

Abstract
Plasmodium falciparum (Pf) prolyl-tRNA synthetase (ProRS) is one of the few chemical-genetically validated drug targets for malaria, yet highly selective inhibitors have not been described. In this paper, approximately 40,000 compounds were screened to identify compounds that selectively inhibit PfProRS enzyme activity versus Homo sapiens (Hs) ProRS. X-ray crystallography structures were solved for apo, as well as substrate- and inhibitor-bound forms of PfProRS. We identified two new inhibitors of PfProRS that bind outside the active site. These two allosteric inhibitors showed >100 times specificity for PfProRS compared to HsProRS, demonstrating this class of compounds could overcome the toxicity related to HsProRS inhibition by halofuginone and its analogues. Initial medicinal chemistry was performed on one of the two compounds, guided by the cocrystallography of the compound with PfProRS, and the results can instruct future medicinal chemistry work to optimize these promising new leads for drug development against malaria.
Keywords: Plasmodium falciparum, drug screening, prolyl-tRNA-synthetase, high-throughput screening, halofuginone, antimalarials
Malaria is a devastating infection that affects hundreds of millions of people worldwide and is concentrated in resource-poor tropical and subtropical regions.1 Resistance has been documented against every antiparasitic drug available,2,3 highlighting the need for new, cost-effective antimalarials that target the most deadly causative agent, Plasmodium falciparum (Pf).
For 2000 years, the roots of blue evergreen hydrangea have been used to treat malaria symptoms, but it was not until 1950 that the bioactive constituent responsible for fever and pain relief was derived from the roots and identified as febrifugine (Figure 1).4 However, observed cytotoxicity, especially targeting the liver, precludes the use of febrifugine as a viable drug treatment option.5,6 During the 1960s efforts by the U.S. Army (Walter Reed Army Hospital) to develop a less toxic alternative resulted in the synthesis of halofuginone (Figure 1), a halogenated form of febrifugine. Although intolerable side effects of halofuginone to humans remained, it was approved for use as an anticoccidiostat in poultry and cattle.7 It was not until 2012 that febrifugine and halofuginone were shown to target P. falciparum prolyl-tRNA-synthetase (PfProRS).8 Thus, PfProRS became one of the few chemical-genetically validated targets, but difficulty in deriving halofuginone for specificity of PfProRS inhibition versus Homo sapiens (Hs)ProRS remained.
Figure 1.

Structures of febrifugine, halofuginone, and N-Boc-halofuginone.
Here we report a high-throughput screen against PfProRS and present evidence of the first highly selective compounds that inhibit PfProRS but not HsProRS. X-ray crystallographic structures demonstrate that these inhibitors bind outside the active site in an area of the enzyme that has not previously displayed a binding pocket. The compounds act as allosteric inhibitors through induced fit into a novel pocket adjacent to the active site.
Results and Discussion
Recombinant Expression of PfProRS
To characterize PfProRS in vitro, the PfProRS open reading frame (ORF) (PF3D7_1213800) was cloned from cDNA isolated from P. falciparum 3D7 strain. After analysis of predicted protein structure and comparison to related structures from the ProRS domain of human EPRS (PDB 4HVC) and the thermophilic bacteria Methanocaldococcus jannaschii ProRS (PDB 1NJ8), we designed four N-terminal truncations of the ORF, which we cloned into the pBG1861 vector along with the full-length ORF. We tested protein expression levels of the five constructs (B1, AA1–746 (full length); B2, AA7–746 (minus 6 amino acids, or 99% of the ORF); B3, AA224–746 (70% ORF); B4, AA249–746 (67% ORF); B5, AA256–746 (66% ORF), Figure S1). The full-length construct (B1) and three truncations (B2, B4, and B5) had soluble protein expression levels sufficient for purification. Recombinant proteins were purified and analyzed for enzyme activity and set up in crystallization experiments.
Determination of ProRS Enzymatic Activity
To assess whether the purified protein had activity, we first determined the relative levels of aminoacylation activity for each of the four soluble recombinant PfProRS proteins. Activity was measured in an aminoacylation assay wherein radioactive proline is linked to bulk tRNA obtained from Saccharomyces cerevisiae. We found that all soluble recombinant proteins except the full-length construct (B1) had enzymatic activity. Figure 2 demonstrates the enzymatic ProRS activity for recombinant proteins B2 and B4. Figure 2 shows the reaction dependence on S. cerevisiae tRNA, and in experiments not shown, enzymatic activity was also shown to be dependent on the presence of proline and ATP. Recombinant protein B2 had only a six amino acid N-terminal deletion compared to the full-length construct, so the B2 purified protein was used for all additional assays, except crystallization and structure determination, for which the B4 purified protein was used (Figure S1).
Figure 2.
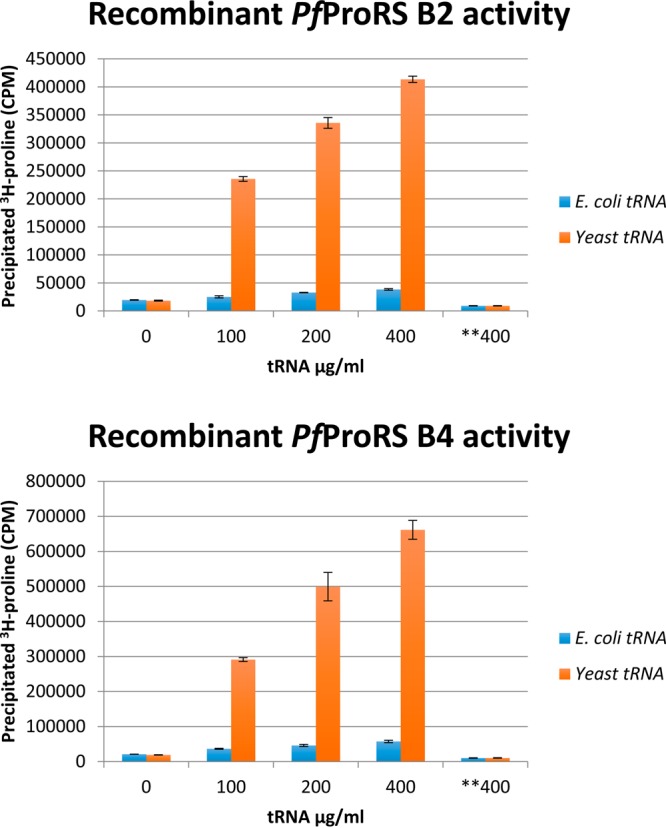
Confirmation of PfProRS activity by aminoacylation assay. Recombinant PfProRS was tested for activity with increasing concentrations of tRNA using the aminoacylation assay and 3H-proline. Activity was dependent on bulk tRNA purified from S. cerevisiae; bulk Escherichia coli tRNA as a substrate did not give any signal. Controls of no tRNA and no protein gave similarly low signal. All soluble recombinant proteins, except for the full-length B1, gave equivalent enzymatic activity (data for B2 and B5 are not shown). Samples were tested in triplicate, error bars indicate standard deviation for each condition. Designation **400 is no enzyme control.
Inhibition of PfProRS Activity and Binding to PfProRS by Halofuginone
Halofuginone is a known antimalarial and a potent inhibitor of PfProRS.8,9 Febrifugine and derivatives, such as halofuginone, have been shown in vivo and in vitro to target both HsProRS and PfProRS.8,9 Halofuginone inhibition of PfProRS was tested by an aminoacylation assay and shown to have an IC50 of 11 nM (Figure S2). N-Boc-halofuginone (Figure 1) was synthesized as a control and displayed <20% inhibition of aminoacylation at the highest concentrations tested (see also Figure S2).
Thermal melt analysis with recombinant protein B2 showed a 6° shift in stability with either the cognate amino acid, proline, or the known inhibitor, halofuginone (data not shown). Increased stability was observed for three of the four soluble recombinant PfProRSs. The full-length recombinant protein B1 showed no increase in stability from halofuginone. This was not surprising because of the disordered structure suggested from the poor melting profile of B1. This disordered folding may be due to the initial stretch of seven asparagine residues at the N-terminus. Recombinant B2 appeared to be properly folded and lacks only the initial seven asparagines, whereas the full-length B1 did not appear to be folded and lacked enzyme activity.
Development of a ProRS Luminescence Assay
As radiolabeled assays are not appropriate for high-throughput screening (HTS), we developed and optimized an ATP depletion luminescence assay. Reactions were incubated for 2 h at 37 °C in the presence of 60 μM proline, 3 μM ATP, and 400 μg/mL yeast tRNA. Under these conditions approximately 85% of the ATP was consumed in the positive control reactions and consumption of ATP continued for another 2 h. The level of ATP consumption was quantified by the addition of luciferase (KinaseGlo, Promega, Madison, WI, USA), which gave a luminescent signal proportional to the remaining ATP. Luminescence is inversely related to the completion of the enzymatic reaction. Inhibitors of luciferase would present as false negatives and are not false positives of this screen, because luciferase inhibitors give rise to a low luminescence signal, whereas inhibitors of ProRS yield a high luminescence signal.
Screening Compounds for Inhibition of ProRS
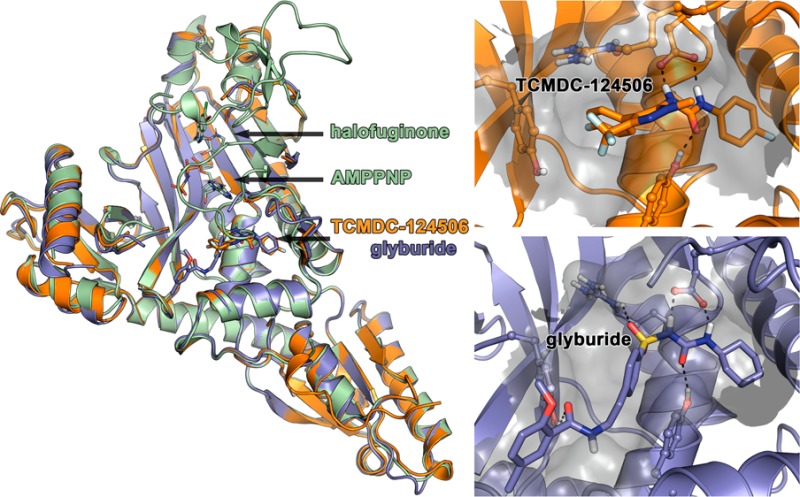
We screened 40,000 compounds in total from five compound libraries (the SPECTRUM Microsource tested at 10 μM, the GlaxoSmithKline Tres Cantos Antimalarial Set tested at 7.5 μM, the DDU small diversity set tested at 30 μM, the Malaria Box tested at 10 μM, and the St. Jude Library tested at 15 μM) for inhibition of ProRS using our ATP depletion assay. Hits were defined as having inhibition >80% or 2 standard deviation units. Statistical analysis was performed using HutchBASE.10 Any hit above this threshold was confirmed by retesting with serial 3-fold dilutions of the compound to establish the IC50. We identified 33 hits for which the estimated IC50 was <100 μM. For these hits, we repurchased compounds and confirmed the IC50 with a complete dose–response curve. Ten hits were reconfirmed (Table S2). Finally, we counter-tested against purified HsProRS, using the luciferase ATP depletion assay (Table S2). Two compounds, glyburide and TCMDC-124506 (Figure 3), stood out as having inhibitory activity (IC50) against PfProRS at 34 μM and 74 μM, respectively, but <40% inhibitory activity for HsProRS up to the limit of solubility (>1 mM). This is in contrast to halofuginone, which showed IC50 values for PfProRS (IC50 = 275 nM) and HsProRS (IC50 = 2 μM) within 8-fold of one another (Figure 4).
Figure 3.

Structures of PfProRS inhibitors glyburide and TCMDC-124506 found by HTS.
Figure 4.

PfProRS inhibition dose response: inhibition of PfProRS enzyme activity at various concentrations of glyburide (blue), halofuginone (green), and TCMDC-124506 (orange). Lines are fitted to Hill equation with a slope = 1, and the calculated IC50 values for the compounds in this assay are PfProRS for 34 μM glyburide, 0.28 μM halofuginone, and 73 μM TCMDC-124506 and HsProRS for >700 μM glyburide, 2.13 μM halofuginone, and >700 μM TCMDC-124506. Compounds were counter-screened against HsProRS to determine specificity. Enzyme activity was determined by luciferase ATP depletion assay (75 nM ProRS enzyme, 60 μM proline, 6 μM ATP, and 400 μg/mL yeast tRNA were incubated for 2 h at 37 °C). Inhibition values shown on the graph are the average of three data points.
Structure Determination
Three-dimensional structural modeling provides insight into areas of the ProRS enzyme that can be exploited for drug development. HsProRS structures were recently solved for both the apo form and the form bound to halofuginone.11,12 The structure of cytoplasmic PfProRS bound to halofuginone has been solved both by our team and independently by another group.13 The crystal structure indicates that the B4 recombinant PfProRS protein forms a dimer, which is further supported by the sieving properties observed through a size exclusion column. There is a disordered loop that folds over the binding site that is poorly resolved, but may provide a promising site for optimizing specific binding of halofuginone PfProRS compared to HsProRS. Halofuginone sits in the active site pocket and can be competitively removed by the addition of excess proline. The PfProRS has striking similarity to the HsProRS orthologue. There is >66% amino acid similarity between PfProRS and HsProRS. One hundred percent identity was observed throughout the binding residues for halofuginone.13 This suggests that achieving specificity for binding of halofuginone analogues to PfProRS compared to HsProRS would be difficult.
On the basis of the results of our compound library screening and identification of their high affinity for PfProRS, glyburide and TCMDC-124506 were used in cocrystallization trials with PfProRS. The compounds are diverse in structure, but share a common urea core, bracketed on each side by a six-membered ring (head) and a longer tail (Figure 3). We determined the structure of PfProRS with glyburide to 2.45 Å and that of PfProRS with TCMDC-124506 to 1.65 Å. Surprisingly, these compounds did not bind in the known active site, neither where the nucleotide nor where halofuginone binds (Figure 5A). Instead, both compounds bound adjacent to the ATP binding site in a pocket formed by α9 (residues 261–272), α5 (residues 513–524), β-hairpin β1-β2 (residues 276–287), and α2 (residues 261–272) (Figure 5B,C). Both compounds bind in a similar fashion, within space normally occupied by the hairpinned end of the loop between residues 389 and 404. In both structures no density was observed for most of this loop. The headgroup of each compound sits in a hydrophobic pocket normally occupied by Phe399 (Figure 7B). Phe399 lies within the hairpin loop that is displaced upon compound binding. The urea group from each compound makes hydrogen bonds to Glu404 and Tyr266 (Figure 6). For both compounds, these are strong hydrogen bonds made between the compound and protein. Moving beyond the head and urea components, the compounds differ in the binding of the tail. TCMDC-124506’s tail occupies the cavity by making hydrophobic interactions with the surrounding residues (Figure 6A). Glyburide’s tail extends through the length of the same cavity to the solvent, predominately making hydrophobic interactions with surrounding residues Tyr266, Arg514, Tyr746, Ser263, and Asn470. There is also a weak hydrogen bond between Arg514 and the sulfoxide group of glyburide (Figure 6B).
Figure 5.

Structures of halofuginone and AMPPNP, TCMDC-124506, or glyburide bound to PfProRS: (A) structures of halofuginone and AMPPNP bound to PfProRS (green), TCMDC-124506 bound to PfProRS (orange), and glyburide bound to PfProRS (blue); (B) magnification of the TCMDC-124506 binding site indicating key polar interactions with the pocket surface in gray; (C) magnification of the glyburide binding site indicating key polar interactions with the pocket surface in gray.
Figure 7.

Binding of TCMDC-124506 and glyburide to PfProRS. (A) The binding of TCMDC-124506 (orange) or glyburide (blue) displaces the PfProRS TXE-Loop from a conserved conformation seen in liganded and unliganded structures of both HsProRS (4HVC (magenta), 4K86 (pink), 4K87(red), 4K88 (maroon)) and PfProRS (4OLF (green), 4TWA (yellow), and 4YDQ (teal)). (B) The TXE-Loop in AMPPNP and halofuginone bound PfProRS (green) forms interacts in the TCMDC-124506 and glyburide binding site and interacts tightly with the ATP mimic. The binding of TCMDC-124506 (orange) or glyburide (blue) in the novel binding site significantly distorts the conformation of the TXE-Loop, disrupting key interactions between ATP and PfProRS.
Figure 6.

Key PfProRS residues that complex with TCMDC-124506 (A, orange) and glyburide (B, blue).
ProRS Function Is Inhibited by ATP Binding Site Distortion
The binding of both glyburide and TCMDC-124506 into the novel binding site significantly distorts the nucleotide binding site (Figure 7). Here we compare the nucleotide binding site observed in the halofuginone/AMPPNP bound structure (green) to the glyburide (blue) and TCMDC-124506 (orange) structures. Movement of the loop between residues 389 and 404 displaces several key residues that participate in nucleotide binding: Phe405 (which forms pi–pi stacking interactions with the adenosine ring), Arg390 (which hydrogen bonds to the α and β phosphates), and Arg401 (which hydrogen bonds to the γ phosphate). Although Arg390 and Arg401 were not seen in the electron densities in either the glyburide or TCMDC-124506 structures, we can hypothesize their approximate positions on the basis of the displacement of their adjacent residues. Additionally, the movement of this loop condenses the nucleotide binding site. We hypothesize that it is the presence of the hydrophobic headgroup in glyburide and TCMDC-124506 that initially brings the compound into this adjacent binding site. It is then the urea group that draws Glu404 toward it, which ratchets the side chain around and draws the C-terminal half of this loop into the nucleotide binding site, which then causes residues important to nucleotide binding to be significantly shifted. To elaborate on these compounds for higher affinity, the head and urea portions of the compounds should be retained and the tails manipulated to further exploit this new binding site, causing distortion to the nucleotide binding site. This novel scaffold and binding site provide a new basis for structure-based drug design as these compounds are not based on halofuginone and, unlike halofuginone, are nucleotide-independent binders.
Pyrazolo Urea Analogues of TCMDC-124506
Pyrazolo urea TCMDC-124506 shows low micromolar inhibition of PfProRS in the ATP consumption assay and selectivity over HsProRS. Additionally, good cellular selectivity is also demonstrated for Pf (3D7) (EC50 = 0.6 μM) over a mammalian cell line (HsHEPG2, EC50 > 50 μM) (data not shown). In addition to its activity and selectivity, it has good physicochemical and DMPK properties. It has physicochemical properties consistent with oral drug-space, with a molecular weight below 400 (MW 396) and logP 4.4. Moreover, the experimental logD is <3 (CHI-logD 2.9), kinetic solubility is good (>250 μM), and mouse microsomal clearance is moderate (Cli = 1.6 mL/min/g).
The parasite-killing activity of TCMDC-124506 (Table 1) is greater than the compound’s activity against PfProRS. Similar pyrazolo amides and ureas have been reported to target the P-type cation-ATPase PfATP4,14 which has been proposed to extrude Na+ ions from the parasite while importing H+ ions.15 TCMDC-124506 was tested in Na+ and pH assays15,16 in P. falciparum. The compound at (when tested at 1 μM) caused the Na+ concentration in the parasite cytosol to increase in the same manner as that seen for a supramaximal concentration (50 nM) of the spiroindolone KAE609,17 which is believed to target PfATP415 (Figure S3). As seen for other compounds that have been proposed to exert their antiplasmodial effects through inhibition of PfATP4,14−16 TCMDC-124506 caused an increase in the parasite cytosolic pH and a reduction in the degree of acidification observed following inhibition of the parasite’s V-type H+ pump with concanamycin A (Figure S3). The antiplasmodial activity of TCMDC-124506 is therefore likely to be attributable to inhibition of PfATP4 rather than inhibition of PfProRS. The lack of correlation between the EC50s for Pf (3D7) growth inhibition and the IC50s for PfProRS inhibition seen for TCMDC-124506 and its analogues (Table 1) provide further evidence that the compounds do not kill parasites via inhibition of PfProRS.
Table 1. Activity of TCMDC-124506 and Analogues.
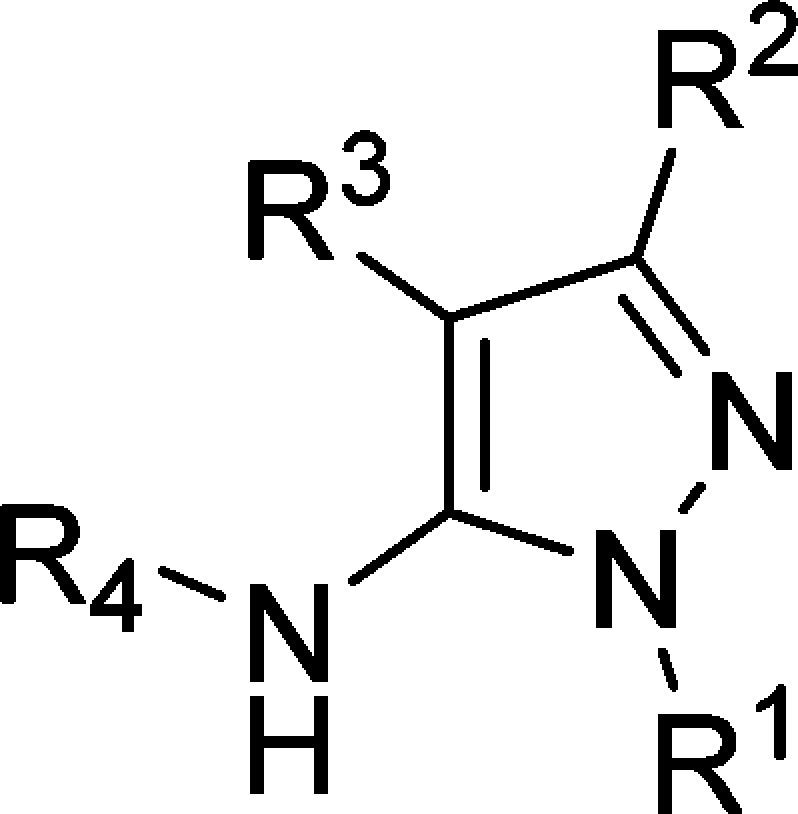

Analogues of TCMDC-124506 were designed, guided by the cocrystal structure with PfProRS, with the aim of increasing binding affinity for PfProRS and potency against Plasmodium. The compound IC50 values against PfProRS enzyme activity were determined by an ATP consumption assay slightly modified from the screening assay. The screening assay was conducted at a higher temperature and with a higher ATP concentration. Inhibition values were determined in the linear range of the assay, such that comparative IC50 values could be reliably measured. ATP and proline compete with TCMDC-124506 for binding (Supporting Information Figure S4) such that higher substrate concentrations yield higher IC50 values. The differences in the two assays explain the difference in the TCMDC-124506 IC50 values determined with the screening assay in Figure 4 versus the lower IC50 determined in Table 1, the latter of which was conducted at lower ATP concentrations. The crystal structure showed that the urea moiety (R4) forms multiple H-bonds with the protein and the aromatic substituents at R4 fit into a hydrophobic pocket (see Table 1 for definitions of R1, R2, R3, and R4) and, in fact, removal of the urea (2) led to an inactive compound. The o,p-diF-substituted phenyl urea derivative (3) retained activity and the m-F substitution on the phenyl ring (4) was tolerated, whereas any other modification around this moiety (see as representative compounds 5 and 6) proved to be detrimental for PfProRS activity. Analysis of the costructure suggested that the displacement of structural water molecules could be achieved by the addition of hydrogen bond acceptors/donors at R1 and R3. Additional expansion to solvent from the R2 (CF3) vector could be used to improve the physicochemical properties of the molecule.
Removal of the R1 pyrazole N-methyl group (7) was not tolerated, leading to an inactive compound. Addition of polar groups with H-bond donors and acceptors (8 and 9) to address Tyr278, Arg403, or Tyr285 was also detrimental for activity (Table 1).
Removal of the p-F-phenyl substituent at R3 (10) or replacement of a bromine (11) led to inactive compounds. Addition of a fluorine atom at the meta position (12) was not tolerated by the enzyme. A water molecule interacting with Glu404 and Arg514 was targeted for replacement, but failed to increase potency. The introduction of a benzylic alcohol at the ortho position of the aromatic ring (13) led to a small (4-fold) drop in potency. Extending the linker to the alcohol from one to two carbons (14) was not tolerated. Moving this hydroxyethyl substituent to a meta position (15) led to a compound with activity similar to that of the hit compound. However, a mesylated benzylic amine (16) was inactive. Finally, m-CF3-phenyl (17) substituent at R3 was also detrimental for PfProRS inhibition (Table 1).
The R2 (CF3) vector points to solvent, and a diversity of polar and basic groups were tested. Removal of the CF3 group (18) was not tolerated. When the CF3 group was replaced by an alcohol (19), a drop in potency was observed. Introduction of primary amides with one (20) and two carbon linkers (21) was tolerated. Modeling showed that amide 21 could interact favorably with Tyr746, and this compound shows levels of PfProRS inhibition similar to that of the initial hit. Nitriles directly linked to the pyrazole (22) and with one carbon (23) and two carbon linkers (24) all inhibit the enzyme in the low micromolar range. Finally, the introduction of basic substituents at R2 (25 and 26) led to the best results in terms of PfProRS inhibition. The medicinal chemistry results described above and the cocrystallography results can help guide future medicinal chemistry approaches to optimize the allosteric hits to a lead for malaria therapy (Figure 8).
Figure 8.

Structure–activity relationship of TCMDC-124506 analogues.
Summary of Findings and Significance
New drugs for malaria are urgently needed due to emerging resistance to artemisinin combination therapy.18 There is a dearth of structural information available on potential drug targets within the organism. PfProRS is a promising new target that is chemically and genetically validated by halofuginone, a synthetic derivative of the natural product febrifugine which has been known for more than 2000 years to possess antimalarial activity.9 However, halofuginone is also a potent inhibitor of HsProRS, which leads to toxicity when human patients are treated.
We have solved the three-dimensional structure of PfProRS, with and without halofuginone. The overlaid structures of PfProRS and HsProRS suggest that biologically it could be difficult to achieve specificity with halofuginone and that halofuginone represents a poor starting point, as the residues binding halofuginone in the active site are identical in PfProRS and HsProRS.
As expected, increasing proline concentrations reduce the inhibition of halofuginone, because halofuginone binds in the area that proline occupies in the active site. Interestingly, increasing ATP concentrations have the opposite effect, and halofuginone becomes more inhibitory at higher concentrations of ATP. One explanation for this observation is that higher ATP concentrations increase the consumption of proline and charges the tRNA, leaving the proline binding pocket accessible for halofuginone to bind, subsequently replacing proline. Another possibility is that ATP binding of PfProRS induces a change in the halofuginone binding site, increasing halofuginone’s affinity for PfProRS.
TCMDC-124506 was identified as a PfProRS inhibitor. The effect on inhibition of PfProRS by this compound was evaluated when the concentrations of proline and ATP were varied (Supporting Information Figure S4). For TCMDC-124506 an increase in IC50 for PfProRS inhibition was observed as the ATP or proline concentration was increased was found. Thus, the inhibitory effect of TCMDC-124506 was reduced by increasing concentrations of ATP or proline, but it was not as marked an effect as for halofuginone. This suggests that proline and ATP binding changes the loop structure of the protein above and adjacent to the active site, making it less accessible for binding of TCMDC-124506. We conclude that TCMDC-124506 is a competitive allosteric inhibitor, because ATP and proline substrates both compete for TCMDC-124506 binding, but yet the TCMDC-124506 binding occurs outside the active site. This also suggests that the parasite could become more resistant to the effects of such an inhibitor by increasing its intracellular concentration of proline, which would be a potential liability. Indeed, this mechanism of resistance to halofuginone analogues, accomplished by the parasite increasing its intracellular proline levels, was described by Hermann et al.9
We synthesized a limited series of variants to TCMDC-124506 to attempt to find more potent inhibitors while retaining specificity. Our initial exploration, detailed above and in Table 1, was unsuccessful in improving potency. However, we hope that the detailed structure of TCMDC-124506 and glyburide binding to PfProRS will encourage others to explore this allosteric pocket in more detail and consider making additional analogues of these inhibitors. Indeed, many more analogues can be envisioned that will fit in the pocket, and it is likely the pocket can move to accommodate additional structural diversity, strengthen interactions with the protein, and improve the potency of the inhibitor. It is possible that further high-throughput screening combined with structure determination will lead to new chemical leads or further elaboration of TCMDC-124506 and glyburide.
Methods
Pf and Hs ProRS Cloning
Recombinant PfProRS was generated using methods described previously.19 The PfProRS sequence was PCR-amplified from P. falciparum 3D7 cDNA; specific primer sequences for each construct are provided in Supporting Information Table S1. Preparative gel electrophoresis was used to isolate the desired band, which was subsequently excised and purified using a gel extraction kit (Zymoresearch, Irvine, CA, USA). The purified PCR product was treated with T4 DNA polymerase (NEB, Ipswich, MA, USA) for ligation-independent cloning (LIC) and annealed to a LIC prepared BG1861 vector, which contains a T7 promoter and a noncleavable N-terminal hexahistidine (6×His) nickel-affinity tag. Purified plasmids were transformed into BL21(DE3)R3 Rosetta Oxford chemically competent E. coli expression strain and screened for expression.
Pf and Hs ProRS Upscale
Starter cultures of PA-0.5G noninducing media with appropriate antibiotics were grown for 18 h at 25 °C.20 Antibiotics were added to 2 L bottles of sterile ZYP-5052 autoinduction media, and the bottles were inoculated with overnight cultures. Inoculated bottles were then placed into a LEX bioreactor and cultures grown for 72 h at 20 °C. To harvest, the medium was centrifuged at 6000 RCF for 30 min at 4 °C. Cell paste was frozen and stored at −80 °C prior to purification.
Pf and Hs ProRS Purification
Frozen cells were resuspended in lysis buffer (25 mM HEPES (pH 7.0), 500 mM NaCl, 5% (v/v) glycerol, 30 mM imidazole, 0.025% (w/v) sodium azide, 0.5% (w/v) CHAPS, 10 mM MgCl2, 1 mM TCEP, 250 ng/mL AEBSF, and 0.05 μg/mL lysozyme) and disrupted on ice for 30 min with a Virtis sonicator using alternating on/off cycles of 15 s. Cell debris was incubated with 20 μL of benzonase nuclease (25 U/mL) at room temperature for 45 min and clarified by centrifugation on a Sorvall SLA-1500 at 30,000 RCF for 60 min at 4 °C. Protein for X-ray crystallography was purified from clarified cell lysate by immobilized metal affinity chromatography. A His Trap FF 5 mL column (GE Healthcare) was equilibrated with binding buffer (25 mM HEPES (pH 7.0), 500 mM NaCl, 5% (v/v) glycerol, 30 mM imidazole, 0.025% (w/v) sodium azide, 1 mM TCEP). The protein was eluted in the same buffer with 250 mM imidazole added. Size exclusion chromatography (SEC) was done using a HiLoad 26/60 Superdex 75 column (GE Healthcare) equilibrated in SEC buffer (20 mM HEPES (pH 7.0), 300 mM NaCl, 2 mM DTT, and 5% (v/v) glycerol). Pure fractions were collected and pooled from a single peak in the chromatogram and concentrated using Amicon Ultra centrifugal filters. The final protein was concentrated to approximately 25 mg/mL, aliquoted into 100 μL tubes, flash frozen in liquid nitrogen, and stored at −80°C.
Crystallography and Structure Determination
ProRS concentration was adjusted to 20–25 mg/mL and incubated with either 5 mM l-proline and ATP; 4 mM halofuginone, AMPPNP, MgCl2, and β-mercaptoethanol; 5 mM TCMDC-124506; or 5 mM glyburide for 5 min at 289 K. Crystals were then grown at 289 K by sitting drop vapor diffusion with 0.4 μL of protein/ligand complex mixed with 0.4 μL of reservoir solution. Each reservoir solution differed. For apo crystals the reservoir solution was 10% PEG-800, 20% ethylene glycol, 0.1 M MES/imidazole, pH 6.5, and 0.03 M each sodium fluoride, sodium bromide, and sodium iodide. For halofuginone/AMPPNP crystals the reservoir solution was 20% PEG-2000 MME and 0.1 M Tris base/HCl, pH 7.0. For TCMDC-124506 crystals the reservoir solution was 10% PEG-800, 20% ethylene glycol, 0.1 M MES/imidazole, pH 6.5, and 0.03 M each magnesium chloride and calcium chloride. For glyburide crystals the reservoir solution was 10% PEG-800, 20% ethylene glycol, 0.1 M MES/imidazole, pH 6.5, and 0.02 M each sodium formate, ammonium acetate, trisodium citrate, sodium potassium tartrate, and sodium oxamate. All crystals were harvested directly into liquid nitrogen without cryoprotection except for halofuginone/AMPPNP crystals, which were cryo-protected in a solution containing 80% reservoir solution and 20% ethylene glycol supplemented with both halofuginone and AMPPNP.
Data for apo PfProRS were collected at 100 K on a Marmosaic 225 mm CCD detector at a wavelength of 0.9786 Å on beamline 21-ID-F at the Advanced Photon Source (APS, Argonne, IL, USA). Data for PfProRS with halofuginone/AMPPNP were collected at 100 K on a Marmosaic 300 mm CCD detector at a wavelength of 0.9786 Å on beamline 21-ID-G at the Advanced Photon Source. Data for PfProRS with TCMDC-124506 and PfProRS with glyburide were collected at 100 K on a Rayonix MX-225 detector at a wavelength of 0.9795 Å on beamline 08ID-1 at the Canadian Light Source (CLS, Saskatoon, SK, Canada). For all data sets, indexing and integration were carried out using XDS and the scaling of the intensity data was accomplished with XSCALE.21 For apo PfProRS, the structure was solved using molecular replacement with Phaser with 4HVC as a starting model.22 All subsequent structures were solved using Fourier synthesis with Refmac5 with the previously solved PfProRS structure providing the phases.23 For the apo PfProRS structure, refinement was carried out using Refmac5, TLS, and manual refinement in Coot.24,25 For all subsequent structures refinement was carried out with Phenix in place of Refmac.26 All structures were quality checked by Molprobity.27 Data statistics and refinement are given in Table 2 and deposited to the PDB (PDB IDs: 4NCX, 4Q15, 4WI1, 5IFU).
Table 2. Structural Data Statistics.
| crystal | apo | halofuginone AMPPNP | TCMDC-124506 | glyburide |
|---|---|---|---|---|
| PDB code | 4NCX | 4Q15 | 4WI1 | 5IFU |
| Data collection | ||||
| resolution range (Å) | 50.00–1.85 | 50.00–2.35 | 50.00–1.65 | 50.00–2.45 |
| (1.90–1.85) | (2.41–2.35) | (1.69–1.65) | (2.51–2.45) | |
| space group | C2 | P212121 | C2 | P43212 |
| unit cell dimensions | 147.96, 91.38, 110.84 | 76.58, 78.09, 167.82 | 148.18, 91.34, 110.91 | 138.31, 138.31, 156.66 |
| 90.00, 129.48, 90.00 | 90.00, 90.00, 90.00 | 90.00, 129.53, 90.00 | 90.00, 90.00, 90.00 | |
| no. of unique reflections | 96638 | 42603 | 133468 | 56139 |
| Rmerge (%)a | 4.9 (49.1) | 6.4 (54.7) | 4.9 (53.9) | 6.6 (53.3) |
| redudancy | 4.5 (4.6) | 5.1 (5.3) | 4.2 (4.3) | 7.3 (7.5) |
| completeness (%) | 99.2 (99.2) | 99.6 (99.9) | 97.2 (96.2) | 99.5 (100.0) |
| l/σI | 19.36 (3.11) | 19.82 (3.24) | 16.31 (3.07) | 18.69 (4.15) |
| Refinement | ||||
| resolution range | 50.00–1.85 | 50.00–2.35 | 50.00–1.65 | 50.00–2.45 |
| (1.90–1.85) | (2.41–2.35) | (1.69–1.65) | (2.51–2.45) | |
| no. of protein atoms | 7084 | 7190 | 7203 | 6942 |
| no. of water molecules | 603 | 273 | 765 | 243 |
| Rcryst (%) | 16.9 | 19.5 | 16.2 | 18.5 |
| Rfree (%) | 20.6 | 20.9 | 18.8 | 21.3 |
| Root-mean-square deviations from ideal stereochemistry | ||||
| bond lengths (Å) | 0.013 | 0.003 | 0.007 | 0.003 |
| bond angles (deg) | 1.511 | 0.757 | 1.047 | 0.668 |
| mean B factor (all atoms) (Å2) | 33.04 | 44.51 | 33.27 | 61.33 |
| Ramachandran plot | ||||
| favored region (%) | 97.08 | 96.90 | 97.79 | 96.74 |
| allowed regions (%) | 2.56 | 2.99 | 2.08 | 2.56 |
| outlier regions (%) | 0.37 | 0.11 | 0.12 | 0.70 |
Compound Libraries for Screening
SPECTRUM Microsource targeted diversity library (2320 compounds) is a commercially available library composed of drug-like components, natural products, and bioactive components. Compounds were tested at a single concentration of 10 μM. GlaxoSmithKline (GSK) Tres Cantos Antimalarial Set (TCAMS) is a collection of 13,533 confirmed inhibitors of P. falciparum cell growth, selected from over 2 million compounds from GSK’s chemical library.28 These compounds have been confirmed to inhibit parasite growth by at least 80% at 2 μM and have antiplasmoidal activity while not inhibiting human cell lines by >20%. The TCAMS compounds were tested at a single concentration of 7.5 μM. The DDU small diversity library composed of 15,667 compounds was tested at 30 μM. The St. Jude Library (gift of Dr. Kip Guy) is a collection of 7798 compounds;29 these were tested at 15 μM. The MMV Malaria Box compounds are a 400 compound subset of the TCAMS, Novartis, and St. Jude’s 14,000 compound hits (http://www.mmv.org/malariabox).
Enzyme Assays
Two different enzyme assays were employed: the aminoacylation of tRNA measuring the conjugation of radioactive proline to tRNA and the consumption of ATP and measurement of ATP consumption in the full reaction using luciferase and KinaseGlo reagent.
The method for the aminoacylation assay employed 10 nM PfProRS enzyme, 100 nM [3H]-l-proline (PerkinElmer), 100 μM ATP (Sigma), and 400 μg/mL S. cerevisiae tRNA (Sigma, R4251) with an incubation for 2 h at 25 °C. At the end of 2 h, the macromolecules including tRNAs precipitated with 10% trichloroacetic acid (Fisher Scientific), and precipitates were collected in 96-well filter plates (Millipore MSHVN4B10). Enzyme PfProRS B2 with truncation of the N-terminal six amino acids was employed for most experiments, although B4 truncation enzyme was used as well in Figure 2. Triplicate values were determined, and the standard error was calculated and displayed.
The ATP consumption assay was run in two different formats. For the HTS, the assay was run with 75 nM PfProRS A2 enzyme, 60 μM proline, 6 μM ATP, and 400 μg/mL yeast tRNA and incubated for 2 h at 37 °C. The signal-to-noise was >10-fold, and the Z′ of the assay was ≥0.67. This assay was also run for Figure 4, but 75 nM human ProRS was included for some of the measurements. Note that consumption of ATP continued for another hour in this assay for both enzymes, so the relative IC50 values determined in Figure 4 are significant, although the absolute IC50 values of compounds differ from the modified ATP consumption assay. The modified ATP consumption assay was in the linear part of the consumption curve for comparing IC50 values reported in Table 1. These conditions were 75 nM ProRS A2 enzyme, 60 μM proline, 4 μM ATP, and 400 μg/mL yeast tRNA and incubated for 3.5 h at 20 °C. The conditions were found to be linear up to 5 h, and measuring the IC50 of halofuginone at different time points in the assay (3, 4, and 5 h) gave IC50 values with <2-fold variation, which is within experimental error. The signal-to-noise of this assay was >5-fold, with Z′ > 0.55.
Acknowledgments
Funding was provided by NIH Contract HHSN272201200025C (Seattle Structural Genomics Center for Infectious Disease, SSGCID). We thank the Structure-guided Drug Discovery Coalition (SDDC) for additional funding to elucidate dose–response curves, insightful direction on compound selection, and funding the SAR work on TCMDC-124506 at Dundee University (http://www.thesgc.org/sddc). We acknowledge the Wellcome Trust (Grant 100476) and the Australian National Health and Medical Research Council (Grant 1042272 to K.K. and an Overseas Biomedical Fellowship 585519 to A.M.L.). We are especially grateful to Kip Guy for generous contribution of the St. Jude screening compound library and GlaxoSmithKline (GSK) for the TCAMS compound library and the Medicines for Malaria Venture for the Malaria Box compound library, as well as Jenni Risler at the Fred Hutch Cancer Research Center High Throughput Screening Core for helpful feedback and responsive assistance running the compound screens. Most importantly, we acknowledge that each member of SSGCID played an important part in solving the structures published in this paper.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.6b00078.
Figures S1–S7, Tables S1 and S2, and supplemental experimental procedures (PDF)
Author Contributions
Conceptualization, W.C.V.V. and S.N.H.; methodology, W.C.V.V. and S.N.H.; investigation, S.N.H., D.M.D., B.G.H., B.F., I.H., B.B., J.A., C.J., and M.D.; writing, original draft, W.C.V.V., S.N.H., D.M.D., and B.B.; writing, review, and editing, W.C.V.V., S.N.H., A.H.F., D.M.D., I.H.G, B.G.H., B.F., I.H., C.D., R.C., K.L.R., M.A.H., D.W.G., K.D.R., A.W., C.W., R.S., L.K.B., A.M.L., and K.K.; visualization, D.M.D., C.J., B.B., and S.N.H.; resources, I.H.G., D.W.G., K.D.R., A.M.L., K.K., and R.C.; supervision, W.C.V.V., I.H.G, D.W.G., K.D.R., A.W., C.W., T.E.E., D.W.G., P.J.M., and J.A.; project administration, L.B., A.W., R.S., and C.W.; funding acquisition, W.C.V.V., P.J.M., and I.H.G.
The authors declare no competing financial interest.
Supplementary Material
References
- Murry C.; Rosenfield L.; Lim S.; Andrews K.; Foreman K.; Haring D.; Fullman N.; Naghavi M.; Lozano R.; Lopez A. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431. 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- Dondorp M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; Ringwald P.; Silamut K.; Imwong M.; Chotivanich K.; Lim P.; Herdman T.; An S. S.; Yeung S.; Singhasivanon P.; Day N. P.; Lindegardh N.; Socheat D.; White N. J. (2009) Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbengue A.; Bhattacharjee S.; Pandharkar T.; Liu H.; Estiu G.; Stahelin R. V.; Rizk S. S.; Njimoh D. L.; Ryan Y.; Chotivanich K.; Nguon C.; Ghorbal M.; Lopez-Rubio J. J.; Pfrender M.; Emrich S.; Mohandas N.; Dondorp A. M.; Wiest O.; Haldar K. (2015) A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 30, 683–687. 10.1038/nature14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coatney G. R.; Cooper W. C.; Culwell W. B.; White W. C.; Imboden C. A. Jr. (1950) Studies in human malaria. XXV. Trial of febrifugine, an alkaloid obtained from Dichroa febrifuga Lour. against the Chesson strain of Plasmodium vivax. J. Natl. Malar. Soc. 9, 183–186. [PubMed] [Google Scholar]
- Ablondi F.; Gordon S.; Morton J. II; Williams J. H. (1952) An antimalarial alkaloid from Hydrangea. II. Isolation. J. Org. Chem. 17, 14–18. 10.1021/jo01135a003. [DOI] [Google Scholar]
- Hewitt R. I.; Wallace W. S.; Gill E. R.; Williams J. H. (1952) An antimalarial alkaloid from Hydrangea. XIII. The effects of various synthetic quinazolones against Plasmodium lophurae in ducks. Am. J. Trop Med. Hyg. 1, 768–772. [PubMed] [Google Scholar]
- Ryley J. F.; Betts M. J. (1973) Chemotherapy of chicken coccidiosis. Adv. Pharmacol. 11, 221–293. 10.1016/S1054-3589(08)60459-7. [DOI] [PubMed] [Google Scholar]
- Keller T. L.; Zocco D.; Sundrud M. S.; Hendrick M.; Edenius M.; Yum J.; Kim Y. J.; Lee H. K.; Cortese J. F.; Wirth D. F.; Dignam J. D.; Rao A.; Yeo C. Y.; Mazitschek R.; Whitman M. (2012) Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 8, 311–317. 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman J. D.; Pepper L. R.; Cortese J. F.; Estiu G.; Galinsky K.; Zuzarte-Luis V.; Derbyshire E. R.; Ribacke U.; Lukens A. K.; Santos S. A.; Patel V.; Clish C. B.; Sullivan W. J. Jr; Zhou H.; Bopp S. E.; Schimmel P.; Lindquist S.; Clardy J.; Mota M. M.; Keller T. L.; Whitman M.; Wiest O.; Wirth D. F.; Mazitschek R. (2015) The cytoplasmic prolyl-tRNA synthetase of the malaria parasite is a dual-stage target of febrifugine and its analogs. Sci. Transl. Med. 7, 288ra77. 10.1126/scitranslmed.aaa3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal L. H., Troein C., Vallon-Christersson J., Gruvberger S., Borg A., and Peterson C. (2002) BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol. 3, SOFTWARE0003. 10.1186/gb-2002-3-8-software0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J.; Lee E. H.; Park M.; Kim J. H.; Kim J.; Kim S.; Jeon Y. H.; Hwang K. Y. (2013) Conformational changes in human prolyl-tRNA synthetase upon binding of the substrates proline and ATP and the inhibitor halofuginone. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 2136–2145. 10.1107/S0907444913020556. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Sun L.; Yang X. L.; Schimmel P. (2013) ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature 494, 121–124. 10.1038/nature11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain V.; Yogavel M.; Oshima Y.; Kikuchi H.; Touquet B.; Hakimi M. A.; Sharma A. (2015) Structure of prolyl-tRNA synthetase-halofuginone complex provides basis for development of drugs against malaria and toxoplasmosis. Structure 23, 819–829. 10.1016/j.str.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Vaidya A. B.; Morrisey J. M.; Zhang Z.; Das S.; Daly T. M.; Otto T. D.; Spillman N. J.; Wyvratt M.; Siegl P.; Marfurt J.; Wirjanata G.; Sebayang B. F.; Price R. N.; Chatterjee A.; Nagle A.; Stasiak M.; Charman S. A.; Angulo-Barturen I.; Ferrer S.; Belén Jiménez-Díaz M.; Martínez M. S.; Gamo F. J.; Avery V. M.; Ruecker A.; Delves M.; Kirk K.; Berriman M.; Kortagere S.; Burrows J.; Fan E.; Bergman L. W. (2014) Pyrazoleamide compounds are potent antimalarials that target Na+ homeostasis in intraerythrocytic Plasmodium falciparum. Nat. Commun. 5, 5521. 10.1038/ncomms6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillman N. J.; Allen R. J.; McNamara C. W.; Yeung B. K.; Winzeler E. A.; Diagana T. T.; Kirk K. (2013) Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13, 227–237. 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehane A. M.; Ridgway M. C.; Baker E.; Kirk K. (2014) Diverse chemotypes disrupt ion homeostasis in the malaria parasite. Mol. Microbiol. 2, 327–339. 10.1111/mmi.12765. [DOI] [PubMed] [Google Scholar]
- Rottmann M.; McNamara C.; Yeung B. K.; Lee M. C.; Zou B.; Russell B.; Seitz P.; Plouffe D. M.; Dharia N. V.; Tan J.; Cohen S. B.; Spencer K. R.; González-Páez G. E.; Lakshminarayana S. B.; Goh A.; Suwanarusk R.; Jegla T.; Schmitt E. K.; Beck H. P.; Brun R.; Nosten F.; Renia L.; Dartois V.; Keller T. H.; Fidock D. A.; Winzeler E. A.; Diagana T. T. (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180. 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien C.; Henrich P. P.; Passi N.; Fidock D. A. (2011) Recent clinical and molecular insights into emerging artemisinin resistance in Plasmodium falciparum. Curr. Opin. Infect. Dis. 24, 570–577. 10.1097/QCO.0b013e32834cd3ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi R.; Kelley A.; Leibly D.; Hewitt S. N.; Napuli A.; Van Voorhis W. C. (2011) Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 67, 998–1005. 10.1107/S1744309111017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 63, 32–41. 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Painter J.; Merritt E. A. (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 439–450. 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465, 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jiménez-Díaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. (2010) Chemical genetics of Plasmodium falciparum. Nature 465, 311–315. 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.