

Abstract
Biased agonism at G protein-coupled receptors constitutes a promising area of research for the identification of new therapeutic molecules. In this study we identified two novel biased ligands for the chemokine receptors CCR2 and CCR5 and characterized their functional properties. We showed that J113863 and its enantiomer UCB35625, initially identified as high affinity antagonists for CCR1 and CCR3, also bind with low affinity to the closely related receptors CCR2 and CCR5. Binding of J113863 and UCB35625 to CCR2 or CCR5 resulted in the full or partial activation of the three Gi proteins and the two Go isoforms. Unlike chemokines, the compounds did not activate G12. Binding of J113863 to CCR2 or CCR5 also induced the recruitment of β-arrestin 2, whereas UCB35625 did not. UCB35625 induced the chemotaxis of L1.2 cells expressing CCR2 or CCR5. In contrast, J113863 induced the migration of L1.2-CCR2 cells but antagonized the chemokine-induced migration of L1.2-CCR5 cells. We also showed that replacing the phenylalanine 3.33 in CCR5 TM3 by the corresponding histidine of CCR2 converts J113863 from an antagonist for cell migration and a partial agonist in other assays to a full agonist in all assays. Further analyses indicated that F3.33H substitution strongly increased the activation of G proteins and β-arrestin 2 by J113863. These results highlight the biased nature of the J113863 and UCB35625 that act either as antagonist, partial agonist, or full agonist according to the receptor, the enantiomer, and the signaling pathway investigated.
Keywords: C-C chemokine receptor type 5 (CCR5), chemokine, drug action, drug discovery, G protein-coupled receptor (GPCR), signaling, biased agonist
Introduction
G protein-coupled receptors (GPCRs)3 are one the largest classes of cell surface receptors in the human genome, and they collectively constitute the targets of ∼30–40% of currently marketed drugs. Over the past years, a new paradigm has emerged in the field, assuming that GPCRs can activate differentially selective signaling pathways according to the specific conformation stabilized by the bound ligand (1, 2). Differences in ligand responses may occur through differential activation of heterotrimeric G proteins or differential recruitment of scaffolding proteins, like arrestins or PDZ-proteins, interacting with a number of signaling proteins. The recruitment of cytosolic proteins can also modify receptor trafficking and compartmentalization that in turn may impact on the nature of cellular signaling (3). This concept of biased or selective signaling has gained a lot of attention due to its potential in drug development, and it is anticipated that biased drugs able to selectively target a specific signaling pathway without affecting others would benefit from an improved efficacy and/or a lack of side effects (4). Some biased molecules are presently in clinical development, including the angiotensin receptor 1 (ATR1) ligand TRV027 for the treatment of acute heart failure and the MOR opioid receptor ligand TRV130 for acute pain relief (5–8). We and others have previously shown that chemokine receptors constitute a good model system for the study of biased agonism (9–11). Chemokines are key regulators of leukocyte migration and function, and they play important roles both in the physiology of immune responses and pathological dysregulations of these responses. The system includes about 40 chemokines, which engage a panel of >20 different chemokine receptors. There is an apparent redundancy in the system as many chemokines bind multiple receptors and more than one receptor can interact with each chemokine (12). However, some studies reported different signaling or trafficking responses of a receptor to individual chemokines, suggesting that redundancy may not be as widespread as previously thought. Binding of CCL22 to CCR4 activates arrestin recruitment and receptor down-regulation, whereas binding of CCL17 does not promote arrestin recruitment and induces a weaker down-regulation (13, 14). CCL17 also induces calcitonin-gene related peptide expression much more efficiently than CCL22 (15). In a more recent study, Karin and co-workers (16) showed that CXCL9/10 binding to CXCR3 biased T cell polarization into Th1/Th17 effector cells, whereas CXCL11 binding drives T cell polarization into IL-10 producing Treg, and they linked this behavior to differences in STATs activation. In contrast, for other receptors such as CCR2 or CCR5, the apparent selectivity of chemokines for one particular pathway appears relatively subtle (9, 10). Nevertheless, CCR5 response may be biased toward the activation of one particular pathway with N-terminally modified forms of chemokines. Binding of AOP-RANTES triggers receptor internalization but not monocyte chemotaxis (17, 18). Similarly, 5P14-RANTES promotes efficient CCR5 internalization but no apparent G protein-mediated signaling (19). The CCR5 response may also be biased with synthetic small molecules. For instance, YM-370749 and ESN-196 promote strong calcium mobilization and receptor internalization but not cell migration (20, 21). However, the signaling pathways activated by synthetic small molecules at chemokine receptors remains often unknown. Here, we identified the small molecule enantiomers J113863 and UCB35625 as biased agonists for the chemokine receptors CCR2 and CCR5. These molecules were originally identified as high affinity bispecific antagonists for the closely related receptors CCR1 and CCR3 (22–25). Our data revealed that these enantiomers also bind to CCR2 and CCR5 with low affinity and display the properties of biased ligands, acting as full agonists, partial agonists, or antagonists according to the molecule, receptor, and functional assay considered. We also showed that amino acid 3.33 in CCR5 TM3 is key for the activation of the receptor by these molecules.
Results
J113863 and UCB35625 Bind to and Activate CCR2 and CCR5
The synthetic enantiomers J113863 and UCB35625 were originally identified as CCR1- and CCR3-bispecific high affinity antagonists (Fig. 1A). Here, we tested whether they also interact with the closely related receptors CCR2 and CCR5 by performing binding competition assays and showed that J113863 and UCB35625 fully competed for the binding of 125I-CCL2 and 125I-CCL4 with IC50 values in the micromolar range (Fig. 1, B and C, Table 1). We next investigated the functional properties of the enantiomers and showed that J113863 induced the migration of L1.2 cells expressing CCR2 with a characteristic bell-shaped curve (Fig. 1D). In contrast, J113863 did not induce the migration of L1.2 cells expressing CCR5 at any concentration tested (Fig. 1E). Yet, J113863 fully antagonized cell chemotaxis triggered by CCL4, confirming its effective binding to CCR5 (Fig. 1E). By comparison, the enantiomer UCB35625 promoted at high concentrations the migration L1.2 cells whether they express CCR2 or CCR5 (Fig. 1, D and E). These results on CCR2 and CCR5 contrast with previous data obtained for CCR1 and CCR3, showing that both J113863 and UCB35625 antagonized the chemotaxis of cells expressing these receptors (22–25). Thus, we further examined the functional properties of UCB35625 and J113863 in a calcium mobilization assay. None of the molecules activated CCR1 and CCR3 (Fig. 2, A and B), but both molecules fully inhibited the activation of CCR1 and CCR3 triggered by chemokines, confirming their antagonist properties on these two receptors (Fig. 2, C and D). In striking contrast, J113863 activated both CCR2 and CCR5, although the maximal response for CCR5 was partial and reached only 20% of the maximal response triggered by CCL4 (Fig. 2, E and F). UCB35625 also behaved as a weak agonist of CCR2 and CCR5, not reaching full activation at 100 μm. Altogether, these data suggested that J113863 and UCB35625 might display the behavior of “biased ligands” acting as antagonists or (partial) agonists according to the tested receptor and the functional assay. These results prompted us to further investigate whether J113863 and UCB35625 activate the same signaling pathways as chemokines, through CCR2 and CCR5.
FIGURE 1.
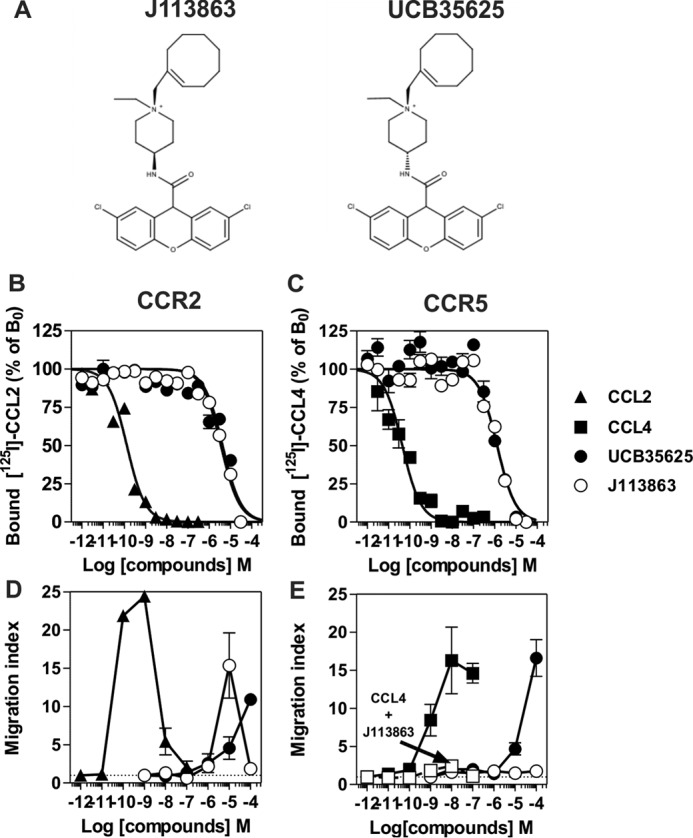
J113863 and UCB35625 activate CCR2 and CCR5. A, structure of the small molecule enantiomers J113863 and UCB35625. B and C, competition binding assays performed on cells expressing CCR2 or CCR5 incubated, respectively, with 0.1 nm 125I-CCL2 or 125I-CCL4 as tracers and increasing concentrations of unlabeled CCL2 (▴), CCL4 (■), UCB35625 (●), or J113863 (○) as competitors. The data were normalized for nonspecific binding (0%) in the presence of 300 nm concentrations of unlabeled chemokine and specific binding in the absence of competitor (100%). D and E, chemotactic response of L1.2 cells stably expressing CCR2 or CCR5 in the presence of increasing concentrations of CCL2 (▴), CCL4 (■), UCB35625 (●), or J113863 (○). L1.2 cells expressing CCR5 were also co-stimulated with 50 μm J113863 and increasing concentrations of CCL4 (□). The data represent the mean values ± S.E. of three independent experiments.
TABLE 1.
Binding and signaling parameters of J113863 and UCB35625
Binding and functional parameters were measured on cells expressing CCR2 or CCR5. The pIC50 and pEC50 and Emax values were obtained from experiments as displayed in Figs. 1 and 2. Values represent the mean ± S.E. of at least three independent experiments. NT, not tested; ND, not determined.
| Ligands | CCR2 |
CCR5 |
||||
|---|---|---|---|---|---|---|
|
125I-CCL2 |
Ca2+ |
125I-CCL4 |
Ca2+ |
|||
| pIC50 | pEC50 | Emax | pIC50 | pEC50 | Emax | |
| CCL2 | 9.7 ± 0.1 | 9.4 ± 0.1 | 99 ± 5 | NT | NT | NT |
| CCL4 | NT | NT | NT | 9,4 ± 0,1 | 8.2 ± 0.1 | 67 ± 5 |
| J113863 | 5.5 ± 0.1 | 6.4 ± 0.1 | 101 ± 4 | 5,9 ± 0.1 | 5.8 ± 0.1 | 16 ± 1 |
| UCB35625 | 5.4 ± 0.1 | ND | ND | 5,6 ± 1,3 | ND | ND |
FIGURE 2.
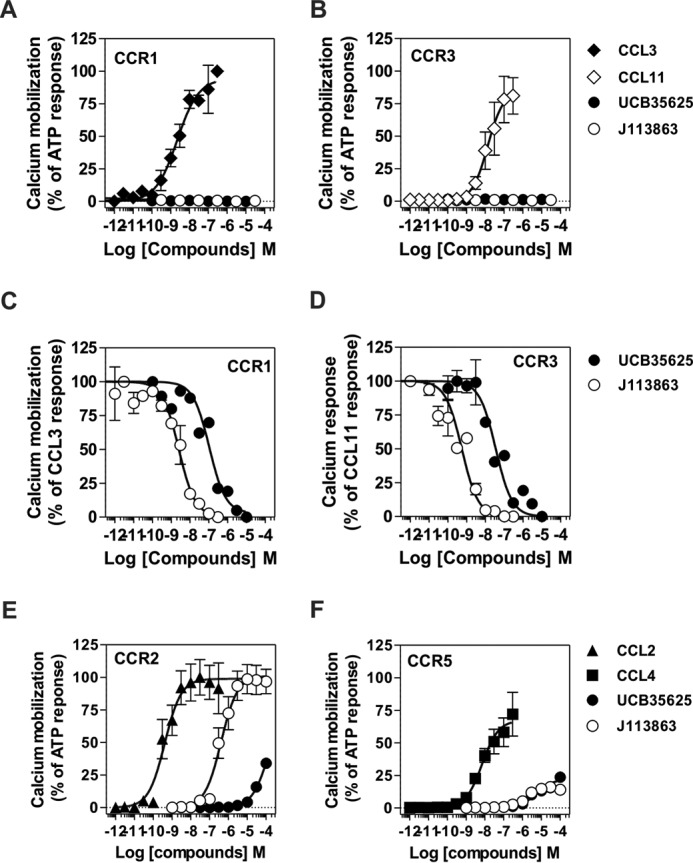
A and B, UCB35625 and J113863 do not activate CCR1 and CCR3 as measured by calcium mobilization. Cells stably expressing apoaequorin and CCR1 (A) or CCR3 (B) were stimulated with CCL3 (♦) CCL11 (♢), UCB35625 (●), or J113863 (○), and luminescence was recorded for 30 s. The results were normalized for basal luminescence in the absence of agonist (0%), and maximal response was obtained with 50 μm ATP (100%). C and D, UCB35625 and J113863 inhibited the activation of CCR1 and CCR3 triggered by chemokines. Cells stably expressing apoaequorin and CCR1 (C) or CCR3 (D) were incubated for 15 min with increasing concentrations of UCB35625 (●) or J113863 (○) before stimulation with 2 nm CCL3 or CCL11, and the luminescence was recorded for 30 s. The results were normalized for basal luminescence in the absence of agonist (0%), and maximal response was obtained with 2 nm chemokine only (100%). E and F, UCB35625 and J113863 activate CCR2 and CCR5 as measured by calcium mobilization. Cells stably expressing apoaequorin and CCR2 (E) or CCR5 (F) were stimulated with CCL2 (■), CCL4 (▴), UCB35625 (●), or J113863 (○), and luminescence was recorded for 30 s. The results were normalized for basal luminescence in the absence of agonist (0%), and maximal response was obtained with 50 μm ATP (100%). All the data represent the mean ± S.E. of three independent experiments.
Activation of G Proteins by J113863 and UCB35625
We first investigated whether J113863 and UCB35625 activate the same G proteins as chemokines by using G protein BRET biosensors, directly monitoring the activation of G protein subtypes. CCR2 and CCR5 were transiently expressed in HEK293T cells with 10 different biosensors belonging to the 4 major classes of G proteins (Gαs, Gαi/o, Gαq/11, and Gα12/13) and stimulated either with chemokines or the chemical enantiomers. In agreement with our previous study, stimulation of CCR2 by CCL2 at 100 nm activated all the Gαi and Gαo isoforms as well as Gα12, whereas stimulation of cells expressing the biosensors without CCR2 generated much weaker signals (Fig. 3) (9). Similarly, stimulation of CCR2 by the inverse agonist TAK-779, used as the negative control, generated very weak signals. Stimulation of CCR2 by J113863 at 100 μm activated all Gαi and Gαo isoforms but not Gα12, indicating that J113863 activates a distinct G protein subset as compared with chemokines (Fig. 3). Stimulation of CCR2 by UCB35625 at 100 μm activated much fewer G proteins, Gαi1, Gαi3, and Gαoa being the only sensors for which activation reached statistical significance (Fig. 3). By comparison, stimulation of CCR5 by J113863 and UCB35625 at 100 μm activated all Gαi and Gαo isoforms with no major differences between the two enantiomers (Fig. 4). No significant BRET signal was detected for the Gαq, Gα11, Gαs, or Gα13 sensors upon stimulation of CCR2 and CCR5 by J113863 or UCB35625 (data not shown). Dose response curves performed with Gαi2 and Gαoa biosensors revealed that activation by J113863 and UCB35625 occurred with potencies in line with their binding affinity (Fig. 5, Table 2). Nevertheless, stimulation of CCR2 by UCB35625 activated Gαi2 with a low efficacy compared with J113863, whereas both enantiomers activated Gαoa with similar efficacies. In contrast, stimulation of CCR5 by J113863 and UCB35625 activated Gαi2 and Gαoa proteins with similar potencies, although with an efficacy lower than CCL4 (Fig. 5, Table 2).
FIGURE 3.

Panel of G proteins activated by CCR2 upon stimulation by CCL2, J113863, or UCB35625. Real-time measurement of BRET signal in HEK293T cells coexpressing CCR2 and G protein biosensors (black bars) or G protein biosensors only (open bars) and stimulated for 1 min with 100 nm CCL2, 1 μm TAK779, 100 μm UCB35625 (UCB), or 100 μm J113863 (J). Results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. Data represent the mean values ± S.E. of six independent experiments. Statistical significance between cells expressing or not CCR2 (***, p < 0.001; **, p < 0.01) and between ligands (###, p < 0.001) was assessed using Tukey's test.
FIGURE 4.

Panel of G proteins activated by CCR5 upon stimulation by CCL4, J113863, or UCB35625. Real-time measurement of BRET signal in HEK293T cells coexpressing CCR5 and G protein biosensors (black bars) or G protein biosensors only (open bars) and stimulated for 1 min with 100 nm of CCL4, 1 μm TAK779, 100 μm UCB35625 (UCB), or 100 μm J113863 (J). Results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. Data represent the mean values ± S.E. of six independent experiments. Statistical significance between cells expressing or not CCR2 (***, p < 0.001; **, p < 0.01; *, p < 0.1) and between ligands (##, p < 0.01; #, p < 0.1) was assessed using Tukey's test.
FIGURE 5.
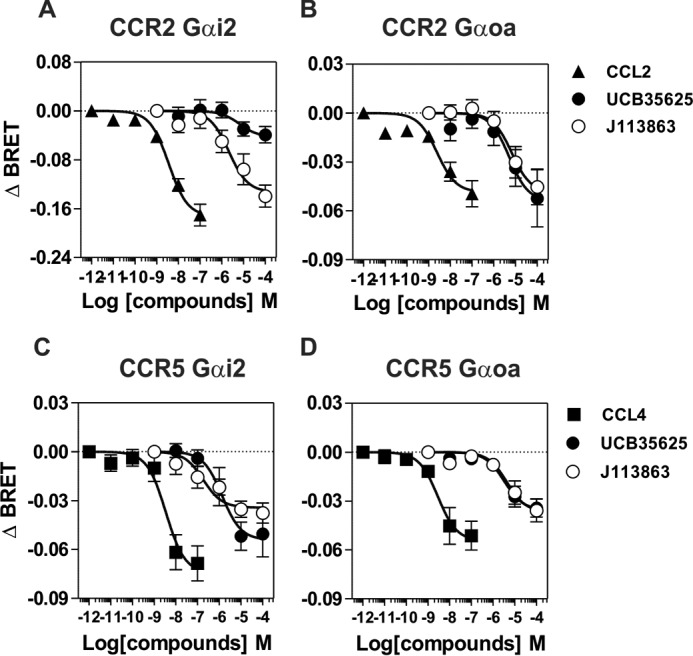
Activation of Gi2 and Goa by CCR2 and CCR5 upon stimulation by J113863 or UCB35625. Real-time measurement of BRET signal in HEK293T cells coexpressing CCR2 (A and B) or CCR5 (C and D) and G protein biosensors and stimulated with increasing concentration of CCL2 (▴), CCL4 (■), UCB35625 (●), or J113863 (○). Results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. Data represent the mean ± S.E. of three independent experiments.
TABLE 2.
Signaling parameters of J113863 and UCB35625
Functional parameters were measured on cells expressing CCR2 or CCR5. The pEC50 and Emax values were obtained from experiments as displayed in Figs. 5 and 6. Values represent the mean ± S.E. of at least three independent experiments. NT, not tested; ND, not determined.
| Ligands | CCR2 |
CCR5 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gαi2 |
Gαoa |
β-Arr2 |
Gαi2 |
Gαoa |
β-Arr2 |
|||||||
| pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | pEC50 | Emax | |
| CCL2 | 8.45 ± 0.13 | 0.17 ± 0.01 | 8.61 ± 0.22 | 0.05 ± 0.01 | 8.50 ± 0.06 | 0.14 ± 0.06 | NT | NT | NT | NT | NT | NT |
| CCL4 | NT | NT | NT | NT | NT | NT | 8.46 ± 0.29 | 0.08 ± 0.01 | 8.55 ± 0.20 | 0.05 ± 0.01 | 7.85 ± 0.11 | 0.10 ± 0.04 |
| J113863 | 5.68 ± 0.27 | 0.14 ± 0.02 | 5.17 ± 0.33 | 0.05 ± 0.01 | 5.17 ± 0.08 | 0.16 ± 0.01 | 6.74 ± 0.29 | 0.03 ± 0.01 | 5.36 ± 0.27 | 0.04 ± 0.02 | 6.80 ± 0.28 | 0.02 ± 0.01 |
| UCB35625 | 5.18 ± 0.56 | 0.04 ± 0.01 | 5.29 ± 0.38 | 0.05 ± 0.01 | ND | ND | 5.87 ± 0.22 | 0.05 ± 0.01 | 5.87 ± 0.22 | 0.04 ± 0.02 | ND | ND |
Activation of β-Arrestin 2 by J113863 and UCB35625
It is well known that ligand binding can trigger the activation of G protein-independent but β-arrestin-dependent signaling pathways. We investigated the ability of J113863 and UCB35625 to recruit β-arrestin 2 by using a BRET proximity assay, which measures the energy transfer between β-arrestin-2-Rluc and a receptor fused to the yellow fluorescent protein Venus (9). J113863 induced the recruitment of β-arrestin 2 to CCR2 with the same kinetics than CCL2 used as control (Fig. 6). Dose-response curves also indicated that the potency of J113863 in this assay is similar to that observed for G protein activation. In contrast, J113863 induced a weak recruitment of β-arrestin 2 to CCR5 as compared with CCL4, and UCB35625 did not promote the recruitment of β-arrestin 2 to CCR2 or CCR5 at any concentration tested (Fig. 6). Collectively, these results show that J113863 and UCB35625, although binding with the same affinity, stabilize different receptor conformations and that CCR2 and CCR5 display a differential selectivity for the enantiomers. The two molecules also possess their own signaling signature compared with chemokines.
FIGURE 6.
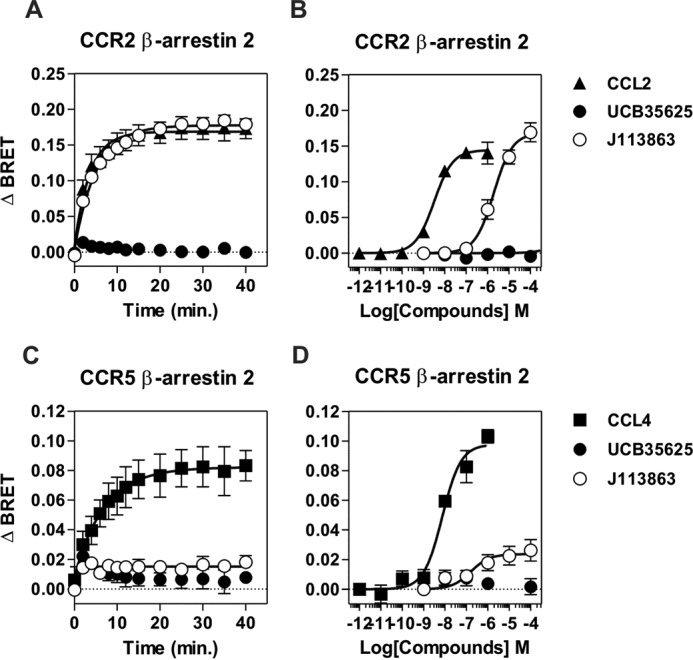
Recruitment of β-arrestin 2 by CCR2 and CCR5 upon stimulation by J113863 or UCB35625. Real-time measurement of BRET signal in HEK293T cells expressing either β-arrestin2-RLuc8 and CCR2-Venus (A and B) or β-arrestin2-RLuc8 and CCR5-Venus (C and D) and stimulated with CCL2 (▴), CCL4 (■), UCB35625 (●), or J113863 (○). For kinetics, BRET signals were measured after the addition of 100 nm of chemokine or 100 μm J113863 or UCB35625. For dose-response curves, BRET was recorded 30 min after stimulation with various concentrations of ligands. Results are expressed as net BRET, corresponding to the difference in BRET signal between cells expressing arrestin plus receptor and cells expressing arrestin only. Data represent the mean ± S.E. of three independent experiments.
Quantification of Ligand Bias at CCR2 and CCR5
We performed a quantitative analysis of bias by using the operational model to derive transduction ratios (Table 3 and 4) and the resulting bias factors Table 5 and 6) (26). Bias factors were determined with the exception of the cases where signaling parameters could not be determined. Not surprisingly, UCB35625 and J113863 showed a reduced efficacy relative to the reference chemokine (Tables 3 and 5). Comparison of bias factors between the different pathways also indicated that J113863 showed at CCR5 a significant bias for Gαi2 relative to Gαoa and calcium mobilization (Table 5). In contrast, J113863 displayed no bias at CCR2 for the pathways tested (Table 6).
TABLE 3.
Transduction ratios log(τ/KA) of CCL4, UCB35625, and J113863 at CCR5
Data were analyzed by non-linear regression using the operational model equation to determine the transduction ratios (log(τ/KA)). Δlog(τ/KA) ratios were calculated from log(τ/KA) values considering CCL4 as the reference ligand. The RE of ligands toward each pathway, relative to CCL4, corresponds to the inverse logarithm of the Δlog(τ/KA) ratio. Data are the mean ± S.E. of 3–6 experiments. ND, not determined.
| Ligand | Gi2 |
Goa |
Ca2+ |
β-Arrestin 2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log | ΔLog | RE | Log | ΔLog | RE | Log | ΔLog | RE | Log | ΔLog | RE | |
| CCL4 | 8.68 ± 0.44 | 0.00 ± 0.63 | 1 | 8.65 ± 0.21 | 0.00 ± 0.29 | 1 | 7.88 ± 0.19 | 0.00 ± 0.27 | 1 | 8.15 ± 0.09 | 0.00 ± 0.12 | 1 |
| UCB35625 | 5.88 ± 0.21 | −2.81 ± 0.49 | 0.0016a | 5.05 ± 0.18 | −3.60 ± 0.27 | 0.0002a | ND | ND | ND | ND | ND | ND |
| J113863 | 7.10 ± 0.72 | −1.58 ± 0.84 | 0.0261 | 5.23 ± 0.32 | −3.42 ± 0.42 | 0.0004a | 4.43 ± 0.44 | −3.46 ± 0.47 | 0.0003a | ND | ND | ND |
a p < 0.05.
TABLE 4.
Transduction ratios (log(τ/KA)) of CCL2, UCB35625, and J113863 at CCR2
Data were analyzed by non-linear regression using the operational model equation to determine the transduction ratios (log(τ/KA)). Δlog(τ/KA) ratios were calculated from log(τ/KA) values considering CCL2 as the reference ligand. The RE of ligands toward each pathway, relative to CCL2, corresponds to the inverse logarithm of the Δlog(τ/KA) ratio. Data are the mean ± S.E. of 3–6 experiments. ND, not determined.
| Ligand | Gi2 |
Goa |
Ca2+ |
β-Arrestin 2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log | ΔLog | RE | Log | ΔLog | RE | Log | ΔLog | RE | Log | ΔLog | RE | |
| CCL2 | 8.24 ± 0.22 | 0.00 ± 0.31 | 1 | 8.40 ± 0.29 | 0.00 ± 0.41 | 1 | 9.41 ± 0.08 | 0.00 ± 0.11 | 1 | 8.41 ± 0.09 | 0.00 ± 0.13 | 1 |
| UCB35625 | ND | ND | ND | 5.40 ± 0.46 | −3.13 ± 0.54 | 0.0007a | ND | ND | ND | ND | ND | ND |
| J113863 | 3.42 ± 0.30 | −4.82 ± 0.37 | 0.00002a | 5.02 ± 0.28 | −3.38 ± 0.40 | 0.0004a | 6.44 ± 0.07 | −2.97 ± 0.11 | 0.0011a | 5.82 ± 0.09 | −3.59 ± 0.13 | 0.003a |
a p < 0.05.
TABLE 5.
Bias factors for CCL4, UCB35625, and J113863 at CCR5
ΔΔlog(τ/KA) ratios between the pathways were calculated from the Δlog(τ/KA) values (Table 3). The ligand bias factors (BF), relative to CCL4, correspond to the inverse logarithm of the ΔΔlog(τ/KA) ratios. Data are the mean ± S.E. of 3–6 experiments. ND, not determined.
| Ligand | Gi2/Goa |
Gi2/Ca2+ |
Gi2/arrest |
Goa/Ca2+ |
||||
|---|---|---|---|---|---|---|---|---|
| ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | |
| CCL2 | 0.00 ± 0.69 | 1 | 0.00 ± 0.40 | 1 | 0.00 ± 0.29 | 1 | 0.00 ± 0.40 | 1 |
| UCB35625 | 0.80 ± 0.56 | 6.2 | ND | ND | ND | ND | ND | ND |
| J113863 | 1.84 ± 0.92 | 69.0a | 1.87 ± 0.61 | 74.8a | ND | ND | 0.03 ± 0.61 | 1.1 |
a p < 0.05.
TABLE 6.
Bias factors for CCL2, UCB35625, and J113863 at CCR2
ΔΔlog(τ/KA) ratios between the pathways were calculated from the Δlog(τ/KA) values (Table 4). The ligand bias factors (BF), relative to CCL2, correspond to the inverse logarithm of the ΔΔlog(τ/KA) ratios. Data are the mean ± S.E. of 3–6 experiments. ND, not determined.
| Ligand | Gi2/Goa |
Gi2/Ca2+ |
Gi2/arrest |
Goa/Ca2+ |
Goa/arrest |
Ca2+/arrest |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | ΔΔLog | BF | |
| CCL2 | 0.00 ± 0.51 | 1 | 0.00 ± 0.44 | 1 | 0.00 ± 0.17 | 1 | 0.00 ± 0.43 | 1 | 0.00 ± 0.43 | 1 | 0.00 ± 0.17 | 1 |
| UCB35625 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| J113863 | −1.44 ± 0.55 | 0.04 | −1.85 ± 0.42 | 0.01 | −1.23 ± 0.17 | 0.06 | −0.41 ± 0.42 | 0.39 | 0.21 ± 0.42 | 1.62 | 0.00 ± 0.17 | 1 |
Role of Aromatic Residues within the TM2-TM3 Interface in Receptor Activation
The ability of J113863 to induce cells chemotaxis varies greatly whether it binds to CCR2 or CCR5. Because CCR2 and CCR5 share >90% of sequence identity within their transmembrane segments, we investigated which amino acid variations might be responsible for the different behavior. We tested CCR5 mutants in which aromatic residues of TM2 and TM3 were substituted by the corresponding amino acids of CCR2. Previously, we showed that these aromatic residues at the TM2-TM3 interface played a key role in the activation of CCR5 by chemokines (27). Here, we showed that the F2.39L, Y2.63S, F3.28L, and F3.36Y substitutions significantly decreased the calcium mobilization triggered by CCL4, in agreement with our previous study (Fig. 7A) (see “Numbering Scheme of GPCRs” under “Discussion”). We also showed that these mutations decreased the activation of CCR5 by J113863, indicating that this aromatic cluster is also important for the activation of CCR5 by this chemical compound (Fig. 7B). The F2.39L and F3.28L substitutions also decreased the activity of UCB35625, whereas Y2.63S and F3.28L had no effect, suggesting that the interaction of the two enantiomers with the CCR5 binding pocket is different (Fig. 7C). However, the impact of these mutations on UCB35625 signaling should be interpreted with caution, as the signal did not saturate at 100 μm. In contrast, we showed that the F3.33H substitution strongly increased the activation of CCR5, converting J113863 from a partial agonist to a full agonist. By comparison, the F3.33H mutation did not impact significantly on the activation of CCR5 by CCL4 and inhibited the response triggered by UCB35625. Competition binding assays showed that the F3.33H substitution did not significantly modify the affinity of UCB35625 and J113863 for CCR5 (Fig. 8). We further investigated the functional consequences of the F3.33H substitution and showed that it also drastically increased the chemotaxis of L1.2 cells toward J113863, converting the molecule from an antagonist to a potent agonist (Fig. 9A). By comparison, the F3.33H substitution inhibited the migration of cells triggered by UCB35625 (Fig. 9B). Then, we investigated the impact of the F3.33H mutation on the activation of G proteins and β-arrestin 2. F3.33H strongly increased the activation of Gαi2 and β-arrestin 2 (Fig. 9, C and E) but had no significant impact on the activation of Gαi2 and β-arrestin 2 in response to UCB35625 (Fig. 9, D and F). Taken together, these results indicate that the nature of the residue at position 3.33 in CCR5 is key for the agonist activity of J113863 and the activation of G proteins and β-arrestins.
FIGURE 7.
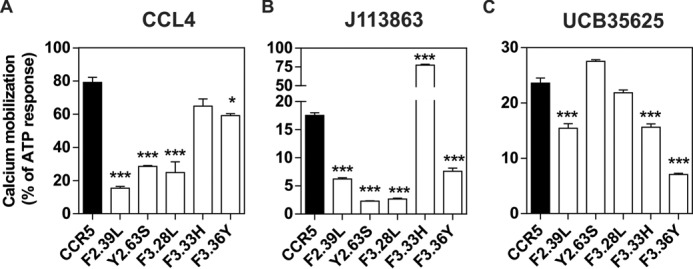
Functional response of CCR5 mutants in which aromatic residues in TM2 and TM3 were substituted by the corresponding amino acids of CCR2. Calcium mobilization measured in cells stably expressing apoaequorin and wild type CCR5 (black bar) or mutant forms of CCR5 bearing F2.39L, Y2.63S, F3.28L, F3.33H, or F3.36Y substitutions. Cells were stimulated either with CCL4 (A), UCB35625 (B), or J113863 (C), and luminescence was recorded for 30 s. The results were normalized for the basal luminescence in absence of agonist (0%) and the maximal response obtained with 50 μm APT (100%). Data represent the mean ± S.E. of three independent experiments. Statistical significance was assessed using Tukey's test (***, p < 0.001; *, p < 0.1).
FIGURE 8.
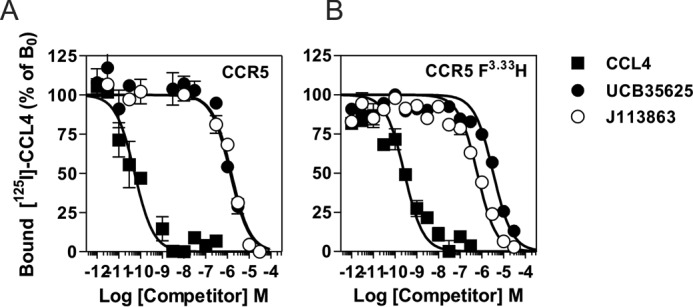
F3.33H substitution does not impact significantly the binding of UCB35625 and J113863 to CCR5. Competition binding assays performed on cells expressing wild type CCR5 (A) or CCR5F3.33H (B) incubated with 0.1 nm 125I-CCL4 as tracer and increasing concentrations of unlabeled CCL4 (■), UCB35625 (●), or J113863 (○) as competitors. The data were normalized for nonspecific binding (0%) in the presence of 300 nm of unlabeled chemokine and specific binding in the absence of competitor (100%).
FIGURE 9.
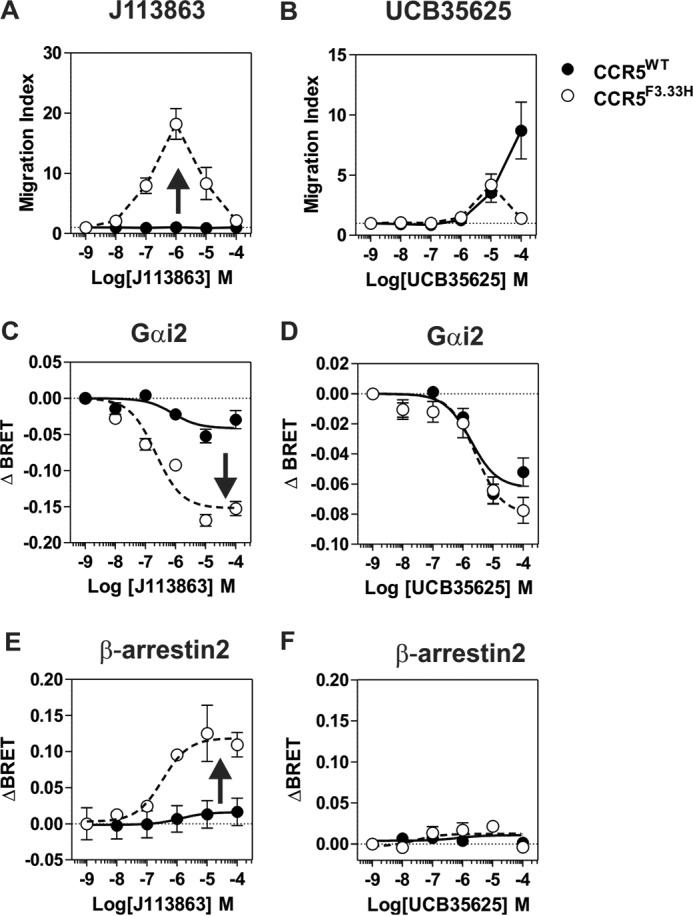
Impact of F3. 33H substitution on cell chemotaxis, G protein activation, and arrestin recruitment. A and B, chemotactic response of L1.2 cells stably expressing CCR5 (●) or CCR5F3.33H (○) in the presence of increasing concentrations of J113863 (A) or UCB35625 (B). The data represent the mean values ± S.E. of three independent experiments. C and D, real-time measurement of BRET signal in HEK293T cells coexpressing the Gi2 biosensor and CCR5 (●) or CCR5F3.33H (○) and stimulated with increasing concentrations of J113863 (C) or UCB35625 (D). Results are expressed as the difference in BRET signal measured in the presence and absence of stimulation. Data represent the mean ± S.E. of three independent experiments. E and F, real-time measurement of BRET signal in HEK293T cells expressing β-arrestin2-RLuc8 and CCR5-Venus (●) or β-arrestin2-RLuc8 and CCR5F3.33H-Venus (○) and stimulated with J113863 (E) or UCB35625 (F). Results are expressed as net BRET, corresponding to the difference in BRET signal between cells expressing arrestin plus the receptor and cells expressing arrestin only. Data represent the mean ± S.E. of three independent experiments.
Discussion
J113863 and its enantiomer UCB35625 are acknowledged as high affinity antagonists of CCR1 and CCR3. By using a combination of assays, we showed here that these enantiomers bind also, although with much lower affinity, to the closely related receptors CCR2 and CCR5. In contrast to CCR1 and CCR3, the enantiomeric ligands were able to activate at least partially CCR2 and CCR5, therefore acting as antagonists or agonists according to the nature of the receptor. Binding of chemokines to CCR2 and CCR5 activated the Gi/o proteins as well as G12, whereas binding of J113863 and UCB35625 only activated Gi/o proteins. Chemokines also induced the recruitment of β-arrestin 2 to CCR2 and CCR5, whereas UCB35625 did not, indicating that the signaling pathways activated by J113863 and UCB35625 differ significantly from those activated by chemokines. We also showed that the efficacy of J113863 as an agonist varied considerably between CCR2 and CCR5. J113863 behaved as a full agonist of CCR2, activating calcium mobilization, β-arrestin 2, and cell migration as efficiently as chemokines, whereas it behaved as a very partial agonist of CCR5, poorly activating calcium mobilization and β-arrestin 2 and not at all cell chemotaxis. Comparison of the bias factors for each ligand relative to a reference chemokine revealed that J113863 showed at CCR5 a significantly bias for Gi2 over Goa or calcium mobilization. It is, however, more difficult to estimate the bias for UCB35625 because of the lack of concentration-response curves at the concentrations of ligand tested. J113863 and UCB35625 activated distinct signaling pathways through CCR2 or CCR5. As said above, binding of J113863 to CCR2 activated Gi2 protein and β-arrestin 2 more efficiently than UCB35625. UCB35625 induced the migration of L1.2 cells expressing CCR5, whereas J113863 antagonized the cell migration triggered by chemokines. These results constitute, to our best knowledge, the first description of chemokine receptor ligands displaying enantioselective properties. Ligands with enantioselective properties have been described for other GPCRs. The μ-opioid receptor ligand levometorphan acts as a narcotic analgesic, whereas dextromethorphan shows no analgesic activity but displays an antitussive action (28). The adrenergic blockers R(+) and S(−) carvedilol are equally effective in blocking α-adrenergic activity, whereas the S(−) enantiomer of carvedilol is 100-fold more potent as an antagonist of β-adrenergic receptors (29–31). More recently, it was also reported that carvedilol antagonizes the activation of Gs protein while stimulating β-arrestin signaling, but detailed properties of S(−) and R(+) enantiomers were not investigated (32). Collectively, these studies show that the racemic nature of compounds targeting GPCRs may be of crucial importance in the frame of biased agonism. This is of therapeutic importance as many drugs, including molecules targeting GPCRs, are marketed as racemates consisting of a mixture of enantiomers (33). The chiral separation of racemic compounds targeting GPCRs might thus be used to identify novel drugs with biased properties and improved therapeutic benefits. Finally, we also showed here that changing a single amino acid of CCR5 by the corresponding amino acid of CCR2 (F3.33H) converts J113863 from an antagonist of cell chemotaxis and a weak agonist in other assays to a full agonist as efficient as chemokines, acting both through G proteins and β-arrestins. In contrast, the F3.33H substitution impacts weakly the activation of G proteins and β-arrestin by UCB35625, suggesting that each enantiomer binds and activates CCR5 in a somehow different manner. However, it is unlikely that the nature of the amino acid at position 3.33 would drive activation in the whole CCR family, as CCR1 and CCR3 contain, respectively, a tyrosine and a histidine in position 3.33. It appears more likely that each chemokine receptor possessed its own set of key residues required for the activation by J113863 and UCB35626. In line with this hypothesis, mutation of tyrosine 3.32 to alanine in CCR1 leads to resistance to UCB35625 blockade, whereas the same mutation in CCR3 converts UCB35625 from an antagonist to a partial agonist (24). Conversion of an antagonist into an agonist has also been reported as a consequence of mutations in TM6 and TM7 of CCR5. Mutation of tryptophan 6.48 to phenylalanine in CCR5 leads to partial resistance to SCH-C blockade, whereas it converts Aplaviroc from an antagonist to an agonist for the Gi pathway (34). Similarly, mutation of glycine 7.42 to phenylalanine also converts Aplaviroc from an antagonist to an agonist (34). Collectively, these data illustrate that the functional properties of synthetic small molecules targeting chemokine receptors are much more complex than initially thought. They also show that the border between agonist and antagonist activities for chemokine receptors is very thin and that single amino acid polymorphisms may drastically change the properties of synthetic molecules.
In summary, we show that J113863 and UCB35625 initially identified as CCR1 and CCR3 antagonists also bind to CCR2 and CCR5 and act as full agonist, partial agonist, or antagonist according to the nature of the receptor, the enantiomer considered, and the signaling pathway investigated. A large number of synthetic molecules targeting GPCRs are currently in use or in clinical trials. However, the classification of these drugs as GPCR blockers or agonists often relies on the use of a restricted number of functional assays, leaving unaddressed their protean nature. Revisiting properties of some of these molecules could, therefore, unveil unexpected biased signaling, with putative influence on their use in vivo.
Experimental Procedures
Materials and Cell Lines
Chemokines and small molecules J113863 and UCB35625 were purchased from BioTechne. TAK779 was provided by the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, NIAID. Plasmids encoding G protein and arrestin constructs were kindly provided by C. Galés (INSERM, Equipe 8, Toulouse, France). Human embryonic kidney cells HEK293T were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). CHO-K1 cells were cultured in Ham's F-12 medium supplemented with 10% fetal bovine serum (GIBCO), 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen). CHO-K1 cells stably expressing apoaequorin, Gα16 and receptors were cultured in presence of 10 μg/ml Zeocin and G418 (Invitrogen). L1.2 cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). L1.2 cells stably expressing receptors were cultured in the presence of 10 μg/ml G418 (Invitrogen).
Numbering Scheme of GPCRs
In this work we used a general numbering scheme identifying residues located at the same position in the transmembrane segments of different receptors (35). Each residue is numbered according to the helix (1–7) in which it is located and to the position relative to the most conserved residue in that helix, arbitrarily assigned to 50. For instance, Phe-3.33 is the phenylalanine in transmembrane helix 3 (TM3) 17 residues before the highly conserved arginine Arg-3.50.
Binding Assays
Binding experiments were performed as previously described (36). CHO-K1 cells were incubated for 45 min at 25 °C in the assay buffer (50 mm HEPES, pH 7.4, 1 mm CaCl2, 5 mm MgCl2, 250 mm sucrose, 0.5% BSA, 0.01% NaN3) with 0.1 nm 125I-CCL2 or -CCL4 as tracers and variable concentrations of unlabeled competitors. Tubes were incubated for 45 min at 25 °C, and bound tracer was separated by filtration through GF/B filters presoaked in 1% polyethyleneimine. Filters were counted in a γ-scintillation counter. Binding parameters were determined with the PRISM software (Graphpad Software) using nonlinear regression applied to a single site model.
G Protein BRET Assay
G protein activation was assayed by BRET as previously described (9). Briefly, plasmids encoding G protein biosensors and receptors of interest were cotransfected into HEK293T cells by using the calcium phosphate method. Forty-eight hours after transfection cells were washed twice with PBS, detached, and resuspended in PBS plus 0.1% (w/v) glucose at room temperature. Cells were then distributed (80 μg of proteins per well) in a 96-well microplate (Optiplate, PerkinElmer Life Sciences). BRET2 between RLuc8 and GFP10 was measured 1 min after the addition of coelenterazine 400a/Deep blue C (5 μm, Gentaur). BRET readings were collected using an Infinite F200 reader (Tecan). The BRET signal was calculated as the ratio of emission of GFP10 (510–540 nm) to RLuc8 (370–450 nm).
β-Arrestin BRET Assay
β-Arrestin recruitment was measured by a BRET proximity assay as previously described (9). Plasmids encoding Rluc-β-arrestin 2 and receptors fused to Venus were cotransfected into HEK293T cells by using the calcium phosphate method. Twenty-four hours post-transfection, cells were collected and seeded in 96-well microplates (165306, Nunc) and cultured for an additional 24 h. Cells were then rinsed once with PBS and incubated in PBS plus 0.1% (w/v) glucose at 25 °C to slow down the kinetics of arrestin recruitment and improve temporal resolution. BRET1 between RLuc and Venus was measured after the addition of coelenterazine h (5 μm, Promega). Ligands were added 5 min after coelenterazine h, and BRET readings were collected using a Mithras LB940 Multilabel Reader (Berthold Technologies). The BRET signal was calculated as the ratio of emission of Venus (520–570 nm) to RLuc (370–480 nm).
Intracellular Calcium Mobilization Assay
Calcium mobilization was measured in CHO-K1 cells stably expressing chemokine receptors. Cells expressing apoaequorin and the receptor of interest were incubated for 4 h in the dark in the presence of 5 μm coelenterazine h (Promega) then diluted before use to reach the appropriate cell density. The cell suspension (25,000 cells/well) was added to wells containing various concentrations of chemokines, and luminescence was recorded for 30 s in an EG&G Berthold luminometer (PerkinElmer Life Sciences).
Chemotaxis Assay
The migration of L1.2 cells stably expressing receptors was performed in 96-well Costar transwell chambers (5 μm pore size, Corning). A suspension of 103 cells in RPMI 1640 supplemented with 10% FCS was added to each insert in a well containing a solution of ligands. Wells containing medium without ligands were used as controls. After 1 h at 37 °C, cells in the bottom of the wells were counted by using the ATPlite luminescence assay kit (PerkinElmer Life Sciences). The results are expressed as chemotaxis index, i.e. the ratio of cells migrating in response to the chemoattractant over cells migrating toward the medium alone.
Bias Analysis
The operational model (26) was used to determine the transduction ratios (τ/KA) of the ligands using Equation 1 as described in van der Westhuizen et al. (37),
| (Eq. 1) |
where E is the effect of the ligand, [A] is the concentration of agonist, Em is the maximal response of the system, Basal is the basal level of the response in the absence of agonist, log KA denotes the logarithm of the functional equilibrium dissociation constant, n is the slope of the transducer function, and log R is the logarithm of the transduction ratio (τ/KA). The relative efficacy (RE) of a ligand (lig) to activate signaling pathway relative to a reference agonist (ref) was calculated by using Equations 2 and 3.
| (Eq. 2) |
and
| (Eq. 3) |
The bias index (BF) of a ligand through pathway a and pathway b relative to a reference agonist was calculated using Equations 4 and 5 as the difference between the Δlog(τ/KA) values derived from Equation 2.
| (Eq. 4) |
and
| (Eq. 5) |
Statistical analyses were performed where appropriate using one-way analysis of variance with Dunnett's post-test test, and statistical significance related to the reference balanced ligand was taken as p < 0.05.
Author Contributions
J.-Y. S. conceived and designed the experiments. J. C., A. H., and J.-Y. S. performed the experiments. J. C., C. G., M. P., and J.-Y. S. analyzed the data. MP and J.-Y. S. wrote the paper.
The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptor
- RANTES
- regulated on activation normal T cell expressed and secreted
- BRET
- bioluminescence resonance energy transfer
- BF
- bias factor
- RE
- relative efficacy.
References
- 1. Whalen E. J., Rajagopal S., and Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kenakin T. (2011) Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302 [DOI] [PubMed] [Google Scholar]
- 3. Luttrell L. M., Maudsley S., and Bohn L. M. (2015) Fulfilling the pROMISE of “biased” G protein-coupled receptor agonism. Mol. Pharmacol. 88, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kenakin T., and Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug. Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 5. DeWire S. M., Yamashita D. S., Rominger D. H., Liu G., Cowan C. L., Graczyk T. M., Chen X. T., Pitis P. M., Gotchev D., Yuan C., Koblish M., Lark M. W., and Violin J. D. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344, 708–717 [DOI] [PubMed] [Google Scholar]
- 6. Soergel D. G., Subach R. A., Cowan C. L., Violin J. D., and Lark M. W. (2013) First clinical experience with TRV027: pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 53, 892–899 [DOI] [PubMed] [Google Scholar]
- 7. Soergel D. G., Subach R. A., Burnham N., Lark M. W., James I. E., Sadler B. M., Skobieranda F., Violin J. D., and Webster L. R. (2014) Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835 [DOI] [PubMed] [Google Scholar]
- 8. Violin J. D., DeWire S. M., Yamashita D., Rominger D. H., Nguyen L., Schiller K., Whalen E. J., Gowen M., and Lark M. W. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 [DOI] [PubMed] [Google Scholar]
- 9. Corbisier J., Galès C., Huszagh A., Parmentier M., and Springael J. Y. (2015) Biased signaling at chemokine receptors. J. Biol. Chem. 290, 9542–9554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rajagopal S., Bassoni D. L., Campbell J. J., Gerard N. P., Gerard C., and Wehrman T. S. (2013) Biased agonism as a mechanism for differential signaling by chemokine receptors. J. Biol. Chem. 288, 35039–35048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steen A., Larsen O., Thiele S., and Rosenkilde M. M. (2014) Biased and g protein-independent signaling of chemokine receptors. Front. Immunol. 5, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonecchi R., Galliera E., Borroni E. M., Corsi M. M., Locati M., and Mantovani A. (2009) Chemokines and chemokine receptors: an overview. Front. Biosci. (Landmark Ed.) 14, 540–551 [DOI] [PubMed] [Google Scholar]
- 13. Ajram L., Begg M., Slack R., Cryan J., Hall D., Hodgson S., Ford A., Barnes A., Swieboda D., Mousnier A., and Solari R. (2014) Internalization of the chemokine receptor CCR4 can be evoked by orthosteric and allosteric receptor antagonists. Eur. J. Pharmacol. 729, 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Viney J. M., Andrew D. P., Phillips R. M., Meiser A., Patel P., Lennartz-Walker M., Cousins D. J., Barton N. P., Hall D. A., and Pease J. E. (2014) Distinct conformations of the chemokine receptor CCR4 with implications for its targeting in allergy. J. Immunol. 192, 3419–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonner K., Pease J. E., Corrigan C. J., Clark P., and Kay A. B. (2013) CCL17/thymus and activation-regulated chemokine induces calcitonin gene-related peptide in human airway epithelial cells through CCR4. J. Allergy Clin. Immunol. 132, 942–950 [DOI] [PubMed] [Google Scholar]
- 16. Zohar Y., Wildbaum G., Novak R., Salzman A. L., Thelen M., Alon R., Barsheