

Abstract
Lung carcinoma is the leading cause of cancer-related death worldwide, and among this cancer, non-small cell lung carcinoma (NSCLC) comprises the majority of cases. Furthermore, recurrence and metastasis of NSCLC correlate well with CD133+ve tumor cells, a small population of tumor cells that have been designated as cancer stem cells (CSC). We have demonstrated for the first time high expression of D2 dopamine (DA) receptors in CD133+ve adenocarcinoma NSCLC cells. Also, activation of D2 DA receptors in these cells significantly inhibited their proliferation, clonogenic ability, and invasiveness by suppressing extracellular signal-regulated kinases 1/2 (ERK1/2) and AKT, as well as down-regulation of octamer-binding transcription factor 4 (Oct-4) expression and matrix metalloproteinase-9 (MMP-9) secretion by these cells. These results are of significance as D2 DA agonists that are already in clinical use for treatment of other diseases may be useful in combination with conventional chemotherapy and radiotherapy for better management of NSCLC patients by targeting both tumor cells and stem cell compartments in the tumor mass.
Keywords: cancer, cancer biology, cancer stem cells, cancer therapy, dopamine, signal transduction, D2 receptors, lung cancer
Introduction
Globally, lung cancers are considered to be the leading causes of cancer-related deaths, and non-small cell lung carcinoma (NSCLC)4 is the predominant type of lung cancer, occurring in 85% of the cases (1, 2). The prognosis of NSCLC is poor, and the survival rate is only 15% after 5 years because many of these patients ultimately do not respond to chemotherapy and radiotherapy due to the presence of CSC (3–5). The CSC have the unique ability to promote tumor growth, recurrence, metastasis, and resistance to treatment (6, 7).
Moreover, CD133, a surface glycoprotein with a five-transmembrane domain, has been widely used as a surface marker to identify CSC in many human tumors including NSCLC (8–12). Although CD133 is expressed in various human tissues, its glycosylated epitopes specific for stem cells may be discordant or sometimes absent, making it difficult to identify this cell population (13). There may be transcriptional or post-translational modifications (14), leading to some degree of alteration in its expression, or variation in its expression in different tumors may be due to the use of antibodies from different clones (12). However, despite these limitations, recent results from different laboratories have shown significant association of CD133 (epitope-1) expression in a population of tumor cells with CSC characteristics in the brain, prostate, liver, and lung (15–18).
There are now several reports that have further strengthened the concept of CD133+ve tumor cells as CSC in NSCLC as these tumor cells possess the characteristics of CSC (18–20). In contrast to the CD133−ve tumor cells, CD133+ve tumor cells are more tumorigenic and resistant to radiation and anticancer drugs (18–20). These cells also have a greater ability to form anchorage-independent floating spheres, proliferation, and tumor mass than the CD133−ve NSCLC cells. The CD133+ve cells like CSC demonstrate significantly increased stemness, adhesion, motility, and expression of drug efflux gene (21–23) than CD133−ve tumor cells. Importantly, the presence of CD133+ve tumor cells correlates well with poor prognosis, decreased survival, and increased lymph node metastasis in NSCLC patients (24–26).
Furthermore, in addition to CD133, aldehyde dehydrogenase 1 (ALDH1) is also used as a CSC marker in NSCLC (12, 27–30). The ALDH1 activity is mainly due to its ALDH1A1 (aldehyde dehydrogenase 1 family, member A1) isoform, an intracellular cytosolic isoenzyme that oxidizes intracellular aldehyde and controls several functions including drug resistance (30). Besides lung cancer, ALDH1A1 has also been used as a marker of CSC in breast and esophageal cancers (31, 32). NSCLC cells expressing high ALDH1 activity show the characteristics of CSC such as increased proliferation, self-renewal, and resistance to chemotherapy in vitro, are highly tumorigenic in immunocompromised mice, and maintain heterogeneity like the parent tumors (27). These ALDH1A1-expressing NSCLC cells are also known for their clone formation ability, proliferation, growth, and migration in vitro (28).
On the contrary, silencing of ALDH1A1 in these cells results in considerable loss of their stem cell-like characteristics (28). Importantly, reports from the clinics have indicated that ALDH1 expression is strongly associated with poor prognosis and lymph node metastasis in NSCLC (28, 29, 33).
DA acts as a neurotransmitter regulating locomotor and behavioral functions (34), and reports from our laboratory have indicated that in addition, DA can inhibit vascular endothelial growth factor-A (VEGF-A), mediating angiogenesis by suppressing VEGFR-2 phosphorylation (35–38). Studies from our laboratory have further reported that D2 DA receptors can regulate different functions of normal progenitor and stem cells such as endothelial progenitor and mesenchymal stem cells (38, 39). Therefore, it will be interesting to study the expression profile of D2 DA receptors in the CD133+ve tumor cell population in NSCLC and investigate the regulatory role of this neurotransmitter, if any, on the biology of this specific tumor cell population having CSC characteristics. Accordingly, we selected adenocarcinoma of the lung, the most common histological type of NSCLC observed in patients, for our present study.
Results
Association of CD133 and D2 DA Receptors in Human NSCLC
NSCLC is the most common type of lung cancer (2). Among NSCLC patients, adenocarcinoma is predominant in both men and women (40). Accordingly, we used human lung adenocarcinoma (NSCLC) patient tissue samples and cell lines A549 and NCI-H23 for our present study.
At first, we determined the expression of D2 DA receptors in the CD133-expressing tumor cell population in NSCLC cell lines A549 and NCI-H23. After initial gating to exclude dead cells and debris (Fig. 1, A and B), our FACS analysis demonstrated that in A549 and NCI-H23 cells, 22 and 20%, respectively, of the total tumor cells expressed D2 DA receptors (Fig. 1, C–F), and 5 and 3.5%, respectively, of total A549 and NCI-H23 cells expressed CD133 (Fig. 1, G–J). Furthermore, in both A549 and NCI-H23 cells, more than 95% of CD133+ve cells expressed D2 DA receptors (Fig. 1, K–N). On the contrary, less than 16 and 15%, respectively, of CD133−ve cells in A549 and NCI-H23 expressed D2 DA receptors (Fig. 1, K–N).
FIGURE 1.

Percentage of cancer cells expressing stem cell marker CD133 and D2 DA receptors in human NSCLC adenocarcinoma cell lines (A549 and NCI-H23). A and B, forward scatter/side scatter (FSC/SSC) plots show the gated population. C, unstained A549 cells as a control. D, percentage of A549 cells expressing D2 DA receptors. E, unstained NCI-H23 cells as a control. F, percentage of NCI-H23 cells expressing D2 DA receptors. G, unstained A549 cells as a control. H, percentage of A549 cells expressing CD133. I, unstained NCI-H23 as a control. J, percentage of NCI-H23 cells expressing CD133. K, unstained A549 cells as a control. L, A549 cells grouped by double staining of CD133 and D2 DA receptors. M, unstained NCI-H23 as a control. N, NCI-H23 cells grouped by double staining CD133 and D2 DA receptors. Results are representative of 3 independent experiments.
