, anti-CD19-V500, anti-CD14-AF488, anti-CD56-PE, anti-CD45-PerCP Cy5.5, anti-CD4-PE Cy7, anti-CD8-APC Cy7 (BD Biosciences, San Jose, CA). Samples were treated with FACSLyse (BD Biosciences) for 10 min at room temperature for red blood cell lysis and cell fixation. Cells were washed once with stain buffer (1× PBS + 1% FCS + 0.1% sodium azide) and then stored in 1% paraformaldehyde at 4°C at least overnight. Following surface staining, cells were permeabilized with 1× Perm Wash (BD Biosciences) for 20 min at 4°C. Cells were incubated for 30 min at room temperature with rabbit anti-glucocorticoid receptor polyclonal antibody no. 57 at a concentration of 0.01 μg/μl. Rabbit IgG (Invitrogen) was used as an isotype control. Antibodies were labeled for 30 min at room temperature with goat anti-rabbit IgG-APC (Invitrogen). Ten thousand cells were acquired on a BD FACS Aria II, standardized with Calibrite APC beads (BD Biosciences), and analyzed using FlowJo software (Treestar).
Purification of peripheral blood mononuclear cells, macrophage cell differentiation and RNA isolation.
Thirty-five milliliters of anticoagulated whole blood was collected from each study participant. Peripheral blood mononuclear cells were then isolated using Histopaque 1077 (Sigma-Aldrich, St. Louis, MO) following standard protocol and resuspended in 10 ml of Hank's balanced salt solution at an average concentration of 6 million cells per milliliter. To differentiate the mononuclear cells to macrophages, 5 million cells were plated in 100-mm dishes at an average concentration of 30 million cells per dish and 5 ml of X-VIVO medium (Lonza, Walkersville, MD) containing 10% human serum, 100 IU of penicillin, and 100 mg/ml streptomycin. The cells were cultured in the X-VIVO medium for 7 days, with the medium being replaced every 3–4 days. On day 8, the cells were treated with 100 nM DEX (Steraloids, Newport, RI) or vehicle (H2O) for 6 h. Total RNA was isolated from cells using the QIAGEN RNeasy minikit (including DNase treatment) following the manufacturer's instructions.
Microarray analysis.
Gene expression analysis was performed on RNA from macrophages isolated from seven control and seven N363S carriers and treated ex vivo with 100 nM DEX or vehicle and using the Whole Human Genome 4 × 44 multiplex format oligo array (014850) (Agilent Technologies). Significant changes in gene expression were defined using Partek Genomics Suite software. A one-way ANOVA was performed followed by Bonferroni post hoc correction to adjust for multiple comparisons. Statistical significance was defined as P < 0.05. Significantly regulated genes were then analyzed using Ingenuity Pathways Analysis (IPA; Ingenuity Systems). The microarray data discussed in this manuscript are deposited in NCBI's Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) and can be accessed through GEO accession number GSE87000.
Real-time PCR analysis.
Real-time PCR was performed using the 7900HT sequence detection system predesigned primer/probe sets available from Applied Biosystems (Foster City, CA) and following the manufacturer's instructions. Each primer/probe set was analyzed with RNA isolated from multiple subjects, as indicated in the figure legends. The signal obtained from each gene primer/probe set was normalized to that of the unregulated housekeeping gene cyclophilin B primer/probe set (Applied Biosystems).
NanoString Human Inflammatory panel RNA analysis.
NanoString analysis was performed by the NIEHS Genomics Core using the NanoString Human Inflammatory panel (NanoString Technologies, Seattle, WA). NanoString technology utilizes the nCounter Analysis System, a novel color-coded barcode technology that is based on direct measurement of mRNA expression without amplification. For our analysis, total RNA isolated from macrophages (40 ng) was used to determine gene expression. Hybridization and nCounter were performed according to the manufacturer's protocol. Significant changes in gene expression were defined using Partek Genomics Suite software. A one-way ANOVA was performed followed by Bonferroni post hoc correction to adjust for multiple comparisons. Statistical significance was defined as P < 0.05. The graphed data represent the raw RNA counts for each gene normalized to the internal reference genes HPRT1 and TUBB. The data were further analyzed using Sidak-Bonferroni multiple comparison (P < 0.05) and are shown as mean values ± SE.
Statistical analysis of clinical data and RT-PCR data.
Statistical analyses were performed using Prism software (GraphPad Software, La Jolla, CA). Unpaired t-test with Welch's correction was used for statistical comparison of clinical data. Grouped analyses with Sidak-Bonferroni multiple comparison correction was applied to real-time PCR data. All data are shown as means ± SE. Comparisons were considered statistically significant at P < 0.05.
RESULTS
Prevalence of GR N363S polymorphism in study cohort.
DNA was isolated from 3,695 EPR participants and was genotyped for the GR N363S SNP (rs6195). This analysis revealed that 3,545 participants were noncarriers (0 copies of the 363S allele, control) and 150 participants were heterozygous (1 copy of the 363S allele, N363S). No homozygotes (2 copies of the 363S allele) were identified in the EPR cohort, as shown in Table 1. Of the 150 participants identified as N363S carriers, 71 were male and 79 were female. In addition, 103 N363S carriers were non-Hispanic Caucasian, 11 were Hispanic Caucasian, and 36 were African American (Table 1).
Table 1.
Genotype results from environmental polymorphism registry (EPR) participants carrying the N363S glucocorticoid receptor single nucleotide polymorphism
| N363S, rs6195 | 0 Copies | 1 Copy | 2 Copies |
|---|---|---|---|
| Total, n | 3,545 | 150 | 0 |
| Sex | |||
| Male, n | 1,589 | 71 | 0 |
| Female, n | 1,956 | 79 | 0 |
| Race | |||
| Caucasian, NonHispanic, n | 1,711 | 103 | 0 |
| Caucasian, Hispanic, n | 317 | 11 | 0 |
| Black/African American, n | 1,505 | 36 | 0 |
| Other, n | 12 | 0 | 0 |
Glucocorticoid receptor N363S carriers exhibit higher levels of insulin.
Demographic and laboratory measurements between non-Hispanic Caucasian control or N363S clinical subjects are shown in Table 2. There was no statistical difference between these two groups in measurements for obesity (BMI, waist circumference), CVD (blood pressure, CRP, total cholesterol, HDL, LDL, and triglycerides), creatinine, or glucose, although CRP, total cholesterol, and LDL displayed an increased trend in the N363S cohort. Interestingly, although it has been reported that N363S carriers are sensitive to DEX (21), we saw only a small, insignificant decrease in cortisol measurements after the administration of the DST in our healthy N363S carriers. Of note, although not statistically significant, were the higher levels of insulin in N363S carriers (18.0 ± 5.7 N363S vs. 7.2 ± 1.0 control, P = 0.08). This finding led us to hypothesize that alterations in global gene expression by N363S might be affecting downstream signaling pathways and predisposing N363S carriers to disease.
Table 2.
Characteristics of clinical study subjects carrying the N363S glucocorticoid receptor single nucleotide polymorphism and noncarrier control
| Demographic | Control | N363S | P Value |
|---|---|---|---|
| Race: Caucasian, non-Hispanic, n | 36 | 18 | |
| Age, yr | 50.0 ± 2.0 | 44.7 ± 3.3 | 0.18 |
| Male, n | 14 | 6 | |
| Female, n | 22 | 12 | |
| BMI | 28.6 ± 1.2 | 30.5 ± 1.8 | 0.40 |
| Waist circumference, cm | 95.4 ± 2.4 | 100.0 ± 4.8 | 0.40 |
| Systolic blood pressure | 131.4 ± 3.2 | 136.2 ± 4.3 | 0.38 |
| Diastolic blood pressure | 75.8 ± 1.7 | 74.0 ± 2.1 | 0.51 |
| Laboratory (normal range) | |||
| CRP (<3.0 mg/l) | 2.6 ± 0.7 | 3.2 ± 0.9 | 0.59 |
| Cholesterol (<200 mg/dl) | 187.3 ± 7.1 | 205.3 ± 10.6 | 0.17 |
| HDL (40–59 mg/dl) | 51.0 ± 2.5 | 54.2 ± 3.7 | 0.47 |
| LDL (100–129 mg/dl) | 110.3 ± 5.6 | 127.2 ± 7.8 | 0.09 |
| Triglycerides (<150 mg/dl) | 130.8 ± 9.8 | 119.1 ± 14.3 | 0.51 |
| Creatinine (0.56–1.16 mg/dl) | 0.79 ± 0.02 | 0.76 ± 0.04 | 0.54 |
| Glucose (70–115 mg/dl) | 88.7 ± 1.4 | 87.6 ± 2.1 | 0.66 |
| Insulin (6–27 mcU/ml) | 7.2 ± 1.0 | 18.0 ± 5.7 | 0.08 |
| Cortisol (5–25 mcg/dl) | 11.6 ± 0.7 | 11.2 ± 1.2 | 0.77 |
| DST cortisol (n 32,15) | 7.6 ± 1.0 | 6.9 ± 1.1 | 0.61 |
Means ± SE are shown.
BMI, body mass index; CRP, C-reative protein; HDL, high-density lipoprotein; LDL, low-density lipoprotein; DST, dexamethasone suppression test.
Any subjects taking the medications metformin, rosiglitazone, or glimepiride for any condition or identified with type 2 diabetes during physical examination were excluded from the data analysis. P value, unpaired t-test with Welch's correction.
Glucocorticoid receptor levels do not differ between control and N363S carriers.
