

Abstract
The neuromuscular junction (NMJ) is enriched with glycoproteins modified with N-acetylgalactosamine (GalNAc) residues, and four nominally GalNAc-specific plant lectins have historically been used to identify the NMJ and the utrophin–glycoprotein complex. However, little is known about the specific glycan epitopes on skeletal muscle that are bound by these lectins, the glycoproteins that bear these epitopes or how creation of these glycan epitopes is regulated. Here, we profile changes in cell surface glycosylation during muscle cell differentiation and identify distinct differences in the binding preferences of GalNAc-specific lectins, Wisteria floribunda agglutinin (WFA), Vicia villosa agglutinin (VVA), soybean agglutinin (SBA) and Dolichos biflorus agglutinin (DBA). While we find that all four GalNAc binding lectins specifically label the NMJ, each of the four lectins binds distinct sets of muscle glycoproteins; furthermore, none of the major adhesion complexes are required for binding of any of the four GalNAc-specific lectins. Analysis of glycosylation-related transcripts identified target glycosyltransferases and glycosidases that could potentially create GalNAc-containing epitopes; reducing expression of these transcripts by siRNA highlighted differences in lectin binding specificities. In addition, we found that complex N-glycans are required for binding of WFA and SBA to murine C2C12 myotubes and for WFA binding to wild-type skeletal muscle, but not for binding of VVA or DBA. These results demonstrate that muscle cell surface glycosylation is finely regulated during muscle differentiation in a domain- and acceptor-substrate-specific manner, suggesting that temporal- and site-specific glycosylation are important for skeletal muscle cell function.
Keywords: GalNAc, muscle glycosylation, neuromuscular junction, utrophin–glycoprotein complex, Wisteria floribunda agglutinin
Introduction
Skeletal muscle fibers are surrounded by an extracellular matrix (ECM) that provides mechanical support and also transmits signals into the cell via interactions with glycoproteins on the muscle cell membrane, or sarcolemma. The sarcolemma has distinct domains that vary by function, anatomic location and the array of membrane proteins concentrated in these specific domains. The neuromuscular junction (NMJ) is the domain that transmits signals from nerve terminals to the muscle fiber (Tintignac et al. 2015). The myotendinous junction (MTJ) is the domain that transmits forces from muscle to tendon (Tidball 1991). The extrasynaptic, or extrajunctional, domain connects the sarcolemma to the ECM to transmit force during muscle contraction. These distinct domains contain discrete sets of membrane molecules required for their different functions.
On the extrasynaptic sarcolemma, a major adhesion complex is the dystrophin–glycoprotein complex (DGC). The DGC connects the intracellular actin cytoskeleton to the ECM via the laminin receptor dystroglycan and the intracellular adaptor protein dystrophin (Ervasti and Campbell 1993). A homologous complex, the utrophin–glycoprotein complex (UGC), which localizes to the NMJ and MTJ domains, also includes dystroglycan but uses utrophin rather than dystrophin to link the ECM to the cytoskeleton (Clerk et al. 1993; James et al. 1995). Dystroglycan is a heterodimer of soluble alpha-dystroglycan (α-DG) and membrane-bound beta-dystroglycan (β-DG). α-DG has N- and C-terminal globular domains linked by an extended mucin domain. The C-terminal globular domain connects α-DG to β-DG, while the highly glycosylated mucin domain bears unique O-mannose-linked glycosaminoglycans that bind to proteins in the ECM (Kanagawa et al. 2005; Yoshida-Moriguchi et al. 2010). While dystroglycan is present in both the DGC and the UGC, different ECM components are bound by dystroglycan at different sites; α-DG in the DGC at extrasynaptic sarcolemma binds laminin-211 (Ervasti and Campbell 1993), while α-DG at the NMJ binds laminin-221, laminin-421 and laminin-521 (Meier and Ruegg 2000). The α7β1 integrin heterodimer is the other major adhesion complex present in skeletal muscle (Song et al. 1992; Burkin and Kaufman 1999). Splice variants of α7-integrin are also specific for either the synaptic or extrasynaptic sarcolemmal domains (Martin et al. 1996) and selectively bind different laminin isoforms (von der Mark et al. 2002).
Proper glycosylation is critical for sarcolemma–ECM interactions and muscle function. The role of integrin glycosylation in cell adhesion and migration has been characterized in many cell types, although the function of skeletal muscle integrin glycans remain poorly understood (Gundry et al. 2009). In contrast, glycosylation of α-DG has been studied extensively. Laminin binds to α-DG via O-mannose glycans modified with glycosylaminoglycans synthesized by the enzyme like-glycosyltransferase (LARGE); the ability of α-DG to bind laminin is dependent upon proper glycosylation (Yoshida-Moriguchi et al. 2010), as hypoglycosylated α-DG shows reduced binding to laminin and perlecan (Kanagawa et al. 2005; Yoshida-Moriguchi et al. 2010). Mutations in DAG1, the gene encoding dystroglycan, or in any of 17 genes involved in α-DG glycosylation that result in hypoglycosylation of α-DG, cause decreased cell binding to ECM and a spectrum of neuromuscular disorders (Live et al. 2013; Endo 2015).
In addition to distinct types of glycosylation on muscle cell glycoproteins, there is differential glycosylation of glycoproteins in specific sarcolemmal domains. In 1982, Sanes and Cheney used Dolichos biflorus agglutinin (DBA), a plant lectin that recognizes terminal N-acetylgalactosamine (GalNAc) residues, to selectively identify the NMJ (Sanes and Cheney 1982). Utilizing a panel of plant lectins, the Sanes group later demonstrated that the NMJ in rodent muscle is enriched with terminal GalNAc residues compared to the extrasynaptic sarcolemma (Scott et al. 1988). Historically, several lectins specific for GalNAc, including Vicia villosa agglutinin (VVA) and Wisteria floribunda agglutinin (WFA) have been used to differentiate the NMJ from extrasynaptic sarcolemma (McDearmon et al. 2001; Nguyen et al. 2002; Marshall et al. 2012). WFA specifically has been used to differentiate between complexes associated with NMJ vs. the extrasynaptic sarcolemma. WFA preferentially binds to α-DG in the UGC, while the sialic acid and N-acetylglucosamine (GlcNAc)-specific lectin wheat germ agglutinin (WGA) binds to α-DG associated with both the DGC and the UGC (McDearmon et al. 2001; Nguyen et al. 2002; Xia et al. 2002; Cabrera et al. 2012; Marshall et al. 2012).
Manipulating muscle glycosylation to improve muscle cell adhesion to ECM is a potential therapeutic option for some muscular dystrophies. A number of therapeutic approaches have been described in cell and animal models, including overexpression of a specific glycosyltransferase enzyme to modify sarcolemmal glycans (Nguyen et al. 2002; Xia et al. 2002), administration of a recombinant scaffolding glycoprotein that binds to the sarcolemma (Amenta et al. 2011; Van Ry et al. 2015) and pharmacologic treatment to enrich for specific saccharide residues on sarcolemmal glycoproteins (Cabrera et al. 2012). However, as there has been no comprehensive quantitative approach to characterizing glycans expressed on skeletal muscle, it is not clear what glycans are being modified by these approaches, or if there are more specific glycan targets that could be modified to improve muscle function.
While plant lectins with nominal specificity for GalNAc have been interchangeably used to distinguish the NMJ-associated UGC from the sarcolemmal DGC, we have found that these different lectins recognize unique types of glycans and discrete sets of glycoproteins that bear these glycans. These results indicate that muscle cell glycosylation is very finely regulated, implying that specific differences in glycoprotein glycosylation are important for distinct functions and that definition of these differences will enable a more precise approach to enhancing muscle cell function through manipulation of glycosylation.
Results
Four lectins that preferentially recognize GalNAc, WFA, VVA, SBA and DBA have historically been used to identify the NMJ (Sanes and Cheney 1982; Scott et al. 1988). WFA and VVA specifically have been used to detect various α-DG glycoforms, including those that associate with the UGC (McDearmon et al. 2001; Nguyen et al. 2002; Xia et al. 2002; Cabrera et al. 2012; Marshall et al. 2012). However, the assumption that these four lectins recognize the same GalNAc-containing glycans on the same glycoproteins has not been confirmed. Therefore we asked if 1) there were differences among the four lectins in specificity for the NMJ, 2) whether reactivity with VVA, SBA and DBA, like WFA, correlated with utrophin distribution and 3) if any of the major adhesion complexes (DGC, UGC or α7β1 integrin) were required for binding of the four lectins.
As previously described, in wild-type mice, DBA, VVA and SBA reactivity were highly specific for the NMJ, staining discretely and minimally in comparison to the extensive sarcolemma. (Figure 1). In contrast, while enriched at the synapse, WFA binding spread slightly beyond the NMJ. Moreover, significant differences were observed in reactivity of the four GalNAc-specific lectins with muscle from mdx mice. As previously described, we observed spreading of WFA reactivity throughout the sarcolemma in muscle from mdx mice, which correlated with utrophin staining. However, VVA, SBA and DBA staining remained highly specific for the NMJ in muscle from mdx mice. Interestingly, we noted similar differences between the four GalNAc-specific lectins when muscle from α7-integrin-null (α7−/−) mice was stained. While VVA, SBA and DBA staining remained highly NMJ specific, WFA staining spread extrasynaptically and correlated with utrophin staining. As WFA staining redistributed concomitantly with utrophin staining, we next asked if expression of utrophin was necessary for staining of WFA or the other GalNAc-specific lectins. Surprisingly, we observed no loss of reactivity for any of the four lectins with muscle from utrophin-null (Utr−/−) mice compared to wild type, indicating that utrophin expression is not required for binding of WFA, VVA, SBA or DBA (Figure 1).
Fig. 1.

The DGC, UGC and α7-integrin are not requisite for binding of GalNAc-specific lectins. Transverse 8-µm serial cryosections of murine quadriceps muscles from 12-week-old wild-type, mdx, utrophin-null (Utr−/−) and alpha-7-integrin-null (α7−/−) mice were stained with hematoxylin and eosin (H&E, top row), anti-utrophin antibody (bottom row), and the following biotinylated reagents: WFA, VVA, SBA and DBA. Bound lectins and antibodies were detected with FITC-avidin D (green) and counterstained with Texas Red conjugated alpha-bungarotoxin (red). Merged images are depicted. Insets depict neuromuscular junctions. Scale bar is 50 µm. This figure is available in black and white in print and in color at Glycobiology online.
As we observed some qualitative differences in patterns of reactivity of WFA, VVA, SBA and DBA with muscle from wild-type and mdx mice, we developed an in vitro lectin binding assay to quantify differences in binding among the lectins. We used a panel of 11 lectins (Table I) to analyze glycosylation of C2C12 murine myoblasts and differentiated myotubes. Fixed cells were incubated with biotinylated lectins and bound lectins were detected colorimetrically. In addition to the GalNAc-specific lectins WFA, VVA, SBA and DBA, the panel included lectins specific for major classes of glycan structures found on glycoproteins. WGA, Sambucus nigra agglutinin (SNA) and Maackia amurensis agglutinin-II (MAA-II) were included for specificity for terminal sialic acid residues (Greenaway and LeVine 1973; Shibuya et al. 1987; Konami et al. 1994). Peanut agglutinin (PNA) and jacalin (Jac) recognize asialo and sialylated core 1 type O-glycans, respectively (Lotan et al. 1975; Wu et al. 2003), while Phaseolus vulgaris leucoagglutinin (PHA-L) and Concanavalin A (Con A) recognize complex and high mannose-type N-glycans, respectively (Cummings and Kornfeld 1982; Mega et al. 1992). Therefore, this panel of 11 lectins allowed us to detect changes in glycosylation across a wide breadth of glycan structures.
Table I.
Panel of lectins with varying specificities utilized to characterize changes in muscle glycosylation following differentiation of C2C12 myotubes
| Lectin | Abbreviation | Nominal sugar specificity |
|---|---|---|
| Wisteria floribunda agglutinin | WFA | GalNAc |
| Vicia villosa agglutinin | VVA | GalNAc |
| Soybean agglutinin | SBA | GalNAc |
| Dolichos biflorus agglutinin | DBA | GalNAc |
| Wheat germ agglutinin | WGA | Sialic Acid, GlcNAc |
| Sambucus nigra agglutinin | SNA | α2,6 linked Sialic acid |
| Maackia amurensis agglutinin-II | MAA-II | α2,3 linked Sialic acid |
| Peanut agglutinin | PNA | Asialo core 1 O-glycans |
| Jacalin agglutinin | Jac | Core 1 O-glycans |
| Phaseolus vulgaris leucoagglutinin | PHA-L | Complex N-glycans |
| Concanavalin A | Con A | High mannose N-glycans |
As shown in Figure 2A, reactivity of C2C12 myoblasts at day 0 with the different lectins varied considerably. Following 7 days of differentiation, we observed little change in reactivity with the sialic acid-specific lectins WGA, SNA and MAA-II. However, we observed a significant increase in PNA reactivity at day 7 compared with day 0, with a concomitant decrease in Jac binding, indicating an increase in asialo core 1 O-glycans following differentiation. We also observed increased PHA-L binding to cells at day 7 compared to day 0, indicating an increase in complex N-glycans following differentiation.
Fig. 2.

Distinct increases in binding of GalNAc-specific lectins following differentiation of C2C12 myotubes. (A) Glycosylation of C2C12 myotubes differentiated for 7 days was detected utilizing a panel of 11 lectins with varying specificities. Increases were seen in the four GalNAc-specific lectins WFA, VVA, SBA and DBA. (B) Changes in binding of the four GalNAc-specific lectins were determined over a time course of 7 days. Lectin binding was performed at the time of differentiation (day 0), and following 2, 4 and 7 days of differentiation. Binding of VVA increased rapidly, WFA increased following 2 days of differentiation, whereas binding of SBA and DBA did not increase until 7 days of differentiation. Data are mean ± SEM (error bars) of triplicates from three pooled independent experiments. * P < 0.05; *** P < 0.001; **** P < 0.0001.
Importantly, we observed increased binding of all four GalNAc-specific lectins, WFA, VVA, SBA and DBA, with C2C12 differentiation. WFA, VVA, SBA and DBA binding increased approximately 70%, 120%, 300% and 130%, respectively. In addition to the quantitative differences in the amplitude of binding of the four GalNAc-specific lectins, we also observed temporal differences in binding during C2C12 differentiation. As shown in Figure 2B, binding of WFA, VVA, SBA and DBA increased at different rates throughout the 7 days. Binding of WFA did not increase until day 4, while increased VVA binding was detected at day 2. In contrast, increased SBA and DBA binding were not observed until day 7 (Figure 2B). The differential rates of change in reactivity of the four GalNAc-specific lectins suggested that these lectins recognize different glycoproteins or different glycoforms of the same protein.
To determine if the GalNAc-specific lectins WFA, VVA, SBA and DBA bind different glycoproteins on muscle cells, we performed lectin precipitations. C2C12 myotubes differentiated for 7 days were biotinylated to label cell surface glycoproteins, and lectin precipitations of cell lysates were performed with the four lectins. Sarcolemmal glycoproteins precipitated by each lectin were detected with SA-HRP (Figure 3A). Multiple cell surface glycoproteins were precipitated by WFA, VVA and SBA, while DBA precipitated a smaller array of glycoproteins, with a predominant component of approximately 250 kDA. While WFA, VVA and SBA all precipitated an array of glycoproteins, we observed significant differences among the lectins in the patterns of glycoprotein reactivity, indicating that these lectins selectively recognize different repertoires of glycoproteins in C2C12 cells. In addition, the four lectins selectively recognize distinct repertoires of glycoproteins in wild-type mouse muscle. We performed lectin precipitation with extracts of skeletal muscle and detected precipitated glycoproteins with SYPRO® Ruby stain (Figure 3B). Distinct glycoprotein receptors were precipitated by each lectin from wild-type skeletal muscle lysates. Thus, WFA, VVA, SBA and DBA react with distinct sets of glycoproteins and/or protein glycoforms. As modification of α-DG with terminal GalNAc residues has been used to identify the UGC (Nguyen et al. 2002; Xia et al. 2002; Marshall et al. 2012), we asked if each of the four GalNAc-specific lectins were capable of precipitating either α-DG or β-DG. WFA, VVA and SBA precipitated both α-DG and β-DG from C2C12 myotubes and total skeletal muscle lysates, while DBA did not precipitate either α-DG or β-DG (Figure 3, A and B lower panels). Thus, while WFA, VVA and SBA precipitated DG, it was not the only sarcolemmal glycoprotein receptor for these GalNAc-specific lectins on C2C12 myotubes or wild-type skeletal muscle, and DBA, the fourth GalNAc-specific lectin, did not recognize DG.
Fig. 3.

GalNAc-specific lectins precipitate different glycoproteins. Equal amounts of lysates from murine C2C12 myotubes differentiated for 7 days (A) or wild-type murine skeletal muscle (B) were precipitated by WFA (W), VVA (V), SBA (S) and DBA (D) following surface biotinylation and cell lysis. Total proteins precipitated were separated by SDS-PAGE from surface biotinylated murine C2C12 myotubes and detected by SA-HRP (A, upper panel), while proteins from wild-type skeletal muscle were detected with SYPRO® Ruby Protein Gel stain (B, upper panel). Parallel blots were probed with mAb against α-DG and β-DG (A and B, lower panels).
These results indicated that different types of glycans were being recognized by the GalNAc-specific lectins. A number of factors control cell-specific glycoprotein glycosylation, including transcription of glycan modifying genes, availability of preferred glycoprotein acceptors, availability of nucleotide-sugar donors, localization of glycosyltransferases in the secretory pathway and rate of transport through the glycosylation machinery. To determine if differential expression of specific glycosyltransferase or glycosidase gene transcripts could be responsible for the differentiation-dependent changes in lectin reactivity that we observed, we performed a glycotranscriptome analysis (Abbott et al. 2008; Gasimli et al. 2014; Magalhães et al. 2015) of C2C12 cells at day 0 and day 2 of differentiation. As we had observed changes in cell surface glycosylation by lectin binding between 2 and 7 days after addition of differentiation media, we assessed transcript expression at day 2 of differentiation. In our analysis of over 580 glycosylation-related transcripts (Supplementary data, Table S1), we found 22 different glycosyltransferases and glycosidases for which relative expression differed at least 2-fold between day 0 and day 2 (Table II).
Table II.
Twenty-three glycosyltransferase and glycosidase transcripts upregulated following C2C12 differentiation and identified via glycotranscriptome analysis
| Gene Name | Fold Change | Function | |
|---|---|---|---|
| Glycosyltransferases | Fut1 | 24.92 | Blood group Antigen synthesis |
| Extl1 | 22.12 | Heparan polymerization | |
| β3Galt5 | 7.51 | Hybrid N-glycan, neo-lac | |
| β3Galt2 | 6.15 | Hybrid N-glycan, neo-lac | |
| Fut7 | 5.81 | Lewis X Antigen | |
| St8sia4 | 4.86 | Poly Sialic acid | |
| St8sia2 | 4.44 | Poly Sialic Acid | |
| Fut2 | 4.04 | Blood group Antigen synthesis | |
| β4Galnt3a | 4.02 | Hormone glycosylation | |
| Sec1 | 2.99 | Blood group Antigen synthesis | |
| Fut9 | 2.87 | Lewis X Antigen | |
| β3Galnt2a , b | 1.46 | α-DG glycosylation | |
| Glycosylhydrolases | Neu2a | 18.6 | Sialidase (muscle, brain, cytosol) |
| Lctl | 9.13 | Lactase-like protein | |
| Slc3a2 | 4.87 | Solute carrier: dibasic/neutral amino acids | |
| Chi3l1 | 4.51 | Chitinase-like, cartilage protein | |
| Al464131 | 3.58 | Glucosidase family | |
| Amy2 | 3.03 | Amylase | |
| Amy1 | 2.82 | Amylase | |
| Neu1 | 2.66 | Sialic acid catabolism (lysosome) | |
| Gaa | 2.26 | Glucosidase | |
| Edem3 | 2.15 | ER degradation enhancer | |
| Hyal1 | 2.05 | Hyaluronic Acid catabolism |
aGenes of interest for further investigation in this study.
bAlthough fold change for β3Galnt2 was less than 2, this gene was included due to its known role in α-DG glycosylation (Stevens et al. 2013; Hedberg et al. 2014; Praissman et al. 2016).
Focusing on glycosyltransferases that could add terminal GalNAc residues or glycosidases that could unmask subterminal GalNAc residues, we identified beta-1,4-N-acetylgalactosaminyltransferase 3 (β4Galnt3) and sialidase 2 (Neu2), increased 4- and 18.6-fold, respectively, as candidates for further interrogation. We also included beta-1,3-N-acetylgalactosaminyltransferase 2 (β3Galnt2), expression of which increased 1.5-fold by day 2, due to the role of this enzyme in α-DG glycosylation and identification of mutations in this gene in some cases of Walker–Warburg syndrome (Stevens et al. 2013; Hedberg et al. 2014). We hypothesized that increased abundance of either β4Galnt3 or β3Galnt2 GalNAc transferases could directly contribute to increased reactivity of GalNAc-specific lectins following differentiation. Neu2 is a cytosolic sialidase expressed in muscle that removes terminal sialic acid residues and has been reported to be essential for myoblast differentiation (Monti et al. 1999). How cytosolic Neu2 would access glycans in the secretory pathway or on the cell surface is not well understood; however, we reasoned that increased Neu2 transcript expression and activity might remove sialic acid to reveal subterminal GalNAc residues, resulting in increased binding of GalNAc-specific lectins.
Changes in transcript abundance for β4Galnt3, β3Galnt2 and Neu2 at day 0 and day 2 of C2C12 differentiation were validated by qRT-PCR, and the fold changes in abundance were 1.85, 1.49 and 14.2, respectively (Figure 4A). To determine if reduced expression of β4Galnt3, β3Galnt2 or Neu2 abrogated the increase in binding of GalNAc-specific lectins following differentiation, we reduced expression of these three enzymes using siRNA. To determine if decreased expression of any of these enzymes reduced binding of GalNAc-specific lectins, C2C12 cells were transfected with the indicated siRNA, plated for qRT-PCR and lectin binding analysis in parallel and differentiated the same day. Following 2 days of differentiation, we achieved 60% or greater reduction in transcript abundance for each specific enzyme by this method (Figure 4B). After an additional 2 days of differentiation (4 total days of differentiation), binding of the four GalNAc-specific lectins to myotubes plated in parallel was quantified as in Figure 2. To our surprise, knockdown of none of the enzymes reduced binding of any of the GalNAc-specific lectins. In fact, following reduction in β4Galnt3 expression, we observed no change in WFA or VVA binding, while binding of both SBA and DBA increased (Figure 4B, upper panel). Similarly, following reduction in β3Galnt2 expression, we observed increased WFA and SBA binding, while no change was seen in VVA or DBA binding (Figure 4B, middle panel). Finally, reducing Neu2 expression resulted in increased binding of all four GalNAc-specific lectins (Figure 4B, lower panel). While not identifying a specific enzyme that regulates exposure or addition of terminal GalNAc residues, these results further highlight the differences among the specificities of these four lectins; WFA, VVA, SBA and DBA, while nominally GalNAc-specific, clearly recognize unique and distinct glycan structures on C2C12 cells.
Fig. 4.

Reduced expression of β4Galnt3, β3Galnt2 and Neu2 drive distinct changes in binding of GalNAc-specific lectins. (A) Transcript expression of β4Galnt3, β3Galnt2 and Neu2 increased between day 0 and day 2 as detected by qRT-PCR. (B) Cells transfected with siRNA for β4Galnt3, β3Galnt2 and Neu2 showed 60% or greater reduction in expression of each enzyme compared to cells transfected with scramble siRNA. Following siRNA knockdown, lectin binding was performed on parallel cells. No decrease was seen in binding of the GalNAc-specific lectins following siRNA knockdown; however, distinct increases were seen in specific lectins. Data are mean ± SEM (error bars) of triplicates from three pooled independent experiments. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001
Previously, we identified complex N-glycans modified with terminal GalNAc residues on C2C12 cells (Cabrera et al. 2012). We also found that complex N-glycans were required for WFA binding to C2C12 myotubes (Cabrera et al. 2012). Thus, since the glycotranscriptome analysis did not pinpoint enzymes responsible for creating GalNAc-containing ligands, we wanted to determine the type of glycan structures required for binding of the four GalNAc-specific lectins. To interrogate the requirement for complex N-glycans in lectin binding, C2C12 myotubes differentiated for 4 days were treated for a further 72 h with deoxymannojirimycin (DMNJ), which blocks ER alpha-mannosidase I activity and thus the elaboration of complex N-glycans from high mannose precursors on glycoproteins (Figure 5A, Fuhrmann et al. 1984). To confirm the efficacy of DMNJ treatment, we determined that binding of PHA-L, specific for complex N-glycans, was nearly completely abrogated in treated cells (Figure 5B).
Fig. 5.

Complex N-glycans are required for binding of WFA. (A) High mannose N-glycans present on glycoproteins are trimmed to truncated mannose structures by mannosidase enzyme activity, a process inhibited by the small molecule DMNJ. Glycosyltransferase activity elongates truncated mannose structures into complex N-glycans. The bacterial enzyme PNGaseF cleaves N-glycans between the asparagine and GlcNAc residues. (B) C2C12 myotubes were treated with DMNJ or vehicle control for 72 h. DMNJ treatment abrogated WFA binding and modestly reduced binding of SBA. (C) Equal amounts of lysates from murine C2C12 myotubes treated with DMNJ or vehicle control for 72 h were precipitated by WFA, VVA, SBA and DBA following surface biotinylation and cell lysis. Precipitated proteins were separated by SDS-PAGE, and blots probed with SA-HRP. (D) Eight-micrometer serial quadriceps cryosections from wild-type mice were incubated with PNGaseF or vehicle control for 4 h to enzymatically remove N-glycans. PNGaseF treatment abrogated binding of PHA-L and reduced binding of WFA. NMJs were co-stained with Texas Red conjugated alpha-bungarotoxin. Scale bar is 50 µm. Data are mean ± SEM (error bars) of triplicates from three pooled independent experiments. Monosaccharide symbols follow the SNFG (Symbol Nomenclature for Glycans) system (PMID 26543186, Glycobiology 25: 1323–1324, 2015) details at NCBI. *** P < 0.001; **** P < 0.0001. This figure is available in black and white in print and in color at Glycobiology online.
As we had previously found, WFA binding to DMNJ-treated differentiated C2C12 myotubes was substantially reduced, approximately 50%. SBA binding following DMNJ treatment was also reduced, but to a lesser extent. In contrast, we observed no change in VVA or DBA binding to DMNJ-treated cells compared to control. As DMNJ treatment reduced binding of WFA and SBA, but not of VVA or DBA, we reasoned that precipitation of sarcolemmal glycoproteins by WFA and SBA might be sensitive to DMNJ treatment, and we performed lectin precipitations from lysates of control or DMNJ-treated cells with the four GalNAc-specific lectins, as in Figure 3. We observed that fewer glycoproteins were precipitated by WFA and SBA from cell lysates when the cells were treated with DMNJ, compared to control-treated cells (Figure 5C), while no differences in the pattern or abundance of glycoproteins precipitated by VVA or DBA from control- or DMNJ-treated lysates were observed. This further indicated that WFA and SBA bind, at least in part, to complex N-glycans on C2C12 myotubes, while complex N-glycans are not required for VVA and DBA binding.
To determine if N-glycans were required for binding of the lectins to murine muscle, tissue sections were treated with the bacterial enzyme Peptide-N-glycosidase F (PNGaseF) which cleaves N-glycans between the innermost GlcNAc and the underlying asparagine residue (Figure 5A, Chu 1986). As expected, PNGaseF treatment abrogated PHA-L staining of wild-type muscle compared to control-treated tissue (Figure 5D). PNGaseF treatment of tissue sections substantially reduced WFA binding, compared to control-treated sections. However, we observed no qualitative change in binding of VVA, SBA or DBA following PNGaseF treatment (Figure 5D).
Discussion
To date, qualitative lectin staining of muscle sections has provided most of our knowledge regarding muscle glycosylation; for example, the rodent NMJ was shown to preferentially bind GalNAc-specific lectins (Sanes and Cheney 1982; Scott et al. 1988). While WFA, VVA, SBA and DBA have been used to identify the NMJ, presumably due to recognition of terminal GalNAc residues, specific glycoreceptors bound by each lectin remain undetermined. Our work demonstrates significant differences in the binding characteristics of these four GalNAc-specific lectins. Thus, studies using these four lectins interchangeably to identify the NMJ most likely identified different glycoproteins or glycoforms of proteins.
While VVA, SBA and DBA staining of muscle sections from wild-type mice and various null mutants were highly NMJ specific, WFA staining differed; WFA staining of wild-type muscle spread extrasynaptically compared to the other lectins, and redistributed throughout the sarcolemma on mdx and α7−/− muscle sections (Figure 1). While WFA staining has been correlated with localization of utrophin, no loss of muscle staining was observed for any of the lectins of tissues from mice lacking utrophin. Similarly, no loss of binding was observed for tissue from mice lacking α7-integrin. Thus, while WFA reactivity may redistribute extrasynaptically in mdx and α7−/− mice in a pattern similar to utrophin localization (Figure 1), utrophin expression is not requisite for WFA binding. Moreover, neither α-DG containing adhesion complex (DGC, UGC) nor α7-integrin are required for binding of these four GalNAc-specific lectins; integrins and α-DG represent only two of the many surface glycoproteins that might be decorated with glycans detected by GalNAc-specific lectins
In addition to different qualitative patterns of binding among the four GalNAc-specific lectins, a quantitative assay showed that binding of the four lectins increased at different rates during differentiation of C2C12 cell in vitro (Figure 2). These two observations led us to postulate that the four GalNAc-specific lectins bind different glycoproteins or different glycoforms of various proteins. In fact, all four GalNAc-specific lectins directly enrich for distinct subsets of cell surface glycoproteins from C2C12 cells or glycoproteins from muscle lysate. We detected no α-DG in DBA precipitates from either C2C12 myotubes or wild-type skeletal muscle lysates, consistent with the previously reported lack of DBA binding to α-DG from rabbit skeletal muscle (Ervasti et al. 1997). Interestingly, DBA has been shown to react with α-DG isolated from rabbit brain, demonstrating the tissue-specific heterogeneity of α-DG glycosylation (Ervasti et al. 1997). A broad band of α-DG was detected in WFA, VVA and SBA precipitates indicating all three lectins recognize multiple glycoforms of α-DG (Figure 3). While our studies demonstrate that VVA binds to α-DG in total skeletal muscle lysate, this differs from a previous result in which VVA did not bind α-DG when the DGC complex was pre-precipitated with WGA; it is possible that the VVA-reactive α-DG we detected was not included in material precipitated by WGA. This would indicate that at least two glycoforms of α-DG, one sialylated and one modified with terminal GalNAc residues, are expressed in muscle (Ervasti et al. 1997).
Several new mechanisms regulating muscle glycosylation are rapidly emerging. For example, an enzyme initially described to transfer a GlcNAc residue to mannosyl O-glycans on α-DG was recently determined to instead act as a primer for LARGE activity and not as a GlcNAc-transferase (Praissman et al. 2014, 2016; Willer et al. 2014). However, the product of the β3Galnt2 gene, that modifies O-mannose glycans on dystrophin, was not required for binding of GalNAc-specific lectins (Figure 4). Also, while we identified increased expression of β4Galnt3 and Neu2 genes during C2C12 differentiation, reduction of enzyme expression differentially affected binding of the lectins; following knockdown of β4Galnt3, no change was observed in binding of WFA or VVA, while binding of SBA and DBA both increased. In contrast, binding of all four lectins increased following a reduction in expression of Neu2. What might cause these unexpected changes in binding following enzyme knockdown? Knockdown of one specific enzyme may shift the balance of glycosyltransferase activity in the Golgi, and therefore alter the glycans ultimately displayed on sarcolemmal glycoproteins. Genetic manipulation of glycosyltransferase expression is known to alter activity of unrelated glycosylation pathways and ultimately drive glycosylation of non-traditional substrates (Lee et al. 1989; Patnaik and Stanley 2005; Cabrera et al. 2006; Zhang and Hu 2012).
We also found that complex N-glycans contribute significantly to binding of WFA, as DMNJ treatment of C2C12 cells and PNGaseF digestion of wild-type muscle substantially reduced WFA binding. In contrast, N-glycans appear to contribute a fraction of binding sites for SBA as DMNJ treatment modestly reduced binding to intact C2C12 as well as C2C12 lysates. PNGaseF digestion did not reduce SBA binding to wild-type muscle tissue sections indicating glycan structures bound by SBA on C2C12 cells may differ from those bound on innervated muscle. N-glycans do not appear to be required for VVA and DBA binding to C2C12 cells or murine muscle (Figure 5).
To further examine which glycan structures were preferentially bound by the four GalNAc-specific lectins, we analyzed glycan array data from the Consortium for Functional Glycomics (CFG) (Supplementary data, Table S2, http://www.functionalglycomics.org/). This identified 23 glycans bound by WFA, 5 glycans bound by VVA, 12 bound by SBA and 3 bound by DBA, which were compared (Supplementary data, Table S2). Of the glycan structures recognized by any of the four GalNAc-specific lectins, 17 contained terminal GalNAc residues (Table III); linkage of terminal GalNAc residues in either the α- or β-confirmation did not affect lectin recgonition. Of those 17 structures, 6 structures were bound by more than one lectin and 2 of these were recognized by three lectins (WFA, VVA and SBA). Interestingly, while the top glycan structures bound by WFA all had terminal GalNAc residues, several glycans also recognized by WFA did not have terminal GalNAc residues. Similarly, the glycan demonstrating the highest affinity binding by SBA was not modified with a terminal GalNAc, though the other 11 structures recognized by SBA were. Thus, these four lectins therefore have overlapping, yet distinct, preferences for glycan structures terminated with GalNAc residues, while WFA and SBA also bound glycan structures lacking terminal GalNAc residues; future work will have to precisely identify glycan structures on cells and tissues to fully understand patterns of lectin binding. While glycan structures on glycan arrays may not completely recapitulate all structures present on a muscle cell, these data clearly demonstrate differences in the binding preferences of these four lectins.
Table III.
Seventeen glycans with terminal GalNAc residues on CFG glycan arrays were identified as binding at least one of the four GalNAc-specific lectins
| Structure | Nomenclature | WFA | VVA | SBA | DBA |
|---|---|---|---|---|---|
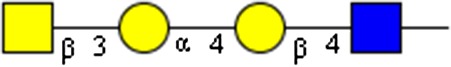
|
GalNAcβ1-3Galα1-4Galβ1-4GlcNAc | x | x | ||
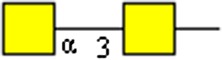
|
GalNAcα1-3GalNAc | x | |||
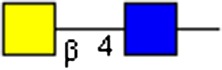
|
GalNAcβ1-4GlcNAc | x | x | x | |
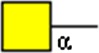
|
GalNAcα | x | x | x | |
|
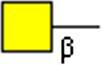
GalNAcβ | x | x | ||
|
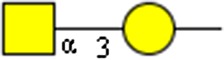
GalNAcα1-3Gal | x | |||
|
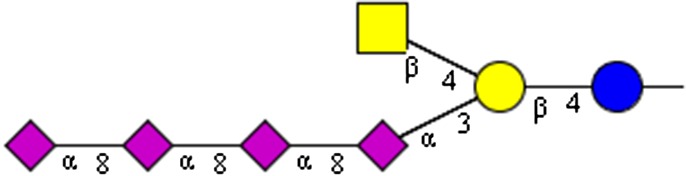
NeuAcα2-8NeuAcα2-8NeuAcα2-8NeuAcα2-8(GalNAcβ1-4)Galβ1-4Glc | x | |||
|
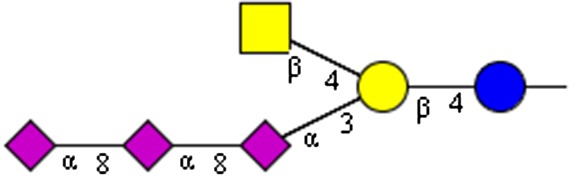
NeuAcα2-8NeuAcα2-8NeuAcα2-8(GalNAcβ1-4)Galβ1-4Glc | x | |||
|
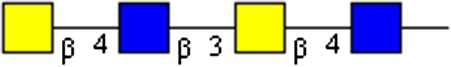
GalNAcβ1-4GlcNAcβ1-3GalNAcβ1-4GlcNAc | x | |||
|
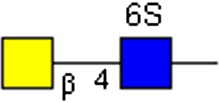
GalNAcβ1-4GlcNAc[6S] | x | |||
|
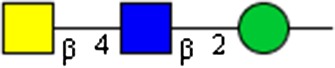
GalNAcβ1-4GlcNAcβ1-2Man | x | x | ||
|
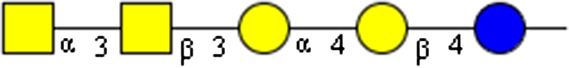
GalNAcα1-3GalNAcβ1-3Galα1-4Galβ1-4Glc | x | x | ||
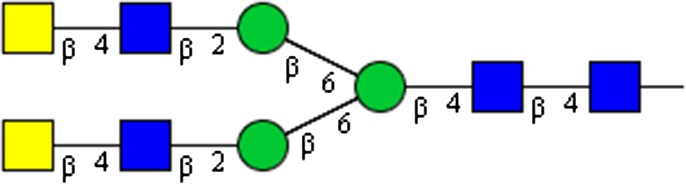
|
GalNAcβ1-4GlcNAcβ1-2Man β1-6 (GalNAcβ1-4GlcNAcβ1-2Man)Manβ1-4GlcNAcβ1-4GlcNAc | x | |||

|
GalNAcβ1-3Galα1-4Galβ1-4GlcNAcβ1-3Galβ1-4Glc | x | |||
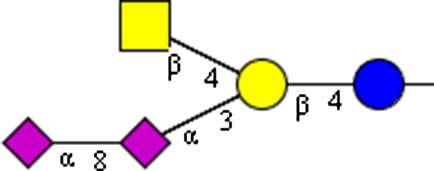
|
NeuAcα2-8NeuAcα2-3(GalNAcβ1-4)Galβ1-4Glcβ | x | |||
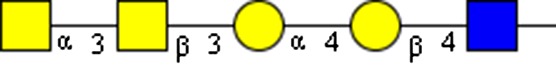
|
GalNAcα1-3GalNAcβ1-3Galα1-4Galβ1-4GlcNAc | x | |||
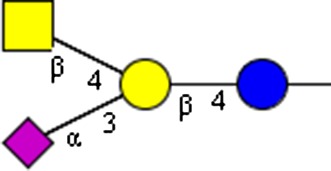
|
NeuAcα2-3(GalNAcβ1-4)Galβ1-4GlcNAcβ | x |
Changes in glycosylation seen in muscle disease could result from multiple aspects of pathophysiology and disease progression. Each protein in an adhesion complex may bear unique glycan structures, so that loss of one protein, or of an entire complex, from the sarcolemma would result in altered sarcolemmal glycosylation. Sarcolemmal adhesion complexes are known to regulate intracellular signaling and subsequent glycosylation of sarcolemmal proteins (Marshall et al. 2012), so that initial loss of sarcolemmal adhesion complexes could further alter glycosylation of sarcolemmal proteins. Loss of adhesion complexes results in destabilization of the sarcolemma and release of intracellular enzymes into the extracellular space. It is conceivable that release of glycosyltransferase enzymes and sugar substrates into the extracellular milieu may also result in modification of glycans on sarcolemmal glycoproteins (Iwata et al. 2013).
While precise changes in muscle glycosylation in human disease are not well described, therapies to alter muscle cell glycosylation to improve muscle cell function have been proposed. These include administration of recombinant galectin-1 (gal-1) and biglycan (Amenta et al. 2011; Van Ry et al. 2015), genetic overexpression of a glycosyltransferase (Nguyen et al. 2002), and treatment with a small molecule (Cabrera et al. 2012). Both gal-1 and biglycan may act to stabilize the sarcolemma by mediating interactions between sarcolemmal glycoproteins and between glycoproteins and the ECM.
Overexpression of the GalNAc transferase β4Galnt2 has been shown to increase abundance of the UGC, increase reactivity of α-DG with WFA and rescue the dystrophic phenotype in mdx mice (Nguyen et al. 2002). However, specific glycan structures modified on α-DG following β4Galnt2 enzyme overexpression remain to be determined. Moreover, overexpression of glycosyltransferases is known to result in modification of atypical substrates in some cases (Patnaik and Stanley 2005; Zhang and Hu 2012), so that the possibility that additional glycoproteins may be modified in cells expressing high levels of β4Galnt2 should be considered. Interestingly, while we saw a significant increase in binding of all four GalNAc-specific lectins following C2C12 differentiation, we did not detect expression of β4Galnt2 (Supplementary data, Table S1).
Any potential therapy for restoring muscle viability and/or function by manipulating muscle glycosylation must address species-specific differences in glycosylation. For example, humans do not produce the sialic acid Neu5Gc due to an inactivating exon deletion in the cytidine monophosphate-N-acetylneuraminic acid hydroxylase (CMAH) gene (Chou et al. 1998). In mouse models of Duchenne muscular dystrophy (DMD) bearing this mutation (CMAH/mdx mice), muscle pathophysiology and disease progression are more severe than in mdx mice, and more accurately reflect the disease progression seen in boys with DMD (Chandrasekharan et al. 2010). Similarly alpha-sarcoglycan (α-SG) deficient mice that also bear the CMAH mutation (CMAH/Sgca−/−) have worse cardiac and skeletal muscle pathophysiology than control mice lacking α-SG (Sgca−/−) (Martin et al. 2013). While this difference in sialic acid usage between humans and rodents is significant, this is most likely not the only difference in muscle glycosylation between mice and humans. In 1988, it was noted that VVA staining of rodent muscle was highly specific for the NMJ, but binding to human muscle spread extrasynaptically (Scott et al. 1988). Furthermore, we found that lobeline treatment did not increase WFA binding to human inducible directly reprogrammable myotubes (Cabrera et al. 2012).
To date, characterization of muscle glycosylation has focused on murine models. While dramatic progress has been made in identification of glycosyltransferases required for creation of O-mannose glycans on α-DG in human muscle, future areas of investigation should focus on 1) characterization of all sarcolemmal glycoproteins and functions of the glycans, 2) identification of glycosylation changes that occur during myogenesis and 3) elucidation of glycosylation differences that occur during progression of muscle disease.
Materials and methods
Animals and cells
Wild-type and mdx breeders were purchased from Jackson Laboratories (Bar Harbor, ME), α7-integrin-deficient (Itga-nulls, α7-/-) mice were a generous gift from D. Burkin (University of Nevada, Reno), and mdx:utrophin−/− mice (mdx:Utr−/−) were a generous gift of J. Sanes (Harvard University, Cambridge) and sent from the mouse colony of J. Chamberlain (University of Washington, Seattle). Tissue sections from 12-week-old wild-type, mdx, Utr-/- and α7-/- mice were analyzed. All procedures involving mice were carried out in accordance with guidelines set by the UCLA Institutional Animal Care and Use Committee. Murine C2C12 cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured and differentiated as previously described (Cabrera et al. 2012), except without antibiotics.
Antibodies and reagents
Antibodies and reagents were purchased: mouse anti-Dystrophin MANDYS1 (3B7), mouse anti-utrophin MANCHO3(8A4) (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), mouse anti-α-DG IIH6-C4 (EMD Millipore, Billerica, MA), goat polyclonal IgG anti-α7-integrin (Santa Cruz Biotechnology, Dallas, TX), goat anti-mouse IgG (H+L)-HRP (Bio-Rad, Hercules, CA), goat anti-mouse IgG+M (H+L)-HRP (Jackson ImmunoResearch, West Grove, PA), streptavidin-conjugated horseradish peroxidase (SA-HRP, Jackson ImmunoResearch) and fluorescein-conjugated avidin D (A-2001, Vector Laboratories, Burlingame, CA).
Biotinylated lectins were bought from Vector Laboratories: WFA (B-1355), VVA (B-1235), SBA (B-1015), DBA (B-1035), PNA (B-1075), Jac (B-1155), SNA (B-1305), MAA-II (B-1265), WGA (B-1025), PHA-L (B-1115) and Con A (B-1005). Biotinylated bovine serum albumin (BSA, B-2007) was purchased for use as a control in lectin binding assays. The following agarose bound lectins were purchased from Vector Labs: WFA (AL-1353), VVA (AL-1233), SBA (AL-1013) and DBA (AL-1033).
Reagents were purchased as indicated: protease inhibitor cocktail (P8340, Sigma-Aldrich), SYPRO® Ruby protein gel stain (S12000), 1-Step Ultra TMB-ELISA Substrate solution (34028), EZ-Link® Sulfo-NHS-LC-Biotin (21335) (Thermo Fisher Scientific, Waltham, MA), DMNJ (EMD Millipore), Avidin/Biotin blocking kit (SP-2001), Mouse on Mouse (M.O.M.) Biotinylated Anti-Mouse Ig Reagent (MKB-2225), VECTASHIELD™ Antifade Mounting Medium (Vector) and Texas Red conjugated α-bungarotoxin (α-BTX; 00015, Biotium, Hayward, CA).
Immunofluorescence and lectin fluorescence assays on tissue sections
Quadricep sections were mounted in 10.2% polyvinyl alcohol, 4.3% polyethylene glycol, flash frozen in liquid nitrogen-cooled isopentane and stored at −80°C. Transverse sections of 8 µm were mounted on positively charged glass slides (Thermo Fisher Scientific). For immunofluorescence assays, sections were brought to room temperature for 15 min, blocked in 3% BSA in 1× phosphate-buffered saline (1× PBS) for 1 h at room temperature, and blocked with Avidin/Biotin blocking kit according to manufacturer's instructions. Non-specific binding of mouse antibodies was blocked with M.O.M. kit. Sections were incubated in primary antibodies against utrophin (MANCHO3 1:5) in M.O.M. diluent solution overnight at 4°C. Following overnight incubation, sections were rinsed with 1× PBS, bound primary antibodies were detected with secondary antibody biotinylated anti-mouse diluted in M.O.M. diluent (Vector Labs) and tertiary fluorescein-conjugated avidin D (10 µg/mL) in 1× PBS for 1 h at room temperature. NMJs were stained with α-BTX conjugated to Texas Red (0.3 µg/mL) during incubation with fluorescein-avidin D. Control sections were incubated in tertiary fluorescein-avidin alone. To prevent photo bleaching, sections were then mounted with VECTASHIELD™. Bound antibodies were visualized on an Olympus BX51 fluorescence microscope and Olympus DP2-BSW software (Olympus America Inc., Center Valley, PA).
For lectin fluorescence assays, sections were brought to room temperature for 15 min, blocked in 3% BSA in 1X PBS for 1 h at room temperature and blocked with Avidin/Biotin blocking kit according to manufacturer's instructions. Biotinylated WFA (4 µg/mL), VVA (4 µg/mL), SBA (10 µg/mL) and DBA (20 µg/mL) were diluted in 1× PBS and added to sections overnight at 4°C. Sections were washed three times with PBS, and bound lectin detected with fluorescein-avidin (10 µg/mL) for 1 h at room temperature; NMJs were stained with α-BTX conjugated to Texas Red (0.3 µg/mL) during incubation with fluorescein-avidin D. Control slides were incubated in fluorescein-avidin alone. Slides were mounted in VECTASHIELD™ to prevent photo bleaching. Bound lectin was visualized as stated above.
N-glycans present on tissue sections were enzymatically removed by treatment with bacterial PNGaseF (New England BioLabs, Ipswich, MA) after sections were brought to room temperature for 15 min. Sections were incubated with 0.5 μL PNGaseF in 20 µL total reaction buffer for 4 h in a 37°C water bath, refreshing reaction buffer when necessary to guarantee sections did not dry out. Control-treated sections were incubated with reaction buffer alone. Following treatment with PNGaseF, sections were blocked and stained with lectins per above, as well as with the lectin PHA-L (2 µg/mL).
Lectin binding assays
Of a 96-well plate, 1.5 × 103 cells were plated per well. Cells were grown for 4 days, refreshing growth media every 2 days until cells were 90–100% confluent. Once confluent, cells were differentiated to form myotubes, refreshing differentiation media every 2 days until harvested. Cells were collected at the time of differentiation (day 0), and 2, 4 and 7 days following differentiation. At the appropriate time point, cells were rinsed with ice-cold 1× PBS and fixed overnight in 2% paraformaldehyde in PBS at 4°C. Non-specific binding to cells was blocked by incubating with 1% BSA in PBS for 1 h at room temperature. Triplicate wells were incubated with the following biotinylated lectins overnight at 4°C: WFA (0.5 µg/mL), VVA (7.5 µg/mL), SBA (0.5 µg/mL), DBA (7.5 µg/mL), PNA (2 µg/mL), Jac (2.5 µg/mL), WGA (0.2 µg/mL), SNA (0.5 µg/mL), MAA-II (1.25 µg/mL), PHA-L (0.1 µg/mL) and Con A (5 ng/mL). Control wells were incubated with biotinylated BSA (7.5 µg/mL). Following overnight incubation, unbound lectins were removed by washing with 1% BSA/0.1% Tween-20/1× PBS solution. Wells were incubated with SA-HRP (50 ng/mL) with bound lectins detected using 1-Step Ultra TMB-ELISA Substrate solution per manufacturer's instructions with a colorimetric spectrophotometer (Bio-Rad Benchmark Plus) at 450 nm.
C2C12 cell surface biotinylation
Per 10 cm dish, 2.5 × 105 cells were plated and grown until 90–100% confluent for approximately 4 days, refreshing growth media every 2 days. Once confluent, cells were differentiated to form myotubes, refreshing differentiation media every 2 days. Seven days following differentiation, cells were rinsed twice with ice-cold 1× PBS (pH 8.0) and incubated in 5 mL of 1 mg/mL EZ-Link® Sulfo-NHS-LC-Biotin in 1× PBS (pH 8.0) for 1 h at room temperature. The biotinylation reaction was quenched by rinsing twice with 5 mL of ice-cold 100 mM glycine in 1× PBS (pH 8.0). Cells were then immediately used for lectin precipitations.
Lectin precipitation
Biotinylated cells were scraped into 200 μL ice-cold Nonidet P-40 lysis buffer (NP-40 lysis buffer) consisting of 50 mM Tris-HCl, pH 7.4, 1% Nonidet P-40, 150 mM NaCl and 5 mM EDTA, homogenized with a dounce homogenizer, rotated for 30 min at 4°C and clarified via centrifugation at 13,000 rpm for 20 min at 4°C. Quadriceps from wild-type murine muscle were snap frozen in liquid nitrogen, ground to a powder using a mortar and pestle and added to NP-40 lysis buffer. Wild-type murine lysates were homogenized, rotated and clarified per above. Protein concentration was quantified by Pierce™ BCA Protein Assay kit (Life Technologies). Lectin-agarose beads were prepared by washing with ice-cold NP-40 lysis buffer. Prepared beads were loaded with 100 µg of whole-cell lysate plus 5 µL protease inhibitor cocktail adjusting the total volume to 350 µL with NP-40 lysis buffer. Beads were rotated top over bottom overnight at 4°C. Following precipitation, beads were washed three times with ice-cold NP-40 lysis buffer and protease inhibitor cocktail (Sigma-Aldrich). Precipitated proteins were denatured in NuPAGE sample buffer and reducing agent (Life Technologies), and half of the precipitated proteins were separated on a NuPAGE 3–8% Tris Acetate polyacrylamide gel (Life Technologies) before transferring to nitrocellulose membrane for immunoblotting or staining with SYPRO Ruby protein gel stain.
UGA glycotranscriptome analysis
2.5 × 105 cells were plated, grown until 90–100% confluent and differentiated as above. Cells were collected at the time of differentiation (day 0) or 48 h post differentiation (day 2) by lifting with Trypsin/EDTA (Life Technologies). Cells were pelleted for 5 min at 1200 rpm, rinsed twice with 1× PBS and snap frozen in liquid nitrogen. RNA isolation, cDNA synthesis and transcript abundance determination for 580 glycan-related genes were performed as previously described (Nairn et al. 2008).
siRNA-mediated knockdown
Cells were used for transfection when they reached approximately 65% confluency at which point 1 × 106 cells were resuspended in 100 μL of Nucleofector™ solution V (Lonza, Basel, Switzerland). Individual batches of cells were transfected via Amaxa™ Nucleofector II™ (Lonza) with optimized concentrations of siRNA to β4Galnt3 (1 μM), β3Galnt2 (0.3 μM) and Neu2 (0.3 μM) (Santa Cruz Biotechnology). Control cells were transfected with 1 μM scramble siRNA (Santa Cruz Biotechnology). Transfected cells were resuspended to a concentration of 1 × 105 cells/mL growth media and plated at 5 × 104 per well for lectin binding assays or at 7.5 × 105 cells/well of a 12-well plate. Cells were switched to differentiation media 4 h after plating. Samples were collected for analysis of transcript expression level via quantitative real-time RT-PCR (qRT-PCR) following 2 days of differentiation while samples for lectin binding assays were collected 4 days after differentiation.
Quantitative real-time RT-PCR
RNA from cells was isolated via RNeasy Mini kit per manufacturer's instructions including digestion of genomic DNA. cDNA was synthesized via qScript™ XLT cDNA SuperMix (Quantabio, Beverly, MA). cDNA was amplified with specific primers via LightCycler® 480 SYBR Green I Master reagent (Roche, Indianapolis, IN) with the LightCycler® 480 (Roche) and its detection software. The following primer sets were ordered from Integrated DNA Technologies (IDT, Coralville, IA): β4Galnt3 (Forward: CCA AGA AAT TGC CCG AAG TAG G; Reverse: GGA GGC CAT AGT TGG TCC A), β3Galnt2 (Forward: GTC TTG GAG TGT TCT ACG ATG C; Reverse: ACT GAA TCG GGC GAT GAA AAG), Neu2 (Forward: CAC AGG CGT CCA TGC TTA CA; Reverse: CTG CGT GCT CAT CCG TCT T), Gapdh (Forward: AGG TCG GTG TGA ACG GAT TTG; Reverse: TGT AGA CCA TGT AGT TGA GGT CA). Expression of the gene of interest was normalized to expression of the housekeeping gene Gapdh.
Glycosylation Inhibitor treatment (DMNJ)
Deoxymannojirimycin (2 mM, DMNJ) or vehicle control was added to differentiation media of cells on day 4 of differentiation. Cells were treated for 72 h and analyzed by lectin binding assay or lectin precipitation as described above.
CFG glycan array analysis
For each lectin, the relative fluorescent units (RFU) for the most highly bound glycan was identified and all glycans with an average RFU of 10% or more of the highest bound glycan were compared. For example, the highest RFU for WFA was 54,879.56, therefore all glycans with an RFU in excess of 5488 were compared. While multiple entries for each lectin may exist in the CFG database, only those manufactured by Vector Laboratories and assayed at 10 µg/mL were included in our analysis. At this lectin concentration, RFU for all four lectins was within 3- to 4-fold of the maximum.
Supplementary data
Supplementary data for this article is available online at http://glycob.oxfordjournals.org/.
Acknowledgements
The authors would like to thank Sandra Thiemann and Katrin Schaefer (UCLA, Baum lab) for their intellectual input and discussion, as well as Grace Hong (UCLA, Crosbie-Watson lab) for muscle tissue sample preparation.
Funding
The Muscular Dystrophy Association (RG 135449 to L.G.B., RG 274143 to R.C.W.); the National Institutes of Health (NIH P30 AR057230 and NIH/CATS UCLA CTSI UL1TR000124 subprojects to L.G.B., and R.C.W. NIH R01 AR048179 to R.C.W., GM103490 to K.M.); the National Research Service Award GM07104, Edith Hyde Fellowship and Eureka Pre-doctoral Training Fellowship (to J.L.M.); Ruth L. Kirschstein National Research Service Award from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (T32AR059033 to J.L.M. and E.M.G.) and the Broad Stem Cell Institute and the Center for Duchenne Muscular Dystrophy at UCLA.
Conflict of interest statement
None declared.
Abbreviations
α7−/−, alpha-7-integrin-null; α-BTX, alpha-bungarotoxin; α-DG, alpha-dystroglycan; α-SG, alpha-sarcoglycan; β3Galnt2, beta-1,3-N-acetylgalactosaminyltransferase 2; β4Galnt3, beta-1,4-N-acetylgalactosaminyltransferase 3; β-DG, beta-dystroglycan; BSA, bovine serum albumin; CFG, Consortium for Functional Glycomics; CMAH, cytidine monophosphate-N-acetylneuraminic acid hydroxylase; Con A, concanavalin A; DBA, Dolichos biflorus agglutinin; DGC, dystrophin–glycoprotein complex; DMD, Duchenne muscular dystrophy; DMNJ, deoxymannojirimycin; ECM, extracellular matrix; gal-1, galectin-1; GalNAc, N-acetylgalactosamine; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GlcNAc, N-acetylglucosamine; Jac, Jacalin; LARGE, like-glycosyltransferase; MAA-II, Maackia amurensis agglutinin-II; MTJ, myotendinous junction; Neu2, sialidase 2; NMJ, neuromuscular junction; PBS, phosphate-buffered saline; PHA-L, Phaseolus vulgaris Leucoagglutinin; PNA, peanut agglutinin; PNGaseF, Peptide-N-glycosidase F; RFU, relative fluorescent unit; SBA, soybean agglutinin; SNA, Sambucus nigra agglutinin; UGC, utrophin–glycoprotein complex; Utr−/−, utrophin-null; VVA, Vicia villosa agglutinin; WFA, Wisteria floribunda agglutinin; WGA, wheat germ agglutinin.
References
- Abbott KL, Nairn AV, Hall EM, Horton MB, McDonald JF, Moremen KW, Dinulescu DM, Pierce M.. 2008. Focused glycomic analysis of the N-linked glycan biosynthetic pathway in ovarian cancer. Proteomics. 8:3210–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta AR, Yilmaz A, Bogdanovich S, McKechnie BA, Abedi M, Khurana TS, Fallon JR.. 2011. Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci USA. 108:762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin DJ, Kaufman SJ.. 1999. The alpha7beta1 integrin in muscle development and disease. Cell Tissue Res. 296:183–190. [DOI] [PubMed] [Google Scholar]
- Cabrera PV, Amano M, Mitoma J, Chan J, Said J, Fukuda M, Baum LG.. 2006. Haploinsufficiency of C2GnT-I glycosyltransferase renders T lymphoma cells resistant to cell death. Blood. 108:2399–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera PV, Pang M, Marshall JL, Kung R, Nelson SF, Stalnaker SH, Wells L, Crosbie-Watson RH, Baum LG.. 2012. High throughput screening for compounds that alter muscle cell glycosylation identifies new role for N-glycans in regulating sarcolemmal protein abundance and laminin binding. J Biol Chem. 287:22759–22770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekharan K, Yoon JH, Xu Y, deVries S, Camboni M, Janssen PM, Varki A, Martin PT.. 2010. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci Transl Med. 2:42ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HH, Takematsu H, Diaz S, Iber J, Nickerson E, Wright KL, Muchmore EA, Nelson DL, Warren ST, Varki A.. 1998. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci USA. 95:11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu FK. 1986. Requirements of cleavage of high mannose oligosaccharides in glycoproteins by peptide N-glycosidase F. J Biol Chem. 261:172–177. [PubMed] [Google Scholar]
- Clerk A, Morris GE, Dubowitz V, Davies KE, Sewry CA.. 1993. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J. 25:554–561. [PubMed] [Google Scholar]
- Cummings RD, Kornfeld S.. 1982. Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J Biol Chem. 257:11230–11234. [PubMed] [Google Scholar]
- Endo T. 2015. Glycobiology of α-dystroglycan and muscular dystrophy. J Biochem. 157:1–12. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Burwell AL, Geissler AL.. 1997. Tissue-specific heterogeneity in alpha-dystroglycan sialoglycosylation. Skeletal muscle alpha-dystroglycan is a latent receptor for Vicia villosa agglutinin b4 masked by sialic acid modification. J Biol Chem. 272:22315–22321. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP.. 1993. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann U, Bause E, Legler G, Ploegh H.. 1984. Novel mannosidase inhibitor blocking conversion of high mannose to complex oligosaccharides. Nature. 307:755–758. [DOI] [PubMed] [Google Scholar]
- Gasimli L, Hickey AM, Yang B, Li G, dela Rosa M, Nairn AV, Kulik MJ, Dordick JS, Moremen KW, Dalton S, et al. 2014. Changes in glycosaminoglycan structure on differentiation of human embryonic stem cells towards mesoderm and endoderm lineages. Biochim Biophys Acta. 1840:1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenaway PJ, LeVine D.. 1973. Binding of N-acetyl-neuraminic acid by wheat-germ agglutinin. Nat New Biol. 241:191–192. [DOI] [PubMed] [Google Scholar]
- Gundry RL, Raginski K, Tarasova Y, Tchernyshyov I, Bausch-Fluck D, Elliott ST, Boheler KR, Van Eyk JE, Wollscheid B.. 2009. The mouse C2C12 myoblast cell surface N-linked glycoproteome: identification, glycosite occupancy, and membrane orientation. Mol Cell Proteomics. 8:2555–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg C, Oldfors A, Darin N.. 2014. B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations. Eur J Hum Genet. 22:707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Suzuki O, Wakabayashi S.. 2013. Decreased surface sialic acid content is a sensitive indicator of muscle damage. Muscle Nerve. 47:372–378. [DOI] [PubMed] [Google Scholar]
- James M, Simmons C, Wise CJ, Jones GE, Morris GE.. 1995. Evidence for a utrophin-glycoprotein complex in cultured cell lines and a possible role in cell adhesion. Biochem Soc Trans. 23:398S. [DOI] [PubMed] [Google Scholar]
- Kanagawa M, Michele DE, Satz JS, Barresi R, Kusano H, Sasaki T, Timpl R, Henry MD, Campbell KP.. 2005. Disruption of perlecan binding and matrix assembly by post-translational or genetic disruption of dystroglycan function. FEBS Lett. 579:4792–4796. [DOI] [PubMed] [Google Scholar]
- Konami Y, Yamamoto K, Osawa T, Irimura T.. 1994. Strong affinity of Maackia amurensis hemagglutinin (MAH) for sialic acid-containing Ser/Thr-linked carbohydrate chains of N-terminal octapeptides from human glycophorin A. FEBS Lett. 342:334–338. [DOI] [PubMed] [Google Scholar]
- Lee EU, Roth J, Paulson JC.. 1989. Alteration of terminal glycosylation sequences on N-linked oligosaccharides of Chinese hamster ovary cells by expression of beta-galactoside alpha 2,6-sialyltransferase. J Biol Chem. 264:13848–13855. [PubMed] [Google Scholar]
- Live D, Wells L, Boons GJ.. 2013. Dissecting the molecular basis of the role of the O-mannosylation pathway in disease: α-dystroglycan and forms of muscular dystrophy. Chembiochem. 14:2392–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotan R, Skutelsky E, Danon D, Sharon N.. 1975. The purification, composition, and specificity of the anti-T lectin from peanut (Arachis hypogaea). J Biol Chem. 250:8518–8523. [PubMed] [Google Scholar]
- Magalhães A, Marcos-Pinto R, Nairn AV, Dela Rosa M, Ferreira RM, Junqueira-Neto S, Freitas D, Gomes J, Oliveira P, Santos MR, et al. 2015. Helicobacter pylori chronic infection and mucosal inflammation switches the human gastric glycosylation pathways. Biochim Biophys Acta. 1852:1928–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JL, Holmberg J, Chou E, Ocampo AC, Oh J, Lee J, Peter AK, Martin PT, Crosbie-Watson RH.. 2012. Sarcospan-dependent Akt activation is required for utrophin expression and muscle regeneration. J Cell Biol. 197:1009–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Camboni M, Xu R, Golden B, Chandrasekharan K, Wang CM, Varki A, Janssen PM.. 2013. N-Glycolylneuraminic acid deficiency worsens cardiac and skeletal muscle pathophysiology in α-sarcoglycan-deficient mice. Glycobiology. 23:833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Kaufman SJ, Kramer RH, Sanes JR.. 1996. Synaptic integrins in developing, adult, and mutant muscle: selective association of alpha1, alpha7A, and alpha7B integrins with the neuromuscular junction. Dev Biol. 174:125–139. [DOI] [PubMed] [Google Scholar]
- McDearmon EL, Combs AC, Ervasti JM.. 2001. Differential Vicia villosa agglutinin reactivity identifies three distinct dystroglycan complexes in skeletal muscle. J Biol Chem. 276:35078–35086. [DOI] [PubMed] [Google Scholar]
- Mega T, Oku H, Hase S.. 1992. Characterization of carbohydrate-binding specificity of concanavalin A by competitive binding of pyridylamino sugar chains. J Biochem. 111:396–400. [DOI] [PubMed] [Google Scholar]
- Meier T, Ruegg MA.. 2000. The role of dystroglycan and its ligands in physiology and disease. News Physiol Sci. 15:255–259. [DOI] [PubMed] [Google Scholar]
- Monti E, Preti A, Nesti C, Ballabio A, Borsani G.. 1999. Expression of a novel human sialidase encoded by the NEU2 gene. Glycobiology. 9:1313–1321. [DOI] [PubMed] [Google Scholar]
- Nairn AV, York WS, Harris K, Hall EM, Pierce JM, Moremen KW.. 2008. Regulation of glycan structures in animal tissues: transcript profiling of glycan-related genes. J Biol Chem. 283:17298–17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HH, Jayasinha V, Xia B, Hoyte K, Martin PT.. 2002. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci USA. 99:5616–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik SK, Stanley P.. 2005. Mouse large can modify complex N- and mucin O-glycans on alpha-dystroglycan to induce laminin binding. J Biol Chem. 280:20851–20859. [DOI] [PubMed] [Google Scholar]
- Praissman JL, Live DH, Wang S, Ramiah A, Chinoy ZS, Boons GJ, Moremen KW, Wells L.. 2014. B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. Elife. 3. DOI:10.7554/eLife.03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praissman JL, Willer T, Sheikh MO, Toi A, Chitayat D, Lin YY, Lee H, Stalnaker SH, Wang S, Prabhakar PK, et al. 2016. The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife. 5. DOI:10.7554/eLife.14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Cheney JM.. 1982. Lectin binding reveals a synapse-specific carbohydrate in skeletal muscle. Nature. 300:646–647. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Bacou F, Sanes JR.. 1988. A synapse-specific carbohydrate at the neuromuscular junction: association with both acetylcholinesterase and a glycolipid. J Neurosci. 8:932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Goldstein IJ, Broekaert WF, Nsimba-Lubaki M, Peeters B, Peumans WJ.. 1987. The elderberry (Sambucus nigra L.) bark lectin recognizes the Neu5Ac(alpha 2-6)Gal/GalNAc sequence. J Biol Chem. 262:1596–1601. [PubMed] [Google Scholar]
- Song WK, Wang W, Foster RF, Bielser DA, Kaufman SJ.. 1992. H36-alpha 7 is a novel integrin alpha chain that is developmentally regulated during skeletal myogenesis. J Cell Biol. 117:643–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens E, Carss KJ, Cirak S, Foley AR, Torelli S, Willer T, Tambunan DE, Yau S, Brodd L, Sewry CA, et al. 2013. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am J Hum Genet. 92:354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG. 1991. Force transmission across muscle cell membranes. J Biomech. 24(Suppl 1):43–52. [DOI] [PubMed] [Google Scholar]
- Tintignac LA, Brenner HR, Rüegg MA.. 2015. Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol Rev. 95:809–852. [DOI] [PubMed] [Google Scholar]
- Van Ry PM, Wuebbles RD, Key M, Burkin DJ.. 2015. Galectin-1 protein therapy prevents pathology and improves muscle function in the mdx mouse model of Duchenne muscular dystrophy. Mol Ther. 23:1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Mark H, Williams I, Wendler O, Sorokin L, von der Mark K, Pöschl E.. 2002. Alternative splice variants of alpha 7 beta 1 integrin selectively recognize different laminin isoforms. J Biol Chem. 277:6012–6016. [DOI] [PubMed] [Google Scholar]
- Willer T, Inamori K, Venzke D, Harvey C, Morgensen G, Hara Y, Beltrán Valero de Bernabé D, Yu L, Wright KM, et al. 2014. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated α-dystroglycan functional glycosylation. Elife. 3. DOI:10.7554/eLife.03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AM, Wu JH, Lin LH, Lin SH, Liu JH.. 2003. Binding profile of Artocarpus integrifolia agglutinin (Jacalin). Life Sci. 72:2285–2302. [DOI] [PubMed] [Google Scholar]
- Xia B, Hoyte K, Kammesheidt A, Deerinck T, Ellisman M, Martin PT.. 2002. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol. 242:58–73. [DOI] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, Oldstone MB, Schachter H, Wells L, Campbell KP.. 2010. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 327:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Hu H.. 2012. Differential glycosylation of α-dystroglycan and proteins other than α-dystroglycan by like-glycosyltransferase. Glycobiology. 22:235–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.