We next validated this association in patient tissue samples. Our confocal data demonstrated significant co-localization of D2 DA receptors with both stem cell markers, CD133 and aldehyde dehydrogenase (ALDH1A1), in human primary lung adenocarcinoma (NSCLC) tissue sections. More than 90% of the CD133 and ALDH1A1+ve tumor cells in human NSCLC tissue sections co-expressed D2 DA receptors in the CSC of human primary lung adenocarcinoma patients (Fig. 2). Appropriate isotype-matched controls were used to determine the specificity of staining (data not shown).
FIGURE 2.

H&E staining and Immunofluorescence staining of CD133, ALDH1A1, and D2 DA receptors in NSCLC patient tissues. A, representative H&E staining of NSCLC lung tissue. B–D and G–I, slides of each patient were co-stained either with DAPI (B) and ALDH1A1 antibody (C) or D2 DA receptor antibody (D) or with DAPI (G) and CD133 antibody (H) and D2 DA receptor antibody (I). After co-staining, images were acquired separately for each marker followed by merging. E, F, J, and K, co-localization of ALDH1A1 and D2 DA receptor (MERGE 1) (E), ALDH1A1, D2 DA receptor and DAP1 (MERGE 2) (F), CD133 and D2 DA receptor (MERGE 1) (J), and CD133 D2 DA receptor and DAP1 (MERGE 2) (K). DAPI is used as nucleus marker. Co-localization appears in yellow and is indicated by arrows in merge pictures. Scale bars, 10 mm. Results are representative of 35 independent experiments.
After that, we isolated and enriched the CD133+ve population from the human NSCLC cell line (A549) to study the significance, if any, of this high expression of D2 DA receptors in these CD133+ve CSC. Accordingly, experiments were undertaken with purified CD133+ve NSCLC tumor cells (A549) to understand the biobehavior of this cell population following activation of D2 DA receptors present in these cells.
D2 DA Receptor Stimulation Inhibits CD133+ve NSCLC Tumor Cell Proliferation in Vitro
The 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) assay showed significant inhibition of proliferation in purified CD133+ve NSCLC tumor cells when these cells were treated with 1 or 10 μm quinpirole, a specific D2 DA receptor agonist (Fig. 3A). However, this action of quinpirole was abolished when these cells were pre-treated with a specific D2 DA receptor antagonist, eticlopride (10 times more than the concentration of quinpirole), thereby further confirming the role of D2 DA receptors (Fig. 3A). On the contrary, 1 or 10 μm quinpirole treatment had no significant effects on the proliferation of CD133−ve tumor cells (Fig. 3A). Furthermore, FACS analysis did not demonstrate a significant number of annexin V- and propidium iodide (PI)-positive tumor cells following treatment with 1 or 10 μm quinpirole treatment, thereby indicating that this low concentration of D2 DA receptor agonist had no effects on the apoptosis of CD133+ve NSCLC tumor cells (Fig. 3B).
FIGURE 3.

Effects of D2 DA receptor stimulation by D2 dopamine agonist quinpirole on proliferation (A) and apoptosis (B) of CD133+ve tumor cells purified (>95%) from A549 NSCLC cell line. A, proliferation measured by the MTT assay following treatment of CD133+ve and CD133−ve tumor cells with 1 or 10 μm quinpirole (Quin) in the presence or absence of eticlopride (Eti), the D2 receptor-specific antagonist (10 times more the concentration of D2 receptor-specific agonist, quinpirole) for 72 h. Results are representative of 3 independent experiments. *, p ≤ 0.05 no treatment (NT) versus 1 μm and no treatment versus 10 μm, O. D., optical density. B, apoptosis was measured by the annexin V-PI staining assay following treatment of CD133+ve tumor cells with 1 or 10 μm quinpirole. Panel a, forward scatter/side scatter (FSC/SSC) plots show the gated population. H indicates height. Panel b, unstained. Panel c, no treatment. Panel d, 1 μm quinpirole. Panel e, 10 μm quinpirole. Lower left (LL), annexin V-PI− (viable cells); lower right (LR), annexin V+PI− (early apoptotic cells); upper left (UL) annexin V-PI+; upper right (UR), annexin V+PI+ (late apoptotic cells). Results are representative of 3 independent experiments. Error bars indicate mean ± S.D.
The Clonogenic Ability of CD133+ve NSCLC Tumor Cells Was Suppressed following Activation of D2 DA Receptors
Results from the colony-forming efficiency assay revealed significant inhibition of the clonogenic ability of CD133+ve A549 NSCLC tumor cells (p ≤ 0.05) when these cells were treated with 1 or 10 μm of specific DA D2 receptor agonist quinpirole (Fig. 4A). Similarly, when these cells were treated with 10 μm AKT inhibitor (LY294002) or 10 μm ERK1/2 inhibitor (U0126), there was also significant inhibition of this characteristic of CD133+ve NSCLC cells when compared with untreated controls. These results thus indicate that these signaling pathways play critical roles in inducing clone formation by CD133+ve NSCLC tumor cells (Fig. 4A). This observation was further validated from the sphere-forming ability of the tumor cells, a surrogate marker for stemness and the self-renewal ability of epithelial cancer cells (41). In addition, CD133+ve tumor cells, when treated with quinpirole (1 or 10 μm) in serum-free medium, tumor cell sphere formation showed a significant decrease in number and size (Fig. 4, B and C).
FIGURE 4.

Effects of D2 DA receptor stimulation by D2 dopamine agonist quinpirole on clonogenicity (A) and tumor sphere formation (B) by CD133+ve tumor cells purified (>95%) from the A549 NSCLC cell line. A, following treatment with Akt inhibitor (LY294002) or ERK inhibitor (U0126) or 1 or 10 μm quinpirole and culturing for 7 days, the colonies were fixed and stained with crystal violet and photographed. Magnification: 20×. Results are representative of 3 independent experiments. B, following treatment with 1 or 10 μm quinpirole and culturing for 7 days, the sphere formation was observed, and randomly chosen fields were then photographed. Magnification: 20×. C, sphere-forming efficacy is expressed as numbers of spheres formed per 100 cells. Results are representative of 3 independent experiments. Editing of all original images was done without obscuring the actual differences. *, p ≤ 0.05 no treatment versus 1 μm and no treatment versus 10 μm. Error bars indicate mean ± S.D.
Inhibition of ERK1/2 and AKT in CD133+ve NSCLC Tumor Cells following D2 DA Receptor Activation
The CD133+ve tumor cell population has been reported to overexpress activated ERK1/2 and AKT, and the inhibition of ERK1/2 and AKT significantly influences their clonogenic potential in colon cancer cells (42). Our results also indicated the same in CD133+ve NSCLC cells (Fig. 4A). To explore D2 DA receptor-mediated inhibition of the clonogenic ability of the CD133+ve NSCLC tumor cells, we examined the effects of D2 DA receptor activation on the status of ERK1/2 and AKT phosphorylation in this population of tumor cells derived from NSCLC cell lines A549 and NCI-H23. Our results indicated significant inhibition of both ERK1/2 and AKT phosphorylation in the CD133+ve population of tumor cells following treatment with either 1 μm or 10 μm of the specific D2 DA receptor agonist, quinpirole (Fig. 5A), in CD133+ve tumor cells from both NSCLC cell lines, A549 and NCI-H23. These results were further validated by using siRNA of D2 DA receptor. Our results indicated that quinpirole had no significant effects on ERK1/2 and AKT phosphorylation when these cells were pretreated with D2 DA receptor siRNA, thereby confirming the role of D2 DA receptors (Fig. 5A). The concentrations of the scramble siRNA and D2 DA receptor siRNA were optimized to ensure that they did not affect cell viability (data not shown), but significantly inhibited D2 DA receptor expressions in the CD133+ve NSCLC cells (Fig. 5B).
FIGURE 5.

Effects of quinpirole on the expression of phospho-AKT and phospho-ERK1/2 in CD133+ve tumor cells derived from A549 and NCI-H23 NSCLC cell line. A, the cell lysates prepared from cultured A549 and NCI-H23 cell lines at specific times after treatment were subjected to Western blotting using phospho-AKT (p-AKT) antibodies or phospho-ERK1/2 (p-ERK1/2) antibodies. For each of the experiments, the purified cells from both the cell lines were treated with 1 or 10 μm quinpirole or D2 DA receptor siRNA plus 1 μm quinpirole or D2 DA receptor siRNA plus 10 μm quinpirole or scrambled siRNA plus 1 μm quinpirole or scrambled siRNA plus 10 μm quinpirole and cultured for 72 h. ERK, extracellular-signal-regulated kinases; Scram, scrambled; Quin, quinpirole. NT, no treatment. B, siRNA-mediated knockdown of D2 DA receptor expression. Results are representative of 3 independent experiments.
Down-regulation of Oct-4 Expression in CD133+ve NSCLC Tumor Cells
Because octamer-binding transcription factor 4 (Oct-4) expression maintains the stem-like properties in CD133+ve NSCLC tumor cells (43), we therefore determined the expression of Oct-4 in CD133+ve NSCLC tumor cells. Untreated CD133+ve NSCLC tumor cells expressed significant Oct-4 mRNA, as evident from real-time PCR (Fig. 6). Interestingly, when this tumor cell population was treated with a specific D2 DA receptor agonist, quinpirole, significant down-regulation of Oct-4 mRNA level was observed following treatment with 1 μm (2.5-fold) or 10 μm (10-fold) quinpirole. However, transfection of these tumor cells with D2 DA receptor siRNA abrogated the inhibitory effect of quinpirole, thereby confirming the inhibitory role of D2 DA receptor activation on OCT-4 expression in CD133+ve NSCLC tumor cells (Fig. 6).
FIGURE 6.

Effect of quinpirole on the expression of OCT-4 in CD133+ve tumor cells derived from A549 NSCLC cell line. The transcripts encoding human OCT-4 were measured by real-time PCR and expressed as the -fold change after normalizing with respect to β-actin. For each of the experiments, the purified cells were treated with 1 or 10 μm quinpirole or D2 DA receptor siRNA plus 1 μm quinpirole or D2 DA receptor siRNA plus 10 μm quinpirole and cultured for 72 h. Results are representative of 3 independent experiments. *, p ≤ 0.05 no treatment versus 1 μm, no treatment versus 10 μm. Error bars indicate mean ± S.D.
Inhibition of CD133+ve NSCLC Tumor Cells Invasion in Vitro following Activation of D2 DA Receptors
As expression of CD133 in NSCLC tumor cells correlates with invasion and metastasis in lymph nodes (25), we therefore investigated whether D2 DA receptor activation in these CD133+ve tumor cells in NSCLC has any effect on invasiveness of these cells. In our in vitro experiment, both the concentrations of quinpirole (1 μm and 10 μm) significantly inhibited the invasion of CD133+ve NSCLC tumor cells through the Matrigel (Fig. 7, A and B).
FIGURE 7.

Effects of quinpirole on the invasiveness of CD133+ve tumor cells derived from A549 NSCLC cell line and secretion of MMP-9 from this cell population. A, in vitro invasion was determined by the Matrigel invasion assay. Purified CD133+ve NSCLC tumor cells were seeded at a density of 10,000 cells/well in stem cell medium in the upper cell culture inserts, with 1 or 10 μm quinpirole, and 20% serum-containing medium was placed in the lower chambers as chemoattractant. Relevant controls were kept. The invasion was measured after 24 h. Magnification: 20×. Results are representative of 3 independent experiments. B, invasion index was determined according to the standard formula. *, p ≤ 0.05, no treatment versus 1 μm and no treatment versus 10 μm. C, MMP-9 concentration in the culture supernatants from the upper chambers was measured by ELISA. Results are representative of 3 independent experiments. *, p ≤ 0.05, no treatment versus 1 μm and no treatment versus 10 μm. Error bars indicate mean ± S.D.
D2 DA Receptor Activation Inhibits MMP-9 Secretion by CD133+ve NSCLC Tumor Cells
Matrix metalloproteinase-9 (MMP-9) plays a critical role in tumor cell invasion and metastasis (44). Our results indicated significant inhibition of MMP-9 concentration in the culture medium of D2 DA receptor agonist quinpirole-treated cells after 24 h in comparison with untreated controls (Fig. 7C).
D2 DA Receptor Agonist, Quinpirole, Inhibited CD133+ve A549 Non-small Cell Lung Cancer Tumor Cell Growth in Vivo
Similar to our in vitro experiments, on day 8, we also observed 47% inhibition of tumor growth in nude mice transplanted with CD133+ve NSCLC tumor cells after treatment with 10 mg/kg i.p. of D2 DA receptor agonist, quinpirole, for 7 days (Fig. 8). We did not observe any significant side effects after treatment with this low dose of quinpirole, which corroborated with previous reports (45).
FIGURE 8.
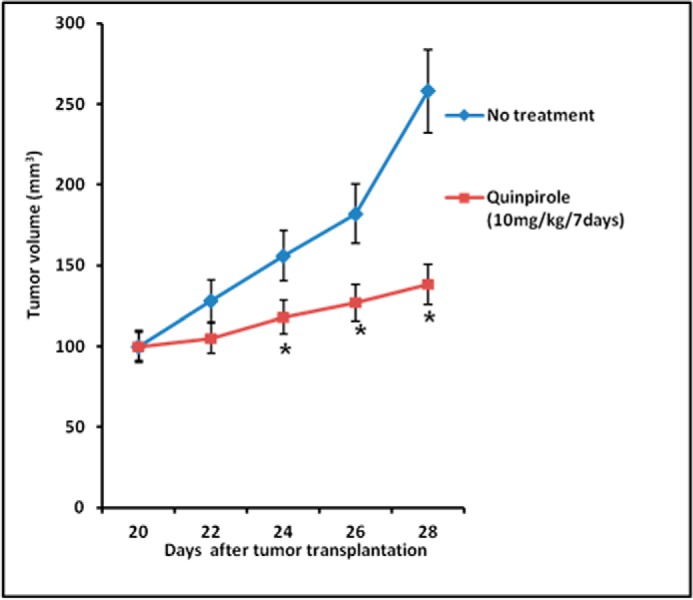
Effect of quinpirole on A549 tumor growth inhibition in nude mice. Freshly isolated CD133+ve tumor cells (104) were mixed with Matrigel (1:1) and inoculated subcutaneously in the right flank of each animal. When the tumor reached 100 mm3, quinpirole (10 mg/kg) was administered intraperitoneally for consecutive 7 days. Control mice received only medium + Matrigel. Thereafter tumor growth inhibition was noted. *, p ≤ 0.05, no treatment versus quinpirole. Error bars indicate mean ± S.D.
Discussion
Taken together, our results for the first time indicate significant expression of D2 DA receptors in CD133+ve tumor cells in human adenocarcinoma of the lung (NSCLC) tumor tissues and cell lines. Importantly, cancer stem cell characteristics of this population of tumor cells are well established (18–23). Only a few other reports had indicated the presence of D2 DA receptors in lung cancer cells (46) and inhibition of tumor growth following treatment with bromocriptine, a D2 DA receptor agonist (47). However, most of the other studies had been undertaken in large cell lung carcinoma and small cell lung carcinoma cells (48, 49). The other study investigated the effects of D2 DA receptor agonist on myeloid-derived suppressor cells and angiogenesis in lung cancer (50). None of these reports mentioned the status of D2 DA receptors and their effects on the treatment-refractory CD133+ve CSC in adenocarcinoma of the lung, the most prevalent histological type of NSCLC (40).
We demonstrate for the first time that stimulation of D2 DA receptors present in the CD133+ve CSC of NSCLC by a D2 DA receptor agonist can inhibit their proliferation, clonogenic ability, and invasiveness in vitro (Figs. 3A, 4, A–C, and 7, A and B), as well as suppressing tumor growth in vivo (Fig. 8). The underlying mechanisms indicate that stimulation of D2 DA receptors suppresses ERK1/2 and AKT phosphorylation (Fig. 5A), resulting in inhibition of the clonogenic ability of these cells. Recent results indicate that this property of CSC is regulated through the ERK1/2 and AKT pathways (42). Accordingly, it would be important to note that we and others had demonstrated D2 DA receptor-mediated inhibition of ERK1/2 and AKT activation or phosphorylation (38, 51–55). Our results indicated that inhibition of ERK1/2 and AKT phosphorylation occurred on activation of D2 DA receptors in progenitor and cancer cells (38, 51). Beaulieu et al. (52) had also demonstrated that activation of D2 DA receptors in neurons forms a signaling complex of adaptor molecule β-arrestin, protein phosphatase 2 (PP2A), and AKT, resulting in dephosphorylation of AKT. Similarly, Chang et al. (54) showed that D2 DA receptor-mediated inhibition of ERK1/2 in adrenal cortical cells is through inhibition of intracellular cAMP.
Clonogenic ability is an important criterion of the stemness of CD133+ve NSCLC tumor cells, and OCT-4 regulates this characteristic of these cells (43). Our results indicate that activation of D2 DA receptors significantly down-regulates OCT-4, and this correlates well with inhibition of tumor sphere formation, a surrogate marker of stemness (41). Interestingly, recent evidence indicates that ERK2 also phosphorylates OCT4 and thus suggests that regulation of OCT4 is through activation of ERK (56). AKT also phosphorylates OCT4 at threonine 235, and this action correlates with resistance to apoptosis and tumorigenesis (57). Therefore, down-regulation of OCT4 in CD133+ve NSCLC stem cells following stimulation of D2 DA receptors in NSCLC cells may be due to inhibition of ERK1/2 and AKT phosphorylation following activation of D2 DA receptors.
MMP-9 synthesis and release by tumor cells are associated with an invasive property of these cells leading to metastasis (44). Reports further indicate an association of CD133+ve NSCLC cells with lymph node metastasis, and high levels of MMP-9 correlate significantly with invasiveness of A549 NSCLC cells (58). Therefore, our results demonstrating significant inhibition of MMP-9 synthesis by CD133+ve tumor cells following treatment with quinpirole may be the cause of inhibition of Matrigel invasion by these cells.
Furthermore, in vivo xeno-transplanted human NSCLC cell line A549 also showed significant inhibition of tumor growth following treatment with D2 DA receptor-specific agonist quinpirole, thereby supporting the in vitro results (Fig. 8). These results, therefore, suggest the possibility of selectively targeting the CD133+ve CSC population in NSCLC with a D2 DA receptor agonist.
Finally, D2 DA receptor agonists are already in use for the treatment of other disorders (59, 60). Therefore, these agonists, together with the current conventional anticancer agents and/or radiotherapy, may be beneficial in the management of NSCLC patients as this combination of therapeutic approaches will not only eradicate the bulk of tumor cells, but may also eliminate the population of CSC that imparts resistance to therapy.
Experimental Procedures
Culture of NSCLC Cell Lines
Human NSCLC cell lines A459 and NCI-H23 procured from the National Center for Cell Sciences (Pune, India) were cultured in DMEM and RPMI 1640 (Life Technologies), respectively, with 10% FBS and penicillin/streptomycin (50 units/ml). These cells were maintained at 37 °C with a supply of 5% CO2. Cell lines were authenticated by short tandem repeat analysis (61).
Patient Tissue Samples and Confocal Imaging
Human NSCLC tissue slides (adenocarcinoma of the lung) were obtained from thirty-five de-identified patients following the Institutional Review Board (IRB) rules of the Ohio State University and Chittaranjan National Cancer Institute. Characteristics of the patients are shown in Table 1.
TABLE 1.
Selection of patients
| Characteristics | No. of patients |
|---|---|
| Male (aged 45–68 years) | 10 |
| Female (aged 58–71 years) | 25 |
| Histological subtypes of adenocarcinoma | 35 |
| Histological staging | |
| Stage 1/II | 29 |
| Stage III | 6 |
Slides were preblocked with 10% donkey serum at room temperature for 1 h. Thereafter, these slides were incubated with unconjugated primary antibodies for D2 DA receptor (Santa Cruz Biotechnology), CD133 (Miltenyi Biotec), or ALDH1A1 (Santa Cruz Biotechnology) overnight at 4 °C. On the next day, the slides were incubated with the respective secondary antibodies conjugated to fluorochrome for 1 h at room temperature, washed in PBS, and mounted with fluorescence mounting gel and DAPI (Electron Microscopy Sciences), and then confocal microscopy was performed (Olympus FBV 1000) (62). All these antibodies have been previously validated by us and others (35, 38, 62–68).
Flow Cytometry
Single cell suspension was stained with phycoerythrin-conjugated CD133/1 (Miltenyi Biotec, validated in several reports (69, 70)) and fluorescein isothiocyanate-conjugated anti-DA D2 antibodies (Santa Cruz Biotechnology, validated in our previous reports (38, 39)), either singly or in combination, for 30 min as per the recommendation of the manufacturer. Following labeling, the cells were washed in FACS buffer (PBS, pH 7.4, with 1% FBS) and fixed in 1% paraformaldehyde in PBS. Then flow cytometry was performed using CellQuest software (BD Biosciences). Appropriate negative isotype controls were used to rule out the background fluorescence. The percentage of each positive population and mean fluorescence intensities were determined using quadrant statistics (38).
Isolation and Purification of CD133+ve Cells by Magnetic-activated Cell Sorting and Generation of Tumor Sphere
The expression of CD133 was determined by flow cytometry (FACSCalibur, BD Biosciences) using CD133/1-phycoerythrin. 1 × 107 cells were labeled with CD133/1 microbeads (Miltenyi Biotec), and the cell suspension was then loaded on a magnetic-activated cell sorting column. The effluent containing the cell population enriched with the CD133+ve cell fraction was collected. The purity of cells (>90%) was checked by flow cytometry after being labeled with CD133/1-phycoerythrin (Miltenyi Biotec). Mouse IgG1 phycoerythrin (Miltenyi Biotec) was used as isotype control. Thereafter, these purified CD133+ve cells were suspended in stem cell medium containing serum-free DMEM/F12 supplemented with penicillin/streptomycin (50 units/ml), 20 ng/ml human recombinant epidermal growth factor (hrEGF), 10 ng/ml human recombinant basic fibroblast growth factor (hrbFGF), and 1% B27 supplement (Invitrogen), and then cultured in 6-well plates (Corning) at different densities as per the requirement varying from 1000 to 50,000 cells/well. 1 or 10 μm quinpirole (Sigma) was added to the medium. The sphere-forming efficiency was monitored following treatment by plating 1000 cells/well in 24-well cell culture plates and culturing them for 7 days with the appropriate controls. Sphere-forming efficiency was calculated by counting the number of tumor spheres formed in a given well and dividing this by the total number of living cells seeded in the well × 100 (21). This and other experiments were carried out in pooled CD133+ve NSCLC cells.
Cell Proliferation by MTT Assay
Proliferation of CD133+ve NSCLC cells in stem cell media was checked by the MTT assay as described previously (35). In brief, 50,000 cells/well were seeded in 96-well round bottom cell plates (Corning) containing stem cell medium in the presence of 1 or 10 μm quinpirole (Sigma) with or without eticlopride (10 times more than the concentration of quinpirole) and cultured for 72 h with the appropriate controls. The cells were pelleted, the medium was aspirated, and 20-μl aliquots of MTT solution (5 mg/ml) (Sigma) were added and incubated for 4 h at 37 °C. The medium was finally removed by aspiration, the purple-colored formazan precipitate was dissolved in DMSO (100 μl), and then absorbance was measured at 550 nm using a microplate reader (BioTek).
In Vitro Tumor Apoptosis Assay
In vitro apoptosis of tumor cells was determined using the apoptosis detection kit (BD Pharmingen) as per the manufacturer's instructions. In brief, 1 × 105 CD133+ve tumor cells were incubated with or without quinpirole (in different doses for 72 h) at 37 °C in a humidified atmosphere with 5% CO2. Cells were then washed with cold PBS, and then suspended in binding buffer and incubated with annexin V-FITC and propidium iodide for 15 min at room temperature in the dark. Apoptotic cells were monitored by flow cytometric analysis (38).
siRNA Knockdown of D2 DA Receptor
Cells were plated at 50% confluence on six-well plates. D2 DA receptor siRNA (Santa Cruz Biotechnology) solution was diluted further with transfection medium (Santa Cruz Biotechnology), and then mixed with siRNA transfection reagent (Santa Cruz Biotechnology) and added to the culture medium to achieve a final siRNA concentration of 50 nmol/liter. Transfected cells were incubated at 37 °C for 48 h. siRNA concentrations were optimized to ensure that they did not affect cell viability and scramble (51). Scramble siRNA (Santa Cruz Biotechnology) was used as a control.
Matrigel Colony Formation Assay
The spheres collected by gentle centrifugation were dissociated by adding trypsin-EDTA followed by repeated pipetting. After determining cell viability by trypan blue staining, single cell suspension was seeded in DMEM with 10% FBS at a density of 4000 cells/well in 6-well plates that were precoated with Matrigel (BD Biosciences) and then treated with 1 or 10 μm quinpirole. The colony formation ability was assessed after 7 days by counting the number of colonies (>70 cells) under a microscope following crystal violet staining (Sigma-Aldrich) (71). To ascertain the effect of ERK and AKT inhibition on the colony-forming ability of CD133+ve NSCLC cells in agarose, cells were treated with 10 μm LY294002, AKT inhibitor (Cell Signaling Technology) or 10 μm U0126, MEK1 inhibitor, and MEK2 inhibitor (42) was procured from Merck Millipore.
Preparation of Whole Cell Protein Lysates and Immunoblotting
To prepare whole cell lysates, CD133+ve tumor cells were rinsed with PBS and disrupted with cell lysis buffer (20 mm Tris-HCl (pH 7.5), 500 mm NaCl, 1% Triton X-100, 1 mm EDTA, 50 mm dithiothreitol, and 2 mm phenylmethylsulfonyl fluoride) and protease inhibitor cocktail (Sigma). The lysate was then spun down, cleared of debris by centrifugation, and assayed for total protein concentration using Bradford reagent (Bio-Rad). For immunoblotting analysis, protein samples were boiled in 1× SDS buffer, separated by SDS-PAGE, and transferred to a PVDF membrane. The PVDF membrane was blocked with 0.2% I-BLOCK (Applied Biosystems) for 2 h at room temperature and incubated with the appropriate primary antibody overnight at 4 °C. Finally, after washing with Tris-buffered saline (containing 0.1% Tween 20), each blot was incubated with the corresponding HRP-conjugated secondary antibody (Sigma), and the immunoblot signal was visualized by chemiluminescence reagents (Pierce) (72). Primary antibodies used in this study were phospho-ERK1/2 from BD Transduction Laboratories (catalogue number 612358; lot number 99125) and phospho-AKT (1:500) from Santa Cruz Biotechnology (catalogue number SC7985-R; lot number AL909). The molecular weight markers were procured from Bio-Rad.
Quantitative Real-time PCR Analysis
Total RNA from treated and untreated CD133+ve tumor cells was extracted using TRIzol reagent (Invitrogen). Total RNA (1 μg) was reversely transcribed into cDNA using cDNA transcription kit (Applied Biosystems) and followed by quantitative real-time PCR analysis using the Universal SYBR Green Master Mix and Step One Plus Real-Time PCR System (Applied Biosystems). β-actin was the endogenous control. The -fold expression of the target host gene was calculated by using the 2−ΔΔCt formula (73). The primers used were as follows: OCT4forward, 5′-TCGAGAACCGAGTGAGAGG-3′; OCT4 reverse, 5′-GAACCACACTCGGACCACA-3′; β-actin forward, 5′-CTGGAGAAGAGCTACGAGC-3′; β-actin reverse, 5′-GGATGCCACAGGACTCCA-3′.
Matrigel Invasion Assay
Invasion assays were performed according to a previously published protocol (74). Briefly, 1 × 104 CD133+ve NSCLC cells in 200 μl of serum-free medium containing 1 or 10 μm quinpirole were added to the upper chamber of a Matrigel insert (BD Biosciences), and 20% serum containing medium or medium without serum was placed in the lower chamber.
The plates were incubated for 24 h at 37 °C in 5% CO2. The cells that did not migrate or invade through the pores were removed with a cotton swab, and cells on the lower surface of the membrane were examined, stained with crystal violet, and counted under a microscope. Supernatant from the upper chamber was collected, stored at −80 °C, and used for the estimation of MMP-9. The invasion index was then calculated (74).
ELISA of MMP-9
To measure the extracellular secretion of MMP-9, cell-free supernatants were quantified by ELISA. In brief, 96-well microtiter plates were coated with cell-free supernatants and incubated overnight at 4 °C. Plates were blocked with BSA (5%) for 2 h. After washing, 50 μl of MMP-9 antibody (R&D Systems) diluted 1:1000 was added to each well and incubated for 45 min. Bound cytokine was detected with secondary IgG peroxidase (Sigma). The color was developed with 3,3′, 5,5′-tetramethylbenzidine (TMB) substrate solution (OptEIATM, BD Pharmingen). The reaction was stopped with 2 n H2SO4 solution, and absorbance was measured (450 nm) using a microplate reader (BioTek) (38).