Before proceeding with global gene expression analysis, we needed to verify that there were no overt changes in glucocorticoid receptor levels between controls and N363S carriers. Therefore, we examined the levels of glucocorticoid receptor protein in the different cellular fractions of peripheral blood mononuclear cells isolated from control or N363S by using a flow cytometry assay developed in our laboratory and also measured glucocorticoid receptor mRNA levels using quantitative real-time PCR. Figure 1A shows that glucocorticoid receptor protein levels were relatively equivalent in CD4+ T cells, CD8+ T cells, CD19+ B cells, and CD56+ natural killer (NK) cells regardless of phenotype. Interestingly, the CD14+ monocytes had significantly increased levels of glucocorticoid receptor compared with other cell types, although there was no statistical significance between control and N363S. Additionally, we observed no difference in the levels of glucocorticoid receptor mRNA between genotypes (Fig. 1B), demonstrating that cellular levels of glucocorticoid receptor were the same in our study cohort.
Fig. 1.

Analysis of glucocorticoid receptor levels in control and N363S subjects. A: glucocorticoid receptor (GR) levels were measured in peripheral blood leukocyte cellular fractions CD4+ T cells, CD8+ T cells, CD19+ B cells, CD14+ monocytes, and CD56+ natural killer (NK) cells isolated from control (n = 36) or N363S (n = 18) carriers using flow cytometry. To accommodate for interassay staining variability, GR median fluorescence intensity (MFI) levels in CD4+, CD8+, CD19+, CD56+, and CD14+ cells are displayed as %total leukocyte (CD45+) GR MFI (relative GR levels). CD14+ monocytes had significantly higher levels of GR than all other cell types, P < 0.001. B: GR mRNA levels were measured in macrophages from control (n = 16) and N363S carriers (n = 15). mRNA levels were normalized to the housekeeping gene cyclophilin B (PPIB) and are expressed as means ± SE.
Gene regulation differs between control and N363S.
To determine whether there was a difference in global gene regulation between controls and N363S carriers, we performed genomewide microarray analysis on RNA isolated from the macrophages of seven control and seven N363S carriers. To assess glucocorticoid responsiveness at the level of mRNA, the macrophages were treated with 100 nM DEX for 6 h. Three-way Venn diagram analysis (Fig. 2A) depicts the differences in gene regulation by N363S compared with control in both the absence and the presence of DEX. In the presence of DEX, 93 genes were significantly regulated by N363S (N363S DEX vs. N363S Veh), and 89 genes were significantly regulated by control (Control DEX vs. Control Veh), respectively. Interestingly, the majority of these genes were upregulated (97% N363S DEX and 89% Control DEX) by glucocorticoid treatment. This upregulation by DEX was not unexpected, as we had previously shown that glucocorticoids could have proinflammatory effects in addition to their anti-inflammatory actions. (4, 29). Finally, literature-based IPA of the unique genes (N363S DEX, 29 genes and Control DEX, 25 genes) indicates that only two of five top diseases and bio functions/diseases and disorders generated from the N363S DEX and Control DEX gene lists are identical with three unique diseases and disorders observed for each (Fig. 2, B and C). These findings demonstrate that there are unique changes in gene regulation between control and N363S in the presence of DEX.
