

Abstract
Elucidating the molecular mechanisms of the host-parasite interaction during red cell invasion by Plasmodium is important for developing newer antimalarial therapeutics. Recently, we have characterized a Plasmodium vivax tryptophan-rich antigen PvTRAg38, which is expressed by its merozoites, binds to host erythrocytes, and interferes with parasite growth. Interaction of this parasite ligand with the host erythrocyte occurs through its two regions present at amino acid positions 167–178 (P2) and 197–208 (P4). Each region recognizes its own erythrocyte receptor. Previously, we identified band 3 as the chymotrypsin-sensitive erythrocyte receptor for the P4 region, but the other receptor, binding to P2 region, remained unknown. Here, we have identified basigin as the second erythrocyte receptor for PvTRAg38, which is resistant to chymotrypsin. The specificity of interaction between PvTRAg38 and basigin was confirmed by direct interaction where basigin was specifically recognized by P2 and not by the P4 region of this parasite ligand. Interaction between P2 and basigin is stabilized through multiple amino acid residues, but Gly-171 and Leu-175 of P2 were more critical. These two amino acids were also critical for parasite growth. Synthetic peptides P2 and P4 of PvTRAg38 interfered with the parasite growth independently but had an additive effect if combined together indicating involvement of both the receptors during red cell invasion. In conclusion, PvTRAg38 binds to two erythrocyte receptors basigin and band 3 through P2 and P4 regions, respectively, to facilitate parasite growth. This advancement in our knowledge on molecular mechanisms of host-parasite interaction can be exploited to develop therapeutics against P. vivax malaria.
Keywords: erythrocyte, malaria, membrane protein, parasite, peptides, protein-protein interaction, receptor, alanine scanning
Introduction
Plasmodium vivax causes malaria in a huge human population in Southeast Asia and South America, thus affecting their socio-economic conditions. Although this parasite causes benign malaria, it can also cause complications, similar to Plasmodium falciparum, leading to death (1, 2). Because of the limited success to cultivate this parasite in the laboratory and very low parasitemia in P. vivax patients, the biology of this parasite is not explored in as much detail as that of P. falciparum. Not only is the parasite becoming more virulent, it is also showing resistance toward commonly used anti-malarial drugs. Furthermore, there is no vaccine available against this parasite. For effective control of this disease, newer anti-malarial drugs and vaccines need to be developed. Thus, identification of drug and vaccine targets is urgently needed. In this regard, the parasite molecules expressed at the merozoite stage and involved in erythrocyte invasion hold their significance as they have been proven to be the most effective drug or vaccine targets (3).
Large numbers of host and parasite molecules need to interact with each other during erythrocyte invasion by the malarial parasite. Identification of all the host erythrocyte receptors and their respective parasite ligands involved in this process is incomplete. P. falciparum exploits lot of erythrocyte receptors, ranging from highly abundant band 3, which has the merozoite surface protein 1 (MSP1) complex (4) as the ligand, to the less abundant basigin, which interacts with P. falciparum reticulocyte-binding homology protein 5 (PfRH5) (5). Several other receptor-ligand interactions are also known for erythrocyte invasion, including the following: P. falciparum erythrocyte-binding antigen 175 (PfEBA175) and glycophorin A (6); P. falciparum erythrocyte-binding ligand 1 (PfEBL1) and glycophorin B (7); P. falciparum erythrocyte-binding antigen 140 (PfEBA140) and glycophorin C (8); P. falciparum reticulocyte-binding homology protein 4 (PfRh4) and complement receptor 1 (CR1); and P. falciparum merozoite thrombospondin anonymous protein and semaphorin 7A (9). Although several erythrocyte receptor molecules have been identified for the P. falciparum merozoite proteins, this number is unfortunately very limited in the case of P. vivax. The most commonly known receptor-ligand interaction in P. vivax is the Duffy antigen/receptor for chemokines being recognized by the parasite ligand Duffy-binding protein 1 (DBP 1) expressed by the merozoite (10). Indications have appeared in the literature that there are other host-receptor molecules recognized by the P. vivax merozoite ligands during the red cell invasion, which may be independent of the Duffy antigen (11). Recently, DBP 1 has been shown to recognize a different receptor for invasion in Duffy null erythrocytes (12).
Although several merozoite proteins of P. vivax, including reticulocyte-binding proteins, are identified, which interact with the erythrocytes during invasion process, their respective receptors have yet to be explored (13). Recently, we have reported that several P. vivax tryptophan-rich antigens (PvTRAgs) belonging to the pv-fam-a family were highly immunogenic in humans, have conserved sequences in parasite population, and bind to host erythrocytes through two receptors (14–17). One of the pv-fam-a family proteins called PvTRAg38, which is expressed at merozoite stage (18), is also highly immunogenic (17), binds to host erythrocytes (14), and promotes the parasite growth in the heterologous P. falciparum culture system (19). We have been able to define two erythrocyte-binding regions, P2 (at amino acid position 167–178) and P4 (at amino acid position 198–208), of this parasite ligand that interact with two different erythrocyte receptors. Among these two erythrocyte receptors, one was sensitive to chymotrypsin and interacts through the P4 region and the other receptor was resistant to this enzyme. The chymotrypsin-sensitive erythrocyte receptor for the P4 region has recently been identified as band 3 (19). Furthermore, multiple residues of the P4 region interact with three different ectodomains of band 3 (20). The second erythrocyte receptor that is resistant to chymotrypsin and is recognized by the P2 region of this parasite ligand remains unidentified. Results of this study indicate that this chymotrypsin-resistant erythrocyte receptor for PvTRAg38 recognized by its P2 region is basigin. Both P2 and P4 peptides interfere with parasite growth, signifying the involvement of both receptors, basigin and band 3, in red cell invasion.
Results
Interacting Erythrocyte Protein Partners for PvTRAg38
Previous studies have shown that the parasite ligand PvTRAg38 interacts with two erythrocyte receptors, where one of them was sensitive to chymotrypsin (14). Subsequently, the chymotrypsin-sensitive human erythrocyte receptor for this parasite protein was identified as band 3 (19). Therefore, in pursuit of identifying the second erythrocyte receptor for PvTRAg38, we used a more sensitive method of LC-MS/MS-based multidimensional protein identification technology (MudPIT)4 analysis (21, 22). This technique identified seven proteins from the erythrocyte membrane extract, including previously identified band 3, as probable interacting partners of PvTRAg38 (Table 1). In a parallel set of experiments, we carried out Label transfer assay (23), using tri-functional cross-linker-tagged PvTRAg38 and erythrocyte membrane lysate (Fig. 1A). As the cross-linker, which had been used to tag the protein before pulldown, was labeled with biotin, streptavidin conjugated to HRP allowed us to detect interacting proteins to which the tag was specifically transferred (Fig. 1B). The corresponding bands, to which the label was transferred, were excised from the colloidal Coomassie Blue-stained gel and identified by mass spectrometric analysis. Except for glycophorin A and calcium-transporting ATPase, the other proteins were commonly identified by both the techniques (Table 1 and Fig. 1). Hence, we excluded these two erythrocyte proteins from further analysis. Furthermore, the 55-kDa erythrocyte membrane protein (EMP) and solute carrier family protein 1 (SCFP1), known to be highly abundant proteins on the erythrocyte surface and frequently found as contaminants in the pulldown assays, were also eliminated as probable interacting partners. Therefore, besides band 3, basigin or CD59 could be promising erythrocyte receptor candidates and were tested further. Presence of basigin in the eluates was confirmed by Western blotting analysis where a single band at the expected position of ∼28 kDa was observed with anti-basigin antibody (Fig. 1C).
TABLE 1.
Identification of putative erythrocyte surface proteins interacting with PvTRAg38 by MudPIT analysis
Proteins were identified after LC-MS/MS analysis following in solution digestion of GST-PvTRAg38 pulled down proteins.
| S. no | Proteins identified by in solution mass spectrometry | Molecular mass | Coverage | Unique peptides |
|---|---|---|---|---|
| kDa | % | |||
| 1 | Anion exchanger-1 (band 3) | 101.7 | 36 | 23 |
| 2 | Basigin | 28 | 39 | 8 |
| 3 | 55-kDa erythrocyte membrane protein | 50.2 | 22 | 7 |
| 4 | Solute carrier family protein glucose-facilitated transporter member 1 | 54 | 14 | 6 |
| 5 | Calcium-transporting ATPase | 138 | 36 | 4 |
| 6 | CD59 glycoprotein | 17 | 24 | 3 |
| 7 | Glycophorin A | 30 | 43 | 2 |
FIGURE 1.
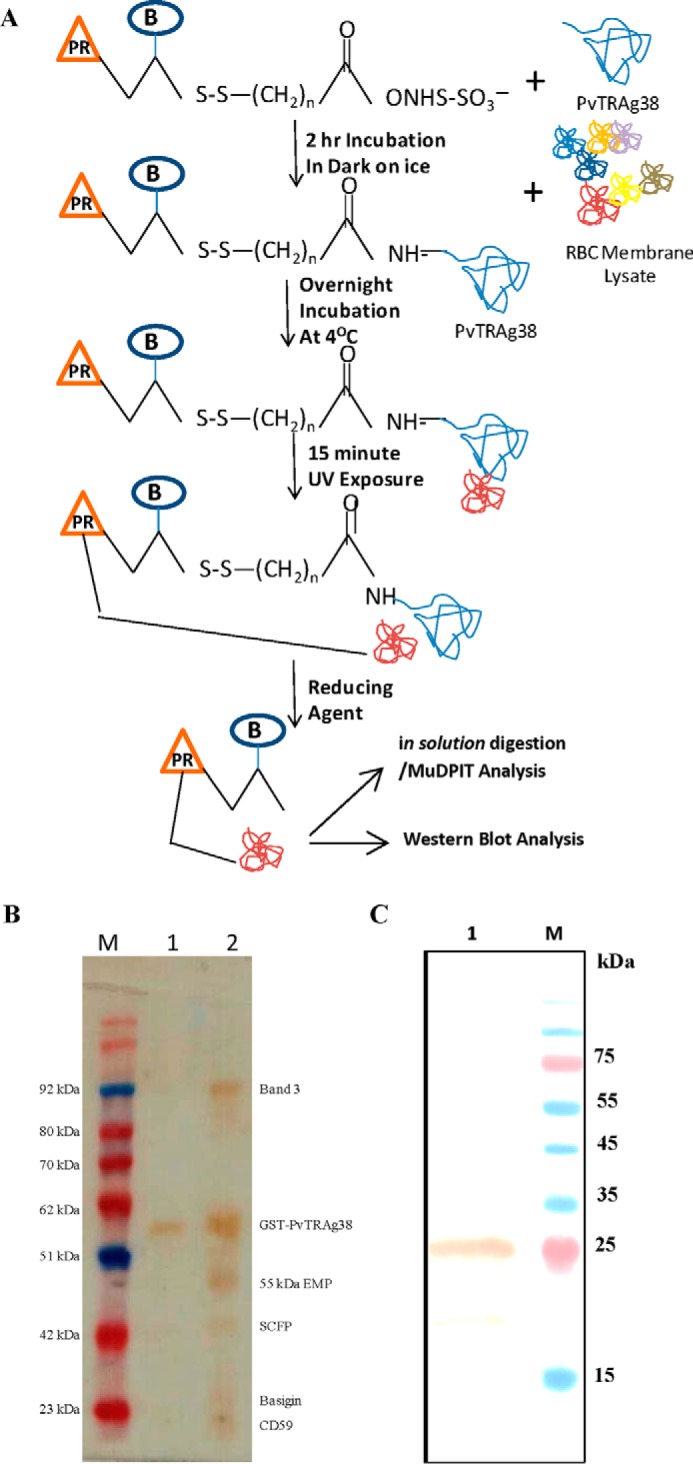
A, schematic diagram depicting the Label transfer method. The GST-tagged PvTRAg38 was incubated with biotin-labeled sulfo-SBED cross-linker. The labeled protein was incubated with erythrocyte membrane proteins, followed by exposure to UV light that photoactivates a photoreactive moiety (represented by orange triangle PR) of trifunctional cross-linker, which will bind to the interacting protein. The Label transfer (represented by blue circle B) was completed by cleaving the spacer arm to release the bait protein by reducing it with DTT. The protein-protein interaction between PvTRAg38 and erythrocyte membrane proteins was detected either by MuDPIT analysis from eluates or by probing eluates with streptavidin-HRP-based Western blot. B, identification of erythrocyte membrane proteins interacting with recombinant PvTRAg38 during Label transfer assay. Erythrocyte membrane proteins extracted with 1% C12E8 were incubated with recombinant GST-tagged PvTRAg38 (labeled with trifunctional Sulfo-SBED cross-linker), washed and resolved on 15% SDS-PAGE, and transferred to nitrocellulose membrane. The blot was probed with streptavidin-HRP. Lane M, molecular weight marker. Lane 1, recombinant GST-tagged PvTRAg38 (labeled with biotin-labeled cross-linker). Lane 2, erythrocyte membrane extract incubated with labeled GST-tagged PvTRAg38. Label transferred to erythrocyte proteins in lane 2 are named on right-hand side. SCFP1, solute-carrier family protein 1. C, detection of basigin in eluates by Western blotting analysis. Eluate was from the Label transfer assay where labeled GST-tagged PvTRAg38 was incubated with reticulocyte-enriched RBCs; membrane lysate was resolved on 15% SDS-PAGE, and protein bands were transferred to nitrocellulose membrane. The blot was probed with anti-basigin antibody (lane 1). Lane M, molecular weight marker lane. Sizes of marker proteins are shown on right-hand side.
Basigin Interacts with PvTRAg38
The above two techniques, MudPIT analysis and Label transfer assay, identified three probable erythrocyte-interacting partners of PvTRAg38. Among these three erythrocyte proteins, band 3 has already been confirmed as one of the erythrocyte receptors for this parasite ligand (19). Therefore, we planned to study direct interaction between PvTRAg38 and each of the other two erythrocyte proteins. We also included 55-kDa EMP as a control for the abundant erythrocyte membrane proteins in this assay. Although CD59 and 55-kDa EMP were obtained commercially, histidine-tagged recombinant basigin was prepared in the laboratory (Fig. 2A) and checked for its proper secondary structure by circular dichroism (CD) studies. The CD spectra showed a dominantly β-pleated sheet for this recombinant basigin (Fig. 2B), which is similar to that of the reported structure of native human basigin (24). Furthermore, the gel permeation chromatography profile of this recombinant basigin indicates that the protein was in a monomeric form (Fig. 2C). Solid phase binding assay results showed a dose-dependent binding of PvTRAg38 to basigin and not with CD59 or 55-kDa EMP (Fig. 3A). These erythrocyte proteins did not show binding to the negative control bacterial thioredoxin (Fig. 3A, inset). The affinity of the basigin and PvTRAg38 interaction was ascertained by surface plasmon resonance (SPR)-based studies. Using basigin as ligand, which was covalently coated to the activated surface of the CM5 chip following the amine coupling method, we flowed different concentrations of analyte, i.e. GST-tagged PvTRAg38. The kinetic analysis sensorgrams (Fig. 3B) show a concentration-dependent increase in the binding of ligand and analyte during the association phase. The binding affinity was found to be fairly significant with a calculated KD value of 3.0 ± 0.68 × 10−6 m. The Label transfer assay using recombinant basigin and labeled recombinant PvTRAg38 further confirmed the interaction between basigin and PvTRAg38 where the label, biotin tag, was transferred from PvTRAg38 to basigin (Fig. 3C).
FIGURE 2.

Expression, purification, and biophysical characterization of recombinant basigin. A, SDS-PAGE analysis of the purified histidine-tagged recombinant basigin. The purified recombinant protein was resolved on 12% SDS-PAGE. Lane 1, purified histidine-tagged basigin. Lane M, molecular weight markers. B, circular dichroism spectra of purified histidine-tagged recombinant basigin. C, gel permeation chromatography profile of purified histidine-tagged basigin. Elution profile of molecular weight markers and purified histidine-tagged basigin are represented by gray and black lines, respectively.
FIGURE 3.

Binding of PvTRAg38 to basigin. A, solid phase ELISA. Increasing concentrations (0–2.5 μm) of histidine-tagged basigin, 55-kDa EMP, or CD59 were added to the wells of an ELISA plate already coated with 50 nm GST-tagged PvTRAg38 or untagged thioredoxin (inset). The plate was developed with anti-His6 monoclonal antibody as described in text. Mean ± S.D. value of absorbance from three experiments is plotted. B, SPR analysis of basigin interaction with PvTRAg38. The recombinant basigin was immobilized on the cell of CM5 chip by amine coupling method. Five different concentrations of recombinant PvTRAg38 (100–500 nm) were injected at a flow rate of 30 μl/min over the surface of immobilized basigin. Curve fit (Langmuir 1:1 model) sensograms show dose-dependent response of PvTRAg38 binding with basigin. C, direct interaction between basigin and PvTRAg38 by Label transfer assay. Recombinant histidine-tagged basigin was incubated with recombinant GST-tagged PvTRAg38 (labeled with trifunctional Sulfo-SBED cross-linker) and processed as in Fig. 1A. The eluate was washed and resolved on 15% SDS-PAGE and transferred to nitrocellulose membrane. The blot was probed with streptavidin-HRP. Lane 1, GST-tagged PvTRAg38 and histidine-tagged basigin after Label transfer. Lane 2, labeled GST-tagged PvTRAg38. Sizes of molecular weight marker proteins are shown on right-hand side. D, specificity of binding of PvTRAg38 to basigin by competition assay. Increasing concentrations of histidine-tagged PvTRAg38 (0–2 μm) were incubated for 2 h with recombinant-untagged basigin immobilized on an ELISA plate well. After washing, a fixed concentration of GST-tagged PvTRAg38 was added to the wells, and the bound protein was detected with GST monoclonal antibody as described in the text. Binding in the absence of histidine-PvTRAg38 was taken as percentage control for the rest of the concentrations. The mean value of three independent experiments is plotted with S.D. E, specificity of binding between basigin and PvTRAg38 by antibody inhibition assay using anti-PvTRAg38 antibodies. Different dilutions of anti-PvTRAg38 antibody were incubated with recombinant GST-tagged PvTRAg38 immobilized on an ELISA plate well for 2 h. After washing, a fixed concentration of histidine-tagged basigin was added to the wells. The bound histidine-tagged basigin was detected using monoclonal anti-His6 antibodies as described in the text. The mean value of three independent experiments is plotted with S.D. F, specificity of binding between basigin and PvTRAg38 by antibody inhibition assay using anti-basigin antibodies. Different dilutions of polyclonal anti-basigin antibody were incubated with recombinant histidine-tagged basigin immobilized on an ELISA plate well for 2 h. After washing, a fixed concentration of GST-tagged PvTRAg38 was added to the wells, and bound recombinant GST-tagged PvTRAg38 was detected by using anti-GST antibodies as described in the text. The mean value of three independent experiments is plotted with S.D.
The specificity of binding of PvTRAg38 with basigin was confirmed by the competition assay where percentage binding of GST-tagged PvTRAg38 to basigin was inhibited with the increased concentration of histidine-tagged PvTRAg38 (Fig. 3D). Specificity of binding between PvTRAg38 and basigin was also evident from the antibody inhibition assay where this interaction was inhibited by the specific antibody in a dilution-dependent manner (Fig. 3, E and F).
Native Basigin Expressed on Eukaryotic Cell Surface Shows Binding to PvTRAg38
The above-mentioned assays used the purified recombinant fusion protein expressed in Escherichia coli. To investigate the binding of PvTRAg38 to the basigin that is expressed in its natural form by the eukaryotic cells, we used two different cell-based assays, e.g. the yeast-two hybrid system and mammalian HEK-293 cell transfections. For yeast two-hybrid assay, basigin has been cloned in bait vector (pGAD) in fusion with the activation domain of the GAL4p promoter, whereas PvTRAg38 was cloned in prey vector (pGBK7) in fusion with the binding domain of the GAL4p promoter. Co-transformed yeast cells (AH109) were able to grow well on various triple dropout media (SD/Leu−His−Trp−), whereas vector alone, pGAD-basigin, pGBK7-PvTRAg38, pGAD-basigin and pGBK7, and pGBK7-PvTRAg38 and pGAD were not showing any growth on the dropout media. These results point toward direct interaction between basigin and PvTRAg38 in this cell-based assay (Fig. 4A). Because PvTRAg38 and basigin interaction was positive on nutritional selection media (SD/Leu−His−Trp−), lacZ reporter gene expression was confirmed by assay using ortho-nitrophenyl-β-d-galactoside (ONPG) as substrate. The AH109 strain of yeast co-transformed with PvTRAg38 and basigin yielded a moderate β-galactosidase activity, whereas the other transformants with combination of empty activation domain or binding domain vectors have low or negligible amounts of β-galactosidase activity as compared with the positive interactions (Fig. 4B).
FIGURE 4.
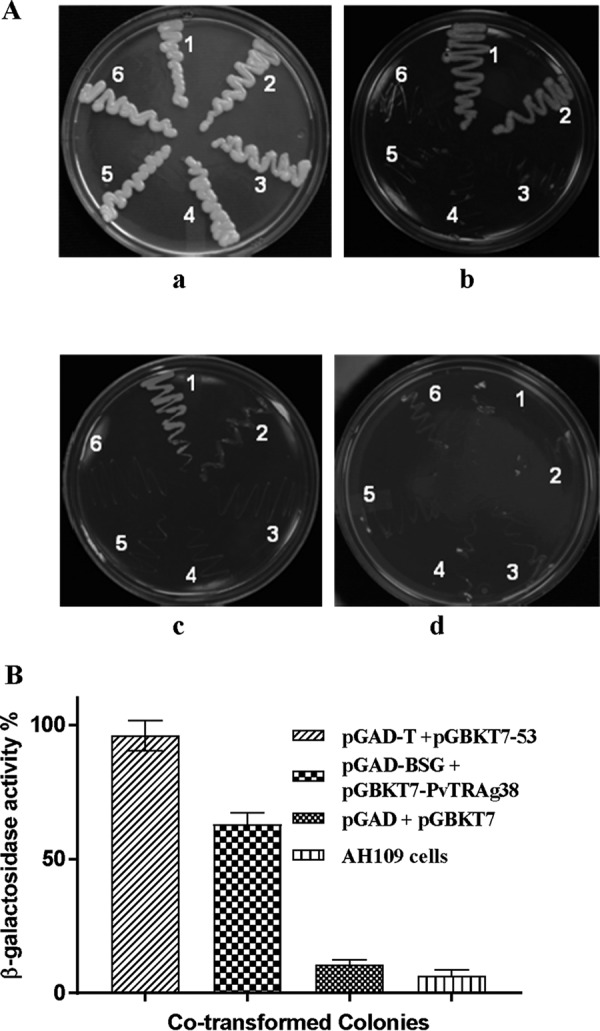
Assessment of interaction between basigin and PvTRAg38 by yeast two-hybrid assay. A, co-transformed yeast lines growing on different dropout media agar plates. Growth of transformed and parent strain yeast cells was on a YPD agar plate (panel a) and on an SD/Trp−Leu−His−Ade− dropout plate with 10 mm AT (panel b), 30 mm AT (panel c), and 50 mm AT (panel d). Zones on the plates are showing growth of yeast AH109 cells co-transformed with positive control pGAD-T + pGBKT7–53 (1), test pGAD-BSG + PGBKT7-PvTRAg38 (2), AH109 cells (3), empty pGAD + PGBKT7 vectors (4), control pGAD-BSG + pGBKT7 (5), and control pGBKT7-PvTRAg38 + pGAD vector (6). B, liquid assay to measure relative β-galactosidase activity. Three independent assays and S.D. are represented by error bars.
For the mammalian system, basigin was expressed on the HEK-293 cell surface, and the transfected cells were used for the interaction with GST-tagged PvTRAg38. The green fluorescence for basigin and red fluorescence for GST tag of PvTRAg38 were overlapping with each other (seen as yellow) across the cell surface (epithelium-like) surrounding the DAPI-stained nucleus (Fig. 5, upper panel). Cells transfected with pRE4 (vector alone) were not recognized by either of the two antibodies (Fig. 5, lower panel). This co-localization confirmed the interaction between basigin and PvTRAg38 (Fig. 5).
FIGURE 5.

Assessment of basigin and PvTRAg38 interaction by mammalian expression system. The pRE4-basigin transfected HEK-293 cells expressing basigin on the surface (upper panel) or HEK-293 cells transfected with pRE4 vector alone (lower panel) were incubated with GST-tagged PvTRAg38 and then co-localized with anti-GST monoclonal and anti-basigin polyclonal antibodies. Secondary antibodies used were Alexa Fluor 488-tagged anti-mouse and Alexa Fluor 594-tagged anti-rabbit. Smears were mounted in DAPI anti-fade and sealed.
Basigin Interacts with PvTRAg38 through its P2 Region
PvTRAg38 interacts with erythrocytes through two peptide regions, P2 and P4 at amino acid positions 161–178 and 197–214, respectively (19). Further studies revealed that last six amino acids of this P4 peptide were not involved in erythrocyte binding, but multiple residues of the remaining 12 amino acid peptides interact with its erythrocyte receptor (20). As it is known that PvTRAg38 recognizes two erythrocyte receptors (14) and peptide P4 binds to one of them, identified as band 3 (19), we investigated here whether peptide P2 of this parasite ligand binds to the above-identified basigin as second erythrocyte receptor. We have used the truncated 12-amino acid-long P2 peptide (positioned at 167–178 of PvTRAg38) in this study because the first six amino acid residues at the 161–166-position of the original 18-amino acid-long peptide were not involved in erythrocyte binding (data not shown). Sequence and position of P2 and P4 peptides are shown in Fig. 6A. Results of the solid phase binding assay suggested that P2 binds to basigin in a dose-dependent manner (Fig. 6B). Peptide P4, which recognizes band 3, did not show binding to the basigin (Fig. 6B, inset). Binding of P2 with basigin was also confirmed by SPR where the binding affinity was significant with a KD value of 1.02 ± 0.12 × 10−9 m (Fig. 6C). The specificity of binding of P2 to basigin was confirmed by the competitive inhibition assay where binding of PvTRAg38 to basigin was increasingly inhibited with increased concentrations of P2 (Fig. 6D). Furthermore, binding of basigin to the P2 region of PvTRAg38 was also inhibited by the polyclonal antibody, raised against P2 peptide, in a dilution-dependent manner (Fig. 6E).
FIGURE 6.

Binding of peptide P2 of PvTRAg38 with basigin. A, schematic diagram depicting domain organization of PvTRAg38 and location of P2 and P4 regions. B, solid phase ELISA. Increasing concentrations (0–2 μm) of recombinant histidine-tagged basigin was added to the wells of an ELISA plate already coated with 1 μm peptide P2 or peptide P4 (inset). The plate was developed with anti-His6 monoclonal antibody as described in text. Mean ± S.D. value of absorbance from three experiments is plotted. C, SPR analysis of basigin interaction with P2 peptide. Recombinant basigin was immobilized on the cell of CM5 chip by the amine coupling method. Four different concentrations of P2 peptide (5–20 μm) were injected at a flow rate of 30 μl/min over the surface of immobilized basigin. Curve fit (Langmuir 1:1 model) sensograms show dose-dependent response of P2 peptide binding with basigin. D, specificity of peptide P2 binding to basigin by competition assay. Increasing concentrations of peptide P2 (0–20 μm) were incubated for 2 h with untagged recombinant basigin immobilized on an ELISA plate well. After washing, a fixed concentration of GST-tagged PvTRAg38 was added to the wells, and bound protein was detected with GST monoclonal antibody as described in the text. Binding in the absence of peptide P2 was taken as percentage control for the rest of the concentrations. The mean value of three independent experiments is plotted with S.D. E, specificity of peptide P2 binding with basigin by antibody inhibition assay. Different dilutions of anti-P2 rabbit antibodies were incubated for 2 h with GST-tagged PvTRAg38 immobilized on an ELISA plate well. After washing, a fixed concentration of histidine-tagged basigin was added to the wells. The bound histidine-tagged basigin was detected by the anti-His6 monoclonal antibodies, as described in the text. The mean value of three independent experiments is plotted with S.D.
Multiple Amino Acid Residues of P2 Region Interact with Basigin
To find out the critical amino acid residues of the P2 region of PvTRAg38 involved in binding to basigin, we synthesized wild type P2 as well as its mutated peptides where we serially replaced the wild type amino acid with alanine (Table 2). These peptides were used as analytes to study their binding affinity with basigin by SPR. The results showed that the binding affinity of peptides P2A5, P2A9, and P2A12 having mutations at 171 (G171A), 175 (L175A), and 178 (Q178A), respectively, was reduced to ∼100 times as compared with the wild type P2 (Table 2). This indicates that Gly-171, Leu-175, and Gln-178 of PvTRAg38 were more critical for interacting with basigin than other residues. However, it may be noted here that the binding affinity of P2A12 toward basigin was less than half of P2A5 or P2A9 (Table 2). E176A of P2A10 had no effect on its basigin binding affinity, and the mutation at other residues of this peptide showed 5–10 times reduction in this binding affinity.
TABLE 2.
Identification of critical amino acid residues of PvTRAg38 from peptide P2 region interacting with basigin through alanine-scanning analysis using SPR and docking studies
Mutated amino acids are shown in bold and underlined. (B) denotes the amino acid position of basigin.
| Peptide name | Peptide sequence (167–178 amino acids) | PvTRAg38 peptide P2-basigin interaction |
|||
|---|---|---|---|---|---|
| SPR, KD value | Docking |
||||
| Hydrogen bonds | Salt bridges | Hydrophobic contacts | |||
| m | |||||
| P2 | IKTEGRKILEAQ | 1.02 ± 0.12 × 10−9 | Gln-178–Asp-77(B) | Arg-172–Asp-79(B) | Arg-172–Asp-80(B) |
| Thr-169–Pro-104(B) | Arg-172–Asp-80(B) | Glu-170–Gln-81(B) | |||
| Gly-171–Trp-82(B) | Arg-172–Asp-77(B) | Lys-173–Gly-103(B) | |||
| Lys-173–Asp-79(B) | Gln-178–Ser-190(B) | ||||
| P2A1 | AKTEGRKILEAQ | 2.81 ± 0.65 × 10−8 | Gln-178–Asp-77(B) | Ala-177–Leu-62(B) | Leu-175–Val-60(B) |
| Arg-172–Asp-77(B) | Lys-168–Leu-67(B) | Ile-174–Leu-62(B) | |||
| Ala-177–Val-61(B) | Lys-168–Lys-63(B) | Lys-168–Glu-64(B) | |||
| Gln-178–Lys-36(B) | Ile-167–Phe-74(B) | ||||
| P2A2 | IATEGRKILEAQ | 1.2 ± 0.22 × 10−8 | Gln-178–Lys-57(B) | Arg-172–Asp-80(B) | Arg–172–Asp-80(B) |
| Gln-178–Asp-79(B) | Arg-172–Asp-79(B) | Arg-172–Asp-79(B) | |||
| Gln-1178–Tyr-85(B) | Arg-172–Asp-77(B) | Arg-172–Asp-77(B) | |||
| Arg-172–Asp-79(B) | |||||
| Arg-172–Asp-77(B) | |||||
| Thr-169–Val-61(B) | |||||
| P2A3 | IKAEGRKILEAQ | 8.59 ± 0.21 × 10−8 | Lys-168–Lys-63(B) | Glu-176–Lys-36(B) | Glu-170–Val-61(B) |
| Lys-173–Asp-79(B) | Leu-175–Glu-73(B) | ||||
| Ile-174–Phe-74(B) | |||||
| P2A4 | IKTAGRKILEAQ | 9 ± 1.54 × 10−8 | Arg-172–Asp-79(B) | Arg-172–Asp-80(B) | Gly-171–Lys-36(B) |
| Gln-178–Asp-77(B) | Glu-176–Lys-36(B) | Ile-174–Val-61(B) | |||
| Lys-173–Asp-77(B) | Lys-173–Asp-77(B) | Leu-175–Phe-74(B) | |||
| P2A5 | IKTEARKILEAQ | 1.03 ± 0.95 × 10−7 | No bonding | Lys-173–Asp-80(B) | Glu-176–Leu-62(B) |
| Lys-173–Asp-79(B) | Leu-175–Phe-74(B) | ||||
| Ile-174–Lys-75(B) | |||||
| Lys-173–Asp-77(B) | |||||
| P2A6 | IKTEGAKILEAQ | 3.2 ± 0.73 × 10−8 | Glu-170–Lys-57(B) | Glu-170–Lys-57(B) | Lys-168–Gly-58(B) |
| Ile-167–Val-60 (B) | Thr-169–Gly-59(B) | ||||
| Glu-171–Val-61(B) | |||||
| Ala-177–Leu-62(B) | |||||
| Gln-178–Lys-63(B) | |||||
| Leu-175–Phe-74(B) | |||||
| Ile-174–Asp-77(B) | |||||
| Ala-172–Asp-80(B) | |||||
| P2A7 | IKTEGRAILEAQ | 1.2 ± 0.21 × 10−8 | Ala-173–Thr-18(B) | Lys-168–Asp-45(B) | Leu-175–Thr-25(B) |
| Ile-174–Thr-29(B) | Glu-176–Lys-71(B) | Ile-167–Phe-27(B) | |||
| Glu-176–Lys-71(B) | Ala-177–Asn-44(B) | ||||
| Arg-172–Asn-98(B) | |||||
| P2A8 | IKTEGRKALEAQ | 5.2 ± 0.95 × 10−8 | Arg-172–Asp-194(B) | Lys-173–Asp-79(B) | Ile-167–Lys-57(B) |
| Leu-175–Pro-104(B) | Arg-172–Asp-194(B) | Thr-169–Asp-80(B) | |||
| Lys-173–Trp-82(B) | |||||
| Ala-174–Pro-104(B) | |||||
| P2A9 | IKTEGRKIAEAQ | 1.2 ± 0.11 × 10−7 | No bonding | No bonding | Gly-171–Asp-79(B) |
| Glu-170–Trp-82(B) | |||||
| Ile-174–Pro-104(B) | |||||
| P2A10 | IKTEGRKILAAQ | 1.2 ± 1.03 × 10−9 | Ile-167–Lys-57(B) | Lys-173–Asp-80(B) | Lys-168–Trp-82(B) |
| Gln-178–Lys-57(B) | Ala-177–Trp-82(B) | ||||
| Lys-173–Pro-105(B) | |||||
| Arg-172–Asp-194(B) | |||||
| P2A12 | IKTEGRKILEAA | 2.23 ± 0.32 × 10−7 | Ala-177–Asp-77(B) | Lys-173–Asp-80(B) | Ile-174–Lys-57(B) |
| Glu-177–Lys-57(B) | Glu-176–Leu-62(B) | ||||
| Glu-176–Lys-36(B) | Lys-173–Lys-57(B) | ||||
| Arg-172–Asp-77(B) | Arg-172–Asp-79(B ) | ||||
| Arg-172–Asp-79(B) | |||||
To further provide insights into the interaction of wild type and mutant P2 peptides with basigin, we carried out molecular docking studies of these peptides with the known crystal structure of human basigin. Despite the large number of degrees of freedom considered to take into account the ligand flexibility, the complexes obtained were characterized by the good sterical complementarities between basigin and peptides (Fig. 7A). To understand the different affinities of mutant peptides, the docking study was extended with all possible interactions. In particular, the P2-basigin complex is stabilized by a network of hydrogen bonds and salt bridges (Fig. 7B and Table 2). This molecular interaction analysis showed peptide P2 to be involved in hydrogen bonding through Thr-169 (Thr-3 in P2), Gly-171 (Gly-5 in P2), Lys-173 (Lys-7 in P2), and Gln-178 (Gln-12 in P2) to basigin (Fig. 7B). Moreover, the complex includes a noticeable salt bridge and hydrophobic interactions between the carboxyl group of the succinic moiety and ammonic group of Arg-172 (Arg-6 in P2), Glu-176 (Glu-10 in P2), Lys-173 (Lys-7 in P2), and Gln-178 (Gln-12 in P2). The mutant peptides, P2A5, P2A9, and P2A12 either have one or no hydrogen bonds with basigin amino acids. They are stabilized with fewer numbers of salt bridges and hydrophobic interaction (Fig. 7B and Table 2) with basigin amino acid residues. But the interaction of mutant peptide P2A12 with basigin is still more stable than that of the mutant peptides P2A5 or P2A9 because it still contains one hydrogen bond and more numbers of salt bridges as well as hydrophobic interactions than those present in these two other mutant peptides (Table 2). From these SPR and molecular docking results, it seems that residues Gly-171 and Leu-175 of peptide P2 are the key residues involved in interaction with the N-terminal domain of basigin. The N-terminal region of basigin is also shown recently to be involved in interaction with PfRH5 (25).
FIGURE 7.

Putative binding sites of PvTRAg38 peptide P2 on human basigin by molecular docking. A, depicted surface model represents P2 peptide (gray) that was docked into the human basigin by computer simulation using Z-DOCK software. Ribbon representation of the human basigin (pink) and stick representation of the peptide P2 (yellow, with transparent electrostatic surface) are shown in zoomed image. B, schematic diagram showing details of interactions of peptide P2 residues with neighboring residues of human basigin. Hydrogen bonds between Gln-12 (corresponding to Gln-178 of PvTRAg38)–Asp-77(basigin), Gly-5 (corresponding to Gly-171 of PvTRAg38)–Trp-82 (basigin), Lys-7 (corresponding to Lys-173 of PvTRAg38)–Asp79 (basigin), and Leu-9 (corresponding to Leu-175 of PvTRAg38)–Pro-104 (basigin) are shown (green dashed lines). Hydrophobic interactions are also depicted with dark red semi-circles. Most of the pictured hydrogen bonds are lost in alanine substitution. The figure was created using LIGPLOT.
Blocking of Basigin by P2 Peptide Leads to Parasite Growth Inhibition
Basigin is a known erythrocyte receptor required for P. falciparum invasion (5). Blockage of this erythrocyte surface protein resulted in invasion inhibition of P. falciparum into the erythrocyte via the parasite ligand PfRH5 (26). So, we made use of this fact and checked for potential P2 to block basigin on the surface of erythrocyte and its role in parasite invasion. Our results on P. falciparum (3D7) culture showed a dose-dependent growth inhibition by peptide P2 up to 20 μm and a plateau thereafter at higher concentrations (Fig. 8A). The parasite culture treated with 20 μm P2 showed ∼18% parasite growth inhibition (Fig. 8B). Peptide P4, which interacts with band 3 (19), was also tested for its parasite growth inhibition potential and was found to inhibit ∼20% of parasite growth at the same concentration (Fig. 8B). The complete protein PvTRAg38 at the same concentration was used as a positive control, which showed ∼35% growth inhibition as reported earlier (19). Furthermore, we assessed the cumulative effect of both P2 and P4 peptides in growth inhibition assay. Indeed, the equimolar mixture of P2 and P4 peptides at 20 μm concentration was able to inhibit ∼32% of the parasite growth, which was almost similar to that of the growth inhibition rate shown by the complete protein PvTRAg38 (Fig. 8). A non-erythrocyte-binding protein PvTRAg38.7 was used as a negative control, which showed an insignificant parasite growth inhibition. The parasite growth inhibition shown by P2, P4, P2 + P4, or PvTRAg38 at 20 μm concentration was significantly higher (p < 0.05) than this negative control. There was no significant difference in parasite growth inhibition between P2 and P4 as well as between P2 + P4 and PvTRAg38.
FIGURE 8.

Inhibition of P. falciparum growth by peptides P2 and P4 of PvTRAg38. Parasite culture at late schizont stage was incubated with peptide P2, peptide P4, peptides P2 + P4, PvTRAg38, PvTRAg38.7, and other controls. After 40 h, parasitemia was determined by ethidium bromide staining and measured by flow cytometry. A, bar diagram shows the percentage of parasite growth in the absence or presence of PvTRAg38 and its derived peptides at different concentrations (0–50 μm). Data shown are the mean ± S.D. for two triplicate experiments. B, representative dot plots showing percent infection. Panel a, infected erythrocytes treated with PBS. Panel b, infected erythrocytes treated with peptide P4 (20 μm). Panel c, infected erythrocytes treated with peptide P2 (20 μm). Panel d, infected erythrocytes treated with 20 μm peptide P2 and peptide P4. Panel e, infected erythrocytes treated with PvTRAg38 (20 μm), and panel f, infected erythrocytes treated with PvTRAg38.7 (20 μm).
Gly-171 and Leu-175 of P2 Are More Critical for Parasite Growth
Because binding affinity of mutant peptides of P2 toward basigin was found to be greatly reduced in SPR, their potential toward parasite growth inhibition was tested to check for the sequence-specific effect. Results of parasite growth inhibition assay using these mutant peptides are shown in Fig. 9A. With the exception of peptides P2A3 and P2A10, others showed a reduction in their ability to inhibit the parasite growth in comparison with wild type P2. For example, binding affinities of P2A5 and P2A9 with basigin were maximally reduced in comparison with others, and similarly, these peptides also lost their parasite growth inhibition potential to the maximum (Fig. 9A and Table 2). There was a correlation between binding affinities of these mutant peptides with basigin and their parasite growth inhibition potential (Pearson's correlation coefficient, r = 0.64, p = 0.02) (Fig. 9B and Table 2). These results, along with SPR data and docking studies, indicate the critical nature of Gly-171 and Leu-175 amino acid residues of P2 involved in interaction with host erythrocyte receptor basigin and thus the red cell invasion.
FIGURE 9.
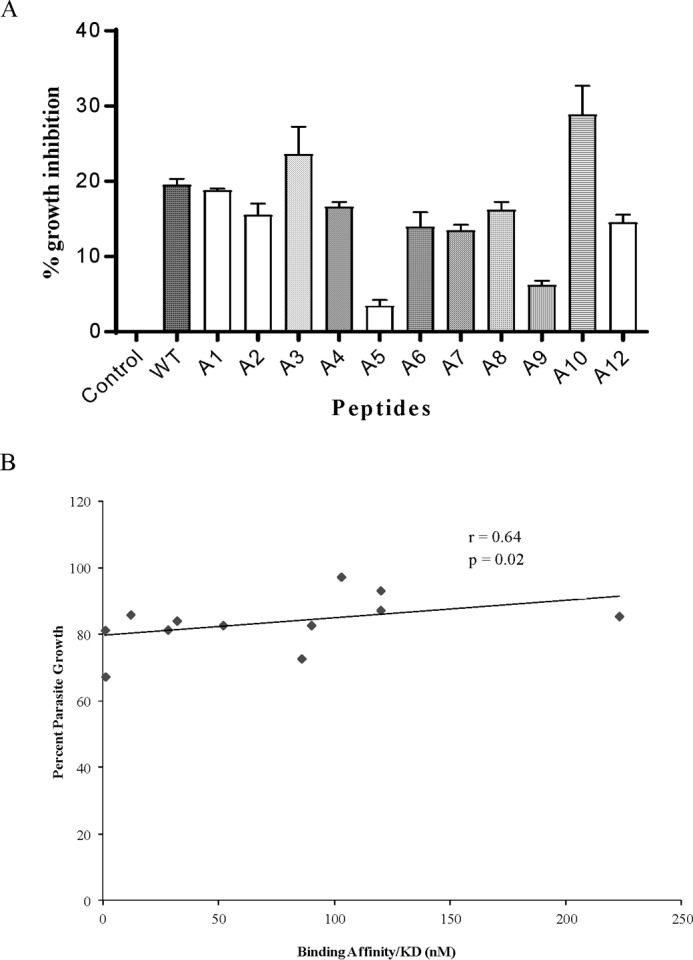
Effect of amino acid mutations in peptide P2 on parasite growth inhibition. A, parasite culture at late schizont stage was incubated with 20 μm wild type P2 or its mutated peptides or PBS alone. After 40 h, parasitemia was determined by ethidium bromide staining and measured by flow cytometry. B, scatter plot was drawn to see the pattern of relationship between parasite growth and basigin-binding affinity of mutant P2 peptides.
Discussion
Identification of the molecules involved in host-parasite interaction and elucidation of the molecular mechanisms that are taking place during host cell invasion are important for developing newer therapeutics to control the disease. The phenomenon of red cell invasion by merozoites of Plasmodium is highly complex because parasites use multiple invasion pathways thus utilizing large numbers of parasite ligands and their respective receptors on host erythrocytes. Complexity is further increased when the individual parasite ligand interacts with more than one erythrocyte receptor, and similarly, each erythrocyte receptor is recognized by more than one parasite ligand (4, 14, 27–29). The numbers of erythrocyte receptors identified for the P. vivax merozoite proteins is very limited (3, 10, 12). Recently, we have identified band 3 as one of the human erythrocyte receptors for a P. vivax merozoite-expressed tryptophan-rich protein called PvTRAg38 and also their binding mechanisms (19). Because this parasite ligand interacts with two erythrocyte receptors, here we have identified basigin as the second erythrocyte receptor using more sensitive techniques.
In our earlier studies, we were able to capture the high copy number protein band 3 on the erythrocyte surface against PvTRAg38 but missed the other receptor, which could be present in low copy numbers (19). Because various other sensitive protein-protein interaction platforms are now widely used for detection of low abundance proteins (3, 5, 23), we utilized here two such techniques, i.e. Label transfer assay and MudPIT analysis, to identify the second erythrocyte receptor of PvTRAg38. These techniques allowed us to identify additional erythrocyte proteins, along with band 3, which could be interacting with this parasite ligand (Table 1 and Fig. 1). After negating the most abundant proteins, and those proteins that were not commonly identified by both techniques, we carried out direct interaction of PvTRAg38 with the remaining two erythrocyte proteins CD59 and basigin along with 55-kDa EMP by solid phase ELISA to show that only basigin was interacting with PvTRAg38. Using various techniques, it was evident that PvTRAg38 was specifically interacting with basigin molecule (Figs. 3–5). Basigin is probably the third erythrocyte receptor identified here so far for any of the P. vivax merozoite proteins.
The resistance of basigin to chymotrypsin treatment further confirms our hypothesis and corroborates our previous results on PvTRAg38-erythrocyte binding, where this interaction was partially affected by chymotrypsin (14), and the other receptor sensitive to this enzyme has already been identified as band 3 (19). Although basigin is highly glycosylated in the extracellular region and its N-glycosylation plays important roles in its function (30), its interaction with PvTRAg38 may not involve the carbohydrate moieties because the non-glycosylated recombinant basigin expressed in E. coli as well as the glycosylated form in HEK-293 were binding to PvTRAg38 in our studies (Figs. 3–5). Basigin has also been identified as an erythrocyte receptor for PfRH5 and shown to be involved in red cell invasion (5). Reticulocytes have a higher abundance of basigin molecules on the surface as compared with mature red blood cells, which shed them off via the exosome-endosome pathway during maturation (31, 32). Therefore, basigin might be acting as the interacting protein with PvTRAg38 and thus playing an important role in the P. vivax merozoite attachment to reticulocytes.
In our earlier studies, host erythrocytes pre-incubated with PvTRAg38 showed parasite growth inhibition in a heterologous P. falciparum culture system (19). The soluble basigin as well as the anti-basigin antibodies have been reported to inhibit the P. falciparum growth in in vitro via PfRH5 (5). Because PvTRAg38 binds to basigin, their interaction might play a role in merozoite entry into the host red cell. Therefore, blocking of basigin molecules on the erythrocyte surface by PvTRAg38 might interfere with invasion. The probable reason of this parasite growth inhibition in the heterologous system could be due to blockade of the basigin receptor at the N terminus by PvTRAg38, which renders this area inaccessible for binding to PfRH5 and thus inhibits the growth, as evident from our molecular docking results (Fig. 7). However, we did not know whether this parasite growth inhibition was due to blockade of only basigin or the previously described band 3 or both of these receptors. Because we have defined the respective binding domain for each of these receptors, P2 for basigin (Fig. 6) and P4 for band 3 (19), we used these synthetic peptides separately as well as in combination to study the parasite growth inhibition rates. Indeed, P2 and P4 peptides showed almost an equal rate of parasite growth inhibition that was almost half if compared with the total protein (Fig. 8). But P2 and P4 together reached the growth inhibition rate that was almost comparable with that of the complete PvTRAg38 protein (Fig. 8). These results indicate that both band 3 and basigin are utilized for the attachment of merozoite to red cell during the invasion process (Fig. 8). These data need to be confirmed by using the P. vivax system as and when available where the blocking of individual P2 and P4 sites of PvTRAg38 by specific antibodies against these domains on the merozoite should be able to inhibit the reticulocyte invasion.
The PvTRAg38 appears to be an important protein in P. vivax biology, as it has only two haplotypes in the parasite population among Indian isolates (15). The variant haplotype has only one amino acid (V270E) change among the field isolates. However, this mutation in the protein was outside the erythrocyte-binding regions of PvTRAg38. The nucleotide sequence as well as the encoded amino acid sequence of P2 and P4 regions of PvTRAg38 binding to basigin and band 3, respectively, were completely conserved. This is expected as these peptides were interacting with the host molecules for biological function, and any variation in parasite ligand in this region could be detrimental for host-parasite interaction. Furthermore, PvTRAg38 has been established as a potent immunogen (16). The presence of memory T cell against PvTRAg38 points toward the importance of this protein as immunogen, which can generate fast and specific immune response during P. vivax infection (17). Also naturally occurring antibodies against PvTRAg38 in sera from P. vivax-infected individuals have shown potential for blocking PvTRAg38-erythrocyte interactions (14), which again points toward important role of this protein in in vivo. Similar to many other potential vaccine candidate antigens of Plasmodium, PvTRAg38 is also a non-glycosylphosphatidylinositol (GPI)-anchored protein with erythrocyte binding ability and high immunogenicity (5, 33). The immunogenic potential of non-GPI-anchored proteins, including PvTRAg38, forms a strong basis for their vaccine candidacy.
Interaction between PvTRAg38 and basigin seems to involve multiple amino acids of the P2 region of PvTRAg38 (Fig. 7 and Table 2). But two amino acid residues (Gly-171 and Leu-175) are more critical in this receptor-ligand interaction. Furthermore, there was a correlation between binding affinities of amino acids of P2 and their potentials for parasite growth inhibition (Fig. 9). These two amino acid positions were found to be more critical in this regard. Earlier, we identified critical amino acid residues of the P4 peptide that are involved in interaction with band 3 (20). Identification of these critical amino acid residues will open new avenues to generate therapeutic measures to control these interactions and thus the parasite growth, similar to various other diseases (32, 33).
In conclusion, identification of basigin along with the previously identified band 3 as interacting partners for PvTRAg38 will provide new directions in P. vivax biology. Currently, inhibition of protein-protein interactions is becoming a popular tool for the development of therapeutics (34, 35). Information generated about the critical residues involved in PvTRAg38-basigin and previously identified PvTRAg38-band 3 interactions can be of utmost importance in developing small molecules that can impede this interaction, as has been done by different studies in Plasmodium (36).
Experimental Procedures
Materials
The following were commercially obtained: human basigin cDNA clone, mammalian expressed recombinant histidine-tagged CD59 (Sino Biological, Beijing, China); human erythrocyte 55-kDa membrane protein (GeneCopoeia, Rockville, MD); Alexa Fluor 488-conjugated goat anti-mouse and anti-rabbit IgG, proofreading Pfx polymerase enzyme (Invitrogen Life Technologies, Inc.); sulfo-SBED-biotin Label transfer cross-linker (sulfosuccinimidyl-2-[6-(biotin amide)-2-(p-azidobenzamido)hexanoamido] ethyl-1,3′-dithiopropionate) (Thermo Scientific, Rockford, IL); GeneJET gel extraction kit (Thermo Scientific, Waltham, MA); HEK-293 cells (American Type Culture Collection (ATCC), Manassas, VA); RPMI 1640 medium, hypoxanthine, penicillin/streptomycin, fetal calf serum, glutamine, glutaraldehyde, poly-l-lysine, monoclonal antibodies against basigin (Clone MEM-M6); His6 and GST (Sigma); Lipofectamine® 2000, synthetic peptides (Thermo Fisher Scientific, GmbH, Germany); mouse monoclonal antibodies DL6 (Santa Cruz Biotechnology, Dallas, TX); HBS-EP buffer (degassed and ready to use 0.01 m HEPES, pH 7.4, 0.15 m NaCl, 3 mm EDTA, 0.005% v/v surfactant P20); protein marker standards for GPC (GE Healthcare); Matchmaker Gold Systems (Clontech); and trypsin (Promega Corp., Madison, WI). Plasmid pRE4 vector was a kind gift from Gary Cohn and Roselyn Eisenberg (University of Pennsylvania, School of Dental Medicine, Philadelphia). Recombinant plasmid pRE4-PvTRAg38, recombinant proteins GST-tagged PvTRAg38, histidine-tagged PvTRAg38, and histidine-tagged thioredoxin from Desulfovibrio desulfuricans were available in the laboratory from previous studies (19, 37).
Pulldown Assay and MudPIT Analysis
Erythrocyte membrane was prepared by hypotonic lysis of the erythrocytes using the method of Dodge et al. (38). To identify the host erythrocyte membrane proteins interacting with PvTRAg38, the pulldown assay was performed as described by Yajima et al. (39). Briefly, the above-mentioned erythrocyte membrane preparation in 5 mm phosphate buffer, pH 7.5, containing protease inhibitor mixture was dissolved in 1% C12E8 by incubating it on ice for 20 min. After centrifugation at 20,000 × g for 30 min at 4 °C, the solubilized erythrocyte membrane preparation was incubated with 1:1 glutathione-Sepharose 4B resin for 1 h. GST-tagged PvTRAg38 or GST alone (25 μg) was incubated with 100 μl of 50% glutathione-Sepharose 4B resin for 1 h at 4 °C, followed by washing with Tris-buffered saline (25 mm Tris, 150 mm NaCl, 2 mm KCl, pH 7.5) containing 0.1% C12E8. The GST-tagged PvTRAg38 (or GST alone) bound to glutathione resin was then incubated overnight at 4 °C with 1 mg of solubilized erythrocyte membrane proteins. To remove unbound material, resin was washed five times with Tris-buffered saline containing 0.1% C12E8 and finally with 10 mm Tris-HCl, pH 7.5. The bound proteins were eluted using reduced glutathione (100 mm). The eluates were reduced with DTT (10 mm), alkylated with iodoacetamide (0.5 mm), and digested with trypsin. The peptide mixture was lyophilized and reconstituted in 50 mm triethylammonium bicarbonate (TEAB), pH 8.0. A pre-packed microtip with C-18 resin was activated with 100% acetonitrile and then conditioned with 50 mm TEAB. Peptides were allowed to bind with this C-18 resin for multiple cycles. The column was washed with 2% acetonitrile in 50 mm TEAB, and the bound peptides were eluted into four fractions with 10–70% acetonitrile gradient. After lyophilization, each fraction was dissolved in 0.1% formic acid and analyzed separately on LC-MS/MS. The peptide mixtures were loaded onto Acclaim PepMap RSLC (50 μm × 15 cm) and separated with a linear gradient of solvent B (95% acetonitrile + 0.1% formic acid) from 5 to 50% in 85 min. The eluting peptides were directly injected into the Orbitrap Velos Pro MS (Thermo Fisher Scientific Inc.). The parent ion spectra were acquired in full-scan mode at a resolution of 60,000. The top 20 precursors were subjected for fragmentation with a collision energy of 35 using collision-induced dissociation in the ion trap. The spectra were analyzed in Proteome Discoverer 1.4 (Thermo Fisher Scientific Inc.) using SEQUEST algorithm.
Label Transfer Assay Using Sulfo-SBED Biotin Label Cross-linker
Sulfo-SBED biotin Label transfer cross-linker (1.12 mg) was dissolved in 25 μl of DMSO immediately before use and was added to the protein vial having 5 mg of GST-tagged PvTRAg38 in PBS and incubated on ice in the dark for 2 h with constant tapping at regular intervals. The reaction mixture was then centrifuged at 10,000 × g for 1 min to remove hydrolyzed cross-linker. The unbound cross-linker was removed by dialyzing the reaction mixture in ice-cold PBS. 40 μg of labeled protein was incubated overnight with 500 μg of the solubilized RBC membrane. For direct interaction, 1 μg of labeled GST-tagged PvTRAg38 was incubated with 2 μg of recombinant histidine-tagged basigin. Reaction mixture was UV (365 nm)-exposed for 15 min using a long wave UV lamp to transfer the biotin label from the recombinant PvTRAg38 to its interacting protein partner. The interacting proteins were pulled down using Sepharose 4B resin, which specifically binds to GST and was incubated for 1 h at room temperature. The beads were washed twice with 1× PBS, and the pulled down complex was boiled at 100 °C for 5 min in 2× SDS loading dye and run on 15% SDS-PAGE. After transferring to nitrocellulose membrane, the blot was blocked with 5% BSA in 1× PBS for 30 min at room temperature. The blot was further incubated with streptavidin-HRP conjugate and was developed using 3,3′-diaminobenzidine/peroxidase substrate solution (0.05% 3,3′-diaminobenzidine, 0.015% H2O2, 0.01 m PBS, pH 7.2). Separately, the blot was developed with rabbit polyclonal anti-basigin antibody.
PCR Amplification, Cloning, Expression and Purification of Recombinant Proteins, and Antibody Generation
Ectodomain of basigin was amplified from the human basigin cDNA clone using primers 5′-GGATCCGGGCCCATGGCGGCTGCGCTGTTCG-3′ and 5′-CTCGAGCGATCGTCAGGAAGAGTTCCTCTGGC-3′. The DNA was denatured at 94 °C for 10 min followed by 35 cycles as follows: denaturation at 94 °C, annealing at 59 °C, and extension at 68 °C for 1 min each. The final extension at 68 °C was carried out for 15 min. The PCR product was resolved on a 1.2% agarose gel, and the corresponding band was excised from the gel. The DNA was eluted using GeneJET gel extraction kit, and subcloned into pET28b expression vector. The expression and purification of histidine-tagged recombinant protein were carried out as described by Song et al. (40). The purity of the recombinant histidine-tagged basigin preparation was checked on 12% SDS-PAGE.
The CD spectra for ∼25 μm histidine-tagged recombinant basigin were acquired in 25 mm sodium phosphate buffer, pH 8.0, using a JASCO J-815 spectropolarimeter (Jasco, Japan). Far-UV CD spectra were monitored from 180 to 300 nm. The path length was 2 mm, and instrument parameters were set to a sensitivity of ∼30 millidegrees, a response time of 1 s, and a scan speed of 100 nm/min. Spectra were recorded as an average of eight scans.
Size-exclusion chromatography of histidine-tagged recombinant basigin was carried out using an ÄKTA fast performance liquid chromatography (FPLC) (GE Healthcare) at 25 °C. Recombinant basigin (∼5 mg) was applied to an HR 10/30 S75 column (GE Healthcare) equilibrated in extraction buffer. Protein was eluted at 0.3 ml/min under maximum pressure of 1 megapascal. Fractions were collected, and the size of eluted proteins was calibrated using protein marker standards.
Antibodies against histidine-tagged PvTRAg38, histidine-tagged basigin, and KLH-tagged peptide P2 were generated in rabbits, following a previously described protocol (41). The animal experiment protocols were approved by the Institutional Animal Ethics Committee (approval number 494/IAEC/09). The CPCSEA guidelines were followed to perform experiments on animals.
Solid Phase Binding Assay
This assay was performed by coating each well of 96-well ELISA plate with 50 nm GST-tagged PvTRAg38 or untagged bacterial thioredoxin in carbonate buffer, pH 9.6, overnight at 4 °C. The coated ELISA plate was blocked with 5% BSA in PBS for 2 h at 37 °C. Furthermore, different concentrations of histidine-tagged proteins (0–2.5 μm) were incubated for 2 h at 37 °C. After washing, the plate was developed with monoclonal anti-His6 antibodies as described earlier (19).
For competition assay, two differently tagged analyte molecules competing for binding sites available on the immobilized ligand (basigin) were used as described previously (19). Briefly, the wells of the 96-well microtiter plate were coated with 50 nm histidine-tagged basigin and then blocked with 5% BSA. The coated wells were then incubated with different concentrations (0–2 μm) of histidine-tagged PvTRAg38 for 2 h at 37 °C, followed by washing with 1× PBS. The same wells were again incubated with 2 μm GST-tagged PvTRAg38 for 2 h at 37 °C. After washing with PBS, the bound protein was detected by monoclonal anti-GST antibody, as described earlier (19).
For antibody inhibition assay, the wells of the 96-well microtiter plate were coated with 250 nm GST-tagged PvTRAg38 or histidine-tagged basigin and then blocked with 5% BSA. The coated wells were then incubated with different dilutions of anti-PvTRAg38, anti-P2, or anti-basigin rabbit antibodies for 2 h at 37 °C, followed by washing with 1× PBS. The same wells were again incubated with 2 μm GST-tagged PvTRAg38 or histidine-tagged basigin for 2 h at 37 °C. After washing with PBS, the bound protein was detected by monoclonal anti-GST or anti-His6 antibody, as described earlier (19).
For peptide-basigin interaction by solid phase binding assay, 1 μm peptide P2 or P4 was coated to each well of a 96-well microtiter plate, pretreated with glutaraldehyde and poly-l-lysine, as described by Sorette et al. (42), in carbonate buffer, pH 9.6, overnight at 4 °C. After washing twice with 1× PBS, the plate was blocked with 5% BSA in PBS for 2 h at 37 °C. Different concentrations (0–2 μm) of histidine-tagged basigin or bacterial thioredoxin were added to the coated wells, and the plate was incubated for 2 h at 37 °C. Plates were developed with monoclonal anti-His6 antibody as described earlier (19).
For competition assay, the histidine-tagged basigin (0.5 μm) was coated to each well and then incubated with increasing concentrations (0–20 μm) of peptide P2 for 2 h. The wells were incubated with 2 μm GST-tagged PvTRAg38 for 2 h at 37 °C. After washing with PBS, the bound protein was detected by monoclonal anti-GST antibody as described earlier (19).
Surface Plasmon Resonance
SPR assay was performed on BIAcore 2000 instrument at 25 °C, using HBS-EP buffer. The recombinant human erythrocyte protein basigin was immobilized (ligand) on CM5 sensor chip using the amine-coupling method. Briefly, the surface of the flow cell was activated by flowing an equivolume mixture of 1-ethyl-3-(3-dimethylamino)propylcarbodiimide hydrochloride and N-hydroxysuccinimide, followed by flow of analyte in sodium acetate buffer, pH 4.5–5.5. Furthermore, 1 m ethanolamine was allowed to flow through the cells to block the unused activated free COO− groups at the surface. Blank surface was also prepared with the same method, except for the flow of ligand protein. Kinetic analysis was performed by flowing different concentrations of analyte (PvTRAg38 or wild/mutated P2) on the immobilized basigin as well as the reference flow cell, at a flow rate of 30 μl/min. The surfaces were regenerated with a pulse of appropriate regeneration buffers at the end of each injection cycle. Reference-subtracted sensorgrams were analyzed using the BIAcore evaluation software 3.2RC1 (BIAcore AB, Uppsala, Sweden). The binding constant, KD, was calculated as kd1/ka1 by using data analysis program BIAevaluation 3.2RC1 (BIAcore AB). Kinetic rate constants were determined by fitting the corrected response data to a simple 1:1 Hill-Langmuir binding isotherm model using BIAevaluation 3.2RC1 software.
Yeast Two-hybrid Assay
Y2H analysis was performed using a GAL4-based Y2H system. The construction of the Y2H library, auto-activation and toxicity test, and the screening of the Y2H library were performed according to the manufacturer's instructions. The bait and prey plasmids were co-transformed into yeast strain AH109. Yeast transformation was performed according to the manufacturer's instructions. The transformants were assayed for growth on synthetic dropout SD/Trp−Leu−His− plates and cultured on liquid synthetic SD/Trp−Leu−His− medium for 36 h before being collected by centrifugation. The concentration of collected yeast cells was adjusted to 106 (cells/ml) using sterile water, and then 5 μl of yeast suspension was assayed for growth on SD/Trp−Leu−His−Ade− plates. The co-transformants growing on SD/Trp−Leu−His−Ade− plates were further plated on SD/Trp−Leu−His−Ade−AT plates to assess their strength of interaction in the presence of 3-amino-1,2,4-triazole (10–50 mm).
β-Galactosidase assay was performed by monitoring the lacZ reporter gene expression. Pellet from the liquid culture was rapidly freeze/thawed as per the manufacturer's instructions. As β-galactosidase accumulates in the medium, it hydrolyzes ONPG to o-nitrophenol, which is spectrophotometrically determined at 420 nm.
Cloning and Expression of Basigin in HEK-293 Cells
The above-mentioned PCR-amplified basigin ectodomain was cloned in PvuII and ApaI sites of pRE4 vector (kindly gifted by Gary Cohn and Roselyn Eisenberg) so as to produce a fusion protein containing the signal sequence at the N terminus and transmembrane region of the herpes simplex virus glycoprotein D (HSVgD) at the C terminus, respectively (43). This recombinant DNA was then used to transfect the HEK-293 cells that were cultured in RPMI 1640 media with 10% fetal calf serum, 4 mm glutamine, 1× penicillin/streptomycin, pH 7.4, in a humidified 5% CO2 incubator at 37 °C as described earlier (16). The transfection was done with cationic lipid Lipofectamine® 2000, following the manufacturer's protocol. DAPI was used for nuclei staining. Images were captured using Nikon confocal microscope A1 (Nikon Corp., Tokyo, Japan).
Direct Immunofluorescence Assay was done to confirm the expression of basigin on the surface of HEK-293 cells and to assess the transfection efficiency. Transfected HEK-293 cells were grown over a coverslip and were subsequently blocked with 5% BSA in 1× PBS for 30 min at room temperature. The transfection was detected with mouse monoclonal antibodies DL6 directed against amino acids 272–279 of the HSVgD sequences as described previously (43, 44). The slide was observed under UV and visible light at ×400 magnification using Nikon Eclipse E600 fluorescent microscope (Nikon, Tokyo, Japan), and images were obtained.
For cell-based studies of protein-protein interactions, the transfected HEK-293 cells expressing basigin were grown over a coverslip, followed by blocking with 5% BSA in 1× PBS for 30 min at room temperature. The recombinant GST-tagged PvTRAg38 (20 μm) was added to the coverslip and incubated for 2 h. Furthermore, the same HEK cells were probed simultaneously with monoclonal anti-GST antibodies (1:500) and monoclonal anti-basigin antibodies (1:100). Secondary antibodies used were Alexa Fluor 488-tagged anti-mouse and Alexa Fluor 594-tagged anti-rabbit. Smears were mounted in DAPI anti-fade and sealed. Vector alone pRE4-transfected cells were used as a control. The slides were observed under UV and visible light at ×400 magnification using Nikon Eclipse E600 fluorescent microscope (Nikon Corporation, Tokyo, Japan), and images were obtained.
Docking of Wild Type and Mutant Peptide P2 on the Basigin Structure
The 3D structures of the peptides P2 and its mutants were predicted by PEPstrMOD (45). To get stable structures of each peptide energy minimization, molecular dynamics simulations and related analyses for each peptide structures were performed separately using GROMACS (46, 47) software suite version 5. We used the AMBER99SB (48) interaction potential along with the TIP4P-EW model of water with a periodic boundary condition; Na+ and Cl− ions were used to keep neutralization of the system. For each simulation, the structures were relaxed by the steepest-descent method and conjugated gradient energy minimization, and then the systems were equilibrated for 100 ps in NVT and NPT ensembles followed by a 20-ns molecular dynamics simulation run. Chain B was taken from human basigin (Protein Data Bank code 4uq0) as the protein structure of basigin and energy minimization were done on the protein structure. The docking was carried out by ZDOCK server (49) for each peptide with the protein structure separately, and the first ranked peptide-protein complex from the cluster of complexes was extracted from each case based on the highest ZDOCK score of docked complex. After docking, energy minimization and molecular dynamics simulations were run again on each topped ranked peptide-protein complexes separately as described above for 100 ns. To visualize the structures of the complexes, chimera was used. Ligplot+ (50) suite was used to find the amino acid interactions between peptide and protein in the topped ranked peptide-protein complexes.
P. falciparum Culture and Growth Inhibition Assay
Growth inhibition assay was performed as described by Persson et al. (51). Briefly, the P. falciparum 3D7 strain was cultured in complete RPMI 1640 medium containing, 0.5 g/liter Albumax I 27.2 mg/liter hypoxanthine, and 2 g/liter sodium bicarbonate, using O+ human erythrocytes (4% hematocrit) under mixed gas (5% O2, 5% CO2, and 90% N2). Parasite culture was synchronized by sorbitol treatment, and synchronous cultures at late schizont stage with parasitemia of 1% were treated with different concentrations (0–50 μm) of the P2, P4, P2 + P4, PvTRAg38, or PvTRAg38.7 in a 96-well culture plate in triplicate. Similarly, the growth inhibition assay was performed for the wild type P2 and its mutated peptides at 20 μm concentration. Uninfected erythrocytes, infected erythrocytes alone, and infected erythrocytes with PBS were taken as controls. Parasites were maintained for 40 h and stained with ethidium bromide. One hundred thousand total events were acquired per sample, using Cell Quest software on a FACSCalibur flow cytometer (BD Biosciences).
Statistical Analysis
Pearson's correlation coefficient between parasite growth and binding affinity of the mutant P2 peptides was computed to quantify the strength of the relationship between two variables. A non-parametric Student's t test was applied to calculate the statistical significance of the difference between the two parameters.
Author Contributions
Y. D. S. and S. R. designed the study, analyzed the data, and wrote the paper. S. R. performed growth inhibition assay, docking, flow cytometry, and surface plasmon resonance. S. D. performed solid phase ELISA, Y2H, HEK cell expression, and immunofluorescence. D. K. performed pulldown assay and Label transfer assay. I. K. performed LC-MS/MS and analyzed the data. M. G. performed molecular stimulations and analyzed the data. All authors reviewed the results and approved the final version of the manuscript
Acknowledgments
Plasmid pRE4 was from Gary Cohn and Roselyn Eisenberg. We thank Pawan Malhotra for providing access to mass facility at ICGEB, Dr. R. M. Pandey for statistical analysis, and Shalini Narang for preparing the manuscript.
This work was supported in part by Indian Council of Medical Research Grant 61/4/2012-BMS and Department of Science and Technology-Indian National Science Academy INSPIRE Faculty Fellow Award IFA12-LSBM42 from the Department of Science and Technology, Government of India (to S. R.). The authors declare that they have no conflicts of interest with the contents of this article.
- MudPIT
- multidimensional protein identification technology
- BSG
- basigin
- EMP
- erythrocyte membrane protein
- ONPG
- ortho-nitrophenyl-glactopyranoside
- SPR
- surface plasmon resonance
- GPI
- glycosylphosphatidylinositol
- HSVgD
- herpes simplex virus glycoprotein D
- TEAB
- triethylammonium bicarbonate
- Y2H
- yeast two-hybrid
- AT
- 3-amino-1,2,4-triazole.
