

Abstract
The VEGF/VEGFR2/Akt/eNOS/NO pathway is essential to VEGF-induced angiogenesis. We have previously discovered a novel role of calpain in mediating VEGF-induced PI3K/AMPK/Akt/eNOS activation through Ezrin. Here, we sought to identify possible feedback regulation of VEGFR2 by calpain via its substrate protein phosphotyrosine phosphatase 1B (PTP1B), and the relevance of this pathway to VEGF-induced angiogenesis, especially in diabetic wound healing. Overexpression of PTP1B inhibited VEGF-induced VEGFR2 and Akt phosphorylation in bovine aortic endothelial cells, while PTP1B siRNA increased both, implicating negative regulation of VEGFR2 by PTP1B. Calpain inhibitor ALLN induced VEGFR2 activation, which can be completely blocked by PTP1B overexpression. Calpain activation induced by overexpression or Ca/A23187 resulted in PTP1B cleavage, which can be blocked by ALLN. Moreover, calpain activation inhibited VEGF-induced VEGFR2 phosphorylation, which can be restored by PTP1B siRNA. These data implicate calpain/PTP1B negative feedback regulation of VEGFR2, in addition to the primary signaling pathway of VEGF/VEGFR2/calpain/PI3K/AMPK/Akt/eNOS. We next examined a potential role of PTP1B in VEGF-induced angiogenesis. Endothelial cells transfected with PTP1B siRNA showed faster wound closure in response to VEGF. Aortic discs isolated from PTP1B siRNA-transfected mice also had augmented endothelial outgrowth. Importantly, PTP1B inhibition and/or calpain overexpression significantly accelerated wound healing in STZ-induced diabetic mice. In conclusion, our data for the first time demonstrate a calpain/PTP1B/VEGFR2 negative feedback loop in the regulation of VEGF-induced angiogenesis. Modulation of local PTP1B and/or calpain activities may prove beneficial in the treatment of impaired wound healing in diabetes.
Keywords: Akt PKB, angiogenesis, calpain, diabetes, vascular endothelial growth factor (VEGF), wound healing, PTP1B, VEGFR2
Introduction
Vascular endothelial growth factor (VEGF)2 is one of the main initiators of angiogenesis. We and others have previously shown that calpain activity is increased within 10 min of VEGF stimulation of endothelial cells, which can be inhibited by antagonist of the VEGF receptor (1–4). In addition, we have demonstrated an important role of calpain in mediating VEGF-induced PI3K/AMPK/Akt activation and consequent eNOS phosphorylation and nitric oxide (NO) production (1). Treatments with calpain 2 siRNA or inhibitors of calpain, calpeptin or ALLN, decreased VEGF-induced Akt phosphorylation and eNOS/NO activation (1). However, whether there is any potential feedback regulation of VEGF/VEGF receptor 2 (VEGFR2) signaling by calpain remains unknown.
Protein phosphotyrosine phosphatase 1B (PTP1B) is a non-receptor phosphotyrosine protein phosphatase. Previous studies have shown that PTP1B is cleaved by calpain at the C terminus, resulting in enhanced phosphatase activity (5–7). Furthermore, PTP1B binds to and de-phosphorylates VEGFR2 through direct protein-protein interaction (8). These studies suggest a possible calpain/PTP1B regulation on VEGFR2 phosphorylation.
It has been reported that mice overexpressing calpastatin, a natural calpain inhibitor, have impaired angiogenesis and delayed wound healing (4, 9). On the other hand, there is evidence showing that PTP1B serves as a negative regulator of VEGF-activated ERK signaling in endothelial cells (8, 10). These observations implicate that calpain/PTP1B may be involved in the regulation of VEGF-induced angiogenesis. Therefore, the present study was designed to identify potential calpain/PTP1B-dependent regulation of VEGFR2 and its roles in angiogenesis. It is known that impaired angiogenesis leads to delayed wound healing and diabetic foot ulcers, which affect 15% of diabetic patients and claim 84% of diabetes-related lower limb amputations (11). We therefore investigated potential impact of PTP1B on diabetic wound healing.
In this study, we demonstrated that calpain inhibitor increased VEGF-induced VEGFR2 phosphorylation through PTP1B, suggesting a negative feedback loop of calpain/PTP1B/VEGFR2. Secondly, we found that PTP1B knockdown significantly enhanced VEGF-induced Akt phosphorylation and angiogenesis. In agreement with these findings, the impairment in wound healing in streptozotocin (STZ)-induced diabetic mice was deteriorated by topical delivery of overexpressed PTP1B or calpain inhibitor. On the other hand, accelerated diabetic wound healing was observed in calpain overexpressed wounds or PTP1B inhibitor treated wounds. Taken together, our data for the first time revealed a negative feedback loop of calpain/PTP1B/VEGFR2 and its role in VEGF signaling and angiogenesis. Our data also implicate the exciting possibility that local therapies targeting calpain or PTP1B may offer new avenues to treat defective wound healing in diabetes.
Results
Effects of PTP1B Overexpression on VEGF-induced Phosphorylation of VEGFR2 and Akt
Our previous data showed robust calpain activation in VEGF-treated bovine aortic endothelial cells (BAECs), which can be abolished by pretreatment with calpain inhibitor calpeptin or ALLN (1) or VEGF receptor inhibitor 4-[(4′-chloro-2′-fluoro) phenylamino]-6,7-dimethoxyquinazoline (2). Knocking down calpain inhibited VEGF-induced NO production in endothelial cells (1). To further investigate the role of calpain in potential feedback regulations of VEGFR2, we treated BAECs with calpain inhibitor, ALLN. Interestingly, we found that pretreatment with ALLN (30 μm, 30 min) increased VEGF-induced VEGFR2 phosphorylation (Tyr-1175) (Fig. 1), implicating a role of calpain in negative feedback regulation of VEGFR2.
FIGURE 1.

PTP1B overexpression inhibits VEGF-induced phosphorylation of VEGFR2 and Akt. ALLN further increased VEGF-induced VEGFR2 phosphorylation via inhibition of calpain/PTP1B axis, which is however absent in the presence of PTP1B overexpression. Bovine aortic endothelial cells (BAECs) were transfected with wild type PTP1B (HA-PTP1B) or empty vector (pCMV-HA). The cells were treated with ALLN (30 μm) for 30 min prior to VEGF stimulation (100 ng/ml, 5 min). A, representative Western blots of indicated proteins. B and C, grouped densitometric data after normalization to total-VEGFR2 or β-actin (n = 4). PTP1B overexpression inhibited VEGF-induced phosphorylation of VEGFR2 and Akt. *, p < 0.05; **, p < 0.01. NS, not significant.