Fig. 2.
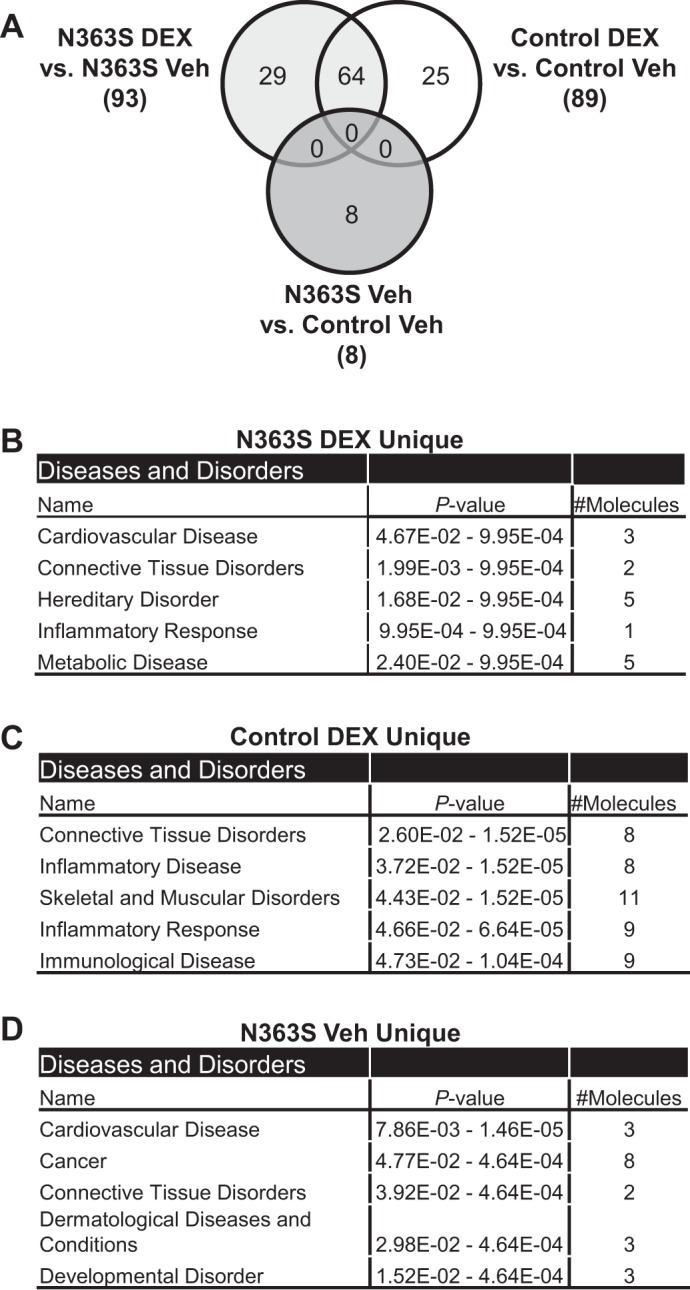
Global gene expression profiles of macrophages from control and N363S carriers. Genomewide microarray analysis was performed on RNA isolated from macrophages of 7 control and 7 N363S carriers treated ex vivo with 100 nM dexamethasone (DEX) or vehicle (Veh) for 6 h. A: Venn diagram analysis compares gene lists from N363S DEX vs. N363S Veh, Control DEX vs. Control Veh, and N363S Veh vs. Control Veh. B, C, D: gene lists were analyzed using Ingenuity Pathways Analysis. Shown are the top 5 diseases and bio functions/diseases and disorders for each gene list.
Interestingly, in the absence of glucocorticoid treatment, we also observed a small but significant difference in gene regulation between N363S carriers compared with control. Three-way Venn diagram analysis illustrates that eight genes total (Fig. 2A) are uniquely regulated by N363S independently of hormone treatment. Analysis of these genes by IPA revealed that N363S was affecting genes involved in CVD and cancer development (Fig. 2D). This observation supports our previous finding in human U-2 OS osteosarcoma cell lines developed to stably express N363S or hGR, where N363S regulates a unique set of genes in the absence of ligand (24).
We then used quantitative real-time PCR analysis to examine two genes identified as being regulated by N363S compared with control by using a different set of mRNA from that used for the microarray analysis. Figure 3 illustrates that the glucocorticoid-converting enzyme 11β-hydroxysteroid dehydrogenase type 1 (HSD1) was upregulated by N363S (HSD11B1, 3.3-fold N363S Veh vs. control Veh). The gene most highly downregulated by N363S was insulin-like growth factor I (IGF1, −9.2-fold N363S Veh vs. control Veh). This differential regulation of IGF1 by N363S led us to further investigate the insulin signaling pathway in our study subjects.
Fig. 3.
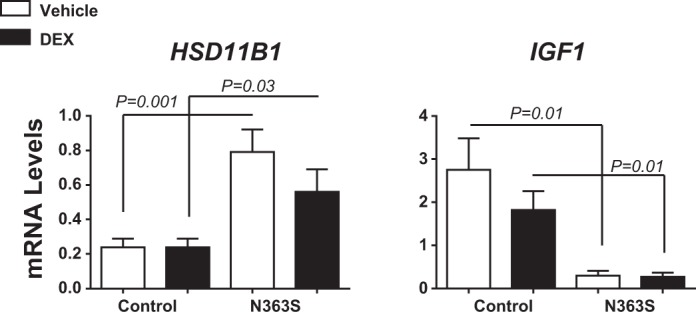
N363S and control gene regulation. RNA isolated from the macrophages of N363S and control treated ex vivo with 100 nM DEX or Veh for 6 h was quantified by RT-PCR. HSD11B1 and IGF1 were two genes differentially regulated by N363S compared with control. mRNA levels were normalized to the housekeeping gene cyclophilin B (PPIB) and are expressed as means ± SE. Thirteen human samples of each genotype were assayed for gene regulation. Statistical significance was determined using grouped analyses with Sidak-Bonferroni multiple comparison correction; P < 0.05 as indicated on the graph.
The insulin signaling pathway is dysregulated in healthy N363S carriers.
Insulin signaling occurs when insulin binds to insulin receptor (INSR) at the cell membrane and signals through the insulin receptor substrate 1 (IRS1). Phosphorylation of insulin receptor substrate 1 leads to translocation of glucose transporter 4 (GLUT4) and subsequent glucose uptake. Therefore, we examined regulation of these three genes by control and N363S (Fig. 4). There was no difference in INSR levels between control and N363S; however, with the addition of DEX, there was significant induction of INSR in N363S. We also observed a statistically significant down regulation of both IRS1 and GLUT4 in the N363S carriers compared with control (Fig. 4). These findings suggest that healthy N363S carriers have a dysregulation of normal insulin signaling at the level of mRNA and that this altered regulation may be a contributor to the decreased insulin sensitivity observed in our N363S subjects.