Xenotransplantation of Tumor in Nude Mice
Mice experiments were undertaken after approval by the Institutional Animal Care and Use Committee. Nude mice (National Institute of Nutrition, Hyderabad, India) 4–6 weeks of age weighing 20–25 g were used for the study. CD133+ve tumor cells from A549 NSCLC tumor cell culture were purified by magnetic bead separation. Purified cells were further evaluated by flow cytometry to confirm ≥90% CD133+ve tumor cells. Freshly isolated CD133+ve tumor cells (104) were mixed with Matrigel (1:1) and inoculated subcutaneously in the right flank of each animal. When the tumor reached 100 mm3, animals were divided into two groups, each consisting of six animals. The control group received tumor cells + Matrigel only, whereas the treated group received D2 DA receptor agonist quinpirole (10 mg/kg) intraperitoneally for consecutive 7 days. Tumor volume was measured (TV (mm3) = d2 × d/2, where d and D are the shortest and longest diameter of the tumor) (62).
Statistics
The results are expressed as mean ± S.D. Statistical significance was determined using an unpaired Student's t test, and the results were considered significant when p ≤ 0.05 (75).
Author Contributions
S. R., K. L., M. K. N., A. B., T. G., S. K., and S. G. performed collection and/or assembly of data; S. R., K. L., R. B., P. S. D., and S. B. performed data analysis and interpretation; S. R., P. S. D., and S. B. wrote the manuscript; P. S. D. and S. B. conceived and designed the study, provided financial support, provided study material or patient tissue samples, and provided final approval of the study.
This study was supported by Council of Scientific and Industrial Research Grant 21(0895)/12/EMR-II (to P. S. D.) and National Institutes of Health Grants R01DK098045, R01CA169158, and R01HL131405 (to S. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NSCLC
- non-small cell lung carcinoma
- CSC
- cancer stem cell(s)
- D2 DA
- D2 dopamine (DA) receptors
- ERK1/2
- extracellular signal-regulated kinases 1/2
- MMP-9
- matrix metalloproteinase-9
- MTT
- 3-(4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide
- PI
- propidium iodide.
References
- 1. Siegel R., Naishadham D., and Jemal A. (2012) Cancer statistics. CA Cancer J. Clin. 62, 10–29 [DOI] [PubMed] [Google Scholar]
- 2. Parsons A., Daley A., Begh R., and Aveyard P. (2010) Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: system review of observational studies with meta analysis. BMJ 340, b5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alberg A. J., Ford J. G., Samet J. M., and American College of Chest Physicians (2007) Epidemiology of lung cancer: ACCP evidence based clinical practice guidelines (2nd edition). Chest 132, (suppl.) 29s–55s [DOI] [PubMed] [Google Scholar]
- 4. Danesi R., Pasqualetti G., Giovannetti E., Crea F., Altavilla G., Del Tacca M., and Rosell R. (2009) Pharmacogenomics in non-small-cell lung cancer chemotherapy. Adv. Drug Deliv. Rev. 61, 408–417 [DOI] [PubMed] [Google Scholar]
- 5. Gomez-Casal R., Bhattacharya C., Ganesh N., Bailey L., Basse P., Gibson M., Epperly M., and Levina V. (2013) Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cells and epithelial mesenchymal transition phenotypes. Mol. Cancer 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dawood S., Austin L., and Cristofanilli M. (2014) Cancer stem cells: implications for cancer therapy. Oncology (Williston Park) 28, 1101–1107, 1110 [PubMed] [Google Scholar]
- 7. Nassar D., and Blanpain C. (2016) Cancer stem cells: basic concepts and therapeutic implications. Annu. Rev. Pathol. 11, 47–76 [DOI] [PubMed] [Google Scholar]
- 8. Li Z. (2013) CD133: a stem cell biomarker and beyond. Exp. Hematol. Oncol. 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singh S. K., Hawkins C., Clarke I. D., Squire J. A., Bayani J., Hide T., Henkelman R. M., Cusimano M. D., and Dirks P. B. (2004) Identification of human brain tumour initiating cells. Nature 432, 396–401 [DOI] [PubMed] [Google Scholar]
- 10. Li H., and Tang D. G. (2011) Prostate cancer stem cells and their potential roles in metastasis. J. Surg. Oncol. 103, 558–562 [DOI] [PubMed] [Google Scholar]
- 11. Zhu Y., Karakhanova S., Huang X., Deng S. P., Werner J., and Bazhin A. V. (2014) Influence of interferon-α on the expression of the cancer stem cell markers in pancreatic carcinoma cells. Exp. Cell Res. 324, 146–156 [DOI] [PubMed] [Google Scholar]
- 12. Roudi R., Korourian A., Shariftabrizi A., and Madjd Z. (2015) Differential expression of cancer stem cell markers ALDH1 and CD133 in various lung cancer subtypes. Cancer Invest. 33, 294–302 [DOI] [PubMed] [Google Scholar]
- 13. Bidlingmaier S., Zhu X., and Liu B. (2008) The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J. Mol. Med. 86, 1025–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fargeas C. A., Huttner W. B., and Corbeil D. (2007) Nomenclature of prominin-1 (CD133) splice variants? An update. Tissue Antigens 69, 602–606 [DOI] [PubMed] [Google Scholar]
- 15. Schiffer D., Mellai M., Annovazzi L., Caldera V., Piazzi A., Denysenko T., and Melcarne A. (2014) Stem cell niches in glioblastoma: a neuropathological view. BioMed Res. Int. 2014, 725921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Steinmetz N. F., Maurer J., Sheng H., Bensussan A., Maricic I., Kumar V., and Braciak T. A. (2011) Two domains of vimentin are expressed on the surface of lymph node, bone and brain metastatic prostate cancer lines along with the putative stem cell marker proteins CD44 and CD133. Cancers 3, 2870–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu H., Zhang W., Jia Y., Yu Q., Grau G. E., Peng L., Ran Y., Yang Z., Deng H., and Lou J. (2013) Single-cell clones of liver cancer stem cells have the potential of differentiating into different types of tumor cells. Cell Death Dis. 4, e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bertolini G., Roz L., Perego P., Tortoreto M., Fontanella E., Gatti L., Pratesi G., Fabbri A., Andriani F., Tinelli S., Roz E., Caserini R., Lo Vullo S., Camerini T., Mariani L., et al. (2009) Highly tumorigenic lung cancer CD133 cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. U.S.A. 106, 16281–16286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eramo A., Lotti F., Sette G., Pilozzi E., Biffoni M., Di Virgilio A., Conticello C., Ruco L., Peschle C., and De Maria R. (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 15, 504–514 [DOI] [PubMed] [Google Scholar]
- 20. Hsu H.-S., Huang P.-I., Chang Y.-L., Tzao C., Chen Y.-W., Shih H.-C., Hung S.-C., Chen Y.-C., Tseng L.-M., and Chiou S.-H. (2011) Cucurbitacin I inhibits tumorigenic ability and enhances radiochemosensitivity in non-small cell lung cancer-derived CD133-positive cells. Cancer 117, 2970–2985 [DOI] [PubMed] [Google Scholar]
- 21. Le H., Zeng F., Xu L., Liu X., and Huang Y. (2013) The role of CD133 expression in the carcinogenesis and prognosis of patients with lung cancer. Mol. Med. Rep. 8, 1511–1518 [DOI] [PubMed] [Google Scholar]
- 22. Eramo A., Haas T. L., and De Maria R. (2010) Lung cancer stem cells: tools and targets to fight lung cancer. Oncogene 29, 4625–4635 [DOI] [PubMed] [Google Scholar]
- 23. Tirino V., Camerlingo R., Franco R., Malanga D., La Rocca A., Viglietto G., Rocco G., and Pirozzi G. (2009) The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur. J. Cardiothorac. Surg. 36, 446–453 [DOI] [PubMed] [Google Scholar]
- 24. Wang W., Chen Y., Deng J., Zhou J., Zhou Y., Wang S., and Zhou J. (2014) The prognostic value of CD133 expression in non-small cell lung cancer: a meta-analysis. Tumour Biol. 35, 9769–9775 [DOI] [PubMed] [Google Scholar]
- 25. Wu H., Qi X. W., Yan G. N., Zhang Q. B., Xu C., and Bian X. W. (2014) Is CD133 expression a prognostic biomarker of non-small-cell lung cancer? A systematic review and meta-analysis. PLoS ONE 9, e100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qu H., Li R., Liu Z., Zhang J., and Luo R. (2013) Prognostic value of cancer stem cell marker CD133 expression in non-small cell lung cancer: a systematic review. Int. J. Clin. Exp. Pathol. 6, 2644–2650 [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang F., Qiu Q., Khanna A., Todd N. W., Deepak J., Xing L., Wang H., Liu Z., Su Y., Stass S. A., and Katz R. L. (2009) Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 7, 330–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X., Wan L., Geng J., Wu C.-L., and Bai X. (2012) Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 7, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 29. Barr M., Macdonagh L., Gray S., O'Byrne K., Cuffe S., and Finn S. (2016) 75P Inhibition and exploitation of aldehyde dehydrogenase 1 (ALDH1) as a cancer stem cell marker in cisplatin resistant NSCLC. J. Thorac. Oncol. 11, (suppl.) S87 [Google Scholar]
- 30. Tomita H., Tanaka K., Tanaka T., and Hara A. (2016) Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 7, 11018–11032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Charafe-Jauffret E., Ginestier C., Iovino F., Tarpin C., Diebel M., Esterni B., Houvenaeghel G., Extra J.-M., Bertucci F., Jacquemier J., Xerri L., Dontu G., Stassi G., Xiao Y., Barsky S. H., et al. (2010) Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 16, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang L., Ren Y., Yu X., Qian F., Bian B.-S.-J., Xiao H.-L., Wang W.-G., Xu S.-L., Yang J., Cui W., Liu Q., Wang Z., Guo W., Xiong G., Yang K., et al. (2014) ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod. Pathol. 27, 775–783 [DOI] [PubMed] [Google Scholar]
- 33. Alamgeer M., Ganju V., Szczepny A., Russell P. A., Prodanovic Z., Kumar B., Wainer Z., Brown T., Schneider-Kolsky M., Conron M., Wright G., and Watkins D. N. (2013) The prognostic significance of aldehyde dehydrogenase 1A1 (ALDH1A1) and CD133 expression in early stage non-small cell lung cancer. Thorax. 68, 1095–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alcaro A., Huber R., and Panksepp J. (2007) Behavioral functions of the mesolimbic dopaminergic system: an affective neuroethological perspective. Brain Res. Rev. 56, 283–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basu S., Nagy J. A., Pal S., Vasile E., Eckelhoefer I. A., Bliss V. S., Manseau E. J., Dasgupta P. S., Dvorak H. F., and Mukhopadhyay D. (2001) The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat. Med. 7, 569–574 [DOI] [PubMed] [Google Scholar]
- 36. Chakroborty D., Sarkar C., Mitra R. B., Banerjee S., Dasgupta P. S., and Basu S. (2004) Depleted dopamine in gastric cancer tissues: dopamine treatment retards growth of gastric cancer by inhibiting angiogenesis. Clin. Cancer Res. 10, 4349–4356 [DOI] [PubMed] [Google Scholar]
- 37. Sarkar C., Chakroborty D., Chowdhury U. R., Dasgupta P. S., and Basu S. (2008) Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 14, 2502–2510 [DOI] [PubMed] [Google Scholar]
- 38. Chakroborty D., Chowdhury U. R., Sarkar C., Baral R., Dasgupta P. S., and Basu S. (2008) Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J. Clin. Invest. 118, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shome S., Dasgupta P. S., and Basu S. (2012) Dopamine regulates mobilization of mesenchymal stem cells during wound angiogenesis. PLoS ONE 7, e31682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Travis W. D., Brambilla E., Noguchi M., Nicholson A. G., Geisinger K. R., Yatabe Y., Beer D. G., Powell C. A., Riely G. J., Van Schil P. E., Garg K., Austin J. H., Asamura H., Rusch V. W., Hirsch F. R., et al. (2011) International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society International multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 6, 244–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., and Wicha M. S. (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes. Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y. K., Zhu Y. L., Qiu F. M., Zhang T., Chen Z. G., Zheng S., and Huang J. (2010) Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133 primary colon cancer cells. Carcinogenesis 31, 1376–1380 [DOI] [PubMed] [Google Scholar]
- 43. Chen Y.-C., Hsu H.-S., Chen Y.-W., Tsai T.-H., How C.-K., Wang C.-Y., Hung S.-C., Chang Y.-L., Tsai M.-L., Lee Y.-Y., Ku H.-H., and Chiou S.-H. (2008) Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 3, e2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu D., McKee C. M., Cao Y., Ding Y., Kessler B. M., and Muschel R. J. (2010) Matrix metalloproteinase-9 regulates tumor cell invasion through cleavage of protease Nexin-1. Cancer Res. 70, 6988–6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bozzi Y., and Borrelli E. (2006) Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 29, 167–174 [DOI] [PubMed] [Google Scholar]
- 46. Sokoloff P., Riou J.-F., Martres M.-P., and Schwartz J.-C. (1989) Presence of dopamine D-2 receptors in human tumoral cell lines. Biochem. Biophys. Res. Commun. 162, 575–582 [DOI] [PubMed] [Google Scholar]
- 47. Sheikhpour M., Ahangari G., Sadeghizadeh M., and Deezagi A. (2013) A novel report of apoptosis in human lung carcinoma cells using selective agonist of D2-like dopamine receptors: a new approach for the treatment of human non-small cell lung cancer. Int. J. Immunopathol. Pharmacol. 26, 393–402 [DOI] [PubMed] [Google Scholar]
- 48. Senogles S. E. (2007) D2 dopamine receptor-mediated antiproliferation in a small cell lung cancer cell line, NCI-H69. Anticancer Drugs 18, 801–807 [DOI] [PubMed] [Google Scholar]
- 49. Ishibashi M., Fujisawa M., Furue H., Maeda Y., Fukayama M., and Yamaji T. (1994) Inhibition of growth of human small cell lung cancer by bromocriptine. Cancer Res. 54, 3442–3446 [PubMed] [Google Scholar]
- 50. Hoeppner L. H., Wang Y., Sharma A., Javeed N., Van Keulen V. P., Wang E., Yang P., Roden A. C., Peikert T., Molina J. R., and Mukhopadhyay D. (2015) Dopamine D2 receptor agonists inhibit lung cancer progression by reducing angiogenesis and tumor infiltrating myeloid derived suppressor cells. Mol. Oncol. 9, 270–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ganguly S., Basu B., Shome S., Jadhav T., Roy S., Majumdar J., Dasgupta P. S., and Basu S. (2010) Dopamine, by acting through its D2 receptor, inhibits insulin-like growth factor-I (IGF-I)-induced gastric cancer cell proliferation via up-regulation of Krüppel-like factor 4 through down-regulation of IGF-IR and AKT phosphorylation. Am. J. Pathol. 177, 2701–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beaulieu J.-M., Sotnikova T. D., Marion S., Lefkowitz R. J., Gainetdinov R. R., and Caron M. G. (2005) An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273 [DOI] [PubMed] [Google Scholar]
- 53. Beaulieu J.-M., Tirotta E., Sotnikova T. D., Masri B., Salahpour A., Gainetdinov R. R., Borrelli E., and Caron M. G. (2007) Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 27, 881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang H.-W., Huang C.-Y., Yang S.-Y., Wu V.-C., Chu T.-S., Chen Y.-M., Hsieh B.-S., and Wu K.-D. (2014) Role of D2 dopamine receptor in adrenal cortical cell proliferation and aldosterone-producing adenoma tumorigenesis. J. Mol. Endocrinol. 52, 87–96 [DOI] [PubMed] [Google Scholar]
- 55. Liu J. C., Baker R. E., Sun C., Sundmark V. C., and Elsholtz H. P. (2002) Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells: a key step in the transcriptional suppression of the prolactin gene. J. Biol. Chem. 277, 35819–35825 [DOI] [PubMed] [Google Scholar]
- 56. Brumbaugh J., Hou Z., Russell J. D., Howden S. E., Yu P., Ledvina A. R., Coon J. J., and Thomson J. A. (2012) Phosphorylation regulates human OCT4. Proc. Natl. Acad. Sci. U.S.A. 109, 7162–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin Y., Yang Y., Li W., Chen Q., Li J., Pan X., Zhou L., Liu C., Chen C., He J., Cao H., Yao H., Zheng L., Xu X., Xia Z., et al. (2012) Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 48, 627–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu F., Cao X., Liu Z., Guo H., Ren K., Quan M., Zhou Y., Xiang H., and Cao J. (2014) Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim. Biophys. Sin. 46, 15–21 [DOI] [PubMed] [Google Scholar]
- 59. Goldberg L. I. (1989) The role of dopamine receptors in the treatment of congestive heart failure. J. Cardiovasc. Pharmacol. 14, Suppl. 5, s19–s27 [PubMed] [Google Scholar]
- 60. Denton M. D., Chertow G. M., and Brady H. R. (1996) “Renal dose” dopamine for the treatment of acute renal failure: scientific rationale, experimental studies and clinical trials. Kidney Int. 50, 4–14 [DOI] [PubMed] [Google Scholar]
- 61. Masters J. R., Thomson J. A., Daly-Burns B., Reid Y. A., Dirks W. G., Packer P., Toji L. H., Ohno T., Tanabe H., Arlett C. F., Kelland L. R., Harrison M., Virmani A., Ward T. H., Ayres K. L., and Debenham P. G. (2001) Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl. Acad. Sci. U.S.A. 98, 8012–8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chakroborty D., Sarkar C., Yu H., Wang J., Liu Z., Dasgupta P. S., and Basu S. (2011) Dopamine stabilizes tumor blood vessels by up-regulating angiopoietin 1 expression in pericytes and Krüppel-like factor-2 expression in tumor endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 20730–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chavali P. L., Saini R. K. R., Zhai Q., Vizlin-Hodzic D., Venkatabalasubramanian S., Hayashi A., Johansson E., Zeng Z.-J., Mohlin S., Påhlman S., Hansford L., Kaplan D. R., and Funa K. (2014) TLX activates MMP-2, promotes self-renewal of tumor spheres in neuroblastoma and correlates with poor patient survival. Cell Death Dis. 5, e1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Al-Lamki R. S., Wang J., Yang J., Burrows N., Maxwell P. H., Eisen T., Warren A. Y., Vanharanta S., Pacey S., Vandenabeele P., Pober J. S., and Bradley J. R. (2016) Tumor necrosis factor receptor 2-signaling in CD133-expressing cells in renal clear cell carcinoma. Oncotarget 7, 24111–24124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang L., Liu Y., Wang X., Tang Z., Li S., Hu Y., Zong X., Wu X., Bu Z., Wu A., Li Z., Li Z., Huang X., Jia L., Kang Q., et al. (2015) The extent of inflammatory infiltration in primary cancer tissues is associated with lymphomagenesis in immunodeficient mice. Sci. Rep. 5, 9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rappa F., Cappello F., Halatsch M.-E., Scheuerle A., and Kast R. E. (2013) Aldehyde dehydrogenase and HSP90 co-localize in human glioblastoma biopsy cells. Biochimie 95, 782–786 [DOI] [PubMed] [Google Scholar]
- 67. Li T., Su Y., Mei Y., Leng Q., Leng B., Liu Z., Stass S. A., and Jiang F. (2010) ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients' outcome. Lab. Invest. 90, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Su Y., Qiu Q., Zhang X., Jiang Z., Leng Q., Liu Z., Stass S. A., and Jiang F. (2010) Aldehyde dehydrogenase 1A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol. Biomarkers Prev. 19, 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mak A. B., Blakely K. M., Williams R. A., Penttilä P.-A., Shukalyuk A. I., Osman K. T., Kasimer D., Ketela T., and Moffat J. (2011) CD133 Protein N-glycosylation processing contributes to cell surface recognition of the primitive cell marker AC133 epitope. J. Biol. Chem. 286, 41046–41056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu S., Li N., Yu X., Xiao X., Cheng K., Hu J., Wang J., Zhang D., Cheng S., and Liu S. (2013) Expression of intercellular adhesion molecule 1 by hepatocellular carcinoma stem cells and circulating tumor cells. Gastroenterology 144, 1031–1041.e10 [DOI] [PubMed] [Google Scholar]
- 71. Cao L., Zhou Y., Zhai B., Liao J., Xu W., Zhang R., Li J., Zhang Y., Chen L., Qian H., and Wu M., Yin Z. (2011) Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 11, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu K., Chakroborty D., Sarkar C., Lu T., Xie Z., Liu Z., and Basu S. (2012) Triphala and its active constituent chebulinic acid are natural inhibitors of vascular endothelial growth factor-A mediated angiogenesis. PLoS ONE 7, e43934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huang C., Yang L., Li Z., Yang J., Zhao J., Dehui X., Liu L., Wang Q., and Song T. (2007) Detection of CCND1 amplification using laser capture microdissection coupled with real-time polymerase chain reaction in human esophageal squamous cell carcinoma. Cancer Genet. Cytogenet. 175, 19–25 [DOI] [PubMed] [Google Scholar]
- 74. Han J., Fujisawa T., Husain S. R., and Puri R. K. (2014) Identification and characterization of cancer stem cells in human head and neck squamous cell carcinoma. BMC Cancer 14, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lu K., and Basu S. (2015) The natural compound chebulagic acid inhibits vascular endothelial growth factor A mediated regulation of endothelial cell functions. Sci. Rep. 5, 9642. [DOI] [PMC free article] [PubMed] [Google Scholar]