

Abstract
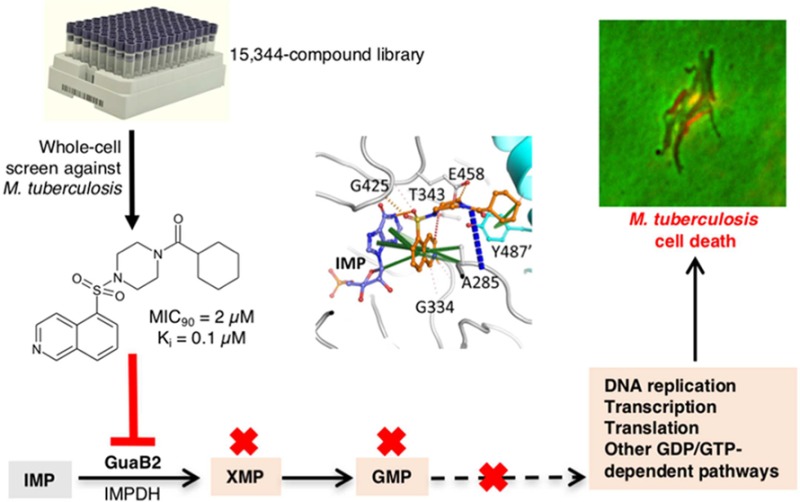
VCC234718, a molecule with growth inhibitory activity against Mycobacterium tuberculosis (Mtb), was identified by phenotypic screening of a 15344-compound library. Sequencing of a VCC234718-resistant mutant identified a Y487C substitution in the inosine monophosphate dehydrogenase, GuaB2, which was subsequently validated to be the primary molecular target of VCC234718 in Mtb. VCC234718 inhibits Mtb GuaB2 with a Ki of 100 nM and is uncompetitive with respect to IMP and NAD+. This compound binds at the NAD+ site, after IMP has bound, and makes direct interactions with IMP; therefore, the inhibitor is by definition uncompetitive. VCC234718 forms strong pi interactions with the Y487 residue side chain from the adjacent protomer in the tetramer, explaining the resistance-conferring mutation. In addition to sensitizing Mtb to VCC234718, depletion of GuaB2 was bactericidal in Mtb in vitro and in macrophages. When supplied at a high concentration (≥125 μM), guanine alleviated the toxicity of VCC234718 treatment or GuaB2 depletion via purine salvage. However, transcriptional silencing of guaB2 prevented Mtb from establishing an infection in mice, confirming that Mtb has limited access to guanine in this animal model. Together, these data provide compelling validation of GuaB2 as a new tuberculosis drug target.
Keywords: drug target, IMPDH, purine nucleotide, purine salvage, Mycobacterium tuberculosis
Tuberculosis (TB), an infectious disease caused by the major human pathogen Mycobacterium tuberculosis (Mtb), ranks alongside HIV/AIDS as a leading cause of death worldwide. Approximately 9.6 million people fell ill with TB in 2014, including 1.2 million people co-infected with HIV. In that year alone, 1.5 million people died from this devastating disease, 400,000 of whom were HIV co-infected.1 Although drug-sensitive (DS)-TB is curable with a 6–9-month regimen comprising the four first-line drugs, isoniazid, rifampicin, ethambutol, and pyrazinamide, the emergence and spread of multi- and extensively drug-resistant (MDR and XDR) strains of Mtb has greatly complicated the control of TB. In 2014, an estimated 480,000 people worldwide developed MDR-TB and 190,000 died from this form of the disease. In addition, in 2015, cases of XDR-TB were reported in 105 countries, and an estimated 9.7% of people diagnosed with MDR-TB developed XDR-TB.1 As a consequence of the inexorable rise in drug resistance over time, reports of “totally” drug-resistant (TDR)-TB, resistant to all first- and second-line antitubercular drugs, have now become increasingly common.2 Against this background, the urgency of the need for new drugs and drug regimens to tackle this global health crisis cannot be overstated.
As in other areas of antimicrobial drug discovery,3 target-based approaches to the development of inhibitors of enzymes that catalyze essential biochemical processes in Mtb have failed to yield compounds with potent and target-selective activity against whole Mtb cells. The formidable challenges associated with target-based approaches have made the discovery of high-quality “hit” compounds to feed the front end of the TB drug pipeline critically reliant upon the use of phenotypic screening to identify small molecules that inhibit the growth and/or survival of Mtb.4 The value of this empiric approach is evidenced by the fact that the clinically approved drugs, bedaquiline (Sirturo) and delamanid (Deltyba), and others, such as PA-824 (Pretomanid), PBTZ169, and Q203, which are currently in clinical development, arose through phenotypic screening.5 Critically, these drugs, drug candidates, and screening hits have also been used to identify a number of new TB drug targets. These include the AtpE subunit of ATP synthase (the target of bedaquiline6); the decaprenylphosphoryl-β-d-ribose 2-epimerase, DprE1 (the target of PBTZ1697 and other pharmacophores8); the trehalose monomycolate transporter, MmpL3 (the target of indolcarboxamides9 and multiple other pharmacophores10,11); QcrB, a component of the cytochrome bc1–aa3 complex (the target of Q20312 and other pharmacophores13); DnaN, the target of griselimycin;14 and FadD32, the target of diarylcoumarins.15 By virtue of their novel mechanisms of action, drugs that are active against these targets have the potential to offer new therapeutic options for the treatment of DS- as well as DR-TB.
During the course of screening a library of compounds for whole-cell activity against Mtb, we identified a molecule, VCC234718, with promising antitubercular activity and limited mammalian cell toxicity. Using a combination of chemical biology, enzymology, and structural biology, we show that the target of VCC234718 in Mtb is the inosine-5′-monophosphate dehydrogenase (IMPDH), GuaB2, an enzyme that catalyzes the NAD+-dependent conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) in the de novo purine biosynthesis pathway. We further show that GuaB2 depletion is bactericidal in Mtb in vitro, in macrophages, and in mouse lung. Together, these data validate GuaB2 as a new TB drug target.
Results
Identification, Antitubercular Activity, and Pharmacological Properties of VCC234718
The compound VCC234718 (Figure 1), first synthesized by J. Pato as early as 2000 as a part of the proprietary molecular library of Vichem Chemie, and known previously as VI-7777, was identified as a phenotypic screening hit with whole-cell activity against Mtb H37Rv, but not against Corynebacterium glutamicum, in the screen described by Magnet et al. in 2010.16 This compound was found to totally inhibit H37Rv growth at 20 μM while not significantly inhibiting nonreplicating, streptomycin-starved Mtb 18b or 18b-Lux when tested at concentrations up to 20 μM.17,18 Initial screening confirmed that VCC234718 had a 90% minimum inhibitory concentration (MIC90) of 5 μM against replicating H37Rv and at least an 8-fold selectivity index (TD50/MIC) over a panel of human cell lines (TD50 values for Huh7, HepG2, A549, and THP-1 were 100, 42, 100, and 62 μM, respectively). Subsequent analysis of resynthesized VCC234718 showed that it had an MIC90 of 2 μM and displayed time- and concentration-dependent kill of Mtb H37Rv with a 99% minimum bactericidal concentration (MBC99) of 16–32 μM over 5–7 days (Figure S1A). The intracellular activity of VCC234718 was assessed by evaluating its ability to protect MRC-5 lung fibroblasts and activated THP-1 macrophages from the cytolytic effects of Mtb infection.19 VCC234718 was completely inactive in the MRC-5 fibroblast anticytolytic assay at a concentration up to 50 μM (Figure S1B). However, this compound did display anticytolytic activity in activated THP-1 macrophages at concentrations >1 μM (Figure S1C).
Figure 1.
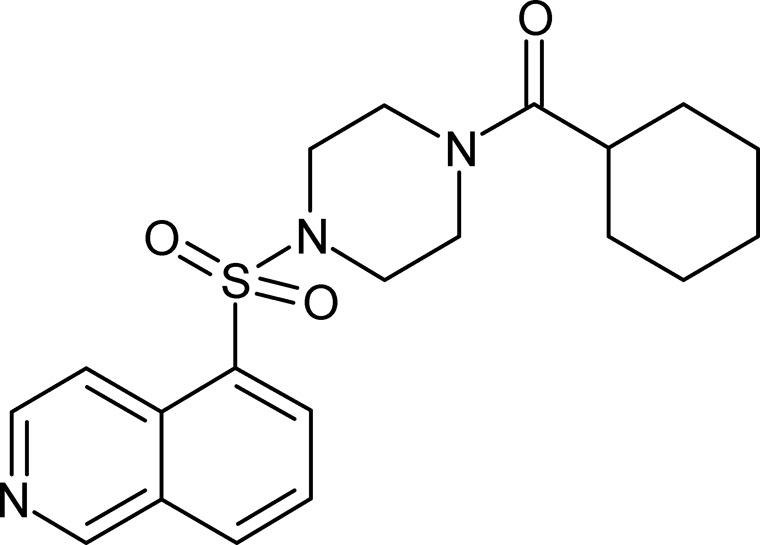
Chemical structure of VCC234718.
To evaluate the ADMET profile of VCC234718, permeability, metabolism, CYP, and ERG channel inhibition were assessed in vitro. VCC234718 exhibited a very high permeability value, well above the threshold value of 20 × 10–7 cm s–1, suggesting that it should be completely absorbed in vivo in humans after oral administration, as long as it is well solubilized in the gastrointestinal tract20 (Table S1). This compound showed possible drug–drug interaction issues, as CYP3A4 inhibition was observed in human liver microsomes (IC50 ∼ 1 μM; Table S1). VCC234718 was highly metabolized in both human and rodent liver microsomes and human liver primary hepatocytes (Table S1), with no contribution from CYP3A4 to its metabolism in human primary hepatocytes. Furthermore, VCC234718 antagonist activity on human ERG channel was moderate as measured in an automated patch clamp assay21 (IC50 = 13.7 μM; Table S1).
