

Abstract
Intrinsically disordered protein regions (IDRs) lack a well defined three-dimensional structure but often facilitate key protein functions. Some interactions between IDRs and folded protein domains rely on short linear motifs (SLiMs). These motifs are challenging to identify, but once found they can point to larger networks of interactions, such as with proteins that serve as hubs for essential cellular functions. The stress-associated plant protein radical-induced cell death1 (RCD1) is one such hub, interacting with many transcription factors via their flexible IDRs. To identify the SLiM bound by RCD1, we analyzed the IDRs in three protein partners, DREB2A (dehydration-responsive element-binding protein 2A), ANAC013, and ANAC046, considering parameters such as disorder, context, charges, and pI. Using a combined bioinformatics and experimental approach, we have identified the bipartite RCD1-binding SLiM as (DE)X(1,2)(YF)X(1,4)(DE)L, with essential contributions from conserved aromatic, acidic, and leucine residues. Detailed thermodynamic analysis revealed both favorable and unfavorable contributions from the IDRs surrounding the SLiM to the interactions with RCD1, and the SLiM affinities ranged from low nanomolar to 50 times higher Kd values. Specifically, although the SLiM was surrounded by IDRs, individual intrinsic α-helix propensities varied as shown by CD spectroscopy. NMR spectroscopy further demonstrated that DREB2A underwent coupled folding and binding with α-helix formation upon interaction with RCD1, whereas peptides from ANAC013 and ANAC046 formed different structures or were fuzzy in the complexes. These findings allow us to present a model of the stress-associated RCD1-transcription factor interactome and to contribute to the emerging understanding of the interactions between folded hubs and their intrinsically disordered partners.
Keywords: Arabidopsis thaliana, intrinsically disordered protein, nuclear magnetic resonance (NMR), protein-protein interaction, thermodynamics, SLiM, affinity, coupled folding and binding, interactome, plant stress
Introduction
Intrinsically disordered protein regions (IDRs),3 which lack a well defined three-dimensional structure, are being recognized as key facilitators of protein function (1–3). This means that the understanding of protein-protein interactions is changing. Short linear motifs (SLiMs) are central to IDRs. They are responsible for interactions with globular protein domains and mediate a wide range of important cellular tasks (4). They are short, typically between 3 and 11 residues (5, 6), and are most often found in IDRs or surface-accessible regions (7, 8). Their limited binding surface may result in low affinity, often in the low micromolar Kd range, with transient and promiscuous interactions (5, 9, 10), which are well suited for dynamic binding typical of intracellular signaling. Molecular recognition features (MoRFs) represent another type of ID-associated interaction element. They are also short, usually less than 20 residues, and are located within longer IDRs (11). Unlike SLiMs, MoRFs are not defined on the basis of sequence, but as interaction-prone disordered segments that may form secondary structures upon binding. MoRFs may themselves contain SLiMs. More than 2000 domain-interacting SLiMs have already been identified (12), but because the human proteome was estimated to encode more than 100,000 binding motifs (4), the identification of many more motifs may be expected. However, motif prediction is difficult mostly due to the challenge of obtaining robust statistical assessments (13), which means that experimental approaches are still needed for de novo SLiM identification.
SLiMs are especially relevant in signaling networks in which the proteins referred to as hubs have high connectivity. Hub proteins are essential for interaction network functionality, and their disruption is therefore frequently associated with diseases. Knock-out of the cellular plant hub protein radical induced cell death1 (RCD1) also resulted in severe phenotypic changes, explained by the pleiotropic roles of the RCD1 gene during both stress responses and plant development (14, 15). RCD1 interacts with at least 30 proteins, including 21 transcription factors representing several different transcription factor families (14), using its small C-terminal α-helical RCD1-SRO-TAF4 (RST) domain (14, 16). The rcd1 knock-out mutant showed altered expression of more than 500 genes, of which several are target genes of the RCD1-interaction partners (14). However, recent studies suggest that RCD1 does not transcriptionally regulate genes encoding its interaction partners (17). Instead, RCD1 was suggested to negatively regulate the stability of stress-associated dehydration-responsive element-binding protein 2A (DREB2A), belonging to the AP2/ERF plant-specific transcription factor family. Upon stress, RCD1 was rapidly degraded, promoting the proper DREB2A function (18).
RCD1 and its interaction partners represent an obvious model system for studies of interactions between folded hubs and intrinsically disordered proteins (IDPs). Several of the transcription factors interacting with RCD1 play significant roles in plant biology. For example, DREB2A modulates gene expression in response to various abiotic stress exposures (19), and the membrane-bound no apical meristem, ATAF, cup-shaped cotyledon (NAC) transcription factor Arabidopsis thaliana (A)NAC013 is involved in mitochondrial retrograde regulation in response to oxidative stress (20). Furthermore, the A. thaliana plant system allows transformation of in vitro results to the organismal level (21). The ability of RCD1 to interact with many proteins was explained by intrinsic disorder (ID)-associated flexibility (21). For ANAC046, the RCD1-interacting site coincides with the only MoRF within its large transcription regulatory domain (TRD) (16). However, most RCD1-interacting transcription factors, e.g. ANAC013 and DREB2A, have complex order-disorder profiles making prediction of protein interaction sites difficult (16). Using yeast two-hybrid analysis, the RCD1-interacting motif FDXXELLXXLN was identified for DREB2A. Although the RCD1-interacting regions of DREB2A and ANAC046 show some compositional similarities (16), the DREB2A RCD1-interacting motif could not be identified in ANAC046 or in the other RCD1 partners (18). In this study, we combined bioinformatics and experimental approaches to identify a loose SLiM present in several RCD1 interaction partners. Based on detailed thermodynamic and biophysical characterizations, a model for interactions between RCD1 and its target transcription factors is presented. This study also provides a novel framework for experimental determination of new SLiMs that, together with the rest of the work, will contribute to the emerging understanding of the complex interactions between folded hubs and their disordered partners.
Results
Identification of the RCD1-binding Region in ANAC013
Discovering new functional motifs using both computational and experimental techniques is of great interest. Because an RCD1-interacting motif has not been identified from previous studies, and because bioinformatics has come short in this endeavor, an experimental strategy is still needed for motif identification. In a previous study, the C-terminal ANAC013 TRD was shown to be responsible for the interaction with RCD1 (16). This 368-residue-long region is mostly disordered and contains nine short regions of predicted secondary structure, four MoRFs, and three MEME motifs (Fig. 1A). It was also shown that the region 161–299 of ANAC013 is sufficient for interactions with RCD1 (16). This region contains two predicted MoRFs and two predicted α-helical regions. Interestingly, the MEME motif E(KE)(ED)(DEM)(YF)(IL)E(MI)(ND)DL(LM), present in a small sub-group of NAC proteins (22), coincides with one of the predicted MoRFs and α-helices. In this study, the ANAC013 TRD was truncated further from both the N- and C-terminal ends and analyzed for its ability to interact with the RST domain of RCD1 in yeast (Fig. 1B). The N-terminal truncation to generate fragment 205–299 and to remove one of the predicted MoRFs did not affect the binding ability. Likewise, the C-terminal truncation of fragment 205–299 to generate a fragment ending with residue 266 and lacking an acidic region (Fig. 1A) did not destroy RCD1 binding. By contrast, further C-terminal truncation to residue 232, removing the MEME motif, abolished detectable binding. ANAC013(232–299), ANAC013(232–274), ANAC013(254-299), and ANAC013(254–274) all interacted with RCD1. However, ANAC013(266–299) had no binding activity. In conclusion, the experimental analysis showed that the RCD1-interacting region maps to one of two TRD regions predicted to contain a MoRF, a secondary structure, and a sequence motif. This demonstrates the strength of combining different prediction methods to identify interaction determinants in IDRs. Based on these results, region 254–274 of ANAC013 may contain the RCD1-interacting SLiM (Fig. 1C).
FIGURE 1.

ANAC013 domain structure, function, and key residues for interactions with RCD1. A, top, schematic structure of ANAC013 showing the N-terminal DNA-binding domain (indicated by NAC) and the C-terminal transcription regulatory domain (TRD). Middle, regions of the C-terminal transcription regulatory domain of ANAC013 predicted by PONDR-FIT to be disordered are shown by black boxes; positions of β-strands and α-helices predicted by PSIPRED are shown by β and α, respectively; MoRFs predicted by MoRFpred are shown by gray boxes, and positions of MEME motifs are shown by white boxes. Bottom, sequence of RCD1-interacting region with MEME motif in bold italics and acidic region underlined. B, directed yeast two-hybrid assays for analysis of ANAC013 and RCD1 interactions. Left, fusions of GAL4 DBD and the RST domain of RCD1 (residues 498–573; DBD-RST) and of GAL4 activation domain (AD) and the ANAC013 fragments shown were expressed in yeast and screened for interactions through the ability to activate the reporter genes HIS3 and ADE2. Empty pDEST32 expressing GAL4 DBD and pDEST22 expressing GAL4-AD served as negative controls. Right, schematic outline of the fragments analyzed with the approximate positions of predicted α-helix and MoRF regions. C, peptides shown were examined for RCD1 binding affinity using ITC in this study (Tables 1–3) or for the ANAC046 peptides in Ref. 16. Residues for which substitution into alanine resulted in more than 40-fold decrease in or no detectable affinity for RCD1, more than 5-fold decrease in affinity for RCD1, or less than 5-fold decrease in affinity for RCD1 are shown in bold italics, bold, and italics, respectively. Only residues marked this way were analyzed by substitution. Residues that, when substituted simultaneously, resulted in abolishment of RCD1 binding are marked by gray shading (18). The peptides were of comparable sizes, but sequence conservation in closely related proteins, secondary structure predictions, and disorder profiles were also considered when defining the peptides. ANAC046(319–338) is at the C terminus of ANAC046 and therefore is defined only based on size. Asterisks at the top of the alignment indicate the core of the RCD1-binding regions. α-Helix regions predicted by PSIPRED are underlined.
Thermodynamic Characterization of the RCD1·ANAC013 Interaction
The interaction between ANAC013 and RCD1 was also analyzed using isothermal titration calorimetry (ITC). ITC directly determines the Kd, the stoichiometry, and the change in enthalpy (ΔH) of binding, and the change in Gibbs free energy (ΔG) and in entropy (ΔS) is derived from these values (23). The affinity of the ANAC013 TRD, ANAC013(161–498), for the RST domain, RCD1-RST(499–572), was previously determined and showed a dissociation constant (Kd) of 537 nm (16). To analyze further the binding of ANAC013 to RCD1, truncated fragments of ANAC013(161–498) were made in this study. Several attempts to truncate ANAC013(161–498) from the C terminus were unsuccessful with no or only insoluble protein being produced. However, the recombinant fragment ANAC013(232–299), which showed binding to the RST domain in the yeast two-hybrid assays (Fig. 1), could be purified in amounts for analysis. This truncation resulted in a significant increase in affinity with the Kd decreasing from 537 to 93 nm (Table 1; Fig. 2). The affinity increased further by additional truncation resulting in a Kd of 32 nm for ANAC013(254–299), and removal of most of the negatively charged fragment (Fig. 1) to generate ANAC013(254–274) resulted in a further decrease of Kd to 9.0 nm (Fig. 2A). This is a relatively low Kd value for an interaction involving a putative SLiM and a globular domain (10), and the results indicate that the ANAC013 context of the SLiM has negative allosteric effects on binding. However, somewhat unexpected, the additional removal of the six C-terminal residues of ANAC013(254–274), including three proline residues, significantly decreased the affinity to a Kd of 595 nm. For the fragments tested, the relative contribution of enthalpy and entropy to binding varied, most markedly with a significant positive entropy contribution to binding for ANAC013(254–299). Furthermore, the decrease in enthalpy for binding of ANAC013(254–268) reflected in a less negative value of ΔH, and compared with binding of ANAC013(254–274), it is partly compensated for by a more negative value of −TΔS. In conclusion, the results suggested that ANAC013 contains a single binding site for RCD1 and that the binding stoichiometry for complex formation is 1:1, as revealed by an N value of ∼1. Region 254–274 of ANAC013 binds the RST domain with high affinity, again suggesting that this region contains a RCD1-interacting SLiM.
TABLE 1.
Thermodynamic analysis by ITC of interactions between ANAC013 wild-type and mutant fragments and RCD1-RST(499–572)
All experiments were performed as described under “Experimental Procedures.” RCD1 was in the syringe, and the ANAC013 peptides were in the cell. The standard errors for ΔH, Kd, and N were obtained from Origin when fitting the data to a one set of sites binding model.
| Protein | Kd | N | ΔH | −TΔS | ΔG |
|---|---|---|---|---|---|
| nm | kJ/mol | kJ/mol | kJ/mol | ||
| ANAC013(161–498)a | 537 ± 105 | 0.96 ± 0.03 | −41.0 ± 2.0 | 5.4 | −35.6 |
| ANAC013(232–299) | 92 ± 13 | 0.90 ± 0.02 | −51.9 ± 1.3 | 11.7 | −40.3 |
| ANAC013(254–299) | 32 ± 12 | 0.80 ± 0.01 | −27.2 ± 0.6 | −15.6 | −42.6 |
| ANAC013(254–274) | 9 ± 4 | 0.80 ± 0.01 | −45.0 ± 0.8 | −0.6 | −45.6 |
| ANAC013(254–268) | 595 ± 117 | 1.02 ± 0.04 | −27.6 ± 1.4 | −7.9 | −35.5 |
| ANAC013(254–274; E256A) | 43 ± 24 | 0.94 ± 0.02 | −25.5 ± 1.2 | −16.5 | −42.0 |
| ANAC013(254–274; D258A) | 61 ± 21 | 0.92 ± 0.02 | −32.8 ± 1.0 | −8.4 | −41.2 |
| ANAC013(254–274; D258P) | 110 ± 35 | 0.78 ± 0.02 | −33.4 ± 1.0 | −6.3 | −40.1 |
| ANAC013(254–274; M259A) | 98 ± 45 | 0.86 ± 0.04 | −26.8 ± 1.6 | −13.2 | −40.0 |
| ANAC013(254–274; Y260A) | 1114 ± 346 | 1.05 ± 0.07 | −16.5 ± 1.6 | −17.5 | −34.0 |
| ANAC013(254–274; L261A) | 885 ± 272 | 1.04 ± 0.07 | −13.6 ± 1.2 | −20.9 | −34.5 |
| ANAC013(254–274; E262A) | 436 ± 100 | 0.94 ± 0.03 | −30.7 ± 1.5 | −5.5 | −36.2 |
| ANAC013(254–274; I263A) | 43 ± 22 | 0.83 ± 0.02 | −35.2 ± 1.3 | −6.8 | −42.0 |
| ANAC013(254–274; N264A) | 64 ± 10 | 0.74 ± 0.01 | −34.5 ± 0.5 | −6.6 | −41.1 |
| ANAC013(254–274; N264K) | NBb | ||||
| ANAC013(254–274; D265A) | NB | ||||
| ANAC013(254–274; D265N) | NB | ||||
| ANAC013(254–274; L266A) | NB | ||||
| ANAC013(254–274; M267A) | 97 ± 26 | 0.87 ± 0.02 | −35.4 ± 1.5 | −4.6 | −40.0 |
a Data were determined as described by O'Shea et al. (16).
b NB means no detectable binding.
FIGURE 2.

ITC measurements of the RCD1 interactions with ANAC013 and DREB2A. ITC data show titrations that are representative for the ITC experiments in this study. A, titration of RCD1-RST(499–572) into ANAC013(254–274). B, RCD1-RST(499–572) into ANAC013(254–274; Y260A). C, RCD1-RST(499–572) into DREB2A(255–272). D, RCD1-RST(499–572) into DREB2A(255–272; F259A). In each panel, the upper portion shows baseline-corrected raw data from the titration, and the lower portion shows the normalized integrated binding isotherms together with the fitted binding curves. The data were fitted to a “one set of sites” binding model. Parameters obtained from the non-linear fits are presented in Tables 1 and 3.
Contribution of Specific ANAC013 Residues to RCD Binding
The interaction between ANAC013(254–274) and the RST domain of RCD1 was also analyzed at the residue level (Fig. 2B; Table 1). Of the residues that constitute a SLiM, only a fraction is fully conserved, whereas other positions are under less or no selective constraints (24). The importance of specific positions of the RCD1-interacting region of ANAC013 was analyzed by substituting all residues with alanine (Fig. 1C; Table 1). The ITC data showed that some positions of the motif were essential for binding. When Asp-265 and Leu-266 were changed into alanine, no detectable binding was observed, and when Tyr-260 was changed into alanine, the Kd value increased 124-fold mainly due to a decreased binding enthalpy (Fig. 2B). Changing Leu-261 and Glu-262 into alanine also resulted in drastic changes in affinity, corresponding to 98- and 48-fold decreases in affinity, respectively. As for the Tyr-260 substitution, the substitution of Leu-261 resulted in a dramatic decrease in ΔH. By contrast, the effects of changing Asp-258, Met-259, Ile-263, Asn-264, and Met-267 were more modest. The importance of a negative charge to binding was analyzed by changing Asp-265 to the corresponding amide asparagine, which also abolished binding. In conclusion, two hydrophobic leucine residues, an aromatic tyrosine residue, and two acidic residues played essential roles in ANAC013 binding to RCD1. The emerging RCD1-binding motif of ANAC013 is bipartite with the essential Tyr-Leu-Glu tripeptide separated from the essential Asp-Leu dipeptide by two residues of less importance to binding.
Comparison of the minimal RCD1-binding regions of ANAC013 and ANAC046 revealed similarities (Fig. 1C). Thus, both regions contain an essential aromatic residue and an aspartic acid residue, of which the latter is followed by a leucine, which was also shown to be essential for binding of ANAC013. Furthermore, an acidic residue is present at one or two positions N-terminal of the aromatic residue and a hydrophobic residue is found at two positions N-terminal of the Asp-Leu motif.
NAC Sub-family Conservation of the RCD1-interacting Sequence Motif
Motif conservation in IDRs may indicate functional importance. This aspect was analyzed for ANAC013 and its close relatives. The disorder pattern of ANAC013 shows similarities with the disorder profiles of ANAC016 and ANAC017 (22). These three membrane-bound NAC transcription factors, which are functionally related by mediating oxidative stress signaling (20, 25, 26), belong to the same sub-group of the NAC family (27). They share the MEME sequence motif E(KE)(ED)(DEM)(YF)(IL)E(MI)(ND)DL, encompassing the RCD1-interacting core of ANAC013 and coinciding with a MoRF in all three proteins. All three proteins also have a dip in the disorder profile in the same region (22). Based on these characteristics, this region of ANAC016 and ANAC017 is also likely to mediate biochemical interactions with RCD1. However, so far ANAC016 and ANAC017 have not been identified as RCD1 interaction partners in screening experiments (14). Therefore, we examined whether ANAC016 and ANAC017 would be able to interact with RCD1. In fact, ANAC016(325–367) and ANAC017(296–339), corresponding to ANAC013(254–299), both interacted with RCD1-RST(499–572) with Kd values of 200 and 37 nm, respectively (Table 2). This identified ANAC016 and ANAC017 as biochemical interaction partners of RCD1. It also showed how ID profiling together with motif and other sequence analyses can be used to identify novel interaction partners of hub proteins without an identified SLiM.
TABLE 2.
Thermodynamic analysis by ITC of interactions between RCD1-RST(499–572) and different transcription factors
All experiments were performed as described under “Experimental Procedures.” Syringe/cell indicates whether RCD1-RST or the transcription factor is the titrant in syringe or the titrant in cell. The standard errors for ΔH, Kd, and N were obtained from Origin when fitting the data to a one set of sites binding model.
| Protein | Kd | N | ΔH | −TΔS | ΔG | Syringe/cell |
|---|---|---|---|---|---|---|
| nm | kJ/mol | kJ/mol | kJ/mol | |||
| ANAC016(325–367) | 200 ± 76 | 1.02 ± 0.04 | −42.5 ± 2.3 | 4.2 | −38.3 | ANAC016/RCD1 |
| ANAC017(296–339) | 37 ± 9 | 0.89 ± 0.02 | −54.2 ± 1.0 | 11.7 | −42.5 | ANAC017/RCD1 |
| bZIP23(15–36) | 128 ± 40 | 0.94 ± 0.05 | −30.6 ± 2.1 | −8.7 | −39.3 | RCD1/bZIP23 |
| STO(229–248) | 90 ± 90 | 1.16 ± 0.12 | −3.8 ± 0.5 | −36.3 | −40.1 | RCD1/STO |
| COL10(175–208) | 418 ± 201 | 1.04 ± 0.08 | −9.2 ± 1.0 | −27.2 | −36.4 | RCD1/COL |
| bHLH19(271–295) | NBa | RCD1/bHLH19 | ||||
| IRL3(69–100) | NB | RCD1/IRL3 | ||||
| MYB91(100–291) | NB | RCD1/MYB91 | ||||
| MYB(100–230) | NB | RCD1/MYB91 | ||||
| MYB92(239–267) | NB | RCD1/MYB91 | ||||
| MYB91(239–291) | NB | RCD1/MYB91 |
a NB means no detectable binding.
Thermodynamic Characterization of the RCD1-DREB2A Interaction
The sequence features of relevance for RCD1 binding were analyzed for additional members of the RCD1 interactome. DREB2A is a significant RCD1 interaction partner (14), and studies in yeast have shown that simultaneous substitution of Phe-259 and Asp-260 and of Glu-263, Leu-264, and Leu-265 of the DREB2A RCD1-interacting motif abolished its ability to interact with RCD1 (18). The motif shows compositional similarity to the RCD1-interacting regions of ANAC013 and ANAC046 (Figs. 1C and 3A). To determine the binding parameters of the RCD1·DREB2A interactions, the DREB2A fragments, DREB2A(150–335) and DREB2A(250–287), spanning regions with alternating structure and disorder (Fig. 4), were produced and purified. The interaction between DREB2A(150–335) or DREB2A(250–287) and RCD1-RST(499–572) were of high affinity with Kd values of 27 and 51 nm, respectively (Table 3). The affinity of the interaction between RCD1-RST(499–572) and a short peptide of DREB2A, DREB2A(255–272), containing a predicted α-helix, was significantly lower, corresponding to a Kd of 117 nm, but with a large enthalpic contribution to binding of −51 kJ/mol (Fig. 2C). The shorter peptide, DREB2A(260–269), containing a predicted α-helical region (Figs. 1C and 3A), but lacking the phenylalanine, was without detectable binding affinity. Thus, in contrast to ANAC013, the DREB2A context conferred positive allosteric effects on binding.
FIGURE 3.

RCD1-binding SLiM and RCD1 interactome. A–C, sequences of transcription factors from the RCD1 interactome that contain the loose RCD1-binding consensus sequence (DE)X(0,2)(YF)X(1,6)(DE)L. A, verified RCD1-binding regions (Tables 1–3) with the consensus sequence (DE)X(1,2)(YF)X(1,4)(DE)L. B, predicted RCD1-binding regions not experimentally analyzed, and C, predicted RCD1-binding regions experimentally shown to be non-binders. i- indicates a gap introduced in the sequences to fit the consensus sequence. α-Helix and β-strands predicted by PSIPRED are underlined by lines and broken lines, respectively. D, RCD1 interactome obtained from the BAR Arabidopsis Interactions Viewer using RCD1 as query and information from the IntAct Molecular Interaction Database. Transcription factors are shown as black circles, and RCD1 as a gray circle. Common names are shown in addition to the gene codes, when known. E, calculated pI values of the consensus sequence region of binding, putatively binding, and non-binding peptides.
FIGURE 4.

Disorder predictions for transcription factors analyzed for interactions with RCD1. Disorder was predicted using PONDR-FIT. The positions of the family-designating DNA-binding domain and the RCD1-binding SLiM are shown as light and dark gray bars, respectively. A threshold was applied with disorder assigned to values greater than or equal to 0.5 (black bar). Profiles above and below the black line are for transcription factors with a functional and non-functional (DE)X(0,2)(YF)X(1,6)(DE)L consensus sequence. Res, residue.
TABLE 3.
Thermodynamic analysis by ITC of interactions between DREB2A wild-type and mutant-substituted fragments and RCD1-RST(499–572)
All experiments were performed as described under “Experimental Procedures.” Syringe/cell indicates whether RCD1-RST(499–572) or DREB2A is the titrant in syringe or the titrant in cell. The standard errors for ΔH, Kd, and N were obtained from Origin when fitting the data to a one set of sites binding model.
| Protein | Kd | N | ΔH | −TΔS | ΔG | Syringe/cell |
|---|---|---|---|---|---|---|
| nm | kJ/mol | kJ/mol | kJ/mol | |||
| DREB2A(150–335) | 27 ± 11 | 0.72 ± 0.02 | −42.3 ± 1.4 | −0.9 | −43.2 | DREB2A/RCD1 |
| DREB2A(250–287) | 51 ± 16 | 1.21 ± 0.01 | −48.7 ± 1.0 | 7.1 | −41.6 | DREB2A/RCD1 |
| DREB2A(255–272) | 117 ± 26 | 1.05 ± 0.03 | −51.5 ± 2.3 | 12.0 | −39.0 | DREB2A/RCD1 |
| DREB2A(260–269) | NBa | DREB2A/RCD1 | ||||
| DREB2A(255–272; F259A) | NB | DREB2A/RCD1 | ||||
| DREB2A(255–272; E263A) | 176 ± 49 | 0.85 ± 0.02 | −41.6 ± 1.3 | 3.1 | −38.5 | RCD1/DREB2A |
| DREB2A(255–272; L264A) | 917 ± 278 | 1.07 ± 0.05 | −16.4 ± 1.1 | −18.0 | −34.5 | RCD1/DREB2A |
| DREB2A(255–272; E263P) | NB | DREB2A/RCD1 | ||||
| DREB2A(255–272; V261A/L265A) | 260 ± 47 | 0.95 ± 0.02 | −41.6 ± 1.3 | 4.0 | −37.6 | DREB2A/RCD1 |
| DREB2A(316–335) | NB |
a NB means no detectable binding.
For comparison, the DREB2A·RCD1 interaction was also analyzed at the residue level. Substitution of the central phenylalanine, Phe-259, with alanine in DREB2A(255–272) was sufficient to abolish detectable binding (Fig. 2D; Table 3). Curiously, alanine substitution of Glu-263 or Leu-264, putatively analogous to the essential Asp-Leu dipeptide in ANAC013 (Fig. 1C), resulted in only 1.5- and 7.8-fold increase in Kd values, respectively. However, introduction of an α-helix breaking proline in the middle of the predicted α-helix of DREB2A (Fig. 1C) to generate DREB2A(255–272; E263P) completely abolished binding. This is in contrast to the effects of introducing a proline in the RCD1-binding ANAC046 peptide (16) and in the predicted α-helix of the RCD1-binding ANAC013(254–274) peptide (Fig. 1C; Table 1), suggesting that the RCD1-binding regions depend on α-helix structure to different degrees. Assuming α-helix formation in the DREB2A·RCD1 complex, the effect of changing two residues on the same side of a putative α-helix, Val-261 and Leu-265, into alanine was analyzed. However, the double substitutions in DREB2A(255–272; V261A/V265A) only affected binding affinity slightly (Table 3). Based on the results presented here and in a previous study (18), DREB2A also has a bipartite RCD1-binding site with Phe-259 as a central residue of one of the sites and Leu-264 as a central residue of the other site (Fig. 1C). For DREB2A, binding is also likely to depend on α-helix formation.
DREB2B and DREB2C also belong to the DREB2 subfamily of the AP2/ERF transcription factor family and interact with RCD1 (14). Motif residues can be distinguished from their sequence neighborhood on the basis of higher evolutionary conservation and from their propensity to form ordered secondary structures upon partner binding (7). Confirmatory sequence alignments of the three DREB2 proteins showed how the large C-terminal IDRs have a low degree of sequence similarity with only a few short conserved sequence regions, one of which corresponds to the RCD1-interacting regions of DREB2A (Fig. 3A). Conclusively, these data so far suggest that the RCD1-interacting motif is bipartite and most likely not limited to a certain structural context conserved in NAC sub-groups.
RCD1-interacting SLiM
Based on the above results, the additional members of the RCD1 interactome were analyzed for the presence of a putative RCD1-binding motif. Transcription factors are over-represented among the RCD1 interaction partners, and 22 of the 30 members of the known RCD1 interaction network are transcription factors (Fig. 3D). Sequence analysis of these suggested that 19 of the RCD1-interacting transcription factors, including ANAC016 and ANAC017 identified as RCD1 interaction partners in this study, contain a sequence region fitting the loose consensus sequence (DE)X(0,2)(YF)X(1,6)(DE)L that was derived from the analyses described above (Figs. 1C and 3A). Our results showed that four NAC transcription factors and one AP2/ERF transcription factor, representing two different transcription factor families, use this SLiM for interactions with RCD1. The RCD1 interactome was experimentally exploited for further validation of the appearing RCD1-binding SLiM. Peptide bZIP23(15–36), derived from bZIP23, a member of the basic ZIP transcription factor family, interacted with RCD1 with an affinity corresponding to a Kd of 128 nm, comparable with that of the DREB2A·RCD1 interaction (Table 2). This demonstrates that it is possible to predict the RCD1-interacting region of a protein from the RCD1 interactome on the basis of sequence analysis. Putative RCD1-binding motifs were also identified for the B-box transcription factors STO and COL10 (Fig. 3A), which bound RCD1 both in vivo and in vitro (14). Here, the Kd values for the interactions of RCD1 with the STO and COL10 peptides were determined to 90 and 418 nm, respectively. Interestingly, in both cases the change in binding enthalpy was low, −3.8 kJ/mol and −9.2 kJ/mol, respectively, which could be explained by entropy-driven interactions (Table 2). Recently, another AP2/ERF-type transcription factor, Rap2.4a, was shown to interact with RCD1 (28). Rap2.4a also contains a region with similarity to the RCD1 SLiM (Fig. 3B).
Non-binding sequences are also of interest in attempting to understand SLiM-binding determinants. DREB2A has an additional putative SLiM at the very C terminus. However, no binding between this region, contained in DREB2A(316–335), and RCD1 was detected (Table 3; Fig. 3C). Likewise, the peptides bHLH019(271–295) and IRL3(69–100), from the basic helix-loop-helix transcription factors bHLH019 and IRL3, which also encompass the loose RCD1 SLiM, were also non-binders. This was also the case for MYB91(239–267). Likewise, the longer peptides, MYB91(239–291) and MYB91(100–291), responsible for interactions with RCD1 in yeast (14, 29), did not bind in vitro (Table 2). This discrepancy may be explained either by MYB91 being a false-positive identified in the yeast two-hybrid screening or by the need for a post-translational modification in the interaction surface. These transcription factors do not contain any other regions with a SLiM, neither in the parallel nor in the antiparallel orientation (Fig. 3C). In fact, analysis for putative SLiMs in the antiparallel orientation revealed that of the putative binders only WRKY47 contains a reverse SLiM. ANAC013 as a binder also contains a reverse SLiM (Fig. 3B). To analyze whether the antiparallel motif in ANAC013 is functional, Glu-256 of the Leu-Glu (Glu-Leu in the antiparallel orientation) dipeptide was changed to alanine (Table 1). This did not significantly affect binding affinity, suggesting that the region is only functional when bound in a parallel orientation.
The transcription factor regions with the loose (DE)X(0,2)(YF)X(1,6)(DE)L SLiM were divided into RCD1 binders, putative binders, and non-binders (Fig. 3, A–C). The flexibility associated with ID was previously proposed to be important for the interaction between RCD1 and the partner proteins (21). For the binders, the SLiM region is either predicted to be disordered or to map to a larger IDR (Fig. 4). This is also the case for the non-binding C-terminal DREB2A peptide, whereas most of the bHLH019(271–295) and MYB91(243–262) peptides were predicted to be structured (Fig. 3C), and the ILR3(69–100) peptide maps to the folded DNA-binding domain (Fig. 4). The RST domain of RCD1 is dominated by positive charges, and the apparent dominance of negative charges in the peptides is therefore not surprising. In fact, none of the verified core binding regions contains basic residues (Fig. 3A). The calculated pI values of the core consensus sequence and two additional residues on each end range from 3.3 to 3.9 for the binders, whereas calculations suggested that two of the non-binding peptides have a significantly higher pI value. To analyze the effect of a basic residue, ANAC013(254–274; N264K) was generated. This peptide has an increased pI (from 3.3 to 3.8) and a positive residue in a position with limited sensitivity to alanine substitution (Table 1). No detectable binding was observed for the substituted peptide (Table 1). In conclusion, on the basis of current experimental data, the consensus sequence (DE)X(1,2)(YF)X(1,4)(DE)L describes the loose RCD1-interacting SLiM. However, additional features such as low pI, lack of basic residues, and ID in the SLiM context should be considered when identifying putative new RCD1 interaction partners. Based on these criteria, the inability of DREB2A(323–335) to bind RCD1 cannot be explained, reflecting the complexity of the interactions.
Identification of Putative RCD1-interacting Transcription Factors
Because Fig. 3D may not represent an exhaustive picture of the RCD1-transcription factor interactome, the non-redundant protein database was searched for additional transcription factors putatively binding to RCD1 using two different BLAST programs (30), pattern hit Initiated (PHI)-BLAST, which combines matching of regular expressions with local alignments surrounding the match, and position-specific iterated (PSI)-BLAST, in which a general profile generated from closely related sequences is used as query. For the PHI-BLAST searches, (DE)X(0,2)(YF)(1,6)(DE)L was used as the regular expression, and the transcription factor regions shown in Fig. 3A were used for the local alignments. This resulted in the identification of 44 new transcription factors putatively interacting with RCD1 (Table 4), when hits with core regions containing the basic residues arginine or lysine and having pI values above 4.5 were excluded from the list. NAC, ERF/AP2, and ZnF transcription factors were over-represented, probably reflecting the transcription factors used for the alignments. PSI-BLAST searches did not result in any additional hits. To analyze for unifying functional features of the RCD1-interacting transcription factors, the Gene Ontology database was searched using AmiGO2 (31) and all the transcription factors of the RCD1 interactome (Fig. 3D) and the predicted RCD1-interacting transcription factors (Table 4) as queries. The Gene Ontology terms for biological function were broad, including heat acclimation, response to abiotic stimulus, ethylene-activated signaling pathway, hormone-mediated signaling pathway, positive regulation of transcription, response to water deprivation, leaf development, and gene expression. As expected, no single biological function dominates among the verified and predicted RCD1-interacting transcription factors, but several of these transcription factors were involved in abiotic stress responses.
TABLE 4.
PHI-BLAST searches for putative RCD1-interacting transcription factors using (DE)X(0,2)(YF)X(1,6)(DE)L as regular expression
The biological function of GO terms for known and predicted RCD1-interacting transcription factors are as follows: heat acclimation (GO:0010286); response to abiotic stimulus (GO:0009628); ethylene-activated signaling pathway (GO:0009873); hormone-mediated signaling pathway (GO:0009755); positive regulation of transcription (GO:0045893); response to water deprivation (GO:0009414); leaf development (GO:0048366); and gene expression (GO:0010467). The putative binders are listed according to TF families.

Structure of RCD1-binding Peptides
Based on predictions, some of the RCD1-interacting peptides form α-helix structures (Fig. 3A). However, the helicity per residue predicted using Agadir (32) was generally low, except in the case of DREB2A(255–272) (Fig. 5A). Experimental analysis for secondary structure content by far-UV CD indicated low α-helical contents of 12, 18, 10, and 13% in the ANAC013(254–274), DREB2A(255–272), COL10(175–208), and bZIP23(15–36) peptides, respectively (Fig. 5, B–E). Structure formation in IDPs is often studied as a function of solvent conditions, and trifluoroethanol (TFE) was thus used to investigate whether the RCD1 SLiM peptides were prone to structure induction (33). Addition of TFE to 10% (v/v) resulted in limited increases in α-helical content to 13, 24, 11, and 16%, respectively, whereas addition of 40% TFE markedly changed the CD spectra, especially of DREB2A(255–274), to spectra characteristic of an α-helix with minima at 222 and 208 nm and estimated α-helical contents of 20, 52, 17, and 31% for ANAC013(254–274), DREB2A(255–272), COL10(175–208), and bZIP23(15–36), respectively (Fig. 5, B–E). A similar pattern was previously observed for ANAC046(319–338) (16). DREB2A(255–272), ANAC046(319-338), and bZIP23(15–36) showed an increase in α-helix content of 189, 150, and 138%, respectively, when comparing samples with 40% TFE and without TFE. By contrast, ANAC013(254–274) and ANAC013(254–274;D258P), with a very low α-helix content (Fig. 5, A and F), and COL10(175–208) only increased their α-helix content with 67, 20, 70%, respectively. This indicates that TFE does not induce α-helical structure equally well in all peptides and that ANAC013(254–274) and COL10(175–274) are less likely to undergo coupled folding and binding involving α-helix formation than DREB2A(255–272).
FIGURE 5.
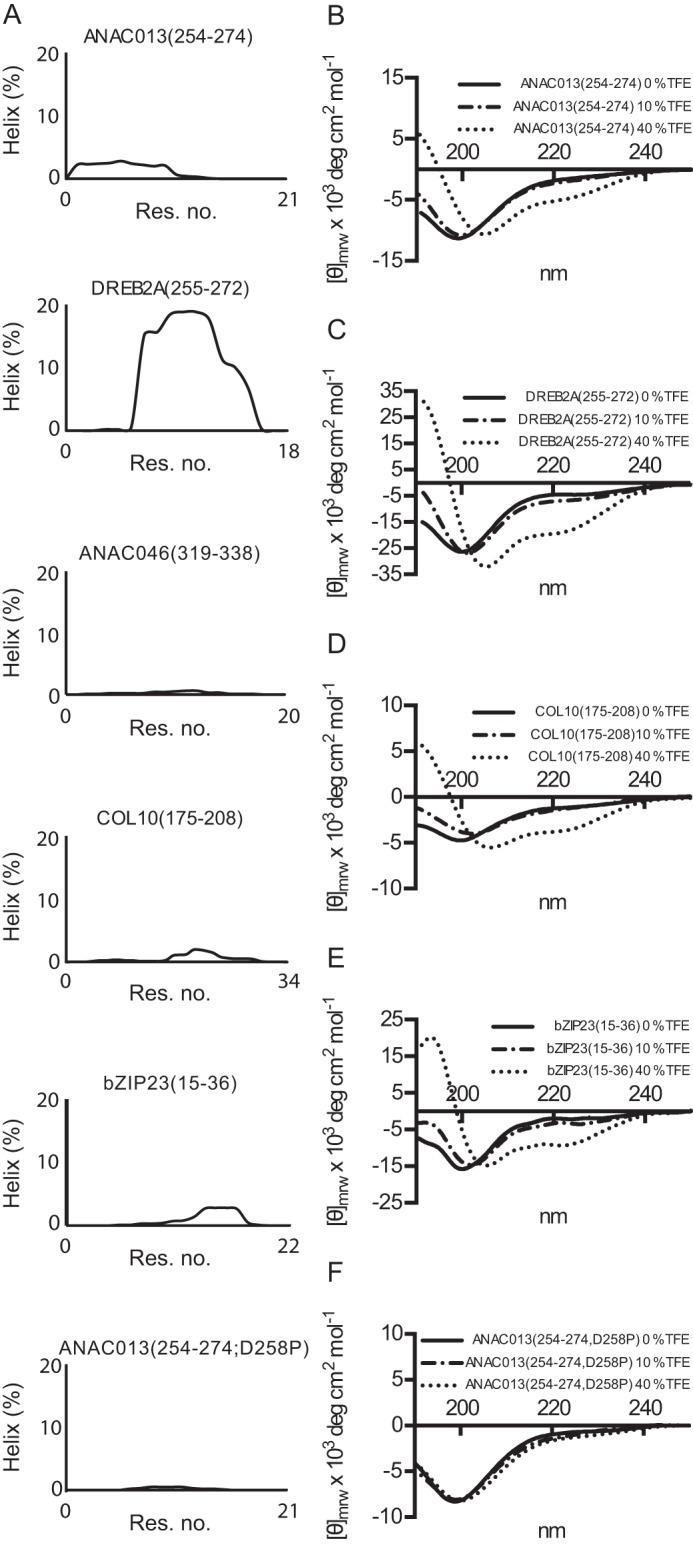
α-Helix propensity of RCD1-interacting peptides. A, helicity per residue of the RCD1-binding peptides predicted using Agadir. B–F, far-UV CD spectra of 15–20 μm ANAC013(254–274), DREB2A(255–272), bZIP23(15–36), and ANAC013(254–274; D258P) in 10 mm Na2HPO4/NaH2PO4, pH 7.0, and 0–40% (v/v) TFE, as indicated, were recorded. Molar ellipticity is shown on the y axis. Res, residue.
CD spectroscopy was also used to analyze for structural changes occurring upon interactions between ANAC013(254–274), DREB2A(255–272), or ANAC046(319–338) and RCD1-RST(499–572) (Fig. 6, A–D). The far-UV CD spectrum of RCD1-RST(499–572) had pronounced minima at 222 and 208 nm indicating that the domain is dominated by α-helical structure (Fig. 6A). A far-UV CD spectrum of each of the peptides mixed in a 1:1 molar ratio with RCD1-RST(499–572) was compared with a theoretical spectrum derived from adding the individual spectra. The spectrum of RCD1-RST(499–572) in complex with DREB2A(255–272) showed an absolute minimum shifted toward a higher wavelength compared with the theoretical spectrum. Moreover, the overall α-helical content in the complex was calculated to 39% compared with 31% in the theoretical complex. This indicated induction of structure upon complex formation (Fig. 6B). For ANAC013(254–274) in complex with RCD1-RST, the wavelength of the absolute minimum changed slightly compared with the theoretical spectra, and the α-helical content of 32% in the complex was minimally higher than in the theoretical complex (29%) (Fig. 6C). The ANAC046(319–338)·RCD1-RST(499–572) complex had an overall α-helical content of 38%, compared with 34% in the theoretical complex. However, in this case a shift toward a lower wavelength in the complex was observed (Fig. 6D). Together, the results suggested that complex formation involving ANAC046(319–338) and ANAC013(254–274) resulted in structure induction, although to a lower degree than that of the DREB2A(255–272)·RCD1-RST(499–572) complex. From the CD data, it was not possible to decipher whether the changes were in both of the binding partners.
FIGURE 6.

Structural analysis of transcription factor SLiM peptide·RCD1 complexes by CD spectroscopy. A, far-UV CD spectra of ANAC013(254–274), ANAC046(319–338), DREB2A(255–272), and RCD1-RST(499–572). Each spectrum was recorded on 6–8 μm protein in 10 mm Na2HPO4/NaH2PO4, pH 7.0, and a total of 10 scans were averaged between 250 and 190 nm. Mean residue molar ellipticity is shown on the y axis. B–D, far-UV CD spectra of the interaction complex of the indicated transcription factor peptides and RCD1-RST(499–572) mixed 1:1 (black lines). For comparison, the predicted additive CD spectrum based on spectra of the individual proteins is included (broken lines). Conditions as in A.
To further assess the structural aspects for the transcription factor peptides and to explore, if possible, what kind of structure they adopt in complex with RCD1-RST(499–572), they were investigated by NMR spectroscopy. Triple resonance NMR spectra of double-isotope 13C,15N-labeled peptides in the absence and presence of stoichiometric amounts of unlabeled RCD1-RST(499–572) were assigned, and secondary Cα chemical shifts for DREB2A(255–272), ANAC013(254–274), and ANAC046(319–338), which reports on the secondary structure (34), were calculated.
The 1H,15N HSQC spectrum of free DREB2A(255–272) showed limited dispersion in the proton dimension, indicating a lack of structure (Fig. 7A, black spectrum) (35). Consistent with this and with its far-UV CD spectrum (Fig. 6A), most residues have near-random coil secondary Cα chemical shifts (ΔδCα ≈0, Fig. 7D, top figure). Positive ΔδCα values (≈0.8) were seen in the region Asp-262–Arg-266, indicating the presence of a transient helical turn structure in free DREB2A(255–272), populated to around 25%. In support of this AGADIR predicts an average helical content of ∼18% for these residues (Fig. 5A). The 1H,15N HSQC was dramatically changed upon addition of RCD1-RST(499–572) with a substantial increase in the 1H resonance dispersion (Fig. 7A, red spectrum). Furthermore, the Cα resonances for residues Val-261–Asp-267 experienced a significant downfield shift (ΔδCα >0), thus confirming the formation of an almost fully formed helical structure in this region (Fig. 7D, bottom figure) (34). Notably, the HSQC as well as the different triple resonance spectra lacked peaks for Phe-259 and Asp-260. This is consistent with the abolished binding of the F259A mutant (Fig. 2D; Table 3) and suggests that these residues are involved in the interaction.
FIGURE 7.

Secondary structure analyzed by NMR spectroscopy. 1H,15N HSQC spectra of 13C,15N-labeled DREB2A(255–272) (A), ANAC013(254–274) (B), and ANAC046(319–338) (C) in the free state (black spectra) and in complex with unlabeled RCD1-RST(499–572) (red spectra). Weak peaks with large changes in chemical shifts in the ANAC013(254–274) spectra are indicated with arrows. Secondary Cα chemical shifts for DREB2A(255–272) (D), ANAC013(254–274) (E), and ANAC046(319–338) (F) in the free state (top figures) and in complex with unlabeled RCD1-RST(499–572) (bottom figures). Consecutive positive and negative values indicate α-helical and β-sheet/extended structures, respectively, whereas values close to zero indicate coil-like structures. SLiM residues are indicated with red letters. Because of the degeneracy of the sequence resulting in almost identical chemical shifts for residues Glu-267–Glu-270 in ANAC013, the glutamate and proline residues were each assigned an average value. Error bars indicate the largest and smallest possible values for each residue.
Similar to DREB2A(255–272), free ANAC013(254–274) and ANAC046(319–338) had poorly dispersed HSQC spectra (Fig. 7, B and C, black spectra), and ΔδCα values close to zero (Fig. 7, E and F, top figures) again confirming a lack of preformed structure. A region with slightly positive ΔδCα values was seen in ANAC013(254–274) for residues Leu-255–Tyr-260 (Fig. 7E, top figure), which, together with low average helical content of <3% predicted by AGADIR (Fig. 5A), did not support the presence of structure. In relation to where the motif is centered (Fig. 3A), it is clear that the transient helix turn structure, albeit lowly populated in ANAC013(254–274), is positioned differently from that of DREB2A, suggesting that formation of helical structure in itself might not be important for the interaction, but instead it reflects the physiochemical nature of their sequences. In support of this, no indications of transiently formed structure were apparent from the secondary Cα chemical shifts (Fig. 7F, top figure) or AGADIR predictions (Fig. 5A) for ANAC046. Comparison of the HSQC spectra of free and bound ANAC013(254–274) and ANAC046(319–338) confirmed their interaction with RCD1-RST(499–572) (Fig. 7, B and C), but in contrast to those of DREB2A(255–272), the dispersion in the proton dimension appeared immediately unaffected. For ANAC013(254–274), we instead observed a set of weak signals with a much larger dispersion (Fig. 7B, indicated by arrows) which, unfortunately, could not be detected in the triple-resonance spectra and hence could not be assigned. The ANAC046(319–338) spectrum showed substantial peak broadening, and peak intensities were generally significantly reduced in the bound state of both transcription factors (Fig. 7, B and C, red spectra). Consequently, only residues outside the motif could be assigned. However, the observation that the ΔδCα values of residues Met-267 and Glu-268 of ANAC013(254–274) and Tyr-336 and Thr-337 of ANAC046(319–338) are clearly not helical is in contrast to residues at similar SLiM positions in DREB2A(255–272) and fully in line with the proline substitutions studies done by ITC. Although we cannot establish the exact structural nature of the residues of the motif in the NAC peptides, these data, together with the different dispersion in the proton dimension, suggest that ANAC013(254–274) and ANAC046(319–338) may form structures that are less helical in their bound state. This dramatic change in the 15N-dimension was not observed for any residues of the DREB2A(255–272) peptide.
The reduced peak intensities observed for these transcription factors most likely occur due to exchange between bound and free states and/or between multiple conformations at a rate comparable with the difference in resonance frequency between the different states (36). Accordingly, regions that experience peak broadening in ANAC013(254–274) and ANAC046(319–338) corresponded well with the proposed SLiMs, whereas residues outside of the SLiMs appeared to have limited secondary Cα chemical shift perturbations upon interaction (Fig. 7, E and F, bottom figures). This suggests that interactions take place in the proposed regions accompanied by helical, non-helical, and even fuzzy structure in complex with RCD1-RST(499–572). Other possibilities are that the three transcription factors, despite having similar motifs, bind to different, possibly overlapping, binding sites, or that RCD1-RST(499–572) adopts slightly different conformations in the three complexes.
Discussion
Only 2% of the ELM database entries are derived from plant proteins (37) making efforts for motif identification in plants urgent. In this study, we defined the SLiM (DE)X(1,2)(YF)X(1,4)(DE)L for transcription factor binding to RCD1. The functional importance of this SLiM is supported by its conservation in IDRs with low degrees of sequence similarities (22) and by its in vivo role for DREB2A·RCD1 interactions (18). A high risk of false-positives exists for motif identification (38), and not all of the regions shown in Fig. 3, A and B, may be biochemically and physiologically relevant. Six transcription factors from the RCD1 interactome do not contain the RCD1 SLiM. They may either contain a variant or a different binding sequence (10).
Several known SLiMs are compositionally similar to the RCD1 SLiM. For example, the region DFDLDMLGD from the herpes simplex virus VP16 and several additional transcriptional regulators are implicated in essential interactions, and these regulatory proteins all interact with the general co-activator TAF9 (39). Interestingly, another general co-activator, Arabidopsis TAF4, contains an RST domain (40), making the RCD1 SLiM of relevance not only to gene-specific transcription factors but also to the general transcriptional apparatus. The RCD1 SLiM also shows similarity to the acidic class EDLL activation motif found in a sub-group of AP2/ERF transcription factors (41, 42).
Substitution analyses of the RCD1 SLiM (Tables 1 and 3) (16) support the observation that substitution of a single residue can abolish SLiM binding due to the short length of SLiMs (10). The results are also in accordance with additional typical features of SLiMs in IDRs. Thus, hydrophobic amino acid residues are over-represented in SLiMs (11); polycation-π interactions involving an aromatic residue can be significant components of interactions involving IDPs (43), and electrostatic interactions involving charged residues are key components of interactions involving IDPs (44). Furthermore, truncation of side chains in a SLiM almost always impairs binding, because SLiM contacts are nearly optimal (24). In conclusion, the RCD1 SLiM characteristics are typical of SLiMs in IDRs and of transcriptional activation motifs.
The affinity of the interactions between the RCD1-RST domain and the transcription factor peptides varied ∼70-fold (Tables 1–3) (16). Although the differences in affinity may have biological consequences, our thermodynamic characterization demonstrated that peptide length and context also significantly affect affinity (Tables 1 and 3). Curiously, removal of the PEPEPT sequence from ANAC013(254–274) resulted in a 66-fold decrease in affinity due to a marked decrease in binding enthalpy. Computational studies suggested that the core of a SLiM contributes about 80% of the binding energy (24). This study revealed a more complex pattern. Whereas the results suggested a negative allosteric effect on binding by the contexts of ANAC013(254–274), the contexts of DREB2A(255–272) had an opposite positive effect. For ANAC013, truncation reversed the entropic contribution to binding from negative to positive, whereas the opposite was observed for the DREB2A truncation series. This is in accordance with the recent finding that the sequences of IDRs are more than passive scaffolds for motifs and contribute to regulation of functions (45). It is generally assumed that IDRs pay an entropic cost upon binding because of conformational restriction (46). However, IDRs may also use entropy for binding (Tables 1–3) through solvent-mediated entropic interactions or by release of charge-charge interactions in the unbound state. It was recently suggested that interactions may increase conformational flexibility to induce favorable entropic changes (47) or lead to less compaction of the surrounding IDR compared with the unbound state (45). This could explain the thermodynamic profiles of interactions between RCD1 and the COL10 and STO peptides (Table 2). If, and how, these complex affinity and thermodynamics patterns relate to the in vivo concentrations of the transcription factors is currently not known.
Although similarities were observed for binding of the different transcription factor peptides to RCD1, differences were also observed. Thus, the importance of α-helix structure for binding differs. Studies of protein binding to the hub proteins Keap1 (48) and calmodulin (49) suggested that a reduction in the conformational freedom of the free state results in an affinity increase. A similar observation was not apparent for the peptides binding to RCD1. Supportive of this, a pre-formed α-helix in different disordered peptides had a small effect in enhancing the binding affinity for the target proteins (50). For the DREB2A peptide, strong α-helix formation in the complex was demonstrated by NMR spectroscopic analysis (Fig. 7). In contrast, the ANAC013 and ANAC046 peptides may form non-helical or even fuzzy interfaces in the complexes (Figs. 5A and 7, B and C) (11, 51). To this end, kinetic analysis is needed to determine whether binding involves conformational selection, induced fit, or a continuum of the two (52).
Based on primary and secondary structure analyses (Fig. 6A) (40), the RST domain of RCD1 is likely to form a fold similar to that of the human TAF4 TAFH domain. This domain adopts an α-helical fold with a large hydrophobic groove that forms the binding surface for TAF4-interacting transcription factors. In addition to hydrophobic/aromatic residues, the TAFH groove also contains a lysine residue, which plays an important role in ligand recognition. The TAFH-binding transcription factor regions contain conserved acidic and hydrophobic residues, similar to the RCD1 SLiM (53). Fig. 8 shows a model of RST-transcription factor interactions. Our data suggest that the RST domain of RCD1 can accommodate structural diversity in its partners, from fuzziness to folded α-helical structure. The observation that the spacing between the core residues of the RCD1-binding SLiM differs most likely reflects the potential to adapt to different structures in the bound state. Fuzziness in the interaction could minimize the entropic loss upon binding, and it is even conceivable that the ligands may interchange between anchoring points within the potential hydrophobic groove of the RST domain. The bipartite architecture of the SLiM may also allow for elasticity thus making variation in structural context possible. Because no current data can determine whether the interactions are carried out through conformer selection or induced fit, Fig. 8 allows for both possibilities, as indicated in the middle of the model by the equilibrium between different conformations of the free ligand. This type of interaction surface is also observed in the complex between the TAZ1 scaffolding domain of the transcriptional co-activator CBP/p300 and RelA of the NF-κB family (54). Part of the RelA activation domain folds upon binding and spirals through the exposed hydrophobic pockets of TAZ1. In this way, it anchors itself at a number of points. However, distinct peptide-binding pockets in the RST domain of RCD1 cannot be excluded at present. To summarize, although a single SLiM describes several RCD1-interacting peptides, the peptides vary structurally from fuzzy to α-helical in the complexes. The RCD1 SLiM is bipartite, and the transcription factors are likely to use at least two anchoring points for RCD1 interactions (Fig. 8).
FIGURE 8.
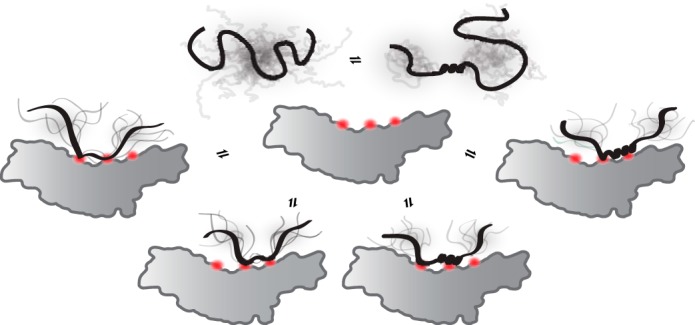
Model for RCD1-transcription factor interactions. Model of RCD1-RST interactions with the IDR of different transcription factors. The gray shadings indicate the ensembles that the IDR can populate both before and after binding. The model is based on the structural similarity between the RCD1-RST and TAF4-TAFH domains (40, 54), and three binding anchor points, shown in orange, are assumed. As suggested in this study, RCD1-RST has the potential to bind both unstructured and α-helical peptides, as illustrated, or possibly peptides with different structures (data not shown). Because current data cannot determine whether the interactions are carried out through conformer selection or induced fit, both possibilities are suggested by the equilibrium between conformations of the free ligand. Furthermore, the RST domain may use one or more binding surfaces, as shown by the different anchor points.
The physiological relevance of most of the RCD1-transcription factor interactions remains elusive. However, in vivo evidence for the RCD1·DREB2A interaction exists (18), and both large scale and specific studies are suggestive of functional RCD1-transcription factor interactions in specific networks. RCD1 plays multiple roles in stress responses, including reactive oxygen species responses, and in the senescence process (14, 15, 18). Accordingly, Gene Ontology analysis of the RCD1-interacting transcription factors revealed enrichment for both abiotic stress responses and developmental processes (Table 4). Recently, the expression profiles of DREB2A, ANAC013, and ANAC046 were shown to be similar in response to various hormone and stress treatments (17), in accordance with previous focused studies demonstrating a role of these transcription factors in regulation of stress responses and senescence (16, 19, 20, 55). In this study, two new RCD1 interaction partners, ANAC016 and ANAC017, were identified. During drought stress, ANAC016 functions as a regulator of leaf senescence through cross-talk with salt and oxidative stress-responsive pathways (56), and ANAC017 mediates H2O2 stress signaling (25). This means that the transcription factors of the RCD1 interactome are involved in stress-related signaling and senescence similarly to RCD1. Future experiments will show whether the transcription factors compete for RCD1 binding under stress conditions.
Many questions remain to be answered for the RCD1-transcription factor interactome. In addition to relating function to evolutionary conservation of SLiMs, the relevance of their occurrence in proteins needs to be characterized in the appropriate model organisms using a combination of high and low throughput methods to assess their functionality. This work has laid the foundation for such studies and has shown that by combining multiple techniques addressing different protein features, important novel SLiMs in IDRs can be discovered.
Experimental Procedures
Bioinformatics Analyses
The RCD1 interactome was obtained from the BAR Arabidopsis Interaction Viewer using RCD1 as query and information from the IntAct Molecular Interaction Database (57). ID was predicted using PONDR-FIT (58); secondary structure was predicted using PSIPRED (59; α-helix propensity was analyzed using Agadir (32); MoRFs were predicted using MoRFpred (60); and sequence motifs were predicted using MEME (61). The Gene Ontology database was searched using AmiGO2 (31), and the non-redundant protein database was searched for additional transcription factors putatively binding RCD1 using two different BLAST programs, PHI-BLAST and PSI-BLAST (30).
Assays in Yeast
Yeast two-hybrid assays were used to detect interactions between the RCD1 RST domain and different fragments of the transcription factor ANAC013. The fragments were amplified using sequence-specific primers and recombined into the pDEST22 vector (Invitrogen). Fusion of GAL4 DNA-binding domain and the RST domain of RCD1 (residues 498–573), named DBD-RST, was as described previously (16). Plasmids were transformed into yeast strain pJ694A.
Production and Purification of Recombinant Proteins and Peptides
Purification of RCD1-RST(499–572) was performed as described previously (16). Gene-specific primers encoding a tobacco etch virus cleavage site were used to amplify transcription factor fragments, which were inserted into pGEX-4T-1 (GE Healthcare) to obtain GST-tagged proteins. The constructs were verified by sequencing. The fragments were amplified from cDNAs obtained from the REGINA TF collection (Paz-Ares and the REGIA Consortium 2002). ANAC016, not present in the REGINA collection, was amplified from genomic DNA purified from Arabidopsis Col0 (Columbia ecotype 0) wild-type seedlings. ANAC017 from the REGIA collection contained a single mutation introducing a stop codon. Back mutation using the QuikChange mutagenesis kit (Stratagene) restored ANAC017. ANAC013 and DREB2A variants were obtained using the QuikChange mutagenesis kit. The GST-tagged proteins were expressed in Escherichia coli strain BL21-(DE3) at 37 °C, induced by 0.5 mm isopropyl β-d-thiogalactopyranoside, harvested after 3 h, resuspended in 20 mm Tris/HCl, pH 8.0, 100 mm NaCl, and sonicated. After centrifugation for 15 min at 15,000 × g, the supernatant was incubated with glutathione-Sepharose 4B (Sigma) resin. Bound GST-tagged recombinant protein was cleaved from the GST tag using tobacco etch virus protease by incubation overnight in resuspension buffer containing 2 mm DTT and 1.0 μg of tobacco etch virus/0.1 mg of protein. This left the peptides with an additional N-terminal glycine residue, which did not affect binding compared with binding of synthetic peptides. N-terminally acetylated and C-terminally amidated peptides were obtained from TAG Copenhagen A/S. Two methods were used to remove the protease after cleavage. Protein fragments that were disordered were boiled for 5 min and centrifuged at 20,000 × g for 10 min. Protein fragments with structure were subjected to TALON resin (Clontech), which removed the protease. To remove salt, the peptides were first freeze-dried before resuspension in 0.1% TFA and purified on a Vydac C18 column (Grace) equilibrated in 20% ethanol, 0.1% TFA and eluted in a linear gradient from 20 to 100% ethanol. Purified peptides were analyzed by MALDI-TOF (Autoflex Bruker) mass spectrometry and SDS-PAGE. For NMR studies, 15N,13C-labeled peptides were produced. Cells were grown in LB media until they reached an A600 of ∼0.6–0.8. Cells were harvested by centrifugation and resuspended in M9 minimal medium with 15NH4Cl and [13C6]glucose as sole sources of nitrogen and carbon, respectively. The cells were grown in the M9 media for 30 min before induction with 0.5 mm isopropyl β-d-thiogalactopyranoside, grown at 37 °C for 4 h before being harvested, and purified as described above.
ITC
ITC was used to determine the thermodynamic parameters, the dissociation constant (Kd), the stoichiometry (N), and the binding enthalpy change (ΔH) from which the binding Gibbs free energy change (ΔG) and the binding entropy change (ΔS) were calculated. The experiments were performed in a MicroCal ITC200 microcalorimeter (GE Healthcare). Protein samples were dialyzed against 50 mm Hepes, pH 7.4, and 100 mm NaCl or as indicated, centrifuged at 15,000 × g for 5 min, and degassed for 10 min by stirring under vacuum. Experiments were performed with a concentration of titrant in the sample cell of 3–6 μm and titrant concentrations of 45–90 μm in the syringe. A total of 19 injections separated by 180 s and with a duration of 4 s each of 2 μl of titrant was injected into the sample at 25 °C. Data from the ITC experiments were analyzed using an Origin 7 software package (MicroCalTM) by fitting data to a “one set of sites” binding model. Standard errors for the thermodynamic parameters ΔH and Kd, as well as the stoichiometry N, were obtained from Origin when fitting the data, and all experiments were repeated at least three times. The heat of dilution was subtracted from the raw data by performing a titration of titrant against buffer or by subtracting the dilution enthalpy obtained in the last injection when the partial enthalpy change had reached a constant level. Similar results were obtained at least twice for all experiments.
CD Spectroscopy
Briefly, the peptides were recorded in 10 mm Na2HPO4/Na2PO4, pH 7.0, at 15–20 μm and increasing amounts of TFE (0–40%) as indicated in the figure legend. To analyze for induced structure by complex formation, the RST domain and the individual peptides were recorded separately in 10 mm Na2HPO4/Na2PO4, pH 7.0, at 6–8 μm, as well as when in a 1:1 molar complex (6–8 μm), all in the absence of TFE. Far-UV CD spectra of the individual components were summed and compared with that of the relevant complex. Recording details were as in Ref. 16.
NMR Spectroscopy
All NMR spectra were recorded at 25 °C on Bruker AVANCE spectrometers operating at 600 or 750 MHz (for 1H) and equipped with cryoprobes. NMR spectra of each sample containing 60–140 μm 13C,15N-labeled DREB2A(255–272), ANAC013(254–274), or ANAC046(319–338) in 100 mm NaCl, pH 7.0, 0.02% (w/v) NaN3, 0.1 mm phenylmethanesulfonyl fluoride, 0.7 mm 4,4-dimethyl-4-silapentane-1-sulfonic acid, 10% D2O (v/v) were recorded in the presence or absence of equivalent molar amounts of unlabeled RCD1-RST(499–572). To prevent cysteine oxidation, the ANAC046(319–338) samples furthermore contained 20 mm Na2HPO4/NaH2PO4, pH 7.0, and 10 mm β-mercaptoethanol. Backbone resonance assignments of the peptides were done by manual analysis of sets of 1H-15N HSQC, HNCACB, HNCA, and CBCA(CO)NH spectra. For the assignment of ambiguous carbon resonances in the 267PEPE270 sequence of ANAC013(254–274), the mean value of each pair of glutamate and proline residues was calculated to account for the almost identical chemical shifts. All spectra were processed using NMRDraw (62) and analyzed using the CcpNMR Analysis software (63). Secondary 13Cα chemical shift values were calculated by subtraction of sequence-dependent random coil values from the experimental values as Δδ13Cα = δ13Cα − δ13CαRC. The random coil values were calculated by the on-line tool made available by Kjaergaard et al. (64), designed from peptides and specifically for IDPs.
Author Contributions
C. O. and L. S. conducted most of the experiments with initial contributions to the DREB2A work from S. K. B. and to the ANAC013 work from A. R. and F. G. T. The manuscript was mainly written by K. S., C. O., and L. S. M. W. and B. B. K. contributed to discussion of the experiments. All authors analyzed the results and discussed and approved the content of the manuscript.
Acknowledgments
We thank Marianne Mortensen for technical help, Dr. Katrine Bugge for NMR advice, and Dr. Caspar Elo Christensen for critical reading of the manuscript. We thank The VELUX Foundations for their generous support for NMR spectrometers.
This work was supported by Danish Research Councils Grant 4181-00344 (to B. B. K. and K. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- IDR
- intrinsically disordered region
- IDP
- intrinsically disordered protein
- ID
- intrinsic disorder
- ITC
- isothermal titration calorimetry
- MoRF
- molecular recognition feature
- NAC
- no apical meristem
- PHI
- pattern hit initiated
- PSI
- position-specific iterated
- RST
- RCD1-SRO-TAF4
- SLiM
- short linear motif
- TFE
- trifluoroethanol
- TRD
- transcription regulatory domain
- DBD
- DNA-binding domain
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Dunker A. K., Lawson J. D., Brown C. J., Williams R. M., Romero P., Oh J. S., Oldfield C. J., Campen A. M., Ratliff C. M., Hipps K. W., Ausio J., Nissen M. S., Reeves R., Kang C., Kissinger C. R., et al. (2001) Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59 [DOI] [PubMed] [Google Scholar]
- 2. Uversky V. N., Gillespie J. R., and Fink A. L. (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41, 415–427 [DOI] [PubMed] [Google Scholar]
- 3. Wright P. E., and Dyson H. J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 [DOI] [PubMed] [Google Scholar]
- 4. Tompa P., Davey N. E., Gibson T. J., and Babu M. M. (2014) A million peptide motifs for the molecular biologist. Mol. Cell 55, 161–169 [DOI] [PubMed] [Google Scholar]
- 5. Neduva V., Linding R., Su-Angrand I., Stark A., de Masi F., Gibson T. J., Lewis J., Serrano L., and Russell R. B. (2005) Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 3, e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diella F., Haslam N., Chica C., Budd A., Michael S., Brown N. P., Trave G., and Gibson T. J. (2008) Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. 13, 6580–6603 [DOI] [PubMed] [Google Scholar]
- 7. Davey N. E., Van Roey K., Weatheritt R. J., Toedt G., Uyar B., Altenberg B., Budd A., Diella F., Dinkel H., and Gibson T. J. (2012) Attributes of short linear motifs. Mol. Biosyst. 8, 268–281 [DOI] [PubMed] [Google Scholar]
- 8. Fuxreiter M., Tompa P., and Simon I. (2007) Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956 [DOI] [PubMed] [Google Scholar]
- 9. Zhou H. X. (2012) Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 37, 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Roey K., Uyar B., Weatheritt R. J., Dinkel H., Seiler M., Budd A., Gibson T. J., and Davey N. E. (2014) Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 114, 6733–6778 [DOI] [PubMed] [Google Scholar]
- 11. Vacic V., Oldfield C. J., Mohan A., Radivojac P., Cortese M. S., Uversky V. N., and Dunker A. K. (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dinkel H., Van Roey K., Michael S., Davey N. E., Weatheritt R. J., Born D., Speck T., Krüger D., Grebnev G., Kuban M., Strumillo M., Uyar B., Budd A., Altenberg B., Seiler M., et al. (2014) The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 42, D259–D266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gould C. M., Diella F., Via A., Puntervoll P., Gemünd C., Chabanis-Davidson S., Michael S., Sayadi A., Bryne J. C., Chica C., Seiler M., Davey N. E., Haslam N., Weatheritt R. J., Budd A., et al. (2010) ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 38, D167–D180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaspers P., Blomster T., Brosché M., Salojärvi J., Ahlfors R., Vainonen J. P., Reddy R. A., Immink R., Angenent G., Turck F., Overmyer K., and Kangasjärvi J. (2009) Unequally redundant RCD1 and SRO1 mediate stress and developmental responses and interact with transcription factors. Plant J. 60, 268–279 [DOI] [PubMed] [Google Scholar]
- 15. Teotia S., and Lamb R. S. (2009) The paralogous genes RADICAL-INDUCED CELL DEATH1 and similar to RCD ONE1 have partially redundant functions during Arabidopsis development. Plant Physiol. 151, 180–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Shea C., Kryger M., Stender E. G., Kragelund B. B., Willemoës M., and Skriver K. (2015) Protein intrinsic disorder in Arabidopsis NAC transcription factors: transcriptional activation by ANAC013 and ANAC046 and their interactions with RCD1. Biochem. J. 465, 281–294 [DOI] [PubMed] [Google Scholar]
- 17. Brosché M., Blomster T., Salojärvi J., Cui F., Sipari N., Leppälä J., Lamminmäki A., Tomai G., Narayanasamy S., Reddy R. A., Keinänen M., Overmyer K., and Kangasjärvi J. (2014) Transcriptomics and functional genomics of ROS-induced cell death regulation by RADICAL-INDUCED CELL DEATH1. PLoS Genet. 10, e1004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vainonen J. P., Jaspers P., Wrzaczek M., Lamminmäki A., Reddy R. A., Vaahtera L., Brosché M., and Kangasjärvi J. (2012) RCD1-DREB2A interaction in leaf senescence and stress responses in Arabidopsis thaliana. Biochem. J. 442, 573–581 [DOI] [PubMed] [Google Scholar]
- 19. Sakuma Y., Maruyama K., Qin F., Osakabe Y., Shinozaki K., and Yamaguchi-Shinozaki K. (2006) Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 18822–18827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Clercq I., Vermeirssen V., Van Aken O., Vandepoele K., Murcha M. W., Law S. R., Inzé A., Ng S., Ivanova A., Rombaut D., van de Cotte B., Jaspers P., Van de Peer Y., Kangasjärvi J., Whelan J., and Van Breusegem F. (2013) The membrane-bound NAC transcription factor ANAC013 functions in mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. Plant Cell 25, 3472–3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kragelund B. B., Jensen M. K., and Skriver K. (2012) Order by disorder in plant signaling. Trends Plant Sci. 17, 625–632 [DOI] [PubMed] [Google Scholar]
- 22. Stender E. G., O'Shea C., and Skriver K. (2015) Subgroup-specific intrinsic disorder profiles of Arabidopsis NAC transcription factors: Identification of functional hotspots. Plant Signal. Behav. 10, e1010967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ladbury J. E. (2010) Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem. Soc. Trans. 38, 888–893 [DOI] [PubMed] [Google Scholar]
- 24. Stein A., and Aloy P. (2008) Contextual specificity in peptide-mediated protein interactions. PLoS One 3, e2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng S., Ivanova A., Duncan O., Law S. R., Van Aken O., De Clercq I., Wang Y., Carrie C., Xu L., Kmiec B., Walker H., Van Breusegem F., Whelan J., and Giraud E. (2013) A membrane-bound NAC transcription factor, ANAC017, mediates mitochondrial retrograde signaling in Arabidopsis. Plant Cell 25, 3450–3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakuraba Y., Kim Y. S., Han S. H., Lee B. D., and Paek N. C. (2015) The Arabidopsis transcription factor NAC016 promotes drought stress responses by repressing AREB1 transcription through a trifurcate feed-forward regulatory loop involving NAP. Plant Cell 27, 1771–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jensen M. K., Kjaersgaard T., Nielsen M. M., Galberg P., Petersen K., O'Shea C., and Skriver K. (2010) The Arabidopsis thaliana NAC transcription factor family: structure-function relationships and determinants of ANAC019 stress signalling. Biochem. J. 426, 183–196 [DOI] [PubMed] [Google Scholar]
- 28. Hiltscher H., Rudnik R., Shaikhali J., Heiber I., Mellenthin M., Meirelles Duarte I., Schuster G., Kahmann U., and Baier M. (2014) The radical induced cell death protein 1 (RCD1) supports transcriptional activation of genes for chloroplast antioxidant enzymes. Front. Plant Sci. 5, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kjaersgaard T., Jensen M. K., Christiansen M. W., Gregersen P., Kragelund B. B., and Skriver K. (2011) Senescence-associated barley NAC (NAM, ATAF1,2, CUC) transcription factor interacts with radical-induced cell death 1 through a disordered regulatory domain. J. Biol. Chem. 286, 35418–35429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 31. Balsa-Canto E., Henriques D., Gábor A., and Banga J. R. (2016) AMIGO2, a toolbox for dynamic modeling, optimization and control in systems biology. Bioinformatics 32, 3357–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muñoz V., Cronet P., López-Hernández E., and Serrano L. (1996) Analysis of the effect of local interactions on protein stability. Fold. Des. 1, 167–178 [DOI] [PubMed] [Google Scholar]
- 33. Lehrman S. R., Tuls J. L., and Lund M. (1990) Peptide α-helicity in aqueous trifluoroethanol: correlations with predicted α-helicity and the secondary structure of the corresponding regions of bovine growth hormone. Biochemistry 29, 5590–5596 [DOI] [PubMed] [Google Scholar]
- 34. Spera S., Ikura M., and Bax A. (1991) Measurement of the exchange rates of rapidly exchanging amide protons: application to the study of calmodulin and its complex with a myosin light chain kinase fragment. J. Biomol. NMR 1, 155–165 [DOI] [PubMed] [Google Scholar]
- 35. Yao J., Dyson H. J., and Wright P. E. (1997) Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 419, 285–289 [DOI] [PubMed] [Google Scholar]
- 36. Kleckner I. R., and Foster M. P. (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dinkel H., Van Roey K., Michael S., Kumar M., Uyar B., Altenberg B., Milchevskaya V., Schneider M., Kühn H., Behrendt A., Dahl S. L., Damerell V., Diebel S., Kalman S., Klein S., et al. (2016) ELM 2016–data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 44, D294–D300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gibson T. J., Dinkel H., Van Roey K., and Diella F. (2015) Experimental detection of short regulatory motifs in eukaryotic proteins: tips for good practice as well as for bad. Cell Commun. Signal. 13, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aguilar X., Blomberg J., Brännström K., Olofsson A., Schleucher J., and Björklund S. (2014) Interaction studies of the human and Arabidopsis thaliana Med25-ACID proteins with the herpes simplex virus. PLoS One 9, e98575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jaspers P., Brosché M., Overmyer K., and Kangasjärvi J. (2010) The transcription factor interacting protein RCD1 contains a novel conserved domain. Plant Signal. Behav. 5, 78–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tiwari S. B., Belachew A., Ma S. F., Young M., Ade J., Shen Y., Marion C. M., Holtan H. E., Bailey A., Stone J. K., Edwards L., Wallace A. D., Canales R. D., Adam L., Ratcliffe O. J., and Repetti P. P. (2012) The EDLL motif: a potent plant transcriptional activation domain from AP2/ERF transcription factors. Plant J. 70, 855–865 [DOI] [PubMed] [Google Scholar]
- 42. Lee C. W., Arai M., Martinez-Yamout M. A., Dyson H. J., and Wright P. E. (2009) Mapping the interactions of the p53 transactivation domain with the KIX domain of CBP. Biochemistry 48, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Song J., Ng S. C., Tompa P., Lee K. A., and Chan H. S. (2013) Polycation-π interactions are a driving force for molecular recognition by an intrinsically disordered oncoprotein family. PLoS Comput. Biol. 9, e1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wong E. T., Na D., and Gsponer J. (2013) On the importance of polar interactions for complexes containing intrinsically disordered proteins. PLoS Comput. Biol. 9, e1003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Das R. K., Huang Y., Phillips A. H., Kriwacki R. W., and Pappu R. V. (2016) Cryptic sequence features within the disordered protein p27Kip1 regulate cell cycle signaling. Proc. Natl. Acad. Sci. U.S.A. 113, 5616–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flock T., Weatheritt R. J., Latysheva N. S., and Babu M. M. (2014) Controlling entropy to tune the functions of intrinsically disordered regions. Curr. Opin. Struct. Biol. 26, 62–72 [DOI] [PubMed] [Google Scholar]
- 47. Heller G. T., Sormanni P., and Vendruscolo M. (2015) Targeting disordered proteins with small molecules using entropy. Trends Biochem. Sci. 40, 491–496 [DOI] [PubMed] [Google Scholar]
- 48. Cino E. A., Killoran R. C., Karttunen M., and Choy W. Y. (2013) Binding of disordered proteins to a protein hub. Sci. Rep. 3, 2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dunlap T. B., Kirk J. M., Pena E. A., Yoder M. S., and Creamer T. P. (2013) Thermodynamics of binding by calmodulin correlates with target peptide α-helical propensity. Proteins 81, 607–612 [DOI] [PubMed] [Google Scholar]
- 50. Bienkiewicz E. A., Adkins J. N., and Lumb K. J. (2002) Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1). Biochemistry 41, 752–759 [DOI] [PubMed] [Google Scholar]
- 51. Tompa P., and Fuxreiter M. (2008) Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8 [DOI] [PubMed] [Google Scholar]
- 52. Kiefhaber T., Bachmann A., and Jensen K. S. (2012) Dynamics and mechanisms of coupled protein folding and binding reactions. Curr. Opin. Struct. Biol. 22, 21–29 [DOI] [PubMed] [Google Scholar]
- 53. Wang X., Truckses D. M., Takada S., Matsumura T., Tanese N., and Jacobson R. H. (2007) Conserved region I of human coactivator TAF4 binds to a short hydrophobic motif present in transcriptional regulators. Proc. Natl. Acad. Sci. U.S.A. 104, 7839–7844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mukherjee S. P., Behar M., Birnbaum H. A., Hoffmann A., Wright P. E., and Ghosh G. (2013) Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 11, e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vermeirssen V., De Clercq I., Van Parys T., Van Breusegem F., and Van de Peer Y. (2014) Arabidopsis ensemble reverse-engineered gene regulatory network discloses interconnected transcription factors in oxidative stress. Plant Cell 26, 4656–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim Y. S., Sakuraba Y., Han S. H., Yoo S. C., and Paek N. C. (2013) Mutation of the Arabidopsis NAC016 transcription factor delays leaf senescence. Plant Cell Physiol. 54, 1660–1672 [DOI] [PubMed] [Google Scholar]
- 57. Kerrien S., Aranda B., Breuza L., Bridge A., Broackes-Carter F., Chen C., Duesbury M., Dumousseau M., Feuermann M., Hinz U., Jandrasits C., Jimenez R. C., Khadake J., Mahadevan U., Masson P., et al. (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res. 40, D841–D846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xue B., Dunbrack R. L., Williams R. W., Dunker A. K., and Uversky V. N. (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bryson K., McGuffin L. J., Marsden R. L., Ward J. J., Sodhi J. S., and Jones D. T. (2005) Protein structure prediction servers at University College London. Nucleic Acids Res. 33, W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Disfani F. M., Hsu W. L., Mizianty M. J., Oldfield C. J., Xue B., Dunker A. K., Uversky V. N., and Kurgan L. (2012) MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 28, i75–i83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W., and Noble W. S. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 63. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 64. Kjaergaard M., Brander S., and Poulsen F. M. (2011) Random coil chemical shifts for intrinsically disordered proteins: effects of temperature and pH. J. Biomol. NMR 49, 139–149 [DOI] [PubMed] [Google Scholar]