Fig. 4.

Regulation of insulin signaling in N363S carriers. Insulin receptor (INSR), insulin receptor substrate 1 (IRS1), and glucose transporter 4 (GLUT4) mRNA levels were measured in macrophages from N363S carriers and compared with control. mRNA levels were normalized to the housekeeping gene cyclophilin B (PPIB) and are expressed as means ± SE. Number of human samples assayed were: INSR, control, 18; N363S, 19; IRS1, control, 10; N363S, 10; GLUT4, control, 6; N363S, 6. Statistical significance was determined using grouped analyses with Sidak-Bonferroni multiple comparison correction; P < 0.05 as indicated on the graph.
N363S carriers exhibit signs of inflammation at the mRNA level.
Inflammation also plays a role in dysregulated insulin signaling and CVD. Specifically, an increase in inflammatory markers in the blood is one of the hallmarks of insulin resistance. Although a subset of immune and inflammatory genes was identified through our genomewide microarray analysis, we wished to delve deeper into the inflammatory status of our N363S carriers. For this analysis we utilized the NanoString nCounter Analysis System, a novel digital color-coded technology that is based on direct measurement of mRNA expression without amplification. RNA isolated from the macrophages of four control and four N363S (different from those used for the microarray analysis) and treated with 100 nM DEX or vehicle for 6 h were hybridized to the NanoString Human Inflammation Panel (249 gene probes). Stringent statistical analysis revealed that three genes, CXCL1, MAP3K5, and RIPK2, were uniquely regulated by N363S compared with control in the absence of hormone. The normalized RNA counts for the most highly regulated of these genes, CXCL1, is graphed on the right (Fig. 5A, N363S Veh). Two genes, CD163 and IL1R1, were uniquely regulated by N363S in the presence of hormone, with CD163 graphed on the right (Fig. 5B, N363S DEX), and one gene, MAPKAPK5, was regulated by control in the presence of Dex (Fig. 5C, Control DEX). In addition, three genes were commonly regulated by N363S and control in the presence of DEX (Fig. 5D, Common, N363S DEX, Control DEX). Of these three commonly regulated genes, TLR2 had a 1.3-fold higher expression in N363S DEX compared with Control DEX. The normalized RNA counts for TLR2 are graphed on the right. Although these findings did not reveal an overwhelming number of inflammatory genes regulated by N363S, the increase in CXCL1 and the hormone-dependent increases in CD163 and TLR2 suggest that inflammation may also play a role in the decreased insulin sensitivity observed in our healthy N363S carriers.
Fig. 5.

NanoString Analysis of N363S regulated inflammatory genes. NanoString Human Inflammation Gene Panel was used to directly measure inflammatory genes regulated by N363S compared with control. A, B, C: genes uniquely regulated by N363S Veh, N363S DEX, and Control DEX. D: genes commonly regulated by N363S DEX and Control DEX are listed in the tables. The most highly upregulated gene for each is graphed to the right of each table. Significant changes in gene expression were defined using Partek Genomics Suite software. A one-way ANOVA was performed followed by Bonferroni post hoc correction to adjust for multiple comparisons. Statistical significance was defined as P < 0.05. Graphed data represent raw RNA counts for each gene normalized to internal reference genes HPRT1 and TUBB. Data were further analyzed using grouped analyses with Sidak-Bonferroni multiple comparison (P < 0.05) and are shown as means ± SE.
DISCUSSION
Previous studies on glucocorticoid receptor SNPs have focused on cohorts of patients with abdominal obesity (11), altered cortisol sensitivity, hypertension, coronary artery disease, and T2D and shown that there is an increase in N363S SNP carriers in these study populations (5, 12, 13, 21, 31–33, 44). In contrast, we undertook a different approach by examining a healthy, asymptomatic population harboring the N363S polymorphism. Clinical measurements showed that there were no significant changes between control, noncarriers, and N363S carriers in BMI, waist circumference, blood pressure, CRP, total cholesterol, HDL, LDL, triglycerides, creatinine, or glucose, although we observed higher, but not statistically significantly higher, levels of insulin in the N363S subjects compared with controls (P = 0.08). More importantly, we discovered that the N363S polymorphism affected global gene regulation and altered both insulin signaling and inflammatory gene expression.
Glucocorticoid sensitivity varies greatly among individuals (22), and glucocorticoid variants have been shown to be associated with increased glucocorticoid sensitivity (N363S, Bcl1) or glucocorticoid resistance (hGR9βA3669G, ER22/23EK) (27) and are thought to be mediated through altered transcription of the NR3C1 gene (27). Increased glucocorticoid sensitivity can lead to the development of Cushing's syndrome-like symptoms, including visceral obesity, hypertension, insulin resistance, and T2D (36), conditions that have been associated with the N363S polymorphism. In the present study, analysis of serum cortisol levels after DST revealed no change in sensitivity between healthy N363S carriers and noncarrier controls. More importantly, we found no difference in glucocorticoid receptor protein levels or glucocorticoid mRNA levels between control and N363S, suggesting that there is no alteration in NR3C1 gene transcription in our N363S carriers.
No studies have shown a link between genomewide gene regulation and the N363S phenotype in primary human cells that contain endogenous receptors. Thus, we employed microarray analysis to examine macrophage RNA from noncarrier controls and N363S carriers in both the absence and the presence of DEX. This analysis revealed that the N363S SNP regulates a unique set of genes distinct from control regardless of glucocorticoid administration.
One of the interesting genes identified as being increased by N363S regardless of hormone treatment was HSD11B1. 11β-HSD1 has been identified as an important participant in obesity, inflammation, insulin resistance, and T2D (25, 40, 46). This microsomal enzyme along with 11β-HSD2 finely regulates glucocorticoids in a tissue specific manner (40, 46). 11β-HSD1 activates cortisone to cortisol and ensures that glucocorticoids have access to the glucocorticoid receptor (40). Several studies have found an increase in HSD11B1 mRNA levels in abdominal subcutaneous adipose tissue (SAT) in obese humans as well as those with T2D and those with features of the metabolic syndrome (25, 30, 37). Interestingly, this rise in HSD11B1 mRNA occurs without a change in systemic circulating glucocorticoid levels similar to our observation in our N363S carriers (40).
The gene most highly down-regulated by N363S regardless of hormone treatment was IGF1. IGF-I is an endocrine, autocrine, and paracrine growth factor synthesized by many tissues (9). IGF-I, growth hormone (GH), and insulin form a tightly regulated axis that informs cells of the nutritional state of an organism (1). IGF-I can act as a potent anti-inflammatory (20), and reports have shown that a deficit of IGF-I is associated with altered lipid metabolism, CVD, and diabetes (9, 41) and recent review articles have associated this hormone with metabolic syndrome (1) T2D, and atherosclerosis (9). Therefore, the decreased IGF1 mRNA in our N363S carriers suggests an alteration in GH/IGF-I signaling that may predispose N363S carriers to development of metabolic syndrome and eventually T2D and CVD.
Insulin resistance is also a hallmark of metabolic syndrome and T2D and occurs many years before the onset of T2D (48). Many complex factors may contribute to insulin resistance, including obesity, inflammation, hyperinsulinemia, hyperlipidemia, and genetic background (48). Decreased sensitivity to insulin has been reported in N363S carriers, but its regulation has not been examined at the level of mRNA (21). The effects of insulin on metabolism initiate when insulin binds to the insulin receptor (INSR) and signals through IRS1, which, upon phosphorylation, triggers downstream signaling cascades by phosphatidylinositide 3-kinase and subsequent GLUT4 translocation to the cell membrane and glucose uptake (28, 45). We examined this pathway in our study subjects and found an increase in INSR mRNA in our N363S carriers and a decrease in IRS1 and GLUT4 mRNAs, suggesting that the insulin signaling pathway is dysregulated at the transcriptional level in our study cohort.
Chronic, low-grade inflammation is involved in the pathogenesis of both obesity and T2D. In particular, CRP, IL-6 (26), and tumor necrosis factor-α (TNFα) (39) were observed to be increased in the plasma of obese patients (23). Inflammatory molecules such as CXCL1 and CXCL5 are associated with increased obesity in db/db mice (38). In our N363S subjects, we identified the mRNA for the chemokine (C-X-C motif) ligand CXCL1 as the gene transcript most highly regulated in the absence of hormone. This ligand-independent gene regulation suggests that the N363S carriers are beginning to exhibit signs of possible obesity-related inflammation without a significant increase in BMI or waist measurement.
Increased levels of serum-soluble CD163 has been shown to be a predictor of T2D in the general population prior to the onset of this disease (35). In addition, the Toll-like receptors, particularly TLR2 and TLR4, have been shown to play a critical role in insulin resistance, inflammation, and diabetes (10). Our analysis shows an increase in CD163 and TLR2 gene transcripts by N363S in the presence of DEX, suggesting that at the level of gene regulation our N363S carriers are beginning to develop metabolic syndrome.
In summary, we have described molecular studies that are supportive of the metabolic syndrome phenotype associated with the glucocorticoid receptor N363S single-nucleotide polymorphism. Provocatively, this regulation occurs prior to the onset of overt disease in healthy individuals. Although there are many factors that contribute to an individual's development of metabolic syndrome and its ensuing complications of T2D and CVD, our data suggests that the glucocorticoid receptor N363S polymorphism acting through selective changes in gene expression in otherwise normal healthy people may impart a predisposition to metabolic syndrome.
GRANTS
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.M.J., S.G., and J.A.C. conception and design of research; C.M.J. and K.S.K. performed experiments; C.M.J., K.S.K., L.M.B., and C.C. analyzed data; C.M.J., K.S.K., S.G., and J.A.C. interpreted results of experiments; C.M.J. and K.S.K. prepared figures; C.M.J. drafted manuscript; C.M.J. and J.A.C. edited and revised manuscript; C.M.J., K.S.K., L.M.B., C.C., S.G., and J.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the members of the National Institute of Environmental Health Sciences (NIEHS) Clinical Research Unit (CRU), with special thanks to the staff of the CRU laboratory. We also thank the NIEHS Genomics Core for their help with the microarray and NanoString analyses.
REFERENCES
- 1.Aguirre GA, De Ita JR, de la Garza RG, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. J Transl Med 14: 3, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray PJ, Cotton RG. Variations of the human glucocorticoid receptor gene (NR3C1): pathological and in vitro mutations and polymorphisms. Hum Mutat 21: 557–568, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Buemann B, Black E, Holst C, Toubro S, Echwald S, Pedersen O, Astrup A, Sorensen T. The N363S polymorphism of the glucocorticoid receptor and metabolic syndrome factors in men. Obes Res 13: 862–867, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem 286: 38703–38713, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cellini E, Castellini G, Ricca V, Bagnoli S, Tedde A, Rotella CM, Faravelli C, Sorbi S, Nacmias B. Glucocorticoid receptor gene polymorphisms in Italian patients with eating disorders and obesity. Psychiatr Genet 20: 282–288, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab 89: 1939–1949, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Chulada PC, Vahdat HL, Sharp RR, DeLozier TC, Watkins PB, Pusek SN, Blackshear PJ. The Environmental Polymorphisms Registry: a DNA resource to study genetic susceptibility loci. Hum Genet 123: 207–214, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 22: 20–32, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cubbon RM, Kearney MT, Wheatcroft SB. Endothelial IGF-1 receptor signalling in diabetes and insulin resistance. Trends Endocrinol Metab 27: 96–104, 2015. [DOI] [PubMed] [Google Scholar]
- 10.Dasu MR, Ramirez S, Isseroff RR. Toll-like receptors and diabetes: a therapeutic perspective. Clin Sci 122: 203–214, 2012. [DOI] [PubMed] [Google Scholar]
- 11.DeRijk RH, Schaaf M, de Kloet ER. Glucocorticoid receptor variants: clinical implications. J Steroid Biochem Mol Biol 81: 103–122, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Di Blasio AM, van Rossum EF, Maestrini S, Berselli ME, Tagliaferri M, Podesta F, Koper JW, Liuzzi A, Lamberts SW. The relation between two polymorphisms in the glucocorticoid receptor gene and body mass index, blood pressure and cholesterol in obese patients. Clin Endocrinol 59: 68–74, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Dobson MG, Redfern CP, Unwin N, Weaver JU. The N363S polymorphism of the glucocorticoid receptor: potential contribution to central obesity in men and lack of association with other risk factors for coronary heart disease and diabetes mellitus. J Clin Endocrinol Metab 86: 2270–2274, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Dulloo AG, Montani JP. Pathways from dieting to weight regain, to obesity and to the metabolic syndrome: an overview. Obes Rev 16, Suppl 1: 1–6, 2015. [DOI] [PubMed] [Google Scholar]
- 15.Echwald SM, Sorensen TI, Andersen T, Pedersen O. The Asn363Ser variant of the glucocorticoid receptor gene is not associated with obesity or weight gain in Danish men. Int J Obes Relat Metab Disord 25: 1563–1565, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Goracy I, Goracy J, Safranow K, Skonieczna-Zydecka K, Ciechanowicz A. Association of glucocorticoid receptor gene NR3C1 genetic variants with angiographically documented coronary artery disease and its risk factors. Arch Med Res 44: 27–33, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109: 433–438, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Halsall DJLJ, Hales N, Wareham NJ, O'Rahilly S. Glucocorticoid receptor variant and body mass index. Rapid Response. Br Med J 7221: 1337–1338, 2000. [Google Scholar]
- 19.Hawkins GA, Amelung PJ, Smith RS, Jongepier H, Howard TD, Koppelman GH, Meyers DA, Bleecker ER, Postma DS. Identification of polymorphisms in the human glucocorticoid receptor gene (NR3C1) in a multi-racial asthma case and control screening panel. DNA Seq 15: 167–173, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharmaceut Design 14: 1225–1230, 2008. [DOI] [PubMed] [Google Scholar]
- 21.Huizenga NA, Koper JW, De Lange P, Pols HA, Stolk RP, Burger H, Grobbee DE, Brinkmann AO, De Jong FH, Lamberts SW. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab 83: 144–151, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Huizenga NA, Koper JW, de Lange P, Pols HA, Stolk RP, Grobbee DE, de Jong FH, Lamberts SW. Interperson variability but intraperson stability of baseline plasma cortisol concentrations, and its relation to feedback sensitivity of the hypothalamo-pituitary-adrenal axis to a low dose of dexamethasone in elderly individuals. J Clin Endocrinol Metab 83: 47–54, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Indulekha K, Surendar J, Mohan V. High sensitivity C-reactive protein, tumor necrosis factor-alpha, interleukin-6, and vascular cell adhesion molecule-1 levels in Asian Indians with metabolic syndrome and insulin resistance (CURES-105). J Diabetes Sci Tech 5: 982–988, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jewell CM, Cidlowski JA. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J Clin Endocrinol Metab 92: 3268–3277, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kannisto K, Pietilainen KH, Ehrenborg E, Rissanen A, Kaprio J, Hamsten A, Yki-Jarvinen H. Overexpression of 11beta-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J Clin Endocrinol Metab 89: 4414–4421, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 89: 2548–2556, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Koper JW, van Rossum EF, van den Akker EL. Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids 92: 62–73, 2014. [DOI] [PubMed] [Google Scholar]
- 28.Kouidhi S, Berrhouma R, Rouissi K, Jarboui S, Clerget-Froidevaux MS, Seugnet I, Bchir F, Demeneix B, Guissouma H, Elgaaied AB. Human subcutaneous adipose tissue Glut 4 mRNA expression in obesity and type 2 diabetes. Acta Diabetol 50: 227–232, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Lannan EA, Galliher-Beckley AJ, Scoltock AB, Cidlowski JA. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFalpha coregulate gene expression in vitro and in vivo. Endocrinology 153: 3701–3712, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Tao T, Wang L, Mao X, Zheng J, Zhao A, Liu W. The expression of 11beta-HSDs, GR, and H6PDH in subcutaneous adipose tissue from polycystic ovary syndrome subjects. Horm Metab Res 45: 802–807, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Lin RC, Wang WY, Morris BJ. High penetrance, overweight, and glucocorticoid receptor variant: case-control study. Br Med J 319: 1337–1338, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin RC, Wang XL, Dalziel B, Caterson ID, Morris BJ. Association of obesity, but not diabetes or hypertension, with glucocorticoid receptor N363S variant. Obes Res 11: 802–808, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Lin RC, Wang XL, Morris BJ. Association of coronary artery disease with glucocorticoid receptor N363S variant. Hypertension 41: 404–407, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann NY Acad Sci 1179: 179–198, 2009. [DOI] [PubMed] [Google Scholar]
- 35.Moller HJ, Frikke-Schmidt R, Moestrup SK, Nordestgaard BG, Tybjaerg-Hansen A. Serum soluble CD163 predicts risk of type 2 diabetes in the general population. Clin Chem 57: 291–297, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Morgan SA, Hassan-Smith ZK, Lavery GG. Mechanisms in Endocrinology: tissue-specific activation of cortisol in Cushing's syndrome. Eur J Endocrinol 175: R83–R89, 2016. [DOI] [PubMed] [Google Scholar]
- 37.Munoz R, Carvajal C, Escalona A, Boza C, Perez G, Ibanez L, Fardella C. 11β-Hydroxysteroid dehydrogenase type 1 is overexpressed in subcutaneous adipose tissue of morbidly obese patients. Obes Surg 19: 764–770, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Nunemaker CS, Chung HG, Verrilli GM, Corbin KL, Upadhye A, Sharma PR. Increased serum CXCL1 and CXCL5 are linked to obesity, hyperglycemia, and impaired islet function. J Endocrinol 222: 267–276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem 271: 13018–13022, 1996. [DOI] [PubMed] [Google Scholar]
- 40.Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab 14: 869–881, 2012. [DOI] [PubMed] [Google Scholar]
- 41.Puche JE, Castilla-Cortazar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J Transl Med 10: 224, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 353: 1711–1723, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Rosmond R. Association studies of genetic polymorphisms in central obesity: a critical review. Int J Obes Relat Metab Disord 27: 1141–1151, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Roussel R, Reis AF, Dubois-Laforgue D, Bellanne-Chantelot C, Timsit J, Velho G. The N363S polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin Endocrinol 59: 237–241, 2003. [DOI] [PubMed] [Google Scholar]
- 45.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278: 14599–14602, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Staab CA, Maser E. 11β-Hydroxysteroid dehydrogenase type 1 is an important regulator at the interface of obesity and inflammation. J Steroid Biochem Mol Biol 119: 56–72, 2010. [DOI] [PubMed] [Google Scholar]
- 47.van Rossum EF, Russcher H, Lamberts SW. Genetic polymorphisms and multifactorial diseases: facts and fallacies revealed by the glucocorticoid receptor gene. Trends Endocrinol Metab 16: 445–450, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Ye J. Mechanisms of insulin resistance in obesity. Front Med 7: 14–24, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]