Isolation and Characterization of VCC234718-Resistant Mutants of Mtb
To investigate the mechanism of action of VCC234718, we raised spontaneous resistant mutants by plating Mtb cells on Middlebrook 7H10 agar containing the compound at 10×MIC90, 20×MIC90, or 50×MIC90. Mutants were obtained only from the 10×MIC plate, at a frequency of approximately 5 × 10–6. Ten individual colonies were picked, grown in 7H9 broth, and retested for VCC234718 susceptibility. Only three of the mutants selected displayed stable phenotypic resistance to the compound, suggesting that the frequency of resistance determined from the original plating overestimated the actual frequency of heritable resistance (Table 1). Whole genome sequence analysis of the three mutants compared to the parental wild type H37Rv strain revealed six different nonsynonymous single-nucleotide polymorphisms (nsSNPs), all of which were confirmed by Sanger sequencing (Table S2). Of the three mutants, SRMV2.6 showed the highest level of resistance to VCC234718 (MIC90 > 100 μM; Table 1). This strain carried a nsSNP in an essential gene, guaB2 (Rv3411c) (Figure S2), which encodes the enzyme, IMPDH, that catalyzes the NAD+-dependent conversion of inosine 5′-monophosphate IMP to XMP in the de novo purine biosynthesis pathway.22−24 The mutation in the guaB2 resulted in a Y487C mutation in the IMPDH domain of GuaB2, suggesting that it may be causal for the resistance phenotype. SRMV2.6 also carried a nsSNP in ppsA, which encodes a type I polyketide synthase involved in phenolphthiocerol and phthiocerol dimycoserosate (PDIM) biosynthesis. The other two mutants carried an identical mutation, which introduced a stop codon in fadD26, a gene located immediately upstream of ppsA, and part of the same virulence gene cluster in the Mtb genome.25,26 Because loss of PDIM biosynthetic capacity is commonly observed during propagation of Mtb in vitro, we concluded that the ppsA and fadD26 mutations were unlikely to contribute significantly to the resistance phenotypes of any of the three mutants. In support of this conclusion, VCC234718 was found to be equally active against a PDIM-deficient H37Rv strain (H37RvJO27) as the PDIM-producing H37Rv strain employed in this study (MIC90 = 2 μM). Of the other genes in which mutations were identified, Rv0678 is notable by virtue of the fact that it encodes a transcriptional regulator which has been implicated in acquired resistance to drugs that include azoles, bedaquiline, and clofazimine, through up-regulation of the multisubstrate efflux pump, MmpL5.28−30 This suggests that the low-level resistance to VCC234718 in SRMV2.10 was likely attributable to drug efflux. In contrast, the association between the VCC234718-resistant phenotypes of SRMV2.3 and SRMV2.10 and the nsSNPs in Rv3479 and Rv3755c, respectively, is unclear. However, as these genes were predicted to be nonessential in vitro by Himar1-based transposon mutagenesis and Bayesian statistical model analysis applied to deep-sequencing data for transposon insertion libraries of Mtb,31 they were not pursued further.
Table 1. Susceptibility of Mtb Strains to VCC234718.
| strain | MIC90 (μM) |
|---|---|
| H37RvMA | 2 |
| SRMV2.3 | 16 |
| SRMV2.6 | >100 |
| SRMV2.10 | 8 |
| SRMV2.6 attB::guaB2 | >200 |
| SRMV2.6 attB::guaB2-guaB3 | >200 |
| H37Rv attB::guaB2 | 4–8 |
| H37Rv attB::guaB2-guaB3 | 4–8 |
| SRMV2.6 attB::guaB2Y487C | >200 |
| H37Rv attB::guaB2Y487C | >100 |
To investigate the association between the guaB2 genotype of SRMV2.6 and its VCC234718-resistance phenotype, we delivered either the wild type guaB2 gene or the mutant allele (guaB2Y487C), under control of the native promoter, into H37Rv or SRMV2.6, via an integration vector. The growth kinetics of both strains were indistinguishable in liquid media, thus arguing against an impact of the guaB2Y487C mutation on the growth of Mtb in axenic culture. Integration of wild type guaB2 in H37Rv or SRMV2.6 conferred a 2–4-fold increase in the MIC90 of VCC234718 relative to the parental strains (Table 1). Because guaB2 is one of three guaB orthologues in Mtb and is operonic with guaB3,22 we also investigated the effect of expression of the wild type guaB2-guaB3 operon, driven by the native promoter, on susceptibility to VCC234718, but found that it conferred a 2–4-fold increase in MIC90 similar to that of guaB2 alone. In contrast, integration of the guaB2Y487C allele in H37Rv raised the MIC90 of VCC234718 >50-fold, to a level equivalent to that of SRMV2.6, but increased the MIC90 in SRMV2.6 only 2-fold. Taken together, these results confirmed that the a1460g mutation in guaB2 was both necessary and sufficient for resistance of Mtb to VCC234718 and, moreover, that the resistance allele was dominant.
VCC234718 Is a Potent Inhibitor of Mtb GuaB2
To establish whether VCC234718 is an inhibitor of GuaB2, we expressed recombinant Mtb GuaB2 in Escherichia coli for use in enzyme activity assays. Active and stable recombinant Mtb GuaB2 was routinely produced with a maximum yield of about 1 mg of pure enzyme per liter of culture by modifying a previously reported protocol.24 Determination of the enzyme kinetics confirmed a hyperbolic behavior and kinetic parameters close to those reported previously,24 and in line with observations for other IMPDH enzymes,32 with IMP showing a KM value 10-fold lower than that of NAD+ (Table S3). VCC234718 was found to inhibit Mtb GuaB2 with a Ki value of 100 nM and an uncompetitive mechanism of inhibition with respect to both IMP and the NAD+ cofactor (Table S3 and Figure S3). To evaluate the impact of the Y487C mutation on the enzymatic activity of Mtb GuaB2, we produced the recombinant variant, Mtb GuaB2Y487C, and determined the inhibitory effect of VCC234718 on this enzyme. Importantly, VCC234718 was found to be inactive against Mtb GuaB2Y487C when tested at a concentration up to 100 μM. Finally, to assess the selectivity of VCC234718 toward Mtb GuaB2, its IC50 against human IMPDH was determined (Figure S4) and found to be 3.35 μM, indicating a selectivity index of >30 for the Mtb enzyme.
Binding of VCC234718 to Mycobacterium thermoresistibile GuaB2
To investigate the structural basis of VCC23718 binding to GuaB2, we attempted to crystallize the full-length Mtb GuaB2. As our initial efforts were unsuccessful, we moved to M. thermoresistible (Mth) GuaB2 to facilitate crystallization. Full-length Mth GuaB2 shares 85% sequence identity with Mtb GuaB2 and is 100% identical in the active site (Figure S2). VCC234718 inhibited this enzyme with a Ki value of 130 nM with IMP as the substrate, which is very similar to the Ki value observed for the Mtb enzyme (120 nM; Table S3). Although we were able to obtain crystals of full-length Mth GuaB2 and solve the structure (A.P., D.A., and T.L.B., unpublished data), the resolution (2.6 Å) was not sufficiently high for an accurate analysis of the enzyme–inhibitor complex. To improve the resolution we designed another construct of Mth GuaB2 with a GG linker replacing the two cystathionine-β-synthase (CBS) domains. This strategy has been used previously to obtain high-resolution structures of other bacterial IMPDHs,33 and this will be henceforth referred to as Mth GuaB2 ΔCBS. As expected, this form of the enzyme retained full IMPDH activity and showed the same susceptibility to VCC234718 as full-length Mtb GuaB2 and Mth GuaB2 (Ki = 130 nM with IMP as the substrate).
Mth GuaB2 ΔCBS crystallized in the I4 space group with one molecule/protomer in the asymmetric unit (Table S4). The complete IMPDH tetramer of 222 symmetry can be seen in the crystal lattice, through the symmetry operations of the space group (Figure 2A). After molecular replacement, observation of a 2F0 – Fc difference map (σ = 3.0) showed clearly visible electron density for IMP, in addition to one molecule of VCC234718 partially occupying the NAD+ binding site (Figure 2B). There is no ambiguity in the interpretation of the density for the molecule of VCC234718 in the structure at this resolution. It could not be NAD+, as the density would look extremely different; for example, there is no density for the phosphates of NAD, and electron densities for the rings are in distinctly different places. Figure 2C is an overlay of the two structures clearly demonstrating differences. Although the kinetic studies of VCC234718 revealed that this compound is uncompetitive with respect to both IMP and NAD+, the binding of the VCC234718 molecule closely resembles that of other previously described uncompetitive inhibitors of Mtb GuaB2.34 The uncompetitive mode of inhibition of VCC234718 with respect to NAD+ is difficult to rationalize as the inhibitor and NAD+ binding sites overlap. IMPDH enzymes have been reported to follow a steady-state ordered bi bi mechanism where IMP binds first, followed by NAD+, and product release proceeds in the order of NADH followed by XMP.32 Moreover, a key feature of the catalytic mechanism is the formation, during the redox step in which NADH is formed, of a covalent enzyme-XMP* intermediate that has been investigated through structural studies of the Vibrio cholerae(33) IMPDH enzyme. Because Mtb GuaB2 follows the same catalytic mechanism, it could be speculated that VCC234718 binds preferentially to the covalent Mtb GuaB2-XMP* intermediate after NADH has been released, thereby explaining the uncompetitive behavior observed toward the NAD+ cofactor. This notion is consistent with the inhibitory mechanism elucidated recently by pre-steady-state kinetics for Bacillus anthracis IMPDH.35
Figure 2.
