

Abstract
P-glycoprotein (P-gp) is a polyspecific ATP-dependent transporter linked to multidrug resistance in cancer; it plays important roles in determining the pharmacokinetics of many drugs. Understanding the structural basis of P-gp, substrate polyspecificity has been hampered by its intrinsic flexibility, which is facilitated by a 75-residue linker that connects the two halves of P-gp. Here we constructed a mutant murine P-gp with a shortened linker to facilitate structural determination. Despite dramatic reduction in rhodamine 123 and calcein-AM transport, the linker-shortened mutant P-gp possesses basal ATPase activity and binds ATP only in its N-terminal nucleotide-binding domain. Nine independently determined structures of wild type, the linker mutant, and a methylated P-gp at up to 3.3 Å resolution display significant movements of individual transmembrane domain helices, which correlated with the opening and closing motion of the two halves of P-gp. The open-and-close motion alters the surface topology of P-gp within the drug-binding pocket, providing a mechanistic explanation for the polyspecificity of P-gp in substrate interactions.
Keywords: ABC transporter, ATPase, crystal structure, glycoprotein, multidrug transporter, ATP-binding, asymmetry, P-glycoprotein, polyspecificity, structure
Introduction
The association of multidrug resistance (MDR)3 in cancer treatment with expression of human ABC transporters on the cell surface has raised the possibility that overcoming MDR might be achieved by inhibiting these transporters (1). One transporter in particular, the human P-glycoprotein (P-gp), has been well characterized; it is able to confer MDR by transporting numerous structurally unrelated drugs at the expense of hydrolyzing ATP (2, 3). In addition, P-gp plays important roles in drug distribution in normal physiology and is an essential component of many physiological barriers (4, 5).
Long-standing efforts have been devoted to understanding the mechanism of P-gp function by various experimental approaches. Among them, structural studies of P-gp from various organisms have been reported (6–8). In particular, a number of structures for the mouse P-gp (mP-gp) were obtained (8–10). However, the diffraction limits of “near-native,” inhibitor-bound, and nanobody-associated mP-gp crystals were relatively low, mostly near 4 Å resolution. The methylated protein gave higher resolution structures (11), but the impact of reductive methylation on the structure and function of P-gp was not addressed. The low resolution diffraction of P-gp crystals has been attributed in part to the intrinsic flexibility of the molecule (12–14).
Mouse P-gp is a 1276-residue polypeptide and bears 87% sequence identity to human P-gp; it consists of two homologous halves connected by a flexible linker of ∼75 residues. Each half is made of a transmembrane domain (TMD) implicated in drug recognition and transport and a nucleotide-binding domain (NBD). Each NBD is able to bind and hydrolyze ATP (15, 16), which is, however, dependent on the other NBD being functional (17, 18). The two NBDs of P-gp are highly homologous, each featuring a consensus nucleotide-binding site with full-fledged Walker A, Walker B, and signature motifs. However, vanadate-induced photooxidative cleavage experiments demonstrated that only one hydrolysis event can occur at a time (19), and one-site inhibition leads to inactivation of both sites (20). This functional asymmetry supports the hypothesis that P-gp functions by coupling alternate ATP hydrolyses at the two NBDs to substrate extrusion (4, 21). Functional asymmetry is better demonstrated by non-equivalent NBDs in the amino acid sequences of a number of eukaryotic ABC transporters, including the human cystic fibrosis transmembrane regulator (22), multidrug resistance-associated protein-1 (23), transporters associated with antigen processing (24), and yeast pdr5 (pleiotropic drug resistance) (25), in which only one NBD has a consensus and the other is a degenerate site. Indeed, the crystal structure of the heterodimeric TM287/TM288 from Thermotoga maritima demonstrated the asymmetric binding of nucleotide to the degenerate site (26). For transporters like P-gp that feature two consensus NBDs, structural support for the observed functional asymmetry has yet to be demonstrated.
The elucidation of the structural basis for P-gp's ability to recognize and transport an ever-increasing number of structurally diverse compounds is another outstanding issue. Crystal structures of P-gp reveal without exception an open-inward conformation with a very large separation between the two NBDs, which inevitably encompasses a large surface area available for drug interaction within the boundary of the membrane bilayer. Attempts to define substrate-interacting residues in this binding pocket by probing with photoaffinity substrates and by cysteine scanning coupled with thioreactive substrates resulted in the identification of residues distributed among nearly all TM helices (27–30). Thus, it appears that there are multiple transport-active sites for a given substrate, and it is difficult to pinpoint interacting residues for specific drugs (31).
Here, by shortening the linker between the two halves of the mP-gp, we effectively reduced the flexibility of the molecule and obtained a 3.3 Å resolution structure of a linker-shortened mP-gp (ΔlnkmP-gp). This structure helped to accurately determine structures of the wild type and methylated full-length mP-gp (FLmP-gp and FLmP-gpMe) and led to a structure with bound nucleotide to the NBD1, demonstrating for the first time asymmetric nucleotide binding for ABC transporters with two consensus NBDs. Analysis of these structures correlates the open-and-close movement of two halves of P-gp with rotation and translation of individual helices in the TM domains of P-gp, suggesting a mechanism of polyspecific substrate recognition by P-gp.
Results
Restricting Conformational Flexibility Leads to Dysfunction of P-gp
The function of P-gp requires the flexibility of its two halves, which are joined by a mobile linker that is disordered in all mP-gp structures reported so far. Shortening of this linker by 34 residues in human P-gp inactivated the function of the transporter (32). We thus generated a mutant mP-gp that has the linker shortened by 34 residues (Δ649–682, ΔlnkmP-gp, and ΔlnkmGlu/GlnP-gp, supplemental Fig. S1). As expected, the ΔlnkmP-gp was expressed normally in P. pastoris and purified to homogeneity from the membrane fraction of disrupted cells (Fig. 1A), and its size was confirmed by tryptic digestion followed by capillary electrophoresis-coupled mass spectrometry. The purified ΔlnkmP-gp in a detergent solution shows no difference in melting temperature (Tm = 42.5 °C) by differential scanning calorimetry compared with FLmP-gp (Fig. 1A), indicating the shortening of the linker does not interfere with the phase transitions of the protein. The ATP hydrolysis assay demonstrates that the linker mutant possesses an elevated basal ATPase activity (8.9 ± 0.7 nmol of Pi/min/mg of protein, ∼20%), compared with the wild-type FLmP-gp (7.4 ± 0.9 nmol of Pi/min/mg of protein, Figs. 1B and supplemental Fig. S2A), which is inhibited to the same extent as the wild type by vanadate (supplemental Fig. S2B). However, the linker mutant exhibits a lack of drug-stimulated or inhibited ATPase activity for drugs known to be P-gp substrates/inhibitors such as verapamil and cyclosporin A (Fig. 1, C and D), although a limited amount of stimulated/inhibited ATPase activity has been detected toward some P-gp substrates such as quinidine (Fig. 1E). HeLa cells transfected with a BacMam baculovirus harboring ΔlnkmP-gp displayed a dramatically reduced rhodamine 123 and calcein-AM transport activity (Fig. 1, G and H, and supplemental Fig. S2, C and D). Taken together, removing a portion of the flexible linker does not appear to affect the folding and the structure of P-gp nor its basal ATPase activity, but it seems to restrict its conformational flexibility which is needed to transport substrates.
FIGURE 1.

Characterization of mP-gp variants. A, differential scanning calorimetry experiment showing the purified ΔlnkmP-gp has an identical melting temperature (Tm) as the wild-type FLmP-gp in a detergent solution. Inset, SDS-PAGE (right) and Western blotting (left) of the purified ΔlnkmP-gp (lane 1) and FLmP-gp (lane 2). Monoclonal antibody C219 was used for the Western blotting. B, basal ATPase activities for wild-type FLmP-gp and ΔlnkmP-gp. The wild type in total soy lipid has a basal ATPase activity of 7.4 nmol of Pi/min/mg of protein, which is set as 100%. C, concentration-dependent stimulation/inhibition of ATPase activity by verapamil for wild type and ΔlnkmP-gp. Inset is a magnified view for ΔlnkmP-gp. D, concentration-dependent ATPase activity by cyclosporin A. E, concentration-dependent ATPase activity by quinidine. F, verapamil concentration-dependent ATPase activity for FLmP-gp that was methylated in the presence or absence of AMP-PNP (1 mm). No AMP-PNP was present during ATPase assay. G, efflux of the fluorescent P-gp substrate rhodamine 123 by FLmP-gp. HeLa cells, which do not express P-gp, were transduced with BacMam baculovirus genetically modified to express FLmP-gp. Non-transduced HeLa cells (red) and virus-transduced HeLa cells were incubated with 1.5 μm rhodamine 123 in the presence (orange) or absence (cyan) of 100 nm elacridar, a P-gp inhibitor, and transport of fluorescent substrates by flow cytometry was carried out. H, efflux of the fluorescent P-gp substrate rhodamine 123 by ΔlnkmP-gp, which was carried out as described in G but with BacMam baculovirus genetically modified to express ΔlnkmP-gp. Accumulation of fluorescent substrate rhodamine 123 in non-transduced HeLa cells was used as control in both G and H. The numbers shown in B represent an average of 21 independent experiments, and in C–H are the results of three duplicates.
Structural Integrity of P-gp Is Maintained in the Linker Mutant
Crystals of ΔlnkmGlu/GlnP-gp diffracted X-rays better than FLmP-gp, leading to a native diffraction data set processed to 3.35 Å resolution (Table 1). Although many heavy-metal derivatives were obtained and a combination of various phasing approaches were tested, the best structure solution for the ΔlnkmGlu/GlnP-gp was obtained from a single mercurial derivative by the single anomalous dispersion method, which resulted in an experimental electron density map of high quality at 3.29 Å resolution (Table 1 and supplemental Fig. S3, A and B). The experimental maps allowed the building of atomic models for both native and mercury-derivatized ΔlnkmGlu/GlnP-gp, which were subsequently refined at 3.35 and 3.29 Å resolution, respectively. Anomalous difference Fourier maps were computed to ascertain correct assignment of cysteine residues in the mercurial derivative crystal (supplemental Fig. S3C) as well as methionine residues in the ΔlnkmGlu/GlnP-gp crystal (supplemental Fig. S3D).
TABLE 1.
Statistics on qualities of diffraction data sets and atomic models
| Data set | ΔlnkmGlu/GlnP-gp (apo) | ΔlnkmGlu/GlnP-gp (ATP) | FLmP-gp (apo) | FLmP-gp (methylated) | ΔlnkmGlu/GlnP-gp (Hg) |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimension (Å) | 98.0, 116.6, 375.6 | 97.5, 116.4, 375.1 | 100.6, 119.5, 383.9 | 88.0, 136.8, 185.9 | 98.2, 114.7, 375.4 |
| Wave length (Å) | 1.0 | 1.0 | 1.0 | 0.89 | 1.0 |
| Resolution (Å) | 29.81–3.35 | 34.72–3.85 | 50.0–4.02 | 50.0–3.50 | 49.11–3.29 |
| (outer shell) | (3.47–3.35)a | (3.987–3.85) | (4.10–4.02) | (3.63–3.50) | (3.41–3.29) |
| Rmergeb | 0.101 (0.46) | 0.131 (0.957) | 0.106 (0.557) | 0.092 (0.449) | 0.150 (0.905) |
| Rmeasc | 0.106 (0.533) | 0.136 (1.005) | 0.113 (0.692) | 0.102 (0.554) | 0.153 (0.934) |
| 〈I〉/σI | 17.5 (1.1) | 12.0 (2.4) | 9.1 (1.4) | 11.5 (1.2) | 13.4 (2.7) |
| Completeness (%) | 0.97 (87.3) | 1.00 (1.00) | 0.93 (0.71) | 0.94 (0.84) | 0.93 (0.86) |
| Average redundancy | 8.9 | 14.6 | 5.0 | 4.3 | 21.6 |
| No. of unique reflections | 58,212 | 41,157 | 37,377 | 27,310 | 60,351 |
| No. free of reflections | 2343 | 1951 | 1881 | 1355 | 1982 |
| Model refinement | |||||
| Resolution (Å) | 22.41–3.35 | 21.30–3.85 | 20.11–4.02 | 21.43–3.50 | 33.54–3.29 |
| No. of molecules/AU | 2 | 2 | 2 | 1 | 2 |
| Rwork/Rfree | 0.2400/0.2826 | 0.2556/0.2910 | 0.2576/0.3026 | 0.2855/0.3149 | 0.2430/0.2848 |
| No. of protein atoms | 18,374 | 18,382 | 18,372 | 9117 | 18,374 |
| No. of residues (% complete) | 2369 (93%) | 2364 (93%) | 2369 (93%) | 1174 (92%) | 2369 (93%) |
| No. of cofactors | 0 | 2 ATP molecules | 0 | 0 | 0 |
| No. of solvents | 0 | 0 | 0 | 2 | 0 |
| r.m.s. deviations | |||||
| Bond length (Å) | 0.008 | 0.007 | 0.007 | 0.005 | 0.005 |
| Bond angle (°) | 1.21 | 1.06 | 1.02 | 1.08 | 0.66 |
| Ramachandran plot (%) | |||||
| Favorable | 95 | 95 | 95 | 96 | 97 |
| Additional allowed | 4.7 | 4.2 | 4.6 | 3.2 | 2.6 |
| Disallowed | 0.3 | 0.8 | 0.4 | 0.8 | 0.4 |
| PDB entry | 5KPD | 5KOY | 5KPI | 5KPJ | 5KO2 |
a Values in parentheses are for the highest resolution shells.
b Rmerge is defined as Σ|Ih,i − 〈Ih〉|/ΣIh,i, where Ih,i is the intensity for ith observation of a reflection with Miller index h, and 〈Ih〉 is the mean intensity for all measured Ih and Friedel pairs.
c Rmeas, redundancy-independent merging R-factor, is defined as Σ(n/(n − 1))1/2|Ih,i − 〈Ih〉|/ΣIh,i, where n is the number of observations for reflection h.
There are two molecules (chains A and B) of ΔlnkmGlu/GlnP-gp in an asymmetric unit (AU), which are oriented nearly orthogonal to each other (supplemental Fig. S4A). Unexpectedly, even with 34 residues removed from the linker, the ΔlnkmGlu/GlnP-gp still takes an open-inward conformation with the two NBDs separated on average by 45.2 Å, as measured from the center of gravity (COG) of NBD1 to that of NBD2 (Table 2). Structure alignment shows a large root-mean-square (r.m.s.) deviation of 2.76 Å for Cα atoms even between the two molecules in the AU (supplemental Fig. S4B and supplemental Table S1), which were non-crystallographic symmetry constrained domain-wise during crystallographic refinement. This is because the two P-gp molecules have a 1 Å difference in the separation between the two NBDs (Table 2). Nevertheless, the overall structure of each domain remains intact, as shown in pairwise alignments of these domains (supplemental Tables S2 and S3). Thus, it can be concluded that the shortening of the linker by 34 residues does not appear to interfere with the structural integrity of P-gp.
TABLE 2.
Observed distances between two NBDs are correlated with magnitude of movement of individual helices
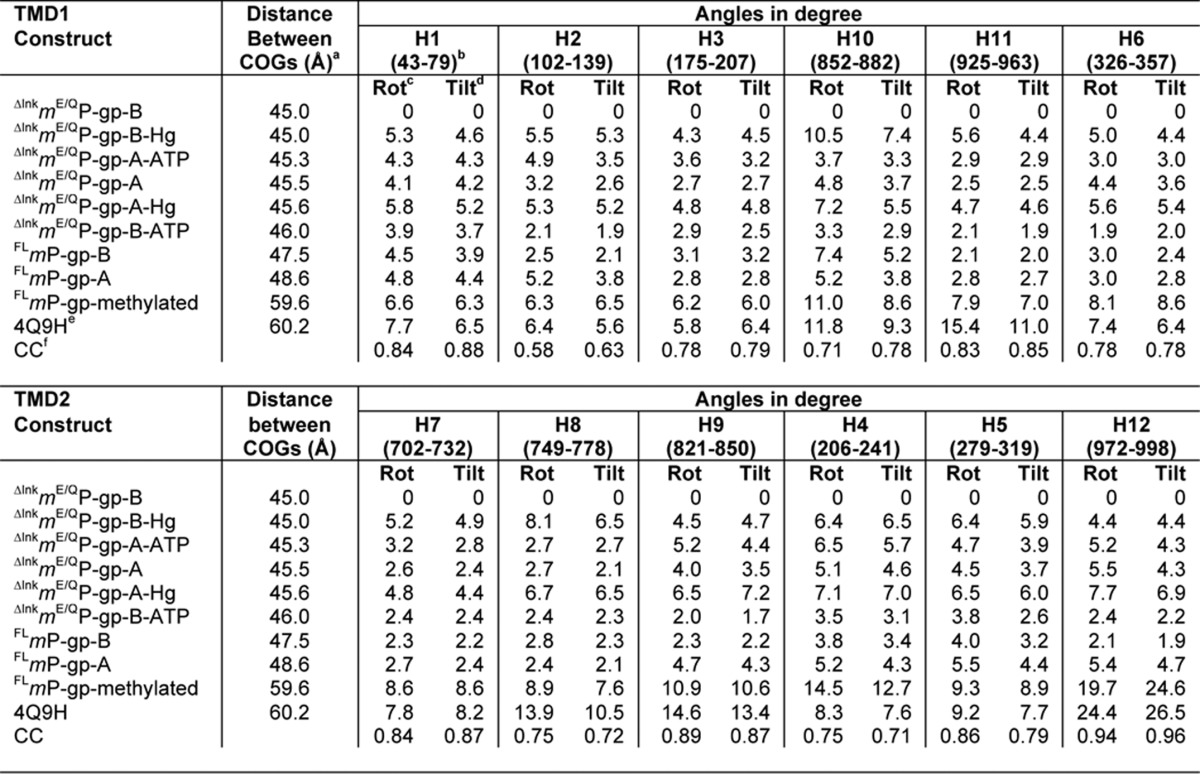
a Distance was measured between centers of gravity of two NBDs; the COG of NBD1 is defined for residues between 378 and 626 and that for NBD2 is defined between 1021 and 1272. The COG for NBD1 is near residue Leu-549 (atom C) and that for NBD2 is near residue Leu-1194 (atom C).
b Numbers in parentheses are designated to the TM part of each helix and were used for calculation.
c Average helix rotation angle (〈HRA〉, °) of each helix in a given structure with respect to that of ΔlnkmGlu/GlnP-gp-B.
d Average helix tilt angle (〈HTA〉, °) of each helix in a given structure with respect to that of ΔlnkmGlu/GlnP-gp-B.
e The coordinate is from fully methylated mP-gp (11).
f The correlation coefficient is calculated between COGs and average helix rotational or tilting angles.
Structural Integrity of ΔlnkmP-gp Is Supported by the Structure of Wild-type FLmP-gp
By molecular replacement using ΔlnkmGlu/GlnP-gp as phasing template, the structure of a wild-type FLmP-gp was determined and refined to 4 Å resolution (Table 1). The structure presented here of wild-type FLmP-gp carries no modifications to cysteine residues by heavy metal compounds and no mutations to the N-glycosylation sites as it does in the previously reported near-native structure (PDB code 4M1M), which bears a number of mutations (C952A, N83Q, N87Q, and N90Q) and has all of its cysteine residues modified with mercury atoms (9). Our structure may therefore be a better model for analysis and molecular dynamics simulations. As in the ΔlnkmGlu/GlnP-gp crystal, there are two molecules per AU, which again superpose with a large r.m.s. deviation of 2.52 Å between them due to the ∼1-Å difference in separation between the two NBDs (47.5 and 48.6 Å for chains B and A, respectively, see Table 2). Thus, for both wild type and linker-shortened mutant mP-gp, an intra-molecular flexibility is displayed. Despite the open-inward conformation for both, the separation between the two NBDs of ΔlnkmGlu/GlnP-gp is only 3 Å smaller than that of FLmP-gp. This is somewhat surprising, as it was expected that the ΔlnkmGlu/GlnP-gp should take a much more closed conformation. The published near-native structure (PDB code 4M1M) also displays shorter separations between the COGs of NBDs, which are 46.2 and 45.7 Å, respectively, for the two molecules in AU. These numbers are closer to those of ΔlnkmGlu/GlnP-gp, suggesting that it may compare structurally better with the ΔlnkmGlu/GlnP-gp than with a true native FLmP-gp.
Structure alignment of both A chains of FLmP-gp and ΔlnkmGlu/GlnP-gp reveals a rather small r.m.s. deviation of 0.70 Å for all Cα atoms (Table S1); a similar result is obtained for both B chains. Thus, under the “Discussion,” we describe mP-gp using the A chains of either FLmP-gp or ΔlnkmGlu/GlnP-gp interchangeably. Our structure model (Fig. 2A) begins at residue Pro-31 and ends at residue Thr-626 for the N-terminal half of the molecule. The linker region is disordered for both FLmP-gp and ΔlnkmGlu/GlnP-gp crystals. The C-terminal half of the molecule starts at residue Asp-687 and ends at residue Gly-1272. Unlike symmetry-preserving homodimeric ABC transporters, the two halves of P-gp are only related by a pseudo 2-fold rotation axis and cannot be superposed. For the convenience of discussion and following previous conventions, the molecule is divided into two regions (helical region or HR and NBD region, Fig. 2A). Helices in the HR are long with 48 residues on average, span across the plasma membrane, and extend into both extra- and intracellular spaces. Thus, we further divided the HR into six domains as defined in Fig. 2A (ECD1, ECD2, TMD1, TMD2, ICD1, and ICD2). The NBD region consists of NBD1 and NBD2. Structure alignments between FLmP-gp and ΔlnkmGlu/GlnP-gp by individual domains reveal no large difference, which is particularly true when alignment was performed for both chains A or B (supplemental Tables S1 and S2). These observations further support the finding that the linker-shortened mP-gp is structurally intact.
FIGURE 2.
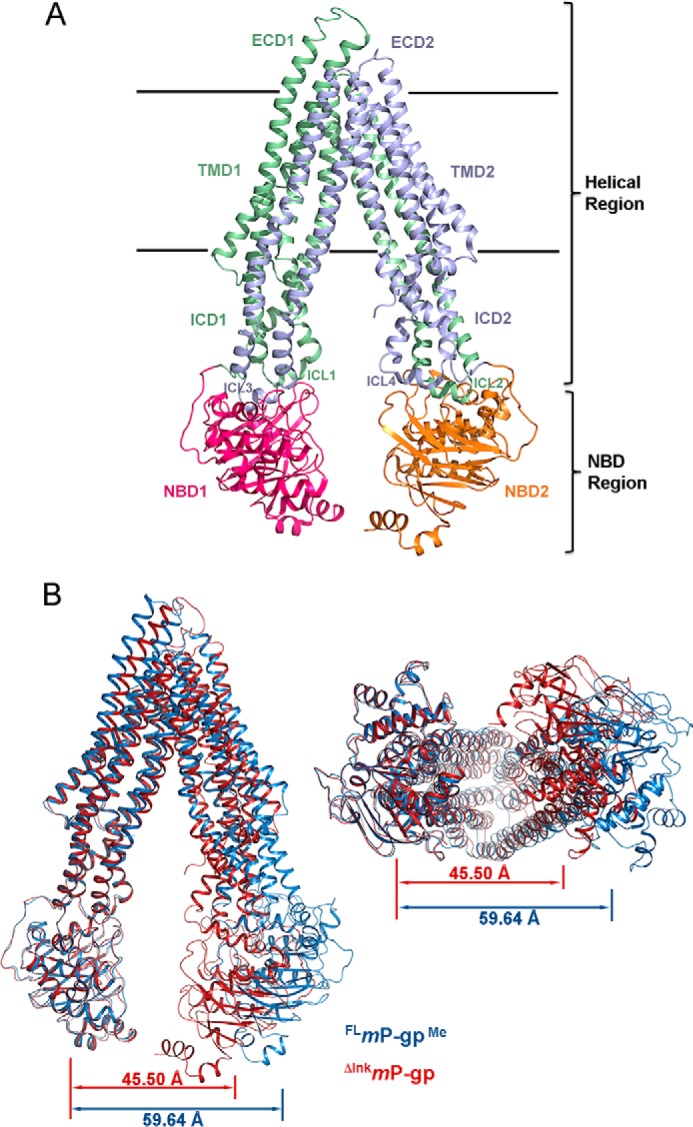
Structures of mP-gp variants and separations between the two halves. A, ribbon representation of mP-gp structure. The structure is divided into two regions and eight domains. The two regions are as follows: the helical region (HR) and NBD region. The HR is further divided into ECD1 (ECL1–2, ECL9–10, and ECL11–12), ECD2 (ECL7–8, ECL3–4, and ECL5–6), TMD1 (TMH1, TMH2, TMH3, TMH6, TMH10, and TMH11), TMD2 (TMH7, TMH8, TMH9, TMH4, TMH5, and TMH12), ICD1 (ICL2–3 and ICL10–11), and ICD2 (ICL4–5 and ICL8–9). The two horizontal lines mark the boundaries for the membrane bilayer. The structure corresponding to residues 31–366 is colored green. NBD1(367–625) is shown in red. The part corresponding to residues 687–1010 is in purple, and the NBD2(1011–1272) is colored orange. B, superposition of the ΔlnkmGlu/GlnP-gp (chain A) with FLmP-gpMe. The N-terminal half of ΔlnkmP-gp was aligned to the corresponding part of the FLmP-gpMe, leaving the C-terminal half misaligned. The ΔlnkmP-gp is shown as ribbon presentation colored in red, and FLmP-gpMe is shown in blue. Two viewing orientations are shown as follows: on the left is the view along the membrane plane and on the right is that perpendicular to the membrane plane from the intracellular side. Separations between the NBDs are also given as distances between COG of the two NBDs.
P-gp Binds ATP Asymmetrically
To gain structural insights into the mechanism of the P-gp catalytic cycle, especially how nucleotides such as ATP bind to the protein, we tested inclusion of ATP in our soaking experiments with crystals of ΔlnkmGlu/GlnP-gp. Only under the conditions in the absence of Mg2+ and in the presence of 18 mm ATP did crystals survive the treatment and diffract X-rays to 3.85 Å resolution (Table 1). The difference Fourier map revealed a large piece of density resembling the shape of an ATP molecule, which was successfully built and refined. Interestingly, ATP was only found in the N-terminal NBD (Fig. 3, A and B). Asymmetric binding of nucleotide to NBD1 is observed in both molecules in the AU, which are in different crystal-packing environment, indicating that the selective binding of nucleotide is not likely influenced by crystal contacts. Since this was the first time a nucleotide bound to P-gp has been experimentally visualized, we repeated the same experiment by collecting several data sets on different ATP-soaked crystals and were able to reproduce the same result (data not shown).
FIGURE 3.

Asymmetric binding of nucleotides. A, van der Waals surface showing asymmetric binding of ATP to the NBD1. Only the surfaces of NBD1 and NBD2 are presented, as viewed from the helical region of the molecule. The bound ATP in NBD1 is rendered as the stick model with carbon atoms in black, oxygen in red, nitrogen in blue, and phosphorus in orange. The two residues descending from ICD1, Arg-901 and Asp-160, are also shown as stick models and labeled. B, stereo pair: the ATP-binding environment in NBD1. The bound ATP is shown as stick model with carbon atoms in black, oxygen in red, nitrogen in blue, and phosphorus in brown. Overlapping the ATP model is the difference electron density shown in gray mesh and contoured at 3σ. Residues in the vicinity of the bound nucleotide are also shown as stick models and labeled. C, stereo pair showing the nucleotide-binding site of NBD2 in the structure of FLmP-gpMe. Difference electron density (green mesh, 3σ) is overlaid with a trimethylated Walker A K1072, shown as a stick model in black. The Walker A P-loop is shown in cartoon representation and is colored in red. Residues that are in the vicinity of the methylated Lys-1072 are shown as stick models and labeled.
The NBD1 ATP-binding site is a shallow surface groove directly facing, but not in contact with, NBD2 (Fig. 3A). Residues that are in the vicinity of bound ATP can be grouped into three categories (Fig. 3B). Those that are near the adenine group are Asp-160, Arg-901, Tyr-397, Ser-399, Tyr-440, and Arg-400, clustering at one end of the groove. Two residues, Thr-431 and Ile-405, are near the ribose group. Residues in the Walker A motif (Ser-425, Gly-426, Cys-427, Gly-428, Lys-429, and Ser-430) and the Walker B motif (Asp-551 and E552Q) form the two ridges contacting the phosphate groups from either side. Residue His-583 is at the other end of the groove. The two charged residues, Asp-160 of ICL1 (intracellular loop 1) and Arg-901 of ICL4, are particularly interesting, as both are descending from ICD1 (Fig. 3, A and B). While in contact with the amine group of the adenine ring, these two residues do not appear to have strong interactions with the bound ATP, which is consistent with the phenotype displayed by D164A or D164C mutations in human P-gp (33). Instead, they form a hydrogen bonding network with Tyr-440 of NBD1. The latter residue may help to position the critical residue Tyr-397 of the A-loop via aromatic-aromatic interactions, thus stabilizing the orientation of the Tyr-397 side chain for nucleotide binding.
Considerable structural variations in the NBDs have been observed in the vicinity of the P-loops (residues 423–429 for NBD1 and 1067–1072 for NBD2). There are two distinct conformations for the P-loops as follows: one adapts to the binding of ATP by unwinding Cys-427 from helix 1 of NBD1 (supplemental Fig. S4C), whereas the other remains in the apo conformation. By contrast, the near-native 4M1M structure was unable to provide insights into crucial aspects of the binding of the triphosphate moiety of ATP, because considerable distortions were introduced into the conformations of both P-loops due to mercury derivatization of the cysteine residues Cys-427 and Cys-1070. Note that the binding of a mercury atom induces a local conformation that is more consistent with a pseudo holo-form of ΔlnkmGlu/GlnP-gpHg.
Methylation of FLmP-gp Supports Asymmetric Nucleotide Binding in Solution
Independent of the work on the linker-shortened mutant P-gp, we also attempted to use of surface entropy reduction for making better diffracting crystals. We purified the FLmP-gp in the presence of AMP-PNP followed by reductive methylation of the protein (FLmP-gpMe) (34). The FLmP-gpMe protein was readily crystallized in the absence of nucleotide. Crystals of FLmP-gpMe diffracted X-rays to 3 Å resolution and a data set to 3.4 Å resolution was obtained (Table 1). Unlike FLmP-gp or ΔlnkmP-gp, there is only one molecule of FLmP-gpMe in the AU. The structure could only be solved by the molecular replacement method with individual N- and C-terminal halves from the ΔlnkmP-gp followed by extensive remodeling, due to the fact that FLmP-gpMe adopts a different conformation in the crystal. Indeed, the FLmP-gpMe has a wider opening between the two NBDs measuring 59.6 Å, compared with 45.5 Å for the ΔlnkmP-gp (Fig. 2B and Table 2), and the structure alignment of the two produced an r.m.s. deviation of 5.80 Å (supplemental Table S1). The wider opening of the two NBDs in the FLmP-gpMe crystal allows extracellular loops of a symmetry-related molecule to be positioned between the two NBDs, producing crystal contacts that are different from the crystal of ΔlnkmP-gp (supplemental Fig. S4D). A similar observation was reported using a fully methylated mP-gp (PDB code 4Q9H) (11).
The difference Fourier calculation showed large pieces of density near a number of lysine residues. Of particular interest is the difference density located in the nucleotide-binding site of NBD2 that can be attributed to a triply modified lysine residue (Lys-1072) of the Walker A motif (Fig. 3C). No difference density was found in the nucleotide-binding site of NBD1 of the FLmP-gpMe. Clearly, the methylation of residue Lys-1072 of NBD2, unlike Lys-429 of NBD1, is a result of the protection afforded by the presence of AMP-PNP during protein purification and subsequent methylation, which is consistent with the model of ATP binding specifically to NBD1 of P-gp.
To test the effect of methylation on P-gp function and how it might be influenced by the presence of nucleotide, we performed methylation of wild-type FLmP-gp in the presence or absence of AMP-PNP. The methylated proteins were repurified by size-exclusion chromatography in the absence of AMP-PNP followed by measuring verapamil-stimulated ATPase activity. In the absence of nucleotide, the resulting methylated protein is catalytically inactive, indicating that both Walker A lysine residues (Lys-429 and Lys-1072) are methylated. By contrast, in the presence of AMP-PNP, the resulting methylated protein (only Lys-1072 of NBD2 is methylated) shows verapamil-stimulated ATPase activity, albeit at a 10-fold lower level than the wild type (Fig. 1F), suggesting that this modified protein is capable of carrying out ATP hydrolysis.
Structure Alignment Suggests Inflexible Coupling Interfaces between NBDs and ICDs
In this study, we have obtained nine independent mP-gp structures from five different crystals, providing an opportunity for structural comparison of the same protein in different conformations. As mentioned previously, the two ΔlnkmGlu/GlnP-gp molecules in an AU display a large r.m.s. deviation when superposed. We further compared all the structures by performing pairwise superposition, which led to the following conclusions (supplemental Table S1). 1) In all cases where there are two molecules per AU, the r.m.s. deviations between the two are high. 2) The structure of the methylated mP-gp aligns poorly with those of wild type and linker-shortened mP-gp. We then decided to align only the NBDs, either NBD1 or NBD2 (Fig. 4, A and B, and supplemental Tables S2 and S4). For example, when the NBD2 domains from mP-gp with different conformations were aligned, the r.m.s. deviation for Cα atoms was in the range between 0.5 and 0.8 Å for about 250 Cα atoms (Fig. 4B and supplemental Table S4). Most interestingly, the helices in the ICD2, which are extensions of TM helices of TMD2, were also aligned very well with r.m.s. deviations in the range of 0.8–1.0 Å for 140 atoms. The alignments get dramatically worse in the TMD2 (r.m.s. deviation of 4.3–8.1 Å for 310 atoms) and ECD2 (r.m.s. deviation of 5.5–13.5 Å for 29 residues); and the entire N-terminal half of the molecule becomes misaligned. Similar observations were made when the NBD1 domains were superposed (Fig. 4A and supplemental Table S4). Because these alignments were performed using the structures with the most dramatic differences in the separation between the two NBDs, the small variations in the coupling or transmission interfaces between NBDs and ICDs, which were observed after superpositions of only the NBDs, indicate conserved interactions and therefore inflexible coupling between the two domains, at least for mP-gp in various open-inward conformations.
FIGURE 4.

Conservation and flexibility revealed by structure alignment. The superposed structures are chain A of ΔlnkmGlu/GlnP-gp in red, chain B of ΔlnkmGlu/GlnP-gp in green, FLmP-gpMe in yellow, and 4Q9H in black. A, conserved interactions between ICD1 and NBD1 based on the superposition of NBD1 of the four structures. The superposed structures are illustrated as Cα tracings. B, conserved interactions between ICD2 and NBD2 are based on superposition of NBD2 of the four structures. C, detailed organization of the coupling/transmission interface for the N-terminal half of P-gp based on superposition of the four structures illustrated in cartoon renditions. A molecular surface is overlaid to NBD1 to show the depth of the coupling site. Interacting residues from ICD1 are shown as the stick models and labeled. D, detailed organization of the coupling site for the C-terminal half of P-gp.
Further inspection of the coupling interfaces shows very conserved arrangements for side chain residues. Conceivably, there are three structural elements for the N-terminal coupling interface (Fig. 4C). The first element is the four-helix bundle descending from the helical region, which is formed by helices H2 and H3 of the N-terminal half and by TMH10 and TMH11 of the C-terminal half. The second element is a 20 Å deep surface depression on the NBD1, into which the helical bundle inserts. The third element is the linker connecting the N-terminal helical region and the NBD1. The C-terminal coupling interface is similarly organized with the four-helix bundle formed by helices H8 and H9 and by helices H4 and H5 (Fig. 4D). In all our structures, the coupling interfaces are well ordered with no indication of flexibility. Consistent with this observation, buried surface areas calculated for the coupling interfaces are 2622 and 2970 Å2, respectively, for the N- and C-terminal half.
Movement of Individual Helices in the TMDs Is Correlated to the Opening and Closing Motion of the Two Halves of P-gp
So far, conformational changes among all the structures determined in this work are described in terms of the changes in the gap between the two NBDs; Table 2 lists the structures in the order of the gap size. However, conformational changes in P-gp are by no means limited to the difference in the separation of the two NBDs. Alignments among these structures demonstrate striking flexibility even for each individual helix in the TMDs. As the molecule assumes different gap sizes, individual TM helices (TMH) experience rotational, translational, and bending movements. Some, such as TMH12, even undergo partial unwinding (Fig. 5A).
FIGURE 5.

Structural flexibility in the TM helices is correlated to the opening and closing movement of the two halves of P-gp. A, movement of helices in the TMD revealed by superposition of mP-gp structures in different conformations. Two structures are superimposed: chain B of ΔlnkmGlu/GlnP-gp (red) and FLmP-gpMe (green). Left panel depicts structural changes of the N-terminal half based on the superposition of NBD1, and the right panel shows the changes of the C-terminal half based on the superposition of NBD2. Each helix is labeled, and a letter a, b, or c is added if breaks exist in the helix. Examples of aromatic residues, Phe-934 of TM11 and Phe-979 of TM12, found in the interior of the TMD, are displayed as the stick models and labeled. B, Cα traces of helices in the TMD1 of superposed structures. The superposition was based on TMH1 of ΔlnkmGlu/GlnP-gp (chain B) as reference molecule. Four structures were used as follows: ΔlnkmGlu/GlnP-gp (chain B, green); ΔlnkmGlu/GlnP-gp (chain A, red); FLmP-gpMe (yellow); and 4Q9H (black). All helices were labeled. C, Cα traces of helices in the TMD2 of superposed structures. The superposition was based on TMH7 of ΔlnkmGlu/GlnP-gp (chain B). D, correlation plots for TMH11 (TMD1) and TMH12 (TMD2) illustrating the relationship between the gap size of two NBDs, expressed as the distance between COGs, and the average helix rotation 〈HRA〉 and tilt 〈HTA〉 angles. Cartoon representations of TMH11 and TMH12 from two superposed structures ΔlnkmGlu/GlnP-gp (chain B, green) and FLmP-gpMe (yellow) are also given. E, changes in the substrate-binding environment shown as van der Waal's surface centered on residue Tyr-949 of TMD1 (top) and on residue Tyr-306 of TMD2 (bottom) in mP-gp structures of different conformation. Four structures are presented: panel I, ΔlnkmGlu/GlnP-gp (chain B); panel II, ΔlnkmGlu/GlnP-gp (chain A); panel III, FLmP-gpMe; and panel IV, 4Q9H. Hydrophobic surface is shown in green, negative charged in red, positive charged in blue, and sulfur in yellow.
Helices in the helical region range in length between 36 and 59 residues with an average of 48 residues. These long helices often contain one or two breaks in hydrogen-bonding pattern along the length of the helix to allow a change in direction. For example, TMH10 features two breaks and thus was labeled as TMH10a, TMH10b, and TMH10c (Fig. 5A). To visualize movement of helices, one way is to align structures of NBDs only and to observe how the individual helix moves. Fig. 5A shows the alignment between ΔlnkmGlu/GlnP-gp (Chain B) and FLmP-gpMe, which have the largest difference in the gap between NBDs. Helices in the TM region become non-overlapping, indicating translational movements, which is much more prominent for the C-terminal half of the helices (Fig. 5A). The rotational movement of a helix is defined as the angle the helix in one conformation has to rotate to superpose to the same helix in a different conformation, which is visualized by the positions of side chains of aromatic residues facing the putative substrate-binding cavity (Fig. 5A).
A second way to show the movement of an individual helix is to superpose only one helix in either the N- or C-terminal helical region. For example, when helix 1 (H1) in one structure (conformation 1) is superposed with the same helix of a different structure (conformation 2), rotational movement can be visualized for other helices (Fig. 5B). Similarly, when helices H7 are aligned, the translational and rotational movement of the other helices in the C-terminal TMD can be demonstrated (Fig. 5C). In particular, there is a partial unwinding of TMH12 between residues Ser-989 and Ala-995, as the protein goes from a larger to a smaller gap between the two NBDs (Fig. 5, A and C). Qualitatively, progressively larger NBD gap distances seem to correlate with increasing magnitudes of individual helix movements. To quantify, we define two parameters to describe the complex motions of each helix. One parameter is the helix rotation angle (HRA, in degree) that is calculated between a pair of vectors from two equivalent helices; each vector joins two consecutive Cα atoms of a helix. The averaged helix rotation angle 〈HRA〉 is the average over all HRAs defining the length of the helix. The second parameter is the helix tilt angle (HTA, in degree), which is computed between a pair of cross-vectors from two equivalent helices; each cross-vector is defined by three consecutive Cα atoms of a helix. The averaged helix tilt angle 〈HTA〉 is the average of all HTAs over the length of the helix. Computed 〈HRA〉 and 〈HTA〉 angles for each helix of each structure with respect to that of ΔlnkmGlu/GlnP-gp (chain B) are listed in Table 2, clearly showing movements of each individual helix as a function of the opening and closing motion of two halves of the P-gp. Indeed, these angles are highly correlated to the gap distances measuring the separation between the two NBDs (Table 2 and Fig. 5D). It is worth pointing out that the same correlation was found when alignment was done using the NBDs only.
Our analysis indicates that the C-terminal TMD is much more conformationally flexible than is the N-terminal TMD. Coupling helices TMH10 and TMH11 of TMD1 and TMHs 4 and 5 of TMD2 have on average bigger 〈HRA〉 and 〈HTA〉, compatible with their roles in P-gp function. By far, the most dramatic movement observed for any helix comes from TMH12 with very large 〈HRA〉 and 〈HTA〉 as the protein steps through different conformations. The combined movement of individual helices with respect to each other brings continuing changes to the surface landscape of the internal cavity. This change of surface can be demonstrated, for example, by the topographic variations in the vicinity of residues Tyr-949 of TMD1 and Tyr-306 of TMD2 (Fig. 5E).
Discussion
Asymmetry of Nucleotide Binding Supports Alternate Site Model for ATP Hydrolysis of P-gp
Because P-gp has two homologous halves and both NBDs are capable of binding and hydrolyzing ATP, various mechanistic models have been proposed as to how the two NBDs coordinate in coupling ATP hydrolysis to substrate translocation. Overwhelming biochemical evidence in the literature supports the asymmetric binding of nucleotides in P-gp, even though it has not been demonstrated structurally. Biochemically, P-gp bearing Walker B mutations (E552A/E552Q and E1197A/E1197Q) occluded ATP or ATPγS at a ratio of 1 ATP per P-gp (35, 36). ADP trapped by vanadate also displayed a stoichiometry of one ADP per P-gp (20). By vanadate-promoted photooxidative cleavage, it was shown that ATP hydrolysis occurs in one or the other ATP site but not both simultaneously (19). These experiments support the alternating site catalytic cycle of ATP hydrolysis model (21). A slightly modified version was proposed to account for the basal ATPase activity, which was shown by thermodynamic consideration to be an integral part of the catalytic mechanism (37). However, the question remains whether the two NBDs are structurally and functionally equivalent, even though hints of their dissimilar behavior have been suggested (38, 39). Our structure with bound ATP provides structural evidence for the asymmetric binding of nucleotide to P-gp, even though it has two consensus NBDs. This structure also shows that in the absence of drug substrate, NBD1 is a preferred site for ATP binding, suggesting there is a considerable difference between the two NBDs in binding affinity for ATP under such conditions. With respect to the P-gp conformation upon ATP binding, although it should be cautioned that the open-inward conformation in the ATP-bound structure might not be representative of what happens in solution because this conformation is preformed in the crystal, an EM study of mP-gp in the presence of ATP did show that most molecules exist in the open-inward conformation (40).
Coupling of Open-and-Close Motion of P-gp Two Halves to Movement of Individual TM Helices Forms the Structural Basis for Polyspecificity
One major unresolved issue in the function of P-gp is the mechanism underlying polyspecificity of P-gp for numerous drug substrates that are structurally unrelated. Using a very limited number of photoaffinity substrates, researchers have attempted to identify the binding sites for specific drugs, yielding results that were far from conclusive and involved both halves of the molecule (27, 28). Cysteine scanning with thioactive substrates helped define a general substrate-binding pocket but failed to pinpoint exact residues for binding specific drugs because of the apparent involvement of almost all TM helices (29, 30, 41). Clearly, using the conventional lock-and-key model, derived from studies of various enzymes that react with a finite number of substrates or inhibitors, is not sufficient to explain the mechanism of polyspecificity conferred by P-gp.
Conformational change has been observed for P-gp driven by drug binding, nucleotide binding, and hydrolysis, as measured by alterations in affinity for antibody interaction (42), by Trp fluorescence changes (43), and by EM (44, 45). Clearly, conformational changes are important in the function of P-gp, and yet crystal structures that provide snapshots of P-gp conformations at high resolution always have the two NBDs widely separated (9, 11), which raised some concerns over their physiological relevance (14, 46). Because P-gp resides in an environment containing 4–5 mm ATP, and structural information from studies of bacterial homologs MsbA and Sav1866 in the presence of nucleotides would suggest an open-outward conformation (47, 48), finding only open-inward conformation for P-gp has been perplexing. Biochemical evidence seems to favor the idea that the two NBDs are likely in a “constant contact” mode (36, 49, 50).
Here, we show that the linker-shortened mutant P-gp is structurally intact and has a slightly elevated basal ATPase activity (Figs. 1 and 2). In the crystal, it adopts an open-inward conformation with the gap between the two NBDs reduced by only 3 Å, compared with the wild-type FLmP-gp. This was unexpected because 1) we had hoped that the deletion of 34 residues (∼130 Å linear distance) would force ΔlnkmP-gp into an open-outward conformation, and 2) the remaining portion of the linker is still disordered. However, this seemingly “perfect” protein has lost the drug-stimulated/inhibited ATPase activity in vitro and is unable to transport substrates in vivo, which suggests that the wider opening between the two halves of P-gp is important for function. The functional defect seems likely to be a result of alteration in substrate interaction because the mutant retains the ability to bind and hydrolyze ATP and has no structural defects to indicate uncoupling. Thus, it seems reasonable to think that the reduced flexibility in the ΔlnkmP-gp mutant as a result of linker truncation is a culprit of its altered ability to interact with substrates. In other words, the opening and closing motion of the two halves is an intrinsic property of P-gp essential for function. One potential benefit for P-gp to open and close, in addition to preventing wasteful expenditure of ATP as implied in the constant contact model, is to provide a mechanism for P-gp to handle extremely large number of substrates, as the motion of opening and closing allows P-gp to change its surface topology within the drug-binding pocket demonstrated in this work (Fig. 5, Table 2, and supplemental Movie S1).
Thus, it seems reasonable to hypothesize that under resting conditions, a P-gp molecule undergoes a constant opening and closing movement of its two halves with most of the time spent in the open conformations. This proposal is consistent with the EM work that shows a distribution of P-gp in different open-inward conformations even in the presence of nucleotide (40). A natural consequence of this hypothesis is the basal ATPase activity, which indicates the frequency of the two NBDs coming close together. Consistently, our ΔlnkmP-gp mutant has an elevated basal ATPase activity (Fig. 1A), and cross-linking between ICDs also led to higher ATPase activity (51).
The open-and-close motion of P-gp is structurally linked to conformational changes of each individual helix of P-gp, as revealed by the structures reported in this work. These changes result in a continuously changing landscape of the substrate-binding pocket (Fig. 5E and supplemental Movie S1). It is tempting to speculate that this continuous change in surface topology creates a drug-binding environment that in principle is able to interact with an unlimited number of substrates. This model is compatible with the “induced fit” model (41), which proposed that a substrate could create its own binding site in the common drug-binding pocket by using a combination of residues from different TMHs, but it differs in one important detail, i.e. the creation of these binding sites is an intrinsic property of the P-gp.
This hypothesis is consistent with a vast amount of experimental data in the literature. For example, the interaction between TMH6 and TMH12 was demonstrated by the cross-linking experiment under drug binding and vanadate trapping conditions (52, 53), in which the authors concluded that either helix or both would have to undergo rotations relative to each other to make cross-linking possible. In our series of structures, as the two NBDs open wider from ΔlnkmP-gp to FLmP-gpMe, TMH12 was rotated and tilted by 24 and 26°, respectively (Table 2), which does not include the effect of rewinding of TMH12 during the movement. This hypothesis is also compatible with the thermodynamics study (37), showing that the basal ATPase activity is an intrinsic mechanistic property of P-gp rather than a result of transporting endogenous lipids.
Experimental Procedures
Materials
n-Dodecyl β-d-maltopyranoside (DDM) was purchased from Anatrace (Anatrace, Pasadena, CA). The Ni-NTA Superflow used for purification of the His-tagged mouse P-gp was purchased from Qiagen (Germantown, MD). Antibiotic Zeocin used for P. pastoris culture was purchased from Invitrogen. Competent cells E. coli strain DH5α, DNA polymerase, restriction endonucleases DpnI and PmeI, and other DNA-modifying enzymes were purchased from New England Biolabs (Ipswich, MA). The vector pPICZ and P. pastoris host strains GS115, X33, KM71H, and SMD1168H were purchased from Invitrogen. The immunoblotting system, including Novex BisTris gels, buffers, and apparatus, was also purchased from Invitrogen. Secondary goat anti-mouse antibody (H&L) with conjugated peroxidase (Min X Human Serum Proteins) was purchased from Rockland (Gilbertsville, PA). Pierce BCA protein assay kit was purchased from Thermo Fisher Scientific (Rockland, IL). All other chemicals were purchased from Sigma unless otherwise noted.
Generation of Recombinant BacMam Baculovirus
The Bac-to-Bac Baculovirus Expression System (Life Technologies, Inc.) was used according to the manufacturer's protocol to generate recombinant BacMam baculovirus. Gateway cloning was used to transfer full-length mdr1a and linker-deleted mdr1a cDNA from bacterial cloning plasmids to the pDest-625 expression vector. Both mdr1a and linker-deleted mdr1a harbored a methionine to leucine substitution at residue 107 to facilitate cloning in E. coli (mouse P-gp carrying M107L is functionally equivalent to wild-type P-gp) (54). Expression vectors were transformed into E. coli DH10Bac cells where site-specific transposition of the gene of interest into a baculovirus shuttle vector (bacmid) occurred. Blue-white screening was carried out; white colonies were selected, and bacmid DNA was purified by alkaline lysis. Bacmid DNA was transfected into High-Five insect cells to generate recombinant BacMam baculovirus.
Transduction of HeLa Cells with BacMam Baculovirus
BacMam transductions were carried out according to a previously optimized protocol (55). In short, 2.5 × 106 cells in 3 ml of DMEM were transduced with BacMam virus at a ratio of 50 virus particles per cell and incubated at 37 °C for 1 h, followed by addition of 23 ml of DMEM and incubation for a further 3 h. Sodium butyrate was added to a final concentration of 10 mm, and cells were incubated for 24 h at 37 °C before harvesting for assays.
Transport of Fluorescent Substrates by Flow Cytometry
HeLa cells were trypsinized and counted, and 2 × 105 cells per assay conditions were transferred into polystyrene flow cytometry tubes. Cells were incubated in Iscove's modified Dulbecco's medium containing either 1.5 μm rhodamine 123 or 1.0 μm calcein-AM with or without P-gp inhibitor elacridar (100 nm) at 37 °C for 45 min. After incubation, cells were centrifuged, and supernatant was removed and resuspended in 200 μl of cold PBS and placed on ice to stop transporter activity. A total of 10,000 individual cell-counting events were recorded using a FACSCanto II flow cytometer. A 488-nm blue laser was used to excite fluorophores, and emission was measured at 530/30. FACS data were analyzed using FlowJo Single Cell Analysis software (FlowJo, Ashland, OR).
Generation of P-gp-expressing Pichia Strains
The Pichia Expression System (Invitrogen) was used to construct vectors for recombinant gene expression in P. pastoris following closely the manufacturer's manual but with small modifications.
The Pichia expression vector pPICZ-A was slightly modified removing the 108-base MCS between restriction sites EcoRI and SalI (including the EcoRI site) and the BamHI site at position 1414. A BamHI site is reintroduced after the SalI site. The full-length mouse P-gp DNA was generated by PCR with 5′-end a SalI site and 3′-end a BglII site. The PCR product was treated with SalI and BglII before being inserted into the modified pPICZ-A plasmid, yielding the Pichia expression construct termed pPICZM-MouseMDR1a. Site-specific mutations were introduced to the pPICZM-MouseMDR1a to produce the plasmid pPICZM-MouseMDR1a-Glu/Gln. These vectors formed the basis for further modification to yield pPICZM-MouseMDR1a-DeltaLinker and pPICZM-MouseMDR1a-E/Q-DeltaLinker, respectively, for expressing ΔlnkmP-gp and ΔlnkmP-gpGlu/Gln. The vectors were transformed into the E. coli strain DH5α competent cells (New England Biolabs) and selected on Low Salt LB medium (1% tryptone, 0.5% yeast extract, 0.5% NaCl, pH 7.5) with 25 μg/ml Zeocin. The sequences of the resulting plasmids were verified to confirm the correct mP-gp constructs.
Pichia expression plasmids carrying mP-gp variants were linearized by PmeI and transformed into four Pichia host strains GS115, KM71H, SMD1168, and X33 competent cells by electroporation according to the manufacturer's manual. Transformants were selected on YPDS (1% yeast extract, 2% peptone, 2% dextrose, 1 m sorbitol) agar plates containing 100 μg/ml Zeocin. After incubation at 30 °C for 5–7 days, 6–10 colonies were picked and streaked on fresh YPDS plates containing 100 μg/ml Zeocin. Colonies formed on the second plates were considered possible positive recombinants. Six to nine recombinants from each host strain were picked for analysis of P-gp expression level in a small-scale screening and for optimization of growth conditions that maximize mP-gp expression. Colonies were seeded in 2 ml of YPG (1% yeast extract, 2% peptone, 1% glycerol) and grown at 30 °C in a shaking incubator (250 rpm) overnight. Cells were transferred to a flask with 100 ml of fresh YPG medium and grown under the same conditions. Cells were collected when A600 reached 15. Expression was induced by resuspending the cell pellets in YPM (1% yeast extract, 2% peptone, 0.5% methanol) to A600 values in the range of 25–30. Methanol was added to the medium to a final concentration of 0.5% every 24 h to maintain the induction for 5 days. Cell samples were taken every 24 h for analysis of mP-gp expression.
Cells were harvested by centrifugation, and the pellets were stored at −80 °C until used. To make crude membrane from small samples, the cell pellets were resuspended in 50 μl of Breaking Solution (50 mm sodium phosphate, pH 7.4, 1 mm EDTA, 5% glycerol, freshly added 2% SDS, 1× protease mixture, and 1 mm DTT) and mixed with acid-washed glass beads. The cell suspension was vortexed for 30 s followed by incubation on ice for 30 s, and the process was repeated 10 times. After removing unbroken cells, cell debris, and glass beads by a low speed centrifugation, the crude membrane was recovered by a high speed centrifugation at 180,000 × g for 30 min. Protein expression levels from the small scale tests were analyzed by NuPAGE 12% BisTris gel (Invitrogen) and Western blotting on Immuno-Blot PVDF membrane sandwiches (Bio-Rad) using P-gp-specific monoclonal antibody C219. Recombinant mP-gp was visualized by chemiluminescent HRP antibody detection reagent on HyBlot ES autoradiography film (Denville Scientific, Metuchen, NJ). Once the recombinants were obtained and optimal expression conditions were established, protein expressions were scaled up to produce large amounts of recombinant proteins.
Recombinant Protein Expression, Membrane Isolation, and Protein Purification
Liquid media and agar plates for P. pastoris cell culture were prepared according to the instructions of the Pichia expression kit manual from Invitrogen. A single colony on YPDS agar plate containing 100 μg/ml Zeocin was inoculated into 5 ml of YPG medium to grow at 30 °C for 22 h. Then, the culture was transferred into 250 ml of YPG medium to grow overnight under the same conditions. The culture was further amplified into 8 liters of YPG in a New Brunswick Scientific BioFlo 3000 fermenter (New Brunswick, Edison, NJ) to grow overnight. Cells were collected by centrifugation at 1500 × g for 8 min. The cell pellets were resuspended in 4 liters of YP medium, and protein expression was induced by adding methanol to a final concentration of 0.5% at 30 °C for 4 days. Samples were taken every 24 h to check expression level by immunoblotting with P-gp-specific monoclonal antibody C219. After harvesting by centrifugation at 3500 × g for 15 min, the cell pellet was washed with a chilled buffer containing 25 mm Tris·HCl, pH 8.0, and 2 mm PMSF. The cells were spun down by centrifugation at 3500 × g for 15 min and stored at −80 °C.
Membrane preparation and purification were performed at 4 °C. All samples were kept on ice during the purification. Frozen Pichia cells were thawed and resuspended in a homogenization buffer containing 100 mm Tris·HCl, pH 8.0, at 4 °C, 100 mm sucrose, 100 mm 6-aminohexanoic acid, 2 mm EDTA, and 2 mm PMSF and stirred on ice for 10 min. Resuspended cells were broken by passage through an APV 2000 homogenizer (APV, Lake Mills, WI) at 900 bars three times with two intervals at no pressure between each pass. Unbroken cells and cell debris were removed by centrifugation at 3500 × g for 20 min. The supernatant was centrifuged at 100,000 × g for 90 min to separate the membrane fraction from the soluble protein fraction. The membrane pellet was homogenized in the same homogenization buffer followed by centrifugation at 100,000 × g for 90 min. The membrane pellet was homogenized again in a wash buffer containing 25 mm Tris·HCl, pH 8.0, at 4 °C, 200 mm NaCl, 5 mm β-mercaptoethanol. After centrifugation at 100,000 × g for 30 min, the pellet was homogenized in a storage buffer containing 25 mm Tris·HCl, pH 8.0, at 4 °C, 30% glycerol, and 5 mm β-mercaptoethanol. The membrane was stored at −80 °C until use. Protein concentration in the membrane was determined with the colorimetric BCA protein assay.
Frozen membrane was thawed on ice and solubilized in a buffer consisting of 10 mm Tris·HCl, pH 8.0, at 4 °C, 30 mm imidazole, 75 mm NaCl, 15% glycerol, 5 mm β-mercaptoethanol, and 2% DDM to a protein concentration of 5 mg/ml. After stirring on ice for 30 min, the admixture was centrifuged at 100,000 × g for 30 min. The supernatant was collected and mixed with a slurry of Ni-NTA Superflow resin (Qiagen) pre-equilibrated with a loading buffer containing 20 mm Tris·HCl, pH 8.0, at 4 °C, 75 mm NaCl, 15% glycerol, 2 mm β-mercaptoethanol, 0.0675% DDM, and 0.04% sodium cholate, supplemented with 30 mm imidazole, and incubated at 4 °C for 2 h with gentle stirring. The slurry was subsequently packed into a column and was washed with the same loading buffer until no protein was detected in the flow-through. P-gp was eluted from the Ni-NTA resin with loading buffer supplemented with 300 mm imidazole. Eluate was concentrated with a 100,000-kDa cutoff Amicon Ultra Concentrator (Millipore, Billerica, MA) to 1 ml. The concentrate was spun at 588,000 × g for 60 min, and the supernatant was injected into a size-exclusion column Superdex S-200 (GE Healthcare) equilibrated with the loading buffer. Fractions of the major peak were combined and concentrated to a protein concentration between 7 and 10 mg/ml.
ATPase Assay
Basal ATPase activity of purified mP-gp was determined by measuring the amount of inorganic phosphate released from ATP hydrolysis, which reacts with molybdate and malachite green (56, 57). Total soy lipid extract was dissolved in 50 mm MOPS buffer, pH 7.0, to a concentration of 20 mg/ml and sonicated until clear. Mouse P-gp was mixed with the lipid solution in a 1:1 ratio (w/w) and incubated on ice for more than 1 h before assay. The activity assay was performed in a buffer containing 50 mm MOPS, pH 7.0, 10 mm MgCl2, and 5 mm DTT. A total of 50 μl of reaction mix containing 2–5 μg of protein and 4 mm buffered ATP was incubated at 37 °C for 15 min. The reaction was immediately stopped by the addition of 800 μl of dye buffer (a fresh mixture of 0.045% malachite green and 1.4% ammonium molybdate tetrahydrate in 4 n HCl in 1:3 ratio) followed by addition of 100 μl of 34% sodium citrate solution after 1 min of incubation. After a further 10-min incubation at room temperature, 16 μl of 10% Tween 20 was added to dissolve precipitate, if any. Absorbance was then measured at 660 nm. The amount of inorganic phosphate released was calculated based on the standard curve established by a known amount of KH2PO4 (10–100 μm) in assay buffer. To test the drug-stimulated ATPase activity, the indicated compounds dissolved in DMSO were mixed with mP-gp to concentrations of 20, 50, 100, 150, 200, and 300 μm and preincubated at 37 °C for 5 min. For ATPase activity assay in the presence of vanadate, mP-gp was pre-incubated with 200 μm pre-boiled sodium orthovanadate in the activity assay buffer at 37 °C for 5 min before starting the reaction.
Methylation of Wild-type FLmP-gp
A 200-μl sample of 8.2 mg/ml protein was dialyzed overnight against a buffer containing 50 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, and 0.068% DDM either in the presence or absence of 1 mm nucleotides. The dialyzed protein was diluted with unused dialysis buffer to give a 1 mg/ml solution. Every time, a freshly prepared solution of 1 m formaldehyde and 1 m dimethylamino borane (DMAB) was used in the following way. The diluted protein solution was treated with 20 μl of DMAB at 4 °C followed by 40 μl of formaldehyde reagent and allowed to react for 2 h. This procedure was repeated twice, and after another 2 h, 10 μl of DMAB was added to drive the reaction to completion overnight at 4 °C. On the next day, the clear solution (no precipitation) was concentrated to 250 μl, centrifuged, and injected into a size-exclusion column for purification. Appropriate fractions were combined and concentrated.
Protein Crystallization
Both native and derivatized proteins were crystallized under similar conditions. Because the need for seeding was anticipated, six identical trials were set up using 4 μl of protein and 4 μl of 18% PEG400, 25 mm HEPES, pH 7.5, 100 mm alkaline metal chlorides, and 0.3 mm Cymal-7 (Anatrace). The reservoir solution was prepared to contain 100 mm HEPES, pH 7.5, 200 mm NaCl, 25% PEG400. After 2 weeks no crystals had formed, and serial micro-seeding starting with a crystal that was crushed with a loop in the first drop. Crystals of 0.1–0.2 mm size formed and could be cryo-cooled at liquid nitrogen temperature directly.
Heavy Metal Derivatization of Wild-type FLmP-gp and ΔlnkmP-gp
A 55-μl sample of the ΔlnkmP-gp protein at 6 mg/ml was treated with 2 μl of 100 mm p-chloromercury phenylsulfonate (sodium salt) yielding a 4 mm solution of the heavy atom compound. The solution was allowed to stand overnight and react at 4 °C.
Diffraction Data Collection and Structure Determination of the ΔlnkmP-gp Mutant
Crystals of derivatized ΔlnkmP-gp that reached a maximal size of 0.2 mm were used to collect diffraction patterns using the inverse beam technique at SER-CAT beam line, Advanced Protein Source (APS). Diffraction data frames were processed with HKL2000 (58). Anomalous signals were used to determine 14 sites. The program Phaser-SAD as implemented in Phenix was used to refine and complete the set of heavy atom contributors followed by calculation of phases (59). Experimental phases were improved by 2-fold molecular averaging with accurate definition of domains in Parrot (60). Manual molecular modeling was performed in Coot (61), and model refinement was done in Phenix.
Analysis of Helix Rotation and Tilt Angles
Coordinates of the structures to be analyzed were superposed in Coot for the TMH1 or TMH7 only. Superposition of TMH1 brought all TM helices together for the N-terminal half of the molecule, whereas superposition of TMH7 did the same for the C-terminal half. Pairwise analysis was carried out using one structure as reference (structure 1). For a helix segment with N residues of structure i, only CA(j,i), (j = 1,N) atoms were used for the calculation. Vectors joining neighboring CA atoms were constructed as follows: v(j,i) = CA(j+1,i) − CA(j,i). Cross-vectors (cv) were also constructed based on two neighboring vectors cv(j,i) = v(j + 1,i) × v(j,i). The helix rotation angle in degree for residue j of structure i, HRA(j,i), is calculated for equivalent residues between structure i and the reference structure 1 based on Equation 1,
| (Eq. 1) |
where j = 1, N − 1. For the average helix rotation angle of a TM helix of structure i, 〈HRA(i)〉 is calculated with Equation 2,
| (Eq. 2) |
To calculate the helix tilt angle of residue j of structure i, we used Equation 3,
| (Eq. 3) |
where j = 1, N − 2. Similarly, the average helix tilt angle for a TM helix of structure i is calculated by Equation 4,
| (Eq. 4) |
PDB Access Codes
Coordinates have been deposited with the RCSB Protein Data Bank with the PDB codes of 5KPD, 5KOY, 5KPI, 5KPJ, and 5KO2, respectively, for the structures of ΔlnkmGlu/GlnP-gp(apo), ΔlnkmGlu/GlnP-gp(ATP), FLmP-gp(apo), FLmP-gp(methylated), and ΔlnkmGlu/GlnP-gp(Hg).
Author Contributions
L. E. did the work and wrote the paper; F. Z. did the work and wrote the paper; K. M. P. did the work; J. S. did the work; J. M. did the work; W. T. did the work; C. G. did the work; A. Z. did the work; S. S. did the work; J. P. M. did the work; T. Z. did the work; P. D. K. wrote the paper; S. V. A. wrote the paper; M. M. G. wrote the paper; D. X. designed the experiments, did the work, and wrote the paper.
Supplementary Material
Acknowledgments
We thank staff of the SouthEastern Region (SER)-Collaborative Access Team (CAT) and the General Medical Sciences and Cancer Institutes Structural Biology Facility (GM/CA) beam lines at Advance Photon Source (APS), Argonne National Laboratory (ANL) for assistance in data collection. We also thank Dr. Lisa Jenkins of NCI, National Institutes of Health, for checking P-gp with mass spectroscopy, and Dr. Alejandro Negrete of NIDDK for large scale fermentation. This study used the DNA Core of NCI and the high performance Biowulf Linux cluster (biowulf.nih.gov) at the National Institutes of Health.
This work was supported by the Intramural Research Program of the NCI, Center for Cancer Research, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.

This article contains supplemental Tables S1–S4, Figs. S1–S4, and Movie 1.
- MDR
- multidrug resistance
- P-gp
- P-glycoprotein
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DDM
- n-dodecyl β-d-maltopyranoside
- AU
- asymmetric unit
- PDB
- Protein Data Bank
- ICD
- intracellular domain
- HRA
- helix rotation angle
- HTA
- helix tilt angle
- DMAB
- dimethylamino borane
- mP-gp
- mouse P-gp
- TM
- transmembrane
- r.m.s.