References
- 1. Kochar D. K., Das A., Kochar S. K., Saxena V., Sirohi P., Garg S., Kochar A., Khatri M. P., and Gupta V. (2009) Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am. J. Trop. Med. Hyg. 80, 194–198 [PubMed] [Google Scholar]
- 2. Mehndiratta S., Rajeshwari K., and Dubey A. P. (2013) Multiple-organ dysfunction in a case of Plasmodium vivax malaria. J. Vector Borne Dis. 50, 71–73 [PubMed] [Google Scholar]
- 3. Hostetler J. B., Sharma S., Bartholdson S. J., Wright G. J., Fairhurst R. M., and Rayner J. C. (2015) A library of Plasmodium vivax recombinant merozoite proteins reveals new vaccine candidates and protein-protein interactions. PLoS Negl. Trop. Dis. 9, e0004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goel V. K., Li X., Chen H., Liu S. C., Chishti A. H., and Oh S. S. (2003) band 3 is a host receptor binding merozoite surface protein 1 during the Plasmodium falciparum invasion of erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 100, 5164–5169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Crosnier C., Bustamante L. Y., Bartholdson S. J., Bei A. K., Theron M., Uchikawa M., Mboup S., Ndir O., Kwiatkowski D. P., Duraisingh M. T., Rayner J. C., and Wright G. J. (2011) basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sim B. K., Chitnis C. E., Wasniowska K., Hadley T. J., and Miller L. H. (1994) Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 264, 1941–1944 [DOI] [PubMed] [Google Scholar]
- 7. Mayer D. C., Cofie J., Jiang L., Hartl D. L., Tracy E., Kabat J., Mendoza L. H., and Miller L. H. (2009) Glycophorin B is the erythrocyte receptor of Plasmodium falciparum erythrocyte-binding ligand, EBL-1. Proc. Natl. Acad. Sci. U.S.A. 106, 5348–5352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lobo C. A., Rodriguez M., Reid M., and Lustigman S. (2003) Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl). Blood 101, 4628–4631 [DOI] [PubMed] [Google Scholar]
- 9. Bartholdson S. J., Bustamante L. Y., Crosnier C., Johnson S., Lea S., Rayner J. C., and Wright G. J. (2012) Semaphorin-7A is an erythrocyte receptor for P. falciparum merozoite-specific TRAP homolog, MTRAP. PLoS Pathog. 8, e1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miller L. H., Mason S. J., Clyde D. F., and McGinniss M. H. (1976) The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N. Engl. J. Med. 295, 302–304 [DOI] [PubMed] [Google Scholar]
- 11. Ménard D., Barnadas C., Bouchier C., Henry-Halldin C., Gray L. R., Ratsimbasoa A., Thonier V., Carod J. F., Domarle O., Colin Y., Bertrand O., Picot J., King C. L., Grimberg B. T., Mercereau-Puijalon O., and Zimmerman P. A. (2010) Plasmodium vivax clinical malaria is commonly observed in Duffy-negative Malagasy people. Proc. Natl. Acad. Sci. U.S.A. 107, 5967–5971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gunalan K., Lo E., Hostetler J. B., Yewhalaw D., Mu J., Neafsey D. E., Yan G., and Miller L. H. (2016) Role of Plasmodium vivax Duffy-binding protein 1 in invasion of Duffy-null Africans. Proc. Natl. Acad. Sci. U.S.A. 113, 6271–6276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan E. R., Barnwell J. W., Zimmerman P. A., and Serre D. (2015) Comparative analysis of field-isolate and monkey-adapted Plasmodium vivax genomes. PLoS Negl. Trop. Dis. 9, e0003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tyagi R. K., and Sharma Y. D. (2012) Erythrocyte binding activity displayed by a selective group of Plasmodium vivax tryptophan rich antigens is inhibited by patients' antibodies. PLoS ONE 7, e50754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeeshan M., Tyagi R. K., Tyagi K., Alam M. S., and Sharma Y. D. (2015) Host-parasite interaction: selective Pv-fam-a family proteins of Plasmodium vivax bind to a restricted number of human erythrocyte receptors. J. Infect. Dis. 211, 1111–1120 [DOI] [PubMed] [Google Scholar]
- 16. Zeeshan M., Tyagi K., and Sharma Y. D. (2015) CD4+ T cell response correlates with naturally acquired antibodies against Plasmodium vivax tryptophan-rich antigens. Infect. Immun. 83, 2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zeeshan M., Bora H., and Sharma Y. D. (2013) Presence of memory T cells and naturally acquired antibodies in Plasmodium vivax malaria-exposed individuals against a group of tryptophan-rich antigens with conserved sequences. J. Infect. Dis. 207, 175–185 [DOI] [PubMed] [Google Scholar]
- 18. Bozdech Z., Mok S., Hu G., Imwong M., Jaidee A., Russell B., Ginsburg H., Nosten F., Day N. P., White N. J., Carlton J. M., and Preiser P. R. (2008) The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 105, 16290–16295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alam M. S., Choudhary V., Zeeshan M., Tyagi R. K., Rathore S., and Sharma Y. D. (2015) Interaction of Plasmodium vivax tryptophan-rich antigen PvTRAg38 with band 3 on human erythrocyte surface facilitates parasite growth. J. Biol. Chem. 290, 20257–20272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alam M. S., Rathore S., Tyagi R. K., and Sharma Y. D. (2016) Host-parasite interaction: multiple sites in the Plasmodium vivax tryptophan-rich antigen PvTRAg38 interact with the erythrocyte receptor band 3. FEBS Lett. 590, 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Florens L., Washburn M. P., Raine J. D., Anthony R. M., Grainger M., Haynes J. D., Moch J. K., Muster N., Sacci J. B., Tabb D. L., Witney A. A., Wolters D., Wu Y., Gardner M. J., Holder A. A., et al. (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419, 520–526 [DOI] [PubMed] [Google Scholar]
- 22. Washburn M. P., Wolters D., and Yates J. R. 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 23. Liu B., Archer C. T., Burdine L., Gillette T. G., and Kodadek T. (2007) Label transfer chemistry for the characterization of protein-protein interactions. J. Am. Chem. Soc. 129, 12348–12349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu X. L., Hu T., Du J. M., Ding J. P., Yang X. M., Zhang J., Yang B., Shen X., Zhang Z., Zhong W. D., Wen N., Jiang H., Zhu P., and Chen Z. N. (2008) Crystal structure of HAb18G/CD147: implications for immunoglobulin superfamily homophilic adhesion. J. Biol. Chem. 283, 18056–18065 [DOI] [PubMed] [Google Scholar]
- 25. Wright K. E., Hjerrild K. A., Bartlett J., Douglas A. D., Jin J., Brown R. E., Illingworth J. J., Ashfield R., Clemmensen S. B., de Jongh W. A., Draper S. J., and Higgins M. K. (2014) Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature 515, 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zenonos Z. A., Dummler S. K., Müller-Sienerth N., Chen J., Preiser P. R., Rayner J. C., and Wright G. J. (2015) basigin is a druggable target for host-oriented antimalarial interventions. J. Exp. Med. 212, 1145–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baldwin M. R., Li X., Hanada T., Liu S. C., and Chishti A. H. (2015) Merozoite surface protein 1 recognition of host glycophorin A mediates malaria parasite invasion of red blood cells. Blood 125, 2704–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baldwin M., Yamodo I., Ranjan R., Li X., Mines G., Marinkovic M., Hanada T., Oh S. S., and Chishti A. H. (2014) Human erythrocyte band 3 functions as a receptor for the sialic acid-independent invasion of Plasmodium falciparum. Role of the RhopH3-MSP1 complex. Biochim. Biophys. Acta 1843, 2855–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Orlandi P. A., Klotz F. W., and Haynes J. D. (1992) A malaria invasion receptor, the 175-kilodalton erythrocyte binding antigen of Plasmodium falciparum recognizes the terminal Neu5Ac(α2–3)Gal-sequences of glycophorin A. J. Cell Biol. 116, 901–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bai Y., Huang W., Ma L. T., Jiang J. L., and Chen Z. N. (2014) Importance of N-glycosylation on CD147 for its biological functions. Int. J. Mol. Sci. 15, 6356–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen K., Liu J., Heck S., Chasis J. A., An X., and Mohandas N. (2009) Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. U.S.A. 106, 17413–17418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffiths R. E., Kupzig S., Cogan N., Mankelow T. J., Betin V. M., Trakarnsanga K., Massey E. J., Parsons S. F., Anstee D. J., and Lane J. D. (2012) The ins and outs of human reticulocyte maturation: autophagy and the endosome/exosome pathway. Autophagy 8, 1150–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams A. R., Douglas A. D., Miura K., Illingworth J. J., Choudhary P., Murungi L. M., Furze J. M., Diouf A., Miotto O., Crosnier C., Wright G. J., Kwiatkowski D. P., Fairhurst R. M., Long C. A., and Draper S. J. (2012) Enhancing blockade of Plasmodium falciparum erythrocyte invasion: assessing combinations of antibodies against PfRH5 and other merozoite antigens. PLoS Pathog. 8, e1002991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mullard A. (2012) Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 11, 173–175 [DOI] [PubMed] [Google Scholar]
- 35. Zinzalla G., and Thurston D. E. (2009) Targeting protein-protein interactions for therapeutic intervention: a challenge for the future. Future Med. Chem. 1, 65–93 [DOI] [PubMed] [Google Scholar]
- 36. Srinivasan P., Yasgar A., Luci D. K., Beatty W. L., Hu X., Andersen J., Narum D. L., Moch J. K., Sun H., Haynes J. D., Maloney D. J., Jadhav A., Simeonov A., and Miller L. H. (2013) Disrupting malaria parasite AMA1-RON2 interaction with a small molecule prevents erythrocyte invasion. Nat. Commun. 4, 2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sarin R., and Sharma Y. D. (2006) Thioredoxin system in obligate anaerobe Desulfovibrio desulfuricans: identification and characterization of a novel thioredoxin 2. Gene 376, 107–115 [DOI] [PubMed] [Google Scholar]
- 38. Dodge J. T., Mitchell C., and Hanahan D. J. (1963) The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch. Biochem. Biophys. 100, 119–130 [DOI] [PubMed] [Google Scholar]
- 39. Yajima A., Urano-Tashiro Y., Shimazu K., Takashima E., Takahashi Y., and Konishi K. (2008) Hsa, an adhesin of Streptococcus gordonii DL1, binds to α2–3-linked sialic acid on glycophorin A of the erythrocyte membrane. Microbiol. Immunol. 52, 69–77 [DOI] [PubMed] [Google Scholar]
- 40. Song F., Zhang X., Li Y., Ru Q., Ren X., Xia B., and Chen Z. N. (2011) An efficient method for refolding the extracellular portion of CD147 from the total bacterial lysate. Acta Biochim. Biophys. Sin. 43, 900–908 [DOI] [PubMed] [Google Scholar]
- 41. Tyagi K., Hossain M. E., Thakur V., Aggarwal P., Malhotra P., Mohmmed A., and Sharma Y. D. (2016) Plasmodium vivax tryptophan-rich antigen PvTRAg36.6 interacts with PvETRAMP and PvTRAg56.6 interacts with PvMSP7 during erythrocytic stages of the parasite. PLoS ONE 11, e0151065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sorette M. P., Galili U., and Clark M. R. (1991) Comparison of serum anti-band 3 and anti-Gal antibody binding to density-separated human red blood cells. Blood 77, 628–636 [PubMed] [Google Scholar]
- 43. Cohen G. H., Wilcox W. C., Sodora D. L., Long D., Levin J. Z., and Eisenberg R. J. (1988) Expression of herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J. Virol. 62, 1932–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chitnis C. E., and Miller L. H. (1994) Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh S., Singh H., Tuknait A., Chaudhary K., Singh B., Kumaran S., and Raghava G. P. (2015) PEPstrMOD: structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct 10, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 47. Abraham M. J. (2011) Performance enhancements for GROMACS nonbonded interactions on BlueGene. J. Comput. Chem. 32, 2041–2046 [DOI] [PubMed] [Google Scholar]
- 48. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 51. Persson K. E., Lee C. T., Marsh K., and Beeson J. G. (2006) Development and optimization of high-throughput methods to measure Plasmodium falciparum-specific growth inhibitory antibodies. J. Clin. Microbiol. 44, 1665–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]