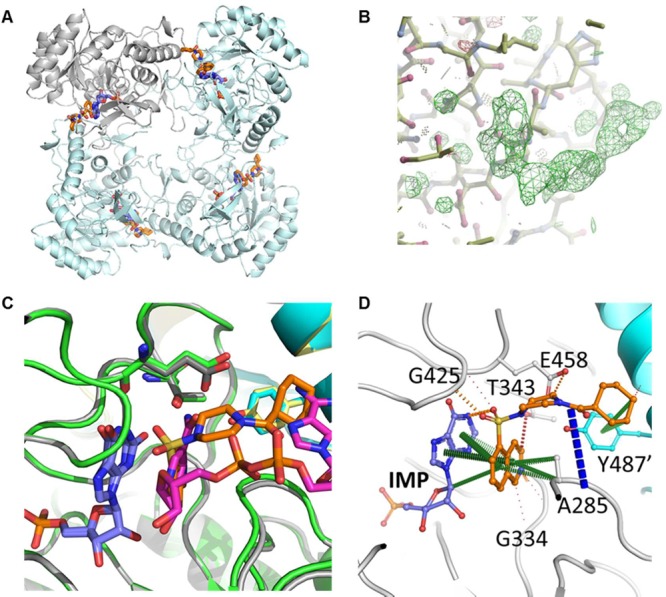
X-ray crystal structure of VCC234718 bound to Mth GuaB2. (A) The GuaB2 tetramer (cyan ribbon, with a representative protomer shown in gray) is shown bound to IMP (blue) and VCC234718 (orange). (B) 2F0-Fc difference map (σ = 3.0) clearly shows visible electron density for VCC234718 in the NAD+ binding site. (C) Overlay of the X-ray crystal structures of the complexes of Mtb GuaB2 (green and yellow) and NAD (magenta) and of Mth GuaB2 (gray and cyan) and VCC234718 (orange) demonstrates the difference in chemical space occupied. XMP and IMP are shown in blue. (D) Interactions were made by VCC234718 (orange) in the X-ray crystal structure of the complex of Mth GuaB2 (gray; and adjacent protomer in cyan) with IMP (blue). Residue numbering is of the corresponding residues in Mtb. Pi interactions are shown in green, amide–amide in blue, hydrogen bonds in red, and polar interactions in orange. PDB ID: 5J5R.
The electron density revealed that VCC234718 sits within the NAD binding pocket of GuaB2, with the isoquinoline group of VCC234718 stacked on top of the hypoxanthine group of IMP, making extensive pi interactions (Figure 2D). The isoquinoline group made further pi interactions with A285 (A269 in the Mth structure), along with weak hydrogen bonds to the side chain of T343 (T327 in the Mth structure) and the main chain atoms of G334, V335, and G336 (G318, V319, and G320 in the Mth structure). An additional polar interaction between the inhibitor and IMP was made by the oxygen of the sulfonamide group of VCC234718 to a nitrogen group of the hypoxanthine IMP ring, with the sulfonamide making further weak polar interactions to the main chain of G425 (G409 in the Mth structure). The cyclohexyl group of VCC234718 was orientated to form strong pi interactions with the Y487′ (Y471′ in the Mth structure) side chain from the adjacent molecule in the tetramer.
VCC234718 Selectively Targets GuaB2 in Mtb
To investigate whether VCC234718 retains target selectivity for GuaB2 in whole Mtb cells, we asked whether conditional depletion of GuaB2 would sensitize Mtb to the growth inhibitory effects of the compound.10 To this end, we constructed anhydrotetracycline (ATc)-regulated GuaB2 knockdown mutants in the Tet-ONM and Tet-OFF configurations, as described previously for other genes.36,37 The guaB2 promoter was replaced by the ATc-regulated promoter-operator element, Pmyc1tetO,38 via single-crossover (SCO) homologous recombination to generate the promoter-replacement mutant, guaB2-guaB3-SCO (Figure S5). A vector carrying the forward (TetR) or reverse tetracycline repressor (rev-TetR) was then introduced into guaB2-guaB3-SCO to produce mutants in the Tet-ONM and Tet-OFF configurations, respectively. The guaB2-guaB3 Tet-ONM and guaB2-guaB3 Tet-OFF mutants showed ATc-dependent growth in liquid culture and on agar (Figure S6). Because both guaB2 and guaB3 appear to be essential for growth of Mtb in vitro,31 the suppression of Mtb growth resulting from guaB2-guaB3 silencing could be due to depletion of GuaB2 and/or GuaB3. To eliminate potentially confounding effects of concomitant guaB3 silencing, a copy of guaB3 under control of the guaB2-guaB3 promoter was thus integrated into the guaB2-guaB3 Tet-OFF strain to generate the complemented derivative, guaB2-guaB3 Tet-OFF attB::guaB3. This mutant retained an ATc-dependent growth phenotype (Figure 3A), but appeared to be slightly (<2-fold) less sensitive to ATc than the parental guaB2-guaB3 Tet-OFF strain (Figure S6B).
Figure 3.

ATc dose-dependent modulation of growth of Mtb correlates with transcriptional silencing of guaB2 and depletion of GuaB2 protein. (A) Growth of guaB2-guaB3 Tet-OFF attB::guaB3 in 7H9 liquid medium supplemented with the indicated concentrations of ATc. Data are from a representative experiment performed in triplicate. (B) Transcriptional response of guaB2 (left) and guaB3 (right) to ATc treatment. Logarithmic phase cultures of the various strains were treated with ATc, as indicated, and the concentrations of guaB2 and guaB3 transcript relative to sigA determined by ddPCR, as described under Methods. All of the data generated by QuantaSoft software included the 95% confidence interval. (C) ATc-dependent depletion of GuaB2 in guaB2-guaB3 Tet-OFF attB::guaB3 confirmed by Western blot analysis. Strains were cultured in Middlebrook 7H9 broth and either left untreated (−) or exposed to ATc (10 ng/mL) (+) for the indicated time. Western blotting was carried out as described under Methods, probing with antibodies against GuaB2 or Wag31.
Gene expression analysis by droplet digital PCR (ddPCR) confirmed that under fully derepressed conditions (i.e., in the absence of ATc) the level of guaB2 transcript in the guaB2-guaB3 Tet-OFF attB::guaB3 strain was ∼35% of that in H37Rv and was further reduced to <1% of the wild type level upon exposure to ATc (10 ng/mL, for 24 h) (Figure 3B). In contrast, the level of expression of guaB3 was unresponsive to ATc and remained at ∼30% of the level of guaB3 transcript in H37Rv. This finding contrasts with the ∼95% reduction in guaB3 transcript level observed following ATc treatment of the parental guaB2-guaB3 Tet-OFF strain (Figure S7) and confirms ATc-independent expression of guaB3 in the complemented strain. However, the level of guaB3 transcript in the complemented mutant was lower than expected (64% of wild type), suggesting that guaB3 expression was partially restored in this strain (Figure 3B). Western blot analysis confirmed that transcriptional silencing of guaB2 resulted in time-dependent depletion of GuaB2 in the guaB2-guaB3 Tet-OFF attB::guaB3 strain, with almost complete disappearance of the protein observed at day 5 (Figure 3C). In contrast, GuaB2 levels in H37Rv were unaffected by ATc treatment over this time course. Together, these results thus confirmed that the ATc-dependent growth inhibition of the guaB2-guaB3 Tet-OFF attB::guaB3 strain correlated with transcriptional silencing of guaB2 and depletion of the cellular level of GuaB2 protein.
The effect of guaB2 silencing on the susceptibility of Mtb to VCC234718 was then examined in a checkerboard assay in which ATc and VCC234718 concentrations were varied in a two-dimensional array (Figure 4A). Transcriptional silencing of guaB2 resulted in hypersensitization to VCC234718 in an ATc dose-dependent manner, with the MIC90 shifting ∼32-fold, from 2.5 to 0.07 μM, with increasing concentration of ATc. In contrast, guaB2 silencing had no effect on susceptibility to other antituberculars with different mechanisms of action (isoniazid, rifampicin, streptomycin, ethambutol, and levofloxacin), providing further evidence that VCC234718 targets GuaB2 selectively in Mtb (data not shown).
Figure 4.

VCC234718 hypersensitivity and growth inhibitory effects of guaB2 silencing can be rescued by guanine supplementation. (A) Effect of guaB2 silencing on susceptibility of Mtb to VCC234718. (B) Guanine rescue of Mtb H37Rv from growth inhibition by VCC234718. (C) Guanine rescue of guaB2-guaB3 Tet-OFF attB::guaB3 from growth inhibition mediated by ATc. These experiments were performed as checkerboard assay, as described under Methods.
Guanine Rescues Mtb from Toxicity Caused by VCC234718 Treatment or GuaB2 Depletion
By blocking the supply of GMP and, hence, other guanine nucleotides, inhibition of de novo biosynthesis at the IMPDH step is expected to be growth inhibitory in Mtb. However, in addition to the de novo pathway, Mtb also possesses a purine salvage pathway.39,40 We thus hypothesized that guanine supplementation might alleviate the toxicity caused by VCC234718 treatment or GuaB2 depletion in Mtb by enabling an alternate route to GMP production via the action of hypoxanthine-guanine phosphoribosyl transferase, Hpt (Rv3624c; hypoxanthine-guanine phosphoribosyl transferase).40,41 Indeed, guanine supplementation showed clear dose-dependent alleviation of VCC234718 toxicity in Mtb, raising the MIC90 of VCC234718 from 2 to >64 μM at a guanine concentration of 200 μM (Figure 4B). However, guanine was unable to rescue Mtb from VCC234718 toxicity at a drug concentration of 128 μM, suggesting that this compound has a secondary target (or targets) in Mtb. In contrast, and as expected from what is known about purine salvage in Mtb,40 xanthine supplementation had no effect on VCC234718 toxicity over the same concentration range (data not shown). Likewise, neither adenine nor guanosine supplementation was able to rescue Mtb from VCC234718 toxicity (data not shown). Guanine supplementation, alone, also rescued guaB2-guaB3 Tet-OFF attB::guaB3 (Figure 4C) and its parental strain, guaB2-guaB3 Tet-OFF (Figure S8), from ATc toxicity in a dose-dependent manner. The response of both strains to guanine supplementation was indistinguishable, with essentially complete rescue observed at a guanine concentration ≥125 μM.
Depletion of GuaB2 Is Bactericidal in Vitro
To assess the impact of guaB2 silencing on the viability of Mtb in vitro, the guaB2-guaB3 Tet-OFF attB::guaB3 mutant was cultured in axenic culture in 7H9 broth, with or without ATc, and aliquots sampled over 288 h were plated on 7H10 agar to score for colony-forming units (CFU) (Figure 5A). All controls behaved as expected: under permissive conditions (i.e., in the absence of ATc), the mutant showed growth comparable to that of the H37Rv control. Furthermore, the ATc-dependent growth phenotype of the mutant was stable over the entire time course, as evidenced by the complete suppression of colony formation on agar containing ATc (Figure 5A). Transcriptional silencing of guaB2 appeared to be rapidly bactericidal, with the conditional mutant showing a 4 log10 decline in CFU at 48 h, whereafter the CFU count dropped to below the limit of detection (10 CFU). Interestingly, inclusion of guanine (200 μM) in the agar media used for CFU enumeration resulted in a partial rescue of culturability of the mutant strain when exposed to ATc in liquid culture prior to plating. However, the guanine-dependent enhancement of culturability was observed only up to 168 h and was no longer detected at the later time points. Moreover, the effect was highly specific, with guanine supplementation of the agar media having no discernible effect on culturability (CFU) in any of the controls. The transient rescue of culturability was indicative of the presence of a progressively declining population of bacilli in the guaB2-silenced culture of the conditional mutant that have impaired culturability on solid medium, but remain poised to detect, transport, and assimilate guanine, that is, organisms that can be defined as “non-growing but metabolically active” (NGMA).42
Figure 5.

Depletion of GuaB2 is bactericidal in Mtb in vitro. (A) The effect of guaB2 silencing on the viability of Mtb was assessed as described under Methods. The guaB2-guaB3 Tet-OFF attB::guaB3 and H37Rv strains were grown in the presence or absence of ATc, and the effect of silencing on viability was assessed by plating serial dilutions at the indicated times on 7H10 agar, with or without ATc (10 ng/mL) and with or without guanine. The results represent the mean ± SD from three biological replicates. The plating conditions are shown in parentheses in the legend. (B) Single-cell analysis of the effect of transcriptional silencing of guaB2 on growth and morphology of Mtb in vitro. The guaB2-guaB3 Tet-OFF attB::guaB3 strain was grown in a microfluidic device in 7H9 medium containing 25 μg/mL kanamycin, 50 μg/mL hygromycin, and 2.5 μg/mL gentamycin (0–74 h). The medium was supplemented with 100 ng/mL ATc between 74 and 379 , and with 125 μM guanine between 334 and 500 h. The bacteria were imaged on the phase channel every 15 min, and representative time series snapshots of two positions in the microfluidic device are shown. The numbers above the snapshots denote the times at which the shots were taken. The white arrows indicate some of the cells that lysed in the presence of ATc. The lower panel represents a microcolony in which some of the surviving cells (indicated with the arrowhead) regrow on guanine supplementation. The last snapshot is a composite image of phase channel and propidium iodide staining (in red, TRITC channel). Cells marked with an asterisk are propidium iodide stained cells and represent cells in which the membrane integrity has been compromised. The yellow horizontal scale bar represents 5 μm. This experiment was performed twice, and representative images from one experiment are shown.
To characterize, in greater detail, the effect of guaB2 silencing on the growth and morphology of Mtb in vitro, we carried out highly temporal time-lapse microscopy experiments on the guaB2-guaB3 Tet-OFF attB::guaB3 strain. The bacteria were grown in standard 7H9 medium (replete conditions) in a flask and seeded into a microfluidic device, as described previously.43,44 The bacteria grew and divided to form small microcolonies, as shown in Figure 5B and in Movies S1 and S2, where the movies show the growth and response to guaB2 silencing of two different microcolonies in the microfluidic device. After approximately 74 h, the bacteria were exposed to 100 ng/mL ATc for about 260 h (between 74 and 334 h, in the experiment shown in Figure 5B). In contrast to the rapid loss of culturability observed in batch culture (Figure 5A), bacteria in the microfluidic device continued to grow and divide normally during the initial phase of exposure to ATc (∼75–125 h). This might be due to differences in the kinetics of response of Mtb to ATc-mediated guaB2 silencing in the two systems. Alternatively, the apparently normal growth and division observed microscopically at the early stages of ATc exposure masked an underlying culturability defect detectable only on solid medium. Subsequently, however, the growth rate of cells in the microcolonies decreased and the cells stopped dividing. After this growth arrest, a major fraction of the cells lysed (white arrows in Figure 5B), visible as phase-bright remnants. However, even after 260 h of ATc exposure, a few intact cells remained (still phase-dark), some of which were able to regrow and divide on supplementation with guanine (125 μM). We initially supplemented with guanine in the presence of ATc to verify if guanine was able to rescue the cells from GuaB2 depletion. Indeed, on guanine supplementation, cells started elongating even in the presence of ATc (frame 379 h, lower panel Figure 5B; Movie S2) and reverted to normal growth rate and division on ATc withdrawal. In the experiment shown in Figure 5B, among the microcolonies exposed to ATc for 260 h and imaged, 42% had at least one survivor that was able to re-initiate growth and division on guanine supplementation. This observation likely reflects cell-to-cell variation in the extent of GuaB2 depletion at the end of period of exposure to ATc, with a small percentage of cells harboring sufficient GuaB2 to remain viable and able to import and assimilate guanine and thereby initiate regrowth.
Depletion of GuaB2 Is Bactericidal ex Vivo
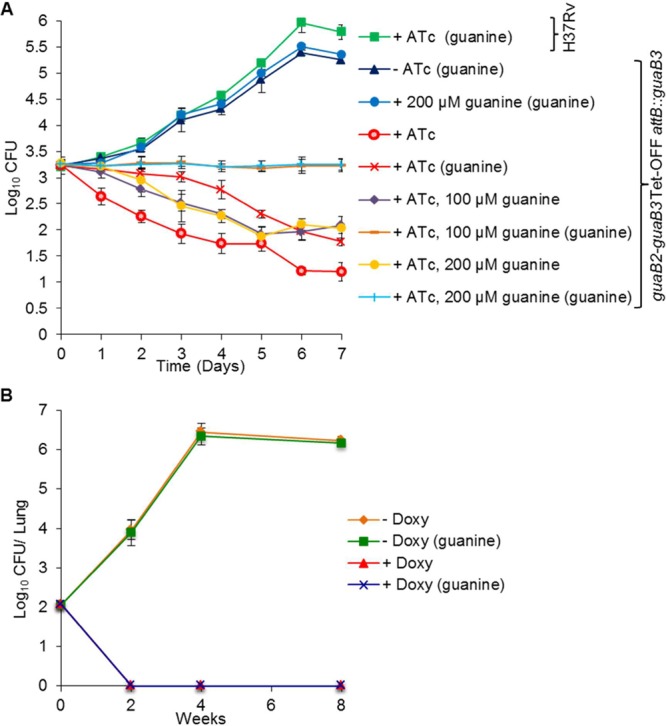
Having established that guaB2 silencing has a bactericidal effect on Mtb in vitro, we then investigated its effect on Mtb in a macrophage infection model. We infected THP-1-derived macrophages with either the guaB2-guaB3 Tet-OFF attB::guaB3 mutant or H37Rv and monitored the viability of Mtb in the presence (200 ng/mL) or absence of ATc, alone or in combination with various concentrations of guanine (0–200 μM) included in the tissue culture medium, by CFU enumeration over a period of 7 days (Figure 6A and Figure S9). On the basis of the guanine-dependent culturability effect observed in vitro, CFUs were scored in all cases by plating samples on agar, either with or without guanine supplement (200 μM). The wild type control grew as expected, and its culturability was not affected by the treatment or plating conditions. The guaB2-guaB3 Tet-OFF attB::guaB3 mutant multiplied efficiently inside macrophages when ATc was not present, albeit slightly less so than H37Rv. Furthermore, at all time points, no CFUs were obtained when this strain was plated on agar containing ATc (with or without guanine), confirming its genetic stability over the course of the experiment. Silencing of guaB2 by ATc treatment resulted in a 2 log10 reduction in the intracellular bacillary load of the mutant strain over the 7-day time course. However, as observed in axenic culture, the inclusion of guanine in the agar media resulted in a 0.5–1 log10 increase in CFU count. In the absence of silencing (no ATc), guanine supplementation of RPMI media gave a slight growth advantage to the conditional mutant, but had no effect on the growth of H37Rv. The rescuing effect of guanine was further enhanced by inclusion of a high concentration of guanine in the tissue culture medium (≥100 μM) as well as in the agar. Under these conditions, guaB2 silencing appeared to be bacteriostatic. Together, these results confirmed that although GuaB2 is required for intracellular growth of Mtb, the organism can be rescued from the bactericidal effects of GuaB2 deficiency if it has access to sufficiently high levels of guanine.
Figure 6.