- root mean square
- HR
- helical region
- COG
- center-of-gravity
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Gottesman M. M., and Pastan I. (1988) The multidrug transporter, a double-edged sword. J. Biol. Chem. 263, 12163–12166 [PubMed] [Google Scholar]
- 2. Sharom F. J. (2011) The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178 [DOI] [PubMed] [Google Scholar]
- 3. Ambudkar S. V., Dey S., Hrycyna C. A., Ramachandra M., Pastan I., and Gottesman M. M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 [DOI] [PubMed] [Google Scholar]
- 4. Ambudkar S. V., Kimchi-Sarfaty C., Sauna Z. E., and Gottesman M. M. (2003) P-glycoprotein: from genomics to mechanism. Oncogene 22, 7468–7485 [DOI] [PubMed] [Google Scholar]
- 5. Gottesman M. M., and Pastan I. (1993) Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 62, 385–427 [DOI] [PubMed] [Google Scholar]
- 6. Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C. J., Fujioka A., Hirokane R., Ikeguchi K., Watanabe B., Hiratake J., Kimura Y., Suga H., Ueda K., and Kato H. (2014) Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U.S.A. 111, 4049–4054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin M. S., Oldham M. L., Zhang Q., and Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aller S. G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P. M., Trinh Y. T., Zhang Q., Urbatsch I. L., and Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J., Jaimes K. F., and Aller S. G. (2014) Refined structures of mouse P-glycoprotein. Protein Sci. 23, 34–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ward A. B., Szewczyk P., Grimard V., Lee C. W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C., Swartz D. J., Falson P. G., Urbatsch I. L., Govaerts C., Steyaert J., and Chang G. (2013) Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 13386–13391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szewczyk P., Tao H., McGrath A. P., Villaluz M., Rees S. D., Lee S. C., Doshi R., Urbatsch I. L., Zhang Q., and Chang G. (2015) Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 71, 732–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wen P. C., Verhalen B., Wilkens S., Mchaourab H. S., and Tajkhorshid E. (2013) On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 288, 19211–19220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones P. M., O'Mara M. L., and George A. M. (2009) ABC transporters: a riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 14. Gottesman M. M., Ambudkar S. V., and Xia D. (2009) Structure of a multidrug transporter. Nat. Biotechnol. 27, 546–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cornwell M. M., Tsuruo T., Gottesman M. M., and Pastan I. (1987) ATP-binding properties of P-glycoprotein from multidrug-resistant KB cells. FASEB J. 1, 51–54 [DOI] [PubMed] [Google Scholar]
- 16. Azzaria M., Schurr E., and Gros P. (1989) Discrete mutations introduced in the predicted nucleotide-binding sites of the mdr1 gene abolish its ability to confer multidrug resistance. Mol. Cell. Biol. 9, 5289–5297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Urbatsch I. L., Beaudet L., Carrier I., and Gros P. (1998) Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry 37, 4592–4602 [DOI] [PubMed] [Google Scholar]
- 18. Hrycyna C. A., Ramachandra M., Germann U. A., Cheng P. W., Pastan I., and Gottesman M. M. (1999) Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry 38, 13887–13899 [DOI] [PubMed] [Google Scholar]
- 19. Hrycyna C. A., Ramachandra M., Ambudkar S. V., Ko Y. H., Pedersen P. L., Pastan I., and Gottesman M. M. (1998) Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J. Biol. Chem. 273, 16631–16634 [DOI] [PubMed] [Google Scholar]
- 20. Urbatsch I. L., Sankaran B., Weber J., and Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 [DOI] [PubMed] [Google Scholar]
- 21. Senior A. E., al-Shawi M. K., and Urbatsch I. L. (1995) The catalytic cycle of P-glycoprotein. FEBS Lett. 377, 285–289 [DOI] [PubMed] [Google Scholar]
- 22. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., and Chou J. L. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 23. Gao M., Cui H. R., Loe D. W., Grant C. E., Almquist K. C., Cole S. P., and Deeley R. G. (2000) Comparison of the functional characteristics of the nucleotide binding domains of multidrug resistance protein 1. J. Biol. Chem. 275, 13098–13108 [DOI] [PubMed] [Google Scholar]
- 24. Procko E., Ferrin-O'Connell I., Ng S. L., and Gaudet R. (2006) Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell 24, 51–62 [DOI] [PubMed] [Google Scholar]
- 25. Rutledge R. M., Esser L., Ma J., and Xia D. (2011) Toward understanding the mechanism of action of the yeast multidrug resistance transporter Pdr5p: A molecular modeling study. J. Struct. Biol. 173, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hohl M., Briand C., Grütter M. G., and Seeger M. A. (2012) Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19, 395–402 [DOI] [PubMed] [Google Scholar]
- 27. Bruggemann E. P., Currier S. J., Gottesman M. M., and Pastan I. (1992) Characterization of the azidopine and vinblastine binding site of P-glycoprotein. J. Biol. Chem. 267, 21020–21026 [PubMed] [Google Scholar]
- 28. Dey S., Ramachandra M., Pastan I., Gottesman M. M., and Ambudkar S. V. (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 10594–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loo T. W., and Clarke D. M. (2001) Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 276, 14972–14979 [DOI] [PubMed] [Google Scholar]
- 30. Loo T. W., and Clarke D. M. (2002) Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 277, 44332–44338 [DOI] [PubMed] [Google Scholar]
- 31. Chufan E. E., Kapoor K., Sim H. M., Singh S., Talele T. T., Durell S. R., and Ambudkar S. V. (2013) Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PLoS One 8, e82463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hrycyna C. A., Airan L. E., Germann U. A., Ambudkar S. V., Pastan I., and Gottesman M. M. (1998) Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry 37, 13660–13673 [DOI] [PubMed] [Google Scholar]
- 33. Kapoor K., Bhatnagar J., Chufan E. E., and Ambudkar S. V. (2013) Mutations in intracellular loops 1 and 3 lead to misfolding of human P-glycoprotein (ABCB1) that can be rescued by cyclosporine A, which reduces its association with chaperone Hsp70. J. Biol. Chem. 288, 32622–32636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y., Quartey P., Li H., Volkart L., Hatzos C., Chang C., Nocek B., Cuff M., Osipiuk J., Tan K., Fan Y., Bigelow L., Maltseva N., Wu R., Borovilos M., et al. (2008) Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat. Methods 5, 853–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tombline G., Bartholomew L. A., Urbatsch I. L., and Senior A. E. (2004) Combined mutation of catalytic glutamate residues in the two nucleotide binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J. Biol. Chem. 279, 31212–31220 [DOI] [PubMed] [Google Scholar]
- 36. Sauna Z. E., Kim I. W., Nandigama K., Kopp S., Chiba P., and Ambudkar S. V. (2007) Catalytic cycle of ATP hydrolysis by P-glycoprotein: evidence for formation of the E.S reaction intermediate with ATP-γ-S, a nonhydrolyzable analogue of ATP. Biochemistry 46, 13787–13799 [DOI] [PubMed] [Google Scholar]
- 37. Al-Shawi M. K., Polar M. K., Omote H., and Figler R. A. (2003) Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 278, 52629–52640 [DOI] [PubMed] [Google Scholar]
- 38. Loo T. W., and Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 [DOI] [PubMed] [Google Scholar]
- 39. Ohnuma S., Chufan E., Nandigama K., Jenkins L. M., Durell S. R., Appella E., Sauna Z. E., and Ambudkar S. V. (2011) Inhibition of multidrug resistance-linked P-glycoprotein (ABCB1) function by 5′-fluorosulfonylbenzoyl 5′-adenosine: evidence for an ATP analogue that interacts with both drug-substrate-and nucleotide-binding sites. Biochemistry 50, 3724–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moeller A., Lee S. C., Tao H., Speir J. A., Chang G., Urbatsch I. L., Potter C. S., Carragher B., and Zhang Q. (2015) Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 23, 450–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Loo T. W., and Clarke D. M. (2000) Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 275, 39272–39278 [DOI] [PubMed] [Google Scholar]
- 42. Mechetner E. B., Schott B., Morse B. S., Stein W. D., Druley T., Davis K. A., Tsuruo T., and Roninson I. B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 12908–12913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sonveaux N., Vigano C., Shapiro A. B., Ling V., and Ruysschaert J. M. (1999) Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis. J. Biol. Chem. 274, 17649–17654 [DOI] [PubMed] [Google Scholar]
- 44. Rosenberg M. F., Velarde G., Ford R. C., Martin C., Berridge G., Kerr I. D., Callaghan R., Schmidlin A., Wooding C., Linton K. J., and Higgins C. F. (2001) Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 20, 5615–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee J. Y., Urbatsch I. L., Senior A. E., and Wilkens S. (2008) Nucleotide-induced structural changes in P-glycoprotein observed by electron microscopy. J. Biol. Chem. 283, 5769–5779 [DOI] [PubMed] [Google Scholar]
- 46. Wilkens S. (2015) Structure and mechanism of ABC transporters. F1000Prime Rep. 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dawson R. J., and Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 48. Ward A., Reyes C. L., Yu J., Roth C. B., and Chang G. (2007) Flexibility in the ABC transporter MsbA: alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Siarheyeva A., Liu R., and Sharom F. J. (2010) Characterization of an asymmetric occluded state of P-glycoprotein with two bound nucleotides: implications for catalysis. J. Biol. Chem. 285, 7575–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verhalen B., and Wilkens S. (2011) P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. J. Biol. Chem. 286, 10476–10482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Loo T. W., and Clarke D. M. (2014) Identification of the distance between P-glycoprotein's homologous halves that triggers the high/low ATPase activity switch. J. Biol. Chem. 289, 8484–8492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Loo T. W., and Clarke D. M. (1997) Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 272, 20986–20989 [DOI] [PubMed] [Google Scholar]
- 53. Loo T. W., and Clarke D. M. (2001) Cross-linking of human multidrug resistance P-glycoprotein by the substrate, tris-(2-maleimidoethyl)amine, is altered by ATP hydrolysis. Evidence for rotation of a transmembrane helix. J. Biol. Chem. 276, 31800–31805 [DOI] [PubMed] [Google Scholar]
- 54. Pluchino K. M., Esposito D., Moen J. K., Hall M. D., Madigan J. P., Shukla S., Procter L. V., Wall V. E., Schneider T. D., Pringle I., Ambudkar S. V., Gill D. R., Hyde S. C., and Gottesman M. M. (2015) Identification of a cryptic bacterial promoter in mouse (mdr1a) P-glycoprotein cDNA. PLoS One 10, e0136396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shukla S., Schwartz C., Kapoor K., Kouanda A., and Ambudkar S. V. (2012) Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 40, 304–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hess H. H., and Derr J. E. (1975) Assay of inorganic and organic phosphorus in the 0.1–5 nanomole range. Anal. Biochem. 63, 607–613 [DOI] [PubMed] [Google Scholar]
- 57. Lanzetta P. A., Alvarez L. J., Reinach P. S., and Candia O. A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 58. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 59. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 60. Zhang K. Y., Cowtan K., and Main P. (1997) Combining constraints for electron-density modification. Methods Enzymol. 277, 53–64 [DOI] [PubMed] [Google Scholar]
- 61. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.