To test the effect of PTP1B on VEGFR2 and Akt phosphorylation, we first overexpressed wild type bovine PTP1B in BAECs. VEGF-induced VEGFR2 and Akt phosphorylation was dramatically reduced by wild type PTP1B overexpression (Fig. 1). PTP1B also potently inhibited ALLN-induced VEGFR2 phosphorylation (Fig. 1B), suggesting that PTP1B may lie downstream of calpain in regulating VEGFR2 phosphorylation.
PTP1B siRNA Increases VEGF-induced VEGFR2 and Akt Phosphorylation
Next, we examined whether RNAi inhibition of endogenous PTP1B can up-regulate VEGF-induced VEGFR2 and Akt activation. Of note, down-regulation of PTP1B expression in BAECs significantly increased VEGF-induced VEGFR2 and Akt phosphorylation (Fig. 2). Limiting PTP1B protein expression by siRNA could not further activate VEGFR2 in the presence of ALLN (Fig. 2B). These data indicate that PTP1B shares the same pathway as ALLN, suggesting that calpain/PTP1B is responsible for the negative feedback regulation of VEGFR2.
FIGURE 2.

PTP1B siRNA increases VEGF-induced VEGFR2 and Akt phosphorylation. ALLN further increased VEGF-induced VEGFR2 phosphorylation via inhibition of calpain/PTP1B axis, while limiting Akt phosphorylation via inhibition of calpain/PI3K/Akt pathway. BAECs were transfected with 50 pmol control siRNA and siRNA targeting bovine PTP1B. The cells were treated with ALLN and VEGF as described in Fig. 1. A, representative Western blots of indicated proteins. B and C, grouped densitometric data after normalization to total VEGFR2 or β-actin (n = 4). *, p < 0.05; **, p < 0.01. NS, not significant.
We also found that ALLN inhibits VEGF-induced phosphorylation of Akt in BAECs (Fig. 2C), which is consistent with our previous report (1). Interestingly, in the presence of PTP1B siRNA, ALLN was able to further decrease Akt phosphorylation (Fig. 2C) while not changing VEGFR2. These data demonstrate that VEGFR2 is downstream of PTP1B while Akt is downstream of calpain.
Calpain Activation Inhibits VEGF-induced VEGFR2 Phosphorylation through PTP1B
To examine calpain activation of PTP1B, we used HEK 293T cells to study the cleavage of endogenous PTP1B by calpain activation. It is reported that full-length PTP1B (50 kD) undergoes cleavage at the C terminus by calpain and releases the active form of PTP1B (42 kD) (5–7). Firstly, we overexpressed plasmids of FLAG-tagged human calpain 1 and calpain 2 (Addgene plasmids #60941 and #60942) in HEK 293T cells. Overexpression of calpain 1 and 2 induced obvious PTP1B cleavage compared to empty vector control (Fig. 3, A and C). At the same time, this cleavage can be blocked by ALLN treatment, indicating that calpain activity is required to induce PTP1B cleavage. The expression level of full-length PTP1B was not significantly changed by calpain overexpression or ALLN treatment (Fig. 3B), suggesting that only a small amount of PTP1B undergoes cleavage. Secondly, we induced endogenous calpain activation by incubating the cells with calcium ionophore A23187 (1 μm) in the presence of 2 mm extracellular calcium (in the form of CaCl2) as previously described (5). As shown in Fig. 3D, calcium/A23187 (Ca/A23) induced robust PTP1B cleavage, which can also be inhibited by ALLN.
FIGURE 3.

Calpain activation induces PTP1B cleavage which can be blocked by ALLN. Cleavage of PTP1B in HEK 293T cells can be induced by either calpain overexpression, or endogenous activation of calpain by calcium and A23187. This cleavage can be blocked by ALLN, indicating that it is calpain dependent. A, HEK 293T cells were transfected with empty vector pCMV or Flag-tagged human calpain 1 and calpain 2 (CAPN1/2) (Addgene plasmids #60941 and #60942). The cells were treated with ALLN (30 μm) for 1.5 h before harvesting. B and C, grouped densitometric data of full-length and cleaved PTP1B expression normalized to β-actin (n = 3). D, confluent HEK 293T cells were treated with ALLN for 30 min, followed by stimulation with CaCl2 (2 mm) and A23187 (1 μm) for 1 h. E and F, grouped densitometric data after normalization to β-actin (n = 3). *, p < 0.05; **, p < 0.01. Arrows indicate full-length PTP1B protein. Asterisks indicate cleaved PTP1B under long exposure condition.
We next applied Ca/A23 to induce calpain activation in BAECs to study the regulation of VEGFR2. BAECs were treated with Ca/A23 at the same concentration as mentioned above. VEGF-induced VEGFR2 phosphorylation was significantly decreased by Ca/A23 treatment (Fig. 4), suggesting that calpain activation decreases VEGF-induced VEGFR2 phosphorylation. However, ALLN pre-treatment abolished the effect of Ca/A23, demonstrating that calpain activity is required to regulate VEGFR2. Of note, PTP1B siRNA also restored VEGFR2 phosphorylation in the presence of Ca/A23, indicating that calpain activation inhibits VEGF-induced VEGFR2 phosphorylation through PTP1B.
FIGURE 4.