GuaB2 is essential for growth and survival of Mtb ex vivo and in vivo. (A) THP-1 cells were infected as described in the Supporting Information and grown in standard RPMI media in the absence or presence of ATc (200 ng/mL) and/or guanine (100 or 200 μM), and the effect of silencing on Mtb viability ws assessed by plating serial dilutions as described under Methods and Supporting Information. The results represent the mean ± SD from three biological replicates. The plating conditions are shown in parentheses in the legend. (B) Female C57BL/6 mice were infected by low-dose aerosol with guaB2-guaB3 Tet-OFF attB::guaB3 and treated as described under Methods. Four mice per group were sacrificed at the indicated time points. Lung homogenates were plated on 7H10 agar with the required antibiotics, and with or without guanine supplementation, as described under Methods.
GuaB2 Is Essential for Mtb To Establish an Infection in Mice
To evaluate the essentiality of guaB2 in vivo, C57BL/6 mice were infected by aerosol with the guaB2-guaB3 Tet-OFF attB::guaB3 mutant strain. Transcriptional silencing of guaB2 was achieved by feeding the animals with doxycycline-containing food from 5 days prior to infection and throughout the 8-week infection time course. Control groups received normal food, without doxycycline, thus allowing expression of guaB2 from the revTetR-regulated promoter. As depicted in Figure 6B, the conditional mutant multiplied normally in the absence of the inducer. Because a control infection with the parental wild type H37Rv strain was not performed in parallel, we could not ascertain whether the reduced level of expression of guaB2 in the conditional mutant in the absence of inducer had an impact on virulence. However, administration of doxycycline at a dose shown in other studies in our45 and other laboratories46,47 to have no effect on the growth of wild type Mtb caused complete clearance of Mtb in the lungs of the infected animals by week 2 post-infection. The lung bacillary load at all later time points was likewise below the limit of detection (1 CFU). Given the data obtained in vitro and ex vivo, where guanine was shown to partly rescue the culturability of Mtb following ATc-mediated silencing of guaB2 up to a week postsilencing (Figures 5A and 6B), the effect of guanine supplementation in the media used to score the lung bacillary loads was also investigated. However, guanine supplementation had no impact on the CFUs recovered from the mouse lung, irrespective of whether the animals had been treated with doxycycline. These results confirm that GuaB2 is essential for Mtb to establish an infection in mice and demonstrate that guaB2 silencing is rapidly bactericidal in this animal model.
Discussion
IMP, which is produced via the de novo purine biosynthesis pathway, is a biosynthetic precursor for both adenine and guanine nucleotides.48 The conversion of IMP to XMP by IMPDH represents the first committed step in the production of guanine nucleotides, which are formed by the subsequent conversion of XMP to GMP and serve as biosynthetic precursors, substrates, regulators, and signaling molecules in multiple essential cellular processes that include DNA and RNA synthesis, protein synthesis, cell envelope biogenesis, vitamin and cofactor biosynthesis, and the stringent response (Figure 7). In addition to the de novo pathway, cells also have recourse to obtaining guanine nucleotides via purine salvage, which allows bypass of biosynthetic enzymes by providing a direct route to GMP (Figure 7). By virtue of its central role in the de novo pathway, IMPDH has emerged as a major target for the development of drugs for the treatment of autoimmune diseases, cancers, and viral infections—conditions associated with rapid cellular proliferation and, hence, a high demand for guanine nucleotides. More recently, IMPDH has also attracted attention as an antimicrobial drug target.32,48 Through a series of elegant studies, focused initially on the protozoan parasite Cryptosporidium parvum, Hedstrom and colleagues have shown that C. parvum IMPDH-selective inhibitors, which do not inhibit the human enzyme, are also active against IMPDH enzymes from a variety of bacterial pathogens, including Mtb.17,34,48−51
Figure 7.

De novo biosynthesis and salvage pathways for purines in Mtb illustrating the impact of GuaB2 inhibition. Solid black arrows denote steps in the de novo pathway, and dashed black arrows denote steps in the salvage pathway. Gray dashed arrows denote steps for which canonical enzymes are absent from Mtb. Abbreviations: add, Rv3313c, adenosine deaminase; adk, Rv0733, adenylate kinase; ADP, adenosine diphosphate; adoK, Rv2202c, adenosine kinase; AMP, adenosine 5′-monophosphate; apt, adenine phosphoribosyltransferase; ATP, adenosine triphosphate; atpA-H, Rv1304-1311, ATP synthase; dADP, deoxyadenosine diphosphate; dATP, deoxyadenosine triphosphate; deoD, Rv3307, purine-nucleoside phosphorylase; dGDP, deoxyguanosine diphosphate; dGTP, deoxyguanosine triphosphate; gmk, Rv1389, guanylate kinase; GMP, guanosine 5′-monophosphate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; guaA, GMP synthase; guaB2, inosine monophosphate dehydrogenase (IMPDH); hpt, Rv3624c, hypoxanthine-guanine phosphoribosyltransferase; IMP, inosine 5′-monophosphate; iunH, Rv3393, purine nucleosidase; ndkA, Rv2445c, nucleoside-diphosphate kinase; PRPP, 5-phosphoribosyl 1-pyrophosphate; purA, Rv0357c, adenylosuccinate synthetase; purB, Rv0777, adenylosuccinate lyase; RNR, ribonucleotide reductase; XMP, xanthosine 5′-monophosphate.
The compound described in this study, VCC234718, adds to the growing list of inhibitors of Mtb GuaB2 with whole-cell activity against the organism.34,52 Making use of a mutant strain of Mtb that conditionally underexpresses guaB2, we obtained direct evidence that VCC234718 retains target selectivity for GuaB2 in whole Mtb cells. As observed for other uncompetitive inhibitors of GuaB2, VCC234718 was shown to bind at the NAD binding site of the enzyme. The X-ray crystal structure also revealed that VCC234718 makes pi interactions with IMP and with the A285 residue and a polar interaction with IMP via the oxygen of the sulfonamide group to a nitrogen group of the hypoxanthine ring. The cyclohexyl group of VCC234718 is orientated to form strong pi interactions with the side chain of the Y487′ residue from the adjacent protomer in the tetramer. Notably, residues A285 and Y487′ comprise the “inhibitor minimal structural motif” in Mtb GuaB2.34 Identified originally in the C. parvum enzyme, this motif has been shown to be required for compound binding to C. parvum IMPDH and to account for inhibitor selectivity versus the human enzyme.49,51
In the X-ray crystal structure of Mtb GuaB2 with XMP and NAD reported recently by Makowska-Grzyska et al. (PDB ID: 4ZQM),34 the purinamine group of NAD makes pi interactions with Y487′, including a donor–pi interaction mediated by the azane. Although the pi–pi and donor–pi interactions are lost upon mutation of this tyrosine to cysteine, the azane group of NAD maintains a polar interaction with Y487′ and additional pi interactions to H286. By contrast, however, the Y487C mutation would result in the loss of all pi interactions to the cyclohexyl group of VCC234718; this would explain the high-level resistance to VCC234718 observed in the mutant strain, SRMV2.6. Using mCSM-Lig,53 the Y487C mutation is predicted to greatly reduce the binding affinity of VCC234718, while having minimal effect on the binding of NAD,54 and was not predicted to destabilize GuaB2 using our in-house methods mCSM, SDM, and DUET.55−57 Consistent with the prediction of a reduced binding affinity for VCC234718, a recombinant form of Mtb GuaB2 carrying the Y487C mutation was refractory to inhibition by VCC234718 at a drug concentration of 100 μM—a value 1000-fold higher than its Ki for wild type Mtb GuaB2. The relatively neutral effect of the Y487C mutation on protein stability and the natural ligand binding affinity and corresponding reduction in VCC234718 binding would make it a good candidate resistance mutation58 and explain its appearance in SRMV2.6. This also suggests that modification of the cyclohexyl group of VCC234718 to take advantage of pi interactions to H286 in addition to Y487′, mimicking the NAD+ natural interactions, could decrease the propensity for resistance to develop.
A potential liability of metabolic drug targets in microbial pathogens is the extent to which metabolite salvage would allow the organism to evade the consequences of target inhibition. When added to culture medium at a concentration of at least 125 μM, guanine was able to completely rescue Mtb from the growth inhibitory effects of VCC234718 exposure or GuaB2 depletion in vitro and in macrophages. The high concentration of guanine required to bypass GuaB2 activity might be due to the absence of canonical nucleobase transporters in Mtb, as noted previously.37 Nonetheless, the rescuing effect, likely mediated by the conversion of guanine to GMP through the action of the Hpt, confirmed the functionality of this component of the purine salvage pathway and established that VCC234718 toxicity was attributable to loss of IMPDH activity. The inability of xanthine to rescue Mtb from the toxic effects of GuaB2 inhibition or depletion is consistent with available information on purine salvage in Mtb.40 However, guanosine also failed to rescue Mtb from GuaB2 inhibition or depletion toxicity even though this organism has both purine nucleoside phosphorylase59 and nucleoside hydrolase activities.60 One possible explanation for this is that Mtb is unable to transport guanosine, a conclusion also consistent with the observation that this organism lacks NCS1 and NCS2 nucleoside cation symporter family homologues.37 Because GuaB2 is the only one of the three GuaB orthologues in Mtb with confirmed IMPDH activity,24 our findings confirm that GuaB2 is the primary target of VCC234718 in Mtb. This conclusion is consistent with the fact that guanine rescue of the noncomplemented conditional mutant, guaB2-guaB3 Tet-OFF, from ATc toxicity, was indistinguishable from that of its guaB3-complemented derivative, guaB2-guaB3 Tet-OFF attB::guaB3; that is, both VCC234718 hypersensitivity and ATc-dependent growth phenotypes of the complemented mutant were attributable exclusively to GuaB2 deficiency.