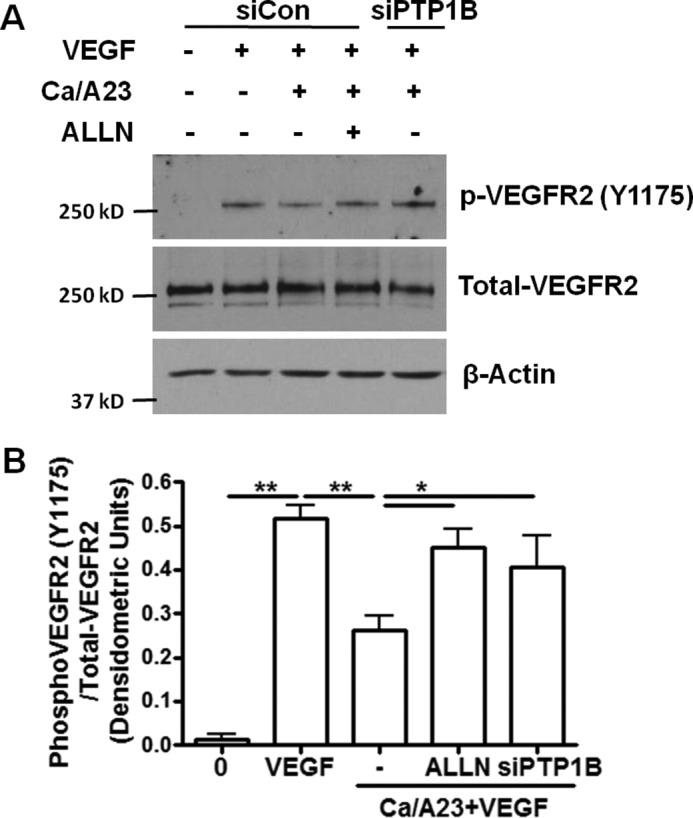
Calpain activation inhibits VEGF-induced VEGFR2 phosphorylation through PTP1B. Activation of endogenous calpain by calcium and A23187 results in diminished VEGF-induced VEGFR2 phosphorylation in BAECs, which can be restored by ALLN and PTP1B siRNA. These results indicate that calpain regulates VEGFR2 through its proteolysis activity and substrate PTP1B. A, BAECs were transfected with control siRNA or siRNA targeting PTP1B. The cells were treated with ALLN (30 μm, 30 min) prior to exposure to CaCl2 (2 mm) and A23187 (1 μm) for 1 h. Cells were harvested after exposure to VEGF for 5 min. B, grouped densitometric data after normalization to total-VEGFR2 (n = 3). *, p < 0.05; **, p < 0.01.
RNAi Inhibition of PTP1B Augments VEGF-induced Angiogenesis: Endothelial Cell Wound Closure Assay
VEGF regulates angiogenesis by activating VEGFR2 in endothelial cells. Knocking down VEGFR2 by siRNA completely blocked VEGF-induced Akt phosphorylation (Fig. 5). To investigate the effects of calpain/PTP1B feedback on VEGFR2 downstream signaling, we examined effects of PTP1B siRNA and ALLN on endothelial cell wound closure. Scratch wounds were made on endothelial monolayers transfected with control siRNA or PTP1B siRNA. PTP1B knockdown promoted wound healing in untreated and VEGF-treated groups (Fig. 6), which is consistent with our results of Akt phosphorylation. ALLN pretreatment diminished the angiogenic effect of VEGF on wound closure in both control and PTP1B inhibited cells. However, PTP1B siRNA treated cells did not show any further increase compared with control siRNA transfected cells in the presence of ALLN. These results showed that PTP1B knockdown improved, while calpain inhibition abolished, VEGF-induced endothelial wound closure. These data confirm an intermediate role of calpain in VEGF-induced angiogenesis (1). Taken together, these data (Figs. 1–4) also reveal a novel role of PTP1B inhibition in promoting VEGF-dependent angiogenesis via VEGFR2.
FIGURE 5.

VEGFR2 siRNA inhibits VEGF activation of Akt. VEGFR2 is required for VEGF-induced phosphorylation of Akt. BAECs were transfected with 50 pmol control siRNA or VEGFR2 siRNA. Then the cells were treated with VEGF (100 ng/ml, 5 min) and harvested. A, representative Western blots of indicated proteins. B and C, grouped densitometric data after normalization to total VEGFR2 or β-actin (n = 4). **, p < 0.01 as indicated.
FIGURE 6.

RNAi inhibition of PTP1B augments VEGF-induced angiogenesis: endothelial cell wound closure assay. RNAi inhibition of PTP1B increased basal and VEGF-induced wound closure in BAECs. ALLN inhibition of wound closure is not affected by PTP1B siRNA, indicating that PTP1B is downstream of calpain. A, scratch wounds were created on confluent BAECs transfected with control siRNA or PTP1B siRNA (50 pmol) at t = 0 h. Cells were then treated with ALLN (10 μm) for 30 min prior to stimulation with VEGF (100 ng/ml). Thirty-two hours later, images were taken at the same locations. B, migrated wound areas were analyzed by Image J. n = 3. *, p < 0.05 compared with control; ##, p < 0.01 compared with VEGF treated; $, p < 0.05; $$, p < 0.01 between indicated groups; NS, not significant.
RNAi Inhibition of PTP1B Augments Angiogenesis: Aortic Disc Assay
We have observed accelerated wound closure in PTP1B inhibited endothelial cells. Next, a mouse aortic disc angiogenesis assay was carried out. In vivo siRNA transfection was achieved by lipid-based transfection kit as previously described (12–14). siRNA targeting mouse PTP1B was found highly effective in attenuating PTP1B expression in mouse aortas (Fig. 7C). As shown in Fig. 7A, freshly isolated aortas were embedded in fibrin gel (converted from fibrinogen by thrombin) and treated with PBS, or VEGF and ALLN. Aortas isolated from mice with scrambled siRNA transfection had moderate capillary-like outgrowth of endothelial sprouts on day 5 (quantified as the areas covered by the migrated endothelial cells), which was increased by VEGF treatment. However, aortas isolated from mice transfected with PTP1B siRNA had significantly enhanced endothelial outgrowth (Fig. 7B). Of note, ALLN significantly inhibited the effects on endothelial outgrowth of VEGF and PTP1B knockdown.
FIGURE 7.

RNAi inhibition of PTP1B augments angiogenesis: aortic disc assay. RNAi inhibition of PTP1B increased growth of aortic sprouts at baseline and in response to VEGF. A, aortic discs (diameter, 2 mm) were prepared using freshly isolated aortas of wild type C57BL6 mice (12 weeks old) that were injected of control scrambled siRNA and mouse PTP1B siRNA. Aortic discs were pretreated with ALLN (10 μm, 30 min) prior to exposure to VEGF (100 ng/ml). Images were taken at day 5. B, areas of migrated sprouts were analyzed by Image J. n = 4. *, p < 0.05 compared with control; ## p < 0.01 compared with VEGF treated; $, p < 0.05; $$, p < 0.01 between indicated groups. C, representative Western blots demonstrating RNAi knockdown of PTP1B in mouse aortas. n = 3. **, p < 0.01 compared with control scrambled siRNA.
Calpain and PTP1B Inhibitor Accelerate Diabetic Wound Healing
In diabetic patients, impaired angiogenesis is associated with defective wound healing. We have found that PTP1B activity was significantly elevated in the skin wound areas of the STZ-induced diabetic mice (Fig. 8), implicating a possible role of PTP1B in diabetic wound healing. To investigate the physiological impact of PTP1B on angiogenesis especially wound healing in diabetes, we examined changes in wound healing of STZ-induced diabetic mice in response to PTP1B overexpression. As shown in Fig. 9, STZ-treated mice had impaired rate of wound healing compared to control mice, which is consistent with previous studies in STZ-induced model (15) and db/db mice (16). VEGF is known to accelerate wound healing, which is also confirmed by our data (Fig. 9, A and B). Five microgram of HA-PTP1B plasmid was mixed with lipid-based transfection reagents and injected intracutaneously near the wound bed to achieve overexpression (Fig. 9C). Interestingly, mice with PTP1B overexpression had delayed wound healing compared to VEGF-treated STZ mice, indicating an inhibitory effect of PTP1B on VEGF-induced angiogenesis. Calpain blockage by ALLN also decreased the rate of wound healing in the presence of VEGF. No further decrease was observed when treated with both PTP1B and ALLN.
FIGURE 8.
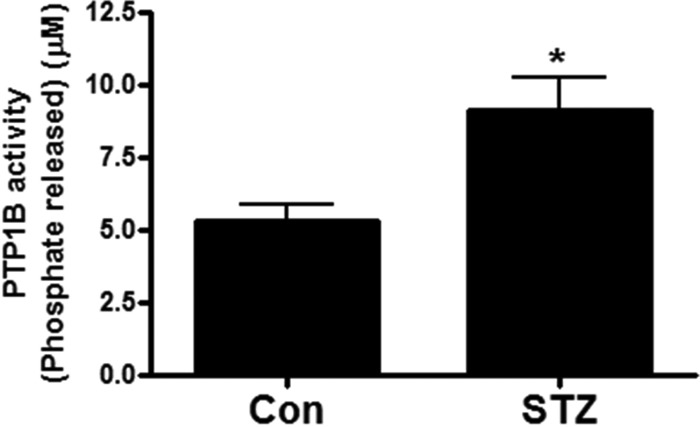
PTP1B activity is increased in diabetic skin wound. Wounded skin areas from STZ-induced diabetic mice have elevated PTP1B activity compared with those from control mice. Wounded skin areas were collected from control and STZ-treated mice on day 12 from wound healing assay in Fig. 9. PTP1B activity was determined by a colorimetric assay based on the cleavage of Tyrosine Phosphatase Substrate I. n = 4 and 6. *, p < 0.05 compared with control.
FIGURE 9.
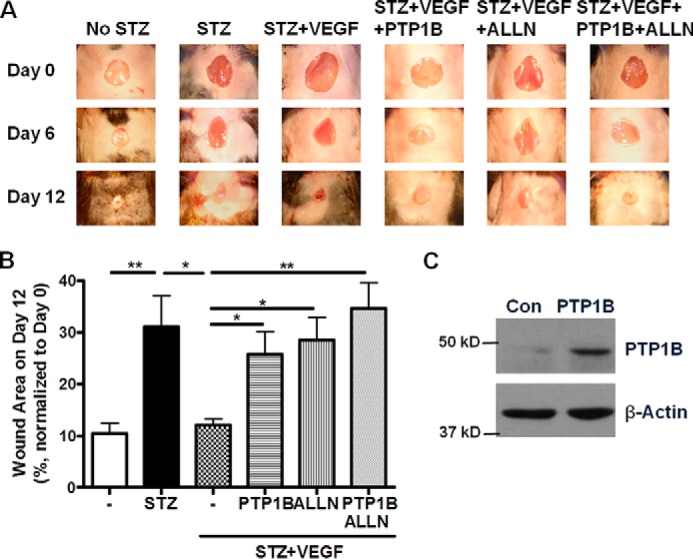
PTP1B or ALLN induces delay in diabetic wound healing. PTP1B overexpression and/or calpain inhibition delayed wound healing in diabetic mice. A, representative images of wound areas on day 0, 6 and 12 of STZ-induced diabetic mice as indicated. STZ-injected mice were treated with VEGF alone or in the presence of PTP1B plasmid and/or ALLN. B, wound areas on day 12 were analyzed and normalized to initial wound area on day 0. n = 3–5. *, p < 0.05; **, p < 0.01 as indicated. C, Western blot analysis of skin sample showing successful PTP1B overexpression by local transfection.
To investigate if PTP1B inhibition and/or calpain activation affect diabetic wound healing, we examined the effects of calpain overexpression and PTP1B inhibitor in the same diabetic wound healing model. Plasmids encoding human calpain 1 and calpain 2 cDNA (2.5 μg each, Addgene plasmids #60941 and #60942) were transfected into the wound area as mentioned above. A PTP1B inhibitor (Calbiochem) was also applied to the wound area with VEGF as described in “Experimental Procedures” (17, 18). Our results showed that both calpain overexpression and PTP1B inhibitor treatment further accelerated VEGF-induced wound healing in STZ induced diabetic mice (Fig. 10).
FIGURE 10.
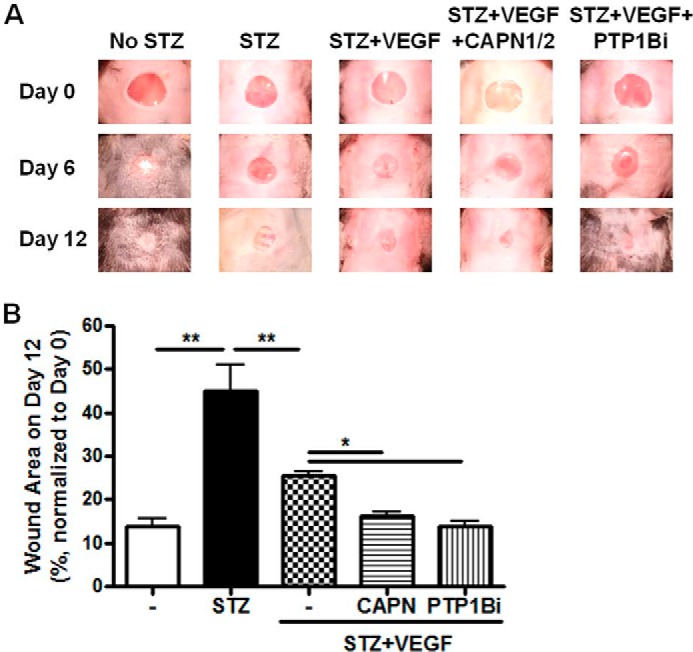
Calpain overexpression or inhibition of PTP1B accelerates wound healing. Calpain activation or PTP1B inhibition promotes diabetic wound healing. A, representative images of wound areas on day 0, 6, and 12 of STZ-induced diabetic mice. STZ-injected mice were treated with VEGF alone or in combination with human calpain plasmids or PTP1B inhibitor. B, wound areas on day 12 were analyzed and normalized to initial wound area on day 0. n = 4–6. *, p < 0.05; **, p < 0.01 as indicated. CAPN: overexpression with human calpain 1 and calpain 2 plasmids (Addgene plasmids #60941 and #60942). PTP1Bi: PTP1B inhibitor (Calbiochem).
Discussion
The present study reveals a novel negative feedback pathway of calpain/PTP1B/VEGFR2 in VEGF signaling, and its potential utilization for improving diabetic wound healing (Fig. 11). We demonstrated that PTP1B is involved in VEGF-dependent angiogenesis and diabetic wound healing. Specifically, 1) calpain/PTP1B mediate negative feedback regulation of VEGFR2 in response to VEGF stimulation; 2) PTP1B down-regulates VEGF-induced VEGFR2 and Akt phosphorylation, as revealed by both gain-of-function and loss-of-function approaches; 3) blocking PTP1B increases VEGF-induced angiogenesis; and 4) topically applied PTP1B inhibitor and/or calpain accelerates diabetic wound healing, indicating local modulation of PTP1B and/or calpain may serve as novel therapeutic solution to improve diabetic wound repair.
FIGURE 11.

Calpain/PTP1B/VEGFR2-feedback axis in VEGF-induced angiogenesis: Utilization for improving diabetic wound healing. Under normal conditions, VEGF activates calpain through VEGFR2, which is negatively regulated by calpain/PTP1B feedback axis. Genetic or pharmacological blockade of the feedback augments VEGF-induced downstream signaling, angiogenesis, and diabetic wound healing. Topical inhibition of PTP1B or activation of calpain may serve as novel therapeutics for impaired diabetic wound healing.
We and others have previously shown evidences of a VEGF/VEGFR2/calpain/PI3K/AMPK/Akt/eNOS/NO signaling pathway (1–4). In this study, we set out to investigate any calpain dependent feedback regulation of VEGFR2. Endothelial cells treated with calpain inhibitor, ALLN, had increased VEGF-induced VEGFR2 phosphorylation, suggesting a negative feedback of calpain on VEGFR2. Next, we found that the feedback regulation of VEGFR2 by calpain was mediated by PTP1B. As one of the substrates of calpain, PTP1B is activated by calpain-catalyzed proteolysis and subsequently de-phosphorylates VEGFR2 (5–8). In the presence of ALLN, PTP1B cleavage (activation) was inhibited (Fig. 3), and this resulted in less de-phosphorylation of VEGFR2 (Figs. 1, 2, and 4). To our knowledge, this is the first evidence of a negative feedback regulation of VEGFR2 by calpain/PTP1B.
VEGF is a crucial regulator of angiogenesis and vascular development during embryogenesis. Therefore, the VEGF signaling is under extremely delicate control. The calpain/PTP1B negative feedback regulation of VEGFR2 could represent a newly identified mechanism that prevents VEGFR2 from over-activation. Along the same line, VEGF/VEGFR2-induced Dll4/Notch signaling is able to down-regulate VEGFR2 expression in endothelial cells (19–21). On the other hand, positive feedback regulation on VEGFR2 has been reported in endothelial cells as well (22). VEGF-activated VEGFR2 nuclear translocation leads to interaction between VEGFR2 and Sp1. The complex subsequently binds to the Sp1-responsive region of the VEGFR2 promoter to enhance VEGFR2 transcription.
Among all the cascades triggered by VEGFR2, the PI3K/Akt pathway is of the most characterized that are important in mediating NO production and angiogenesis. However, the phosphorylation site of VEGFR2 for Akt activation is not well established. Some groups support that VEGF-induced Akt activation is not dependent on the Tyr-1175 site (10, 23). Point mutation of VEGFR2 at Y801F, not Y1175F, completely abolished VEGF-induced Akt-S473 phosphorylation and downstream NO production in BAECs, indicating that phosphorylation of VEGFR2 at Tyr-801 may lie specifically upstream of Akt/eNOS activation (23). However, our Western results showed very clear and robust decrease of VEGFR2-Y1175 and Akt-S473 phosphorylation in PTP1B overexpressed group, and corresponding increase in PTP1B siRNA treated BAECs (Figs. 1 and 2) as well as PTP1B inhibitor treated human aortic endothelial cells (data not shown). Similar effects on VEGFR2 and Akt were also reported with PTP1B inhibitor in human umbilical vein endothelial cells (24). RNAi knocking down Shb, a docking protein directly binding to Tyr-1175, impaired VEGF-induced PI3K activation (25). Therefore, our data and previous literatures demonstrate a PTP1B-dependent negative regulation of VEGFR2-Y1175 phosphorylation and Akt-S473 phosphorylation.
A main finding of this study is that PTP1B is a negative regulator of VEGF-induced angiogenesis in endothelial cells, consistent with the recently unraveled contribution of PTP1B in endothelial cell signaling (8, 10, 26). In a mouse hindlimb ischemic angiogenesis model, PTP1B expression and activity were increased (8). To investigate the angiogenic response to PTP1B inhibition, we employed wound closure assay using BAECs monolayer. In accordance with the results of increased phosphorylation of Akt, knockdown of PTP1B was associated with accelerated wound closure (Fig. 6). Furthermore, we examined sprouting outgrowth of freshly prepared aortic discs of PTP1B siRNA-treated mice, in the presence or absence of VEGF stimulation. Aortic disc assay is considered as an ex vivo angiogenesis assay, providing more complete evaluation of angiogenic process compared with traditional in vitro cellular assays. In agreement with the wound closure assay, aortic discs isolated from PTP1B siRNA-transfected mice showed 1.5-fold increase in VEGF-induced sprouting growth in fibrin gel. These data demonstrate that knockdown of PTP1B enhances the formation of new vessels in mouse aortic explants in a 3-dimensional gel-based environment. Along the same line with our findings, mice deficient in PTP1B in endothelial cells exhibited enhanced capillary growth in response to VEGF in matrigel implants (10). Moreover, PTP1B knock-out mice showed increased cardiac angiogenesis post myocardial infarction (27).
To further investigate the physiological role of PTP1B in angiogenesis, we examined changes in wound healing in diabetic mice. Interestingly, PTP1B activity was increased in the wounded skin in diabetic mice compared to that of control mice (Fig. 8). It is reported that PTP1B expression is increased in mouse aortic endothelial cells of diabetic animals (26). These results indicate that PTP1B may act to negatively counteract endothelial angiogenesis in diabetic environment. To test this hypothesis, we examined wound healing progress in mice with PTP1B activity locally modulated by in situ transfection of overexpressing plasmid or application of inhibitor. Of note, overexpression of PTP1B near the wound bed delayed, while application of PTP1B inhibitor accelerated, VEGF-induced skin wound recovery in diabetic mice (Figs. 9 and 10). Taken together, our results have shown that blocking PTP1B activity enhances VEGF-induced angiogenesis and diabetic wound healing. Another significance of our wound healing data is validation that strategies of topical inhibition of PTP1B may serve as effective treatment for impaired diabetic wound healing. Impaired angiogenesis is a critical component of diabetic foot ulceration, a major complication of diabetes and a leading cause of hospitalizations associated with diabetes. Neglecting ulcers can result in lower leg amputation. Our strategies of local manipulation of PTP1B or calpain activities, however, are on-the-spot, non-chronic and efficient in promoting diabetic wound healing.
In summary, genetic or pharmacological inhibition of PTP1B alleviates calpain/PTP1B-dependent negative feedback regulation of VEGFR2, resulting in increased VEGFR2 and Akt phosphorylation and enhanced angiogenesis. Topical application of PTP1B inhibitor or calpain activation may serve as novel therapeutics for impaired diabetic wound healing.
Experimental Procedures
Cell Culture and Chemicals
Bovine aortic endothelial cells (BAECs, Lonza) were cultured in M199 supplemented with 10% fetal bovine serum (FBS), 1% vitamin and 1% l-glutamine till confluent as previously described (1, 28, 29). All chemicals used in this study were purchased from Sigma in highest purity.
Plasmid Construction
Full-length coding region of bovine PTP1B (NM_001100326.1) was amplified from cDNA of BAECs by PCR and cloned into pCMV-HA using SalI and NotI sites. The construct was confirmed by DNA sequencing. Plasmids encoding FLAG-tagged human calpain 1 and calpain 2 were obtained from Addgene (plasmids #60941 and #60942) (30).
Transfection and Immunoblot
BAECs and HEK 293T cells were grown in 6-well plates until 70–80% confluence. Then, the cells were transfected with 2.5 μg of plasmids of pCMV-HA; HA-tagged wild type bovine PTP1B; or 50 pmol siRNA (control siRNA target sequence: 5′-AATTCTCCGAACGTGTCACGT-3′; bovine PTP1B siRNA target sequence: 5′-CAGGTACAGAGACGTCAGT-3′; bovine VEGFR2 siRNA target sequence: 5′-GGAAATCTGTTACAAGCTA-3′, Dharmacon) according to the instructions of Lipofectamin 2000 and Oligofectamin (Invitrogen), respectively.
Forty-eight hours post transfection, cells were starved overnight with 5% FBS in M199 (BAECs) or DMEM (HEK 293T cells). Cells were then incubated with recombinant human VEGF (100 ng/ml) (R&D System) for 5 min after 30 min pretreatment with N-acetyl-leucyl-leucyl-norleucinal (ALLN, 30 μm, Calbiochem). In some experiments, HEK 293T cells and BAECs were treated with ALLN (30 μm, Calbiochem) for 30 min, followed by incubation with CaCl2 (2 mm) and A23187 (1 μm) for 1 h.
The cells were harvested in cold lysis buffer (20 mmol/liter Tris-HCl pH 7.4, 150 mmol/liter NaCl, 1 mmol/liter EDTA, 1 mmol/liter EGTA, 2.5 mmol/liter sodium pyrophosphate, 1 mmol/liter β-glycerophosphate, 1 mmol/liter sodium orthovanadate, 1% Triton X-100, supplemented with protease inhibitor mixture and phosphatase inhibitor mixture 2 and 3). The cell suspension was incubated on ice for 20 min before centrifugation at 12,000 rpm for 15 min at 4 °C. Then the supernatant was transferred to a new Eppendorf tube, and the protein concentration was determined by DC protein assay (Bio-Rad). The protein lysates were separated in 7.5% SDS-PAGE and detected with anti-phospho-VEGFR2-Y1175 (2478, lot 11), anti-total-VEGFR2 (2479, lot18), anti-phospho-Akt-S473 (9271, lot14) (all used at 1:1000, Cell Signaling Technologies), anti-PTP1B (sc-1718, lot K2912, 1:500, Santa Cruz Biotechnology), and anti-actin (A2066, lot 082M4781, 1:5000, Sigma). The densities of Western blots were analyzed by NIH Image J (National Institutes of Health, Bethesda, MD) and normalized to internal controls as indicated in the figures.
PTP1B Activity Assay
PTP1B activity assay was conducted as previously published with minor modifications (31, 32). Briefly, skin samples of wounded area were collected on day 12 from control and diabetic mice in the wound healing experiments. Tissues were homogenized in cold IP buffer (150 mmol/liter NaCl, 20 mmol/liter Tris-HCl pH 7.5, 1% Nonidet P-40, 5 mmol/liter EDTA, supplemented with protease inhibitor mixture). One milligram of the lysate was incubated with anti-PTP1B antibody at 4 °C overnight. The next morning, PTP1B immunocomplex was precipitated with TrueBlot anti-goat Ig IP beads (Rockland) for another 3 h. Immunoprecipitates were washed three times with PTP assay buffer (100 mm HEPES, 2 mm EDTA, 1 mm dithiothreitol, 150 mm NaCl, 0.5 mg/mg BSA). Then, the beads were incubated with Tyrosine Phosphatase Substrate I (DADEpYLIPQQG, R&D, 200 μm) in 60 μl of PTP assay buffer for 2 h at 30 °C, protected from light. Afterwards, a 50 μl reaction was transferred into a 96-well plate. One-hundred microliters of Malachite Green Solution (Echelon) was added, and absorbance was measured at 620 nm.
Endothelial Cell Wound Closure Assay
Endothelial cells at 70–80% confluence were transfected with siRNA as described above. At 24-h post transfection, the cells were detached with trypsin and seeded into 35-mm dish at 50,000 cells per well. Cells were allowed to grow until confluence, and then starved overnight with 1% FBS in M199. The wound area was generated with a P-1000 tip, followed by washing with PBS to remove debris. Images of the scratch were captured at the locations marked at the bottom of the culture dish, which is considered as t = 0 h. The cells were then pre-treated with ALLN (10 μm) for 30 min prior to VEGF stimulation (100 ng/ml). After incubation for 32 h images were taken at the same locations (t = 32 h). Wound areas were analyzed by NIH Image J software (given as numbers of pixels). Area of migration = Wound area at 0 h − Wound area at 32 h.
In Vivo siRNA Delivery and Mouse Aortic Disc Assay
All animal experiments were approved by the Institutional Animal Care and Usage Committee of University of California Los Angeles. In vivo siRNA delivery was achieved by intravenous injection of 50 nmol in vivo grade siRNA (Control scrambled siRNA target sequence: 5′-GAAAGAACTCCGGACTATT-3′; Mouse PTP1B siRNA target sequence: 5′-TGACCACAGTCGGATTAAA-3′, Dharmacon) following the instruction of lipid-based in vivo transfection kit (Altogen Biosystems). Wild type male C57BL/6 mice (12 weeks old, Charles River) were injected of siRNA on two consecutive days, once every 24 h. Mice were harvested 48 h after the first injection.
Angiogenic assay of mouse aortic discs was carried out as previously described (33). Briefly, aortas from siRNA-injected mice were harvested and cleaned of connective tissues. Then the aortas were cut open, and 2-mm discs were prepared with biopsy punch (Integra Miltex). 12-well plates were coated with 60 μl of thrombin (0.01 units/μl). Freshly prepared aorta discs were placed into the well (endothelium layer facing down) and covered with 120 μl of clotting medium (0.5% 6-aminocaproic acid, 0.3% fibrinogen, 1% fungizone, and 0.04 mg/ml gentamicin in M199). After incubation for 30 min, 2 ml of growth medium (1% FBS, 0.5% 6-aminocaproic acid, 1% fungizone, and 0.04 mg/ml gentamicin in M199) was added to each well with or without treatments (ALLN 10 μm 30 min pretreatment, then add VEGF 100 ng/ml) as indicated. The growth medium was replaced every other day. After 5 days of incubation, images were taken with Nikon Eclipse TE2000-U with NIS-Elements AR software by merging 20 pictures into a final image. The areas covered by migrated cells were analyzed by NIH ImageJ software (given as numbers of pixels) as previously described (34).
Diabetic Mice Wound Healing Assay
Diabetes was induced in wild type male C57BL/6 mice (10–12 weeks old) with two intravenous injections of streptozocin (STZ, Sigma) at 100 mg/kg as previously published (35, 36). Two days after the last injection, blood glucose was measured with a OneTouch blood glucose meter (Ultra). Animals had glucose levels at 343.8 ± 13.5 mg/dl (n = 37). The mice were then anesthetized with isoflurane and dorsum was shaved to remove hair. Then the skin was prepared with betadine and 70% ethanol (three times) and allowed to dry. A full thickness of 6-mm skin punch biopsy (Integra Miltex) was made as previously described (16). Treatments were applied directly on the wound bed in PBS at total volume of 5 μl with or without recombinant human VEGF (200 ng) (37), ALLN (20 nmol), and PTP1B inhibitor (50 nmol, Calbiochem). Five micrograms of HA-tagged PTP1B and human calpain 1 and calpain 2 (2.5 μg each) plasmids were prepared with a lipid-based in vivo transfection kit and intracutaneously injected near the wound bed. The animal was maintained under anesthesia for 15 min before a bioclusive transparent dressing (Johnson & Johnson) was placed on the wound area (16). Control mice were treated with 5 μl of PBS only in the same fashion. Treatments and new dressing were applied every other day till day 12. Digital images of the wound were taken with a scale right before each dressing replacement. Wound sizes at different time points were analyzed by NIH ImageJ software and expressed as percentage of the wound area on day 0.
Statistical Analysis
All data are presented as mean±S.E. Differences were considered statistically significant if p < 0.05 by Student's t test for comparison between two groups, or by one-way ANOVA for multi-group comparison that was followed by a Newman-Keuls post hoc test.
Author Contributions
Y. X. Z. conducted most of the experiments, analyzed the results, and wrote the paper. Y. X. Z., Q. L., and J. Y. Y. conducted experiments of i.v. injection. H. C. conceived the idea, directed the project, analyzed the results, and wrote the paper.
This work was supported by National Heart, Lung and Blood Institute (NHLBI), National Institute of Health Grants HL077440 (to H. C.), HL088975 (to H. C.), HL108701 (to H. C., D. G. H.), HL119968 (to H. C.), an American Heart Association Established Investigator Award (EIA) 12EIA8990025 (to H. C.), and an AHA Postdoctoral Fellowship Award 14POST20380995 (to Y. X. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- VEGF
- vascular endothelial growth factor
- ALLN
- N-acetyl-leucyl-leucyl-norleucinal
- BAECs
- bovine aortic endothelial cells
- NO
- nitric oxide
- PTP1B
- protein phosphotyrosine phosphatase 1B
- STZ
- streptozotocin
- VEGFR2
- VEGF receptor 2.
References
- 1. Youn J. Y., Wang T., and Cai H. (2009) An ezrin/calpain/PI3K/AMPK/eNOSs1179 signaling cascade mediating VEGF-dependent endothelial nitric oxide production. Circ. Res. 104, 50–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Su Y., Cui Z., Li Z., and Block E. R. (2006) Calpain-2 regulation of VEGF-mediated angiogenesis. Faseb. J. 20, 1443–1451 [DOI] [PubMed] [Google Scholar]
- 3. Ma H., Tochigi A., Shearer T. R., and Azuma M. (2009) Calpain inhibitor SNJ-1945 attenuates events prior to angiogenesis in cultured human retinal endothelial cells. J. Ocul. Pharmacol. Ther. 25, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Letavernier B., Zafrani L., Nassar D., Perez J., Levi C., Bellocq A., Mesnard L., Sachon E., Haymann J. P., Aractingi S., Faussat A. M., Baud L., and Letavernier E. (2012) Calpains contribute to vascular repair in rapidly progressive form of glomerulonephritis: potential role of their externalization. Arterioscler. Thromb. Vasc. Biol. 32, 335–342 [DOI] [PubMed] [Google Scholar]
- 5. Frangioni J. V., Oda A., Smith M., Salzman E. W., and Neel B. G. (1993) Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets. EMBO J. 12, 4843–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cortesio C. L., Chan K. T., Perrin B. J., Burton N. O., Zhang S., Zhang Z. Y., and Huttenlocher A. (2008) Calpain 2 and PTP1B function in a novel pathway with Src to regulate invadopodia dynamics and breast cancer cell invasion. J. Cell Biol. 180, 957–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuchay S. M., Kim N., Grunz E. A., Fay W. P., and Chishti A. H. (2007) Double knockouts reveal that protein tyrosine phosphatase 1B is a physiological target of calpain-1 in platelets. Mol. Cell. Biol. 27, 6038–6052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakamura Y., Patrushev N., Inomata H., Mehta D., Urao N., Kim H. W., Razvi M., Kini V., Mahadev K., Goldstein B. J., McKinney R., Fukai T., and Ushio-Fukai M. (2008) Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ. Res. 102, 1182–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nassar D., Letavernier E., Baud L., Aractingi S., and Khosrotehrani K. (2012) Calpain activity is essential in skin wound healing and contributes to scar formation. PLoS ONE 7, e37084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lanahan A. A., Lech D., Dubrac A., Zhang J., Zhuang Z. W., Eichmann A., and Simons M. (2014) PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 130, 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brem H., and Tomic-Canic M. (2007) Cellular and molecular basis of wound healing in diabetes. J. Clin. Invest. 117, 1219–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao L., Siu K. L., Chalupsky K., Nguyen A., Chen P., Weintraub N. L., Galis Z., and Cai H. (2012) Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension 59, 158–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cowan C. E., Kohler E. E., Dugan T. A., Mirza M. K., Malik A. B., and Wary K. K. (2010) Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ. Res. 107, 959–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu C., Wang W., Parchim N., Irani R. A., Blackwell S. C., Sibai B., Jin J., Kellems R. E., and Xia Y. (2014) Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination-preventing isopeptide modification. Hypertension 63, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tie L., Li X. J., Wang X., Channon K. M., and Chen A. F. (2009) Endothelium-specific GTP cyclohydrolase I overexpression accelerates refractory wound healing by suppressing oxidative stress in diabetes. Am. J. Physiol. Endocrinol Metab. 296, E1423–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marrotte E. J., Chen D. D., Hakim J. S., and Chen A. F. (2010) Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J. Clin. Invest. 120, 4207–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H., Wang A. X., Liu Z., and Barrett E. J. (2008) Insulin signaling stimulates insulin transport by bovine aortic endothelial cells. Diabetes 57, 540–547 [DOI] [PubMed] [Google Scholar]
- 18. Lanahan A. A., Hermans K., Claes F., Kerley-Hamilton J. S., Zhuang Z. W., Giordano F. J., Carmeliet P., and Simons M. (2010) VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell 18, 713–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williams C. K., Li J. L., Murga M., Harris A. L., and Tosato G. (2006) Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood 107, 931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harrington L. S., Sainson R. C., Williams C. K., Taylor J. M., Shi W., Li J. L., and Harris A. L. (2008) Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc. Res. 75, 144–154 [DOI] [PubMed] [Google Scholar]
- 21. Jakobsson L., Franco C. A., Bentley K., Collins R. T., Ponsioen B., Aspalter I. M., Rosewell I., Busse M., Thurston G., Medvinsky A., Schulte-Merker S., and Gerhardt H. (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 12, 943–953 [DOI] [PubMed] [Google Scholar]
- 22. Domingues I., Rino J., Demmers J. A., de Lanerolle P., and Santos S. C. (2011) VEGFR2 translocates to the nucleus to regulate its own transcription. PLoS ONE 6, e25668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blanes M. G., Oubaha M., Rautureau Y., and Gratton J. P. (2007) Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J. Biol. Chem. 282, 10660–10669 [DOI] [PubMed] [Google Scholar]
- 24. Yan F., Wang Y., Wu X., Peshavariya H. M., Dusting G. J., Zhang M., and Jiang F. (2014) Nox4 and redox signaling mediate TGF-β-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 5, e1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Holmqvist K., Cross M. J., Rolny C., Hägerkvist R., Rahimi N., Matsumoto T., Claesson-Welsh L., and Welsh M. (2004) The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J. Biol. Chem. 279, 22267–22275 [DOI] [PubMed] [Google Scholar]
- 26. Herren D. J., Norman J. B., Anderson R., Tremblay M. L., Huby A. C., and Belin de Chantemèle E. J. (2015) Deletion of protein tyrosine phosphatase 1B (PTP1B) enhances endothelial cyclooxygenase 2 expression and protects mice from Type 1 diabetes-induced endothelial dysfunction. PLoS ONE 10, e0126866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Besnier M., Galaup A., Nicol L., Henry J. P., Coquerel D., Gueret A., Mulder P., Brakenhielm E., Thuillez C., Germain S., Richard V., and Ouvrard-Pascaud A. (2014) Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 28, 3351–3361 [DOI] [PubMed] [Google Scholar]
- 28. Youn J. Y., Nguyen A., and Cai H. (2012) Inhibition of XO or NOX attenuates diethylstilbestrol-induced endothelial nitric oxide deficiency without affecting its effects on LNCaP cell invasion and apoptosis. Clin. Sci. 123, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao L., Chalupsky K., Stefani E., and Cai H. (2009) Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: a novel HPLC-based fluorescent assay for DHFR activity. J. Mol. Cell Cardiol. 47, 752–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y., and Zhang Y. (2014) Regulation of TET protein stability by calpains. Cell Reports 6, 278–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Suryawan A., and Davis T. A. (2003) Protein-tyrosine-phosphatase 1B activation is regulated developmentally in muscle of neonatal pigs. Am. J. Physiol. 284, E47-E54 [DOI] [PubMed] [Google Scholar]
- 32. Taghibiglou C., Rashid-Kolvear F., Van Iderstine S. C., Le-Tien H., Fantus I. G., Lewis G. F., and Adeli K. (2002) Hepatic very low density lipoprotein-ApoB overproduction is associated with attenuated hepatic insulin signaling and overexpression of protein-tyrosine phosphatase 1B in a fructose-fed hamster model of insulin resistance. J. Biol. Chem. 277, 793–803 [DOI] [PubMed] [Google Scholar]
- 33. Nguyen A., and Cai H. (2006) Netrin-1 induces angiogenesis via a DCC-dependent ERK1/2-eNOS feed-forward mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 6530–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Go R. S., and Owen W. G. (2003) The rat aortic ring assay for in vitro study of angiogenesis. Methods Mol. Med. 85, 59–64 [DOI] [PubMed] [Google Scholar]
- 35. Oak J. H., and Cai H. (2007) Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 56, 118–126 [DOI] [PubMed] [Google Scholar]
- 36. Youn J. Y., Gao L., and Cai H. (2012) The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 55, 2069–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng Y., Watanabe M., Kuraishi T., Hattori S., Kai C., and Shibuya M. (2007) Chimeric VEGF-ENZ7/PlGF specifically binding to VEGFR-2 accelerates skin wound healing via enhancement of neovascularization. Arterioscler. Thromb. Vasc. Biol. 27, 503–511 [DOI] [PubMed] [Google Scholar]