In the absence of guanine supplementation, guaB2 silencing was rapidly bactericidal in vitro, ex vivo, and in mouse lung. Although nonculturable Mtb cells in which growth could be rescued by exogenous guanine persisted for at least 7 days under conditions of guaB2 silencing in vitro and ex vivo, they were not detectable in the lungs of mice sacrificed 14 days post-infection. The rapid loss of viability of Mtb in vivo when guaB2 silencing was initiated at the time of infection confirms that Mtb has limited access to guanine in mouse lung and that GuaB2 is therefore essential for Mtb to establish an infection in mice. These results provide compelling validation for GuaB2 as a TB drug target in this animal model. VCC234718 can be considered as a valuable starting point to develop a compound for treating TB infections in humans: it showed good potential for oral route administration, but liabilities such as potential drug–drug interactions and high metabolism along with an alert on ERG channel interaction will require further chemical optimization. Finally, to our knowledge, the levels of guanine have not been reported in lung tissue from mice infected with Mtb. Although our results suggest that guanine levels are not adequate to bypass the essentiality of GuaB2 in this model, it should be recalled that there are profound differences in disease pathology between mice and humans—and, hence, in the microenvironments encountered by Mtb during the course of infection.61 Further advancement of this target will therefore be contingent upon an assessment of the levels of guanine in human TB lesions.
Methods
Bacterial Culture Conditions
The bacterial strains reported in this study are described in Table S5. Unless otherwise indicated, the strains were grown as described in the Supporting Information.
Identification, Evaluation, and Chemical Synthesis of VCC234718
A total of 15344 small molecules (Vichem Chemie) were screened in 384-well plate format for their antitubercular activity at 20 μM (7 days exposure) against replicating H37Rv and nonreplicating streptomycin-starved 18b as described previously.17 VCC234718 was identified as a hit (percentage inhibition of resazurin reduction of >80% at 20 μM) and re-evaluated to determine its MIC by resazurin microtiter assay (REMA)17 with serial dilutions of the compounds of interest (20–0.04 μM). In parallel to MIC evaluation, VCC234718 was evaluated for its concentration-dependent cytotoxicity (TC50) against four cell lines (HepG2, Huh7, A549, and THP-1) by REMA as described previously.62 The chemical synthesis of VCC234718 is described in the Supporting Information.
Early ADMET
Early ADMET studies were performed as described in the Supporting Information.
Drug Susceptibility Testing
Unless indicated otherwise, MIC testing was carried out by broth microdilution using the AlamarBlue (AB, Invitrogen) assay. For pairwise combination (checkerboard) assays, a two-dimensional array of serial dilutions of two test compounds was prepared in 96-well plates, as previously described.36,37 Time–kill kinetic analyses were carried out as previously described.37 The intracellular activity of VCC234718 was assessed by anticytolytic screening in Mtb-infected MRC-5 fibroblasts or activated THP-1 macrophages, as described by Rybniker et al.19
Isolation and Whole-Genome Sequencing of VCC234718-Resistant Mutants of Mtb
Mutants of Mtb resistant to VCC234718 were isolated as described in the Supporting Information. Genomic DNA from the wild type H37Rv and three VCC234718-resistant mutants was sequenced by Illumina HiSeq, and reads were processed by HTSstation63 to identify single-nucleotide polymorphisms present in the mutants. The mutation found in guaB2 (Rv3411c) in the mutant SRMV2.6 was confirmed by Sanger sequencing following PCR amplification using the oligonucleotides listed in Table S6 and Phusion High-Fidelity DNA Polymerase (Thermo Scientific). The PCR products were purified by using the QIAquick Gel Extraction Kit (Qiagen), and Sanger sequencing was performed by using the BigDye Terminator v3.1 Cycle Sequencing kit and the sequencing primers listed in Table S6.
Construction of Mutant Strains of Mtb with Altered Expression of GuaB2
Mtb strains carrying wild type guaB2, guaB2-guaB3, or guaB2Y487C under the control of the native promoter were constructed using the vectors pTTguaB2, pTTguaB2-guaB3, and pTTguaB2Y487C (Table S5), which were constructed as outlined in the Supporting Information. The conditional mutants, guaB2-guaB3 Tet-OFF and guaB2-guaB3 Tet-ON, and in which guaB2-guaB3 was placed under the control of a Tet-regulated promoter were constructed as described in the Supporting Information. The complemented conditional mutant, guaB2-guaB3 Tet-OFF attB::guaB3, which carries an integrated copy of guaB3 under control of the operon promoter, was constructed by transforming guaB2-guaB3 Tet-OFF with the vector, pTTguaB3 (Table S5).
Western Blot Analysis
Western blotting was performed as described in the Supporting Information according to the protocol reported by Singh et al.37
Gene Expression Analysis by ddPCR
Quantitative gene expression analysis was carried by means of ddPCR using a previously reported method,37 as described in the Supporting Information.
Intracellular Growth and Survival of Mtb
Intracellular growth and survival of wild type and conditional mutant strains of Mtb were assessed using a modification of the method of Manganelli et al.64 as described in the Supporting Information.
Mouse Infection
Female C57BL/6 mice (Charles River Laboratories) were infected by low-dose aerosol with the guaB2 conditional knockdown strain. Mice were fed doxycycline-containing food (2000 ppm, Harlan) starting from 5 days before infection. Control groups were fed regular diet, equal in composition to the doxycycline-containing food except for the antibiotic. Four mice per group and time point were sacrificed at day 0, week 2, week 4, and week 8 post-infection. Lung homogenates were plated on 7H10 plates supplemented with kanamycin (25 μg/mL), hygromycin (50 μg/mL), gentamycin (2.5 μg/mL), and cycloheximide (10 μg/mL). Lung homogenates were plated in parallel on 7H10 plates with and without guanine (250 μM). Experimental procedures involving animals were approved by the Swiss Cantonal and Federal Authorities (authorization no. 2658).
Cloning, Expression, and Purification of Mtb GuaB2, Mtb GuaB2Y487C, and Mth GuaB2
Recombinant forms of Mtb GuaB2, Mtb GuaB2Y487C, and Mth GuaB2 were cloned, expressed, and purified as described in the Supporting Information.
Enzymatic Analyses
The enzymatic characterization and steady-state kinetics of Mtb GuaB2 and the kinetics of inhibition of Mtb GuaB2, Mtb GuaB2Y487C, and human IMPDH by VCC234718 were performed as described in the Supporting Information.
Crystallization, Compound Soaking, and X-ray Data Collection
The Mth GuaB2 ΔCBS protein crystallized in 2 μL hanging drops in 1:1 ratio with 100 mM sodium acetate, pH 5.5, 200 mM calcium chloride, and 8–14% isopropanol. Crystals appeared after 24 h and grew to full size within a week. Crystals were soaked overnight in drops of well solution + 5 mM IMP and 10 mM VCC234718 solubilized in 100% DMSO. The crystals were cryoprotected, passed through drops containing well solution + 25% glycerol, and subsequently flash-frozen in liquid nitrogen. Data were collected from the crystals at Diamond Light Source beamline I24. The VCC234718-soaked crystals diffracted to 1.6 Å resolution.
Structure Solution, Ligand Fitting, and Refinement
Data were processed using XDS65 and Pointless (CCP4). To solve the structure, molecular replacement was performed with Phenix Phaser66 using a previously solved IMP-bound Mth GuaB2 ΔCBS structure as a probe (unpublished data). Refinement was performed using Phenix.refine and manually in Coot.67 IMP and VCC234718 were sequentially fitted into the density using the LigandFit function of Phenix, and the structures were manually refined further using Coot. Final R/Rfree scores obtained were 0.17/0.19, respectively. Information regarding the crystallographic statistics can be found in Table S4. The coordinates for the Mth GuaB2 ΔCBS structure in complex with IMP and VCC234718 have been deposited in the Protein Data Bank under the accession no. 5J5R.
Time-Lapse Microscopy
Time-lapse microscopy of the guaB2-guaB3 Tet-OFF attB::guaB3 strain was carried out using previously described methods.43 Briefly, the cells were cultured in a flask in 7H9 medium supplemented with 25 μg/mL kanamycin, 50 μg/mL hygromycin, and 2.5 μg/mL gentamycin at 37 °C. When the culture reached midexponential phase, the bacteria were concentrated 10-fold and filtered through a 5 μm filter to get rid of clumps. The single-cell suspension of cells was then placed in a custom-made microfluidic chip, which was then assembled on the stage of a Deltavision PersonalDV inverted microscope (GE Healthcare). The entire stage and objectives were enclosed in an incubator chamber maintained at 37 °C. Fresh 7H9 medium (supplemented with either 100 ng/mL ATc or 125 μM guanine) was circulated through the device at a flow rate of 15 μL/min. Imaging was carried out every 15 min using a 100× oil-immersion phase objective (Olympus Plan Semi Apochromat, 1.3 NA) and acquired using a CoolSnap HQ2 camera. Images were acquired on phase-contrast through the entire duration of the experiment. Typically about 50–70 independent points were imaged in each experiment. As an end point assay cells were stained with propidium iodide (1 μg/mL) and imaged on the red channel (excitation filter 555/28, emission filter 617/73) for the last 24 h of the experiment. Images were processed and movies assembled using Softworx 4.1 (Applied Precision, GE HealthCare) or ImageJ v 1.47n.68,69
Acknowledgments
This work was funded by the European Community’s Seventh Framework Programme (Grant 260872); the South African Medical Research Council (to V.M.); the National Research Foundation of South Africa (to V.M.); a Senior International Research Scholar’s grant from the HHMI (Grant 55007649 to V.M.); the Bill and Melinda Gates Foundation (to A.P. and T.L.B.), a Newton Fund RCUK-CONFAP grant awarded by The Medical Research Council (MRC) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) (Grant MR/M026302/1 to T.L.B. and D.B.A.), a NHMRC CJ Martin Fellowship (Grant APP1072476 to D.B.A.); the Swiss National Science Foundation (31003A-162641) and the Fondation Beytout (to S.T.C.); the University of Cambridge and The Wellcome Trust for facilities and support (to T.L.B.).
Glossary
Abbreviations Used
- ATc
anhydrotetracycline
- CFU
colony-forming units
- ddPCR
droplet digital PCR
- GMP
guanosine monophosphate
- IMP
inosine monophosphate
- IMPDH
inosine monophosphate dehydrogenase
- Mtb
Mycobacterium tuberculosis
- MDR
multidrug resistant
- MIC
minimal inhibitory concentration
- NGMA
nongrowing but metabolically active
- TB
tuberculosis
- Tet
tetracycline
- XMP
xanthosine monophosphate
- XDR
extensively drug resistant
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.6b00102.
Author Contributions
V.S. constructed and characterized the mutant strains and performed the drug susceptibility testing, gene expression analysis, Western blotting, and in vitro and ex vivo genetic validation studies; S.D. expressed and purified Mtb GuaB2 and performed the enzymology; A.P. and D.N.A. expressed and purified Mth GuaB2 and performed the X-ray crystallography; C.S., A.L., A.V., and R.S. performed the mouse infection experiments and MIC assays; R.C.H. performed the library screen and genome sequencing of the mutants; N.D. carried out the time-lapse microscopy experiments; G.K. and J.P. synthesized VCC234718; H.V., G.M., and S.D. performed the early ADMET studies; J.B. produced the antibodies; T.L.B., S.T.C., D.F.W., J.D.M., and M.R. contributed to the design of the study, interpretation of the data, and editing of the manuscript; and V.M. coordinated the microbiology and genetics work. V.S. and V.M. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. Global Tuberculosis Report 2015; 2015.
- Parida S. K.; Axelsson-Robertson R.; Rao M. V.; Singh N.; Master I.; Lutckii A.; Keshavjee S.; Andersson J.; Zumla A.; Maeurer M. (2015) Totally drug-resistant tuberculosis and adjunct therapies. J. Intern. Med. 277 (4), 388–405. 10.1111/joim.12264. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Ioerger T. R.; O’Malley T.; Liao R.; Guinn K. M.; Hickey M. J.; Mohaideen N.; Murphy K. C.; Boshoff H. I.; Mizrahi V.; Rubin E. J.; Sassetti C. M.; Barry C. E. 3rd; Sherman D. R.; Parish T.; Sacchettini J. C. (2013) Identification of new drug targets and resistance mechanisms in Mycobacterium tuberculosis. PLoS One 8 (9), e75245. 10.1371/journal.pone.0075245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mdluli K.; Kaneko T.; Upton A. (2015) The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harbor Perspect. Med. 5 (6), a021154. 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W.; Neefs J. M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307 (5707), 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. (2014) Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 6 (3), 372–383. 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterji M.; Shandil R.; Manjunatha M. R.; Solapure S.; Ramachandran V.; Kumar N.; Saralaya R.; Panduga V.; Reddy J.; Prabhakar K. R.; Sharma S.; Sadler C.; Cooper C. B.; Mdluli K.; Iyer P. S.; Narayanan S.; Shirude P. S. (2014) 1,4-Azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 58 (9), 5325–5331. 10.1128/AAC.03233-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S. P.; Lakshminarayana S. B.; Kondreddi R. R.; Herve M.; Camacho L. R.; Bifani P.; Kalapala S. K.; Jiricek J.; Ma N. L.; Tan B. H.; Ng S. H.; Nanjundappa M.; Ravindran S.; Seah P. G.; Thayalan P.; Lim S. H.; Lee B. H.; Goh A.; Barnes W. S.; Chen Z.; Gagaring K.; Chatterjee A. K.; Pethe K.; Kuhen K.; Walker J.; Feng G.; Babu S.; Zhang L.; Blasco F.; Beer D.; Weaver M.; Dartois V.; Glynne R.; Dick T.; Smith P. W.; Diagana T. T.; Manjunatha U. H. (2013) Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 5 (214), 214ra168. 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- Grzegorzewicz A. E.; Pham H.; Gundi V. A.; Scherman M. S.; North E. J.; Hess T.; Jones V.; Gruppo V.; Born S. E.; Kordulakova J.; Chavadi S. S.; Morisseau C.; Lenaerts A. J.; Lee R. E.; McNeil M. R.; Jackson M. (2012) Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 8 (4), 334–341. 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Upadhyay A.; Fontes F. L.; North E. J.; Wang Y.; Crans D. C.; Grzegorzewicz A. E.; Jones V.; Franzblau S. G.; Lee R. E.; Crick D. C.; Jackson M. (2014) Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58 (11), 6413–6423. 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S. A.; Nam J.; Kang H.; Kwon H.; Oh C. T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Oh S.; Oh T.; Nehrbass U.; Han S. J.; No Z.; Lee J.; Brodin P.; Cho S. N.; Nam K. (2013) Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19 (9), 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Arora K.; Ochoa-Montano B.; Tsang P. S.; Blundell T. L.; Dawes S. S.; Mizrahi V.; Bayliss T.; Mackenzie C. J.; Cleghorn L. A.; Ray P. C.; Wyatt P. G.; Uh E.; Lee J.; Barry C. E. 3rd; Boshoff H. I. (2014) Respiratory flexibility in response to inhibition of cytochrome C oxidase in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58 (11), 6962–6965. 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling A.; Lukat P.; Almeida D. V.; Bauer A.; Fontaine E.; Sordello S.; Zaburannyi N.; Herrmann J.; Wenzel S. C.; Konig C.; Ammerman N. C.; Barrio M. B.; Borchers K.; Bordon-Pallier F.; Bronstrup M.; Courtemanche G.; Gerlitz M.; Geslin M.; Hammann P.; Heinz D. W.; Hoffmann H.; Klieber S.; Kohlmann M.; Kurz M.; Lair C.; Matter H.; Nuermberger E.; Tyagi S.; Fraisse L.; Grosset J. H.; Lagrange S.; Muller R. (2015) Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348 (6239), 1106–1112. 10.1126/science.aaa4690. [DOI] [PubMed] [Google Scholar]
- Stanley S. A.; Kawate T.; Iwase N.; Shimizu M.; Clatworthy A. E.; Kazyanskaya E.; Sacchettini J. C.; Ioerger T. R.; Siddiqi N. A.; Minami S.; Aquadro J. A.; Grant S. S.; Rubin E. J.; Hung D. T. (2013) Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. U. S. A. 110 (28), 11565–11570. 10.1073/pnas.1302114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnet S.; Hartkoorn R. C.; Szekely R.; Pato J.; Triccas J. A.; Schneider P.; Szantai-Kis C.; Orfi L.; Chambon M.; Banfi D.; Bueno M.; Turcatti G.; Keri G.; Cole S. T. (2010) Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis (Oxford, U. K.) 90 (6), 354–360. 10.1016/j.tube.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Sala C.; Dhar N.; Hartkoorn R. C.; Zhang M.; Ha Y. H.; Schneider P.; Cole S. T. (2010) Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 54 (10), 4150–4158. 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vocat A.; Hartkoorn R. C.; Lechartier B.; Zhang M.; Dhar N.; Cole S. T.; Sala C. (2015) Bioluminescence for assessing drug potency against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59 (7), 4012–4019. 10.1128/AAC.00528-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybniker J.; Chen J. M.; Sala C.; Hartkoorn R. C.; Vocat A.; Benjak A.; Boy-Rottger S.; Zhang M.; Szekely R.; Greff Z.; Orfi L.; Szabadkai I.; Pato J.; Keri G.; Cole S. T. (2014) Anticytolytic screen identifies inhibitors of mycobacterial virulence protein secretion. Cell Host Microbe 16 (4), 538–548. 10.1016/j.chom.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Hubatsch I.; Ragnarsson E. G. E.; Artursson P. (2007) Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2 (9), 2111–2119. 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- Jones K. A., Garbati N., Zhang H., and Large C. H. (2009) Automated patch clamping using the QPatch. In High Throughput Screening: Methods and Protocols, 2nd ed. (Janzen P. W., and Bernasconi P., Eds.) pp 209–223, Humana Press, Totowa, NJ, USA. [DOI] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Krogh A.; McLean J.; Moule S.; Murphy L.; Oliver K.; Osborne J.; Quail M. A.; Rajandream M. A.; Rogers J.; Rutter S.; Seeger K.; Skelton J.; Squares R.; Squares S.; Sulston J. E.; Taylor K.; Whitehead S.; Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 (6685), 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48 (1), 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Usha V.; Gurcha S. S.; Lovering A. L.; Lloyd A. J.; Papaemmanouil A.; Reynolds R. C.; Besra G. S. (2011) Identification of novel diphenyl urea inhibitors of Mt-GuaB2 active against Mycobacterium tuberculosis. Microbiology 157, 290–299. 10.1099/mic.0.042549-0. [DOI] [PubMed] [Google Scholar]
- Cox J. S.; Chen B.; McNeil M.; Jacobs W. R. Jr. (1999) Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402 (6757), 79–83. 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- Camacho L. R.; Ensergueix D.; Perez E.; Gicquel B.; Guilhot C. (1999) Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol. Microbiol. 34 (2), 257–267. 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- Ioerger T. R.; Feng Y.; Ganesula K.; Chen X.; Dobos K. M.; Fortune S.; Jacobs W. R. Jr.; Mizrahi V.; Parish T.; Rubin E.; Sassetti C.; Sacchettini J. C. (2010) Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192 (14), 3645–3653. 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milano A.; Pasca M. R.; Provvedi R.; Lucarelli A. P.; Manina G.; Ribeiro A. L.; Manganelli R.; Riccardi G. (2009) Azole resistance in Mycobacterium tuberculosis is mediated by the MmpS5-MmpL5 efflux system. Tuberculosis (Oxford, U. K.) 89 (1), 84–90. 10.1016/j.tube.2008.08.003. [DOI] [PubMed] [Google Scholar]
- Hartkoorn R. C.; Uplekar S.; Cole S. T. (2014) Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58 (5), 2979–2981. 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K.; Villellas C.; Coeck N.; Thys K.; Gevers T.; Vranckx L.; Lounis N.; de Jong B. C.; Koul A. (2014) Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9 (7), e102135. 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7 (9), e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L. (2009) IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev. 109 (7), 2903–2928. 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowska-Grzyska M.; Kim Y.; Maltseva N.; Osipiuk J.; Gu M.; Zhang M.; Mandapati K.; Gollapalli D. R.; Gorla S. K.; Hedstrom L.; Joachimiak A. (2015) A novel cofactor-binding mode in bacterial IMP dehydrogenases explains inhibitor selectivity. J. Biol. Chem. 290 (9), 5893–5911. 10.1074/jbc.M114.619767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowska-Grzyska M.; Kim Y.; Gorla S. K.; Wei Y.; Mandapati K.; Zhang M.; Maltseva N.; Modi G.; Boshoff H. I.; Gu M.; Aldrich C.; Cuny G. D.; Hedstrom L.; Joachimiak A. (2015) Mycobacterium tuberculosis IMPDH in complexes with substrates, products and antitubercular compounds. PLoS One 10 (10), e0138976. 10.1371/journal.pone.0138976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Kuzmic P.; Yu R.; Modi G.; Hedstrom L. (2016) Inhibition of IMPDH from Bacillus anthracis: mechanism revealed by pre-steady state kinetics. Biochemistry 10.1021/acs.biochem.6b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams G. L.; Kumar A.; Savvi S.; Hung A. W.; Wen S.; Abell C.; Barry C. E. 3rd; Sherman D. R.; Boshoff H. I.; Mizrahi V. (2012) Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 19 (7), 844–854. 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Brecik M.; Mukherjee R.; Evans J. C.; Svetlikova Z.; Blasko J.; Surade S.; Blackburn J.; Warner D. F.; Mikusova K.; Mizrahi V. (2015) The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem. Biol. 22 (1), 63–75. 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Ehrt S.; Guo X. V.; Hickey C. M.; Ryou M.; Monteleone M.; Riley L. W.; Schnappinger D. (2005) Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33 (2), e21. 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner D. F., Evans J. C., and Mizrahi V.. Nucleotide metabolism and DNA replication. Microbiol. Spectrum 2014, 2 ( (5), ). 10.1128/microbiolspec.MGM2-0001-2013 [DOI] [PubMed] [Google Scholar]
- Ducati R. G.; Breda A.; Basso L. A.; Santos D. S. (2011) Purine salvage pathway in Mycobacterium tuberculosis. Curr. Med. Chem. 18 (9), 1258–1275. 10.2174/092986711795029627. [DOI] [PubMed] [Google Scholar]
- Biazus G.; Schneider C. Z.; Palma M. S.; Basso L. A.; Santos D. S. (2009) Hypoxanthine-guanine phosphoribosyltransferase from Mycobacterium tuberculosis H37Rv: cloning, expression, and biochemical characterization. Protein Expression Purif. 66 (2), 185–190. 10.1016/j.pep.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Manina G.; Dhar N.; McKinney J. D. (2015) Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17 (1), 32–46. 10.1016/j.chom.2014.11.016. [DOI] [PubMed] [Google Scholar]
- Dhar N.; Manina G. (2015) Single-cell analysis of mycobacteria using microfluidics and time-lapse microscopy. Methods Mol. Biol. 1285, 241–256. 10.1007/978-1-4939-2450-9_14. [DOI] [PubMed] [Google Scholar]
- Wakamoto Y.; Dhar N.; Chait R.; Schneider K.; Signorino-Gelo F.; Leibler S.; McKinney J. D. (2013) Dynamic persistence of antibiotic-stressed mycobacteria. Science 339 (6115), 91–95. 10.1126/science.1229858. [DOI] [PubMed] [Google Scholar]
- Boldrin F.; Ventura M.; Degiacomi G.; Ravishankar S.; Sala C.; Svetlikova Z.; Ambady A.; Dhar N.; Kordulakova J.; Zhang M.; Serafini A.; Vishwas K. G.; Kolly G. S.; Kumar N.; Palu G.; Guerin M. E.; Mikusova K.; Cole S. T.; Manganelli R. (2014) The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J. Bacteriol. 196 (19), 3441–3451. 10.1128/JB.01346-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal A.; Trujillo C.; Ehrt S.; Schnappinger D. (2010) Simultaneous analysis of multiple Mycobacterium tuberculosis knockdown mutants in vitro and in vivo. PLoS One 5 (12), e15667. 10.1371/journal.pone.0015667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woong Park S.; Klotzsche M.; Wilson D. J.; Boshoff H. I.; Eoh H.; Manjunatha U.; Blumenthal A.; Rhee K.; Barry C. E. 3rd; Aldrich C. C.; Ehrt S.; Schnappinger D. (2011) Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 7 (9), e1002264. 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L.; Liechti G.; Goldberg J. B.; Gollapalli D. R. (2011) The antibiotic potential of prokaryotic IMP dehydrogenase inhibitors. Curr. Med. Chem. 18 (13), 1909–1918. 10.2174/092986711795590129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollapalli D. R.; Macpherson I. S.; Liechti G.; Gorla S. K.; Goldberg J. B.; Hedstrom L. (2010) Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem. Biol. 17 (10), 1084–1091. 10.1016/j.chembiol.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Makowska-Grzyska M.; Gorla S. K.; Gollapalli D. R.; Cuny G. D.; Joachimiak A.; Hedstrom L. (2015) Structure of Cryptosporidium IMP dehydrogenase bound to an inhibitor with in vivo antiparasitic activity. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 71 (5), 531–538. 10.1107/S2053230X15000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson I. S.; Kirubakaran S.; Gorla S. K.; Riera T. V.; D’Aquino J. A.; Zhang M.; Cuny G. D.; Hedstrom L. (2010) The structural basis of Cryptosporidium-specific IMP dehydrogenase inhibitor selectivity. J. Am. Chem. Soc. 132 (4), 1230–1231. 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usha V.; Hobrath J. V.; Gurcha S. S.; Reynolds R. C.; Besra G. S. (2012) Identification of novel Mt-Guab2 inhibitor series active against M. tuberculosis. PLoS One 7 (3), e33886. 10.1371/journal.pone.0033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E.; Blundell T. L.; Ascher D. B. (2016) mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci. Rep. 6, 29575. 10.1038/srep29575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E.; Blundell T. L.; Ascher D. B. (2015) Platinum: a database of experimentally measured effects of mutations on structurally defined protein-ligand complexes. Nucleic Acids Res. 43 (Database issue), D387–D391. 10.1093/nar/gku966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E.; Ascher D. B.; Blundell T. L. (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 42 (Web Server issue), W314–W319. 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E.; Ascher D. B.; Blundell T. L. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30 (3), 335–342. 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham C. M.; Srinivasan N.; Blundell T. L. (1997) Prediction of the stability of protein mutants based on structural environment-dependent amino acid substitution and propensity tables. Protein Eng., Des. Sel. 10 (1), 7–21. 10.1093/protein/10.1.7. [DOI] [PubMed] [Google Scholar]
- Pires D. E.; Chen J.; Blundell T. L.; Ascher D. B. (2016) In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci. Rep. 6, 19848. 10.1038/srep19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso L. A.; Santos D. S.; Shi W.; Furneaux R. H.; Tyler P. C.; Schramm V. L.; Blanchard J. S. (2001) Purine nucleoside phosphorylase from Mycobacterium tuberculosis. Analysis of inhibition by a transition-state analogue and dissection by parts. Biochemistry 40 (28), 8196–8203. 10.1021/bi010584x. [DOI] [PubMed] [Google Scholar]
- Wink P. L.; Sanchez Quitian Z. A.; Rosado L. A.; Rodrigues Vda S. Jr.; Petersen G. O.; Lorenzini D. M.; Lipinski-Paes T.; Saraiva Macedo Timmers L. F.; de Souza O. N.; Basso L. A.; Santos D. S. (2013) Biochemical characterization of recombinant nucleoside hydrolase from Mycobacterium tuberculosis H37Rv. Arch. Biochem. Biophys. 538 (2), 80–94. 10.1016/j.abb.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Lenaerts A.; Barry C. E. 3rd; Dartois V. (2015) Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev. 264 (1), 288–307. 10.1111/imr.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkoorn R. C.; Sala C.; Neres J.; Pojer F.; Magnet S.; Mukherjee R.; Uplekar S.; Boy-Rottger S.; Altmann K. H.; Cole S. T. (2012) Towards a new tuberculosis drug: pyridomycin – nature’s isoniazid. EMBO Mol. Med. 4 (10), 1032–1042. 10.1002/emmm.201201689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David F. P.; Delafontaine J.; Carat S.; Ross F. J.; Lefebvre G.; Jarosz Y.; Sinclair L.; Noordermeer D.; Rougemont J.; Leleu M. (2014) HTSstation: a web application and open-access libraries for high-throughput sequencing data analysis. PLoS One 9 (1), e85879. 10.1371/journal.pone.0085879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganelli R.; Voskuil M. I.; Schoolnik G. K.; Smith I. (2001) The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol. Microbiol. 41 (2), 423–437. 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9 (7), 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband W. S. ImageJ, http://imagej.nih.gov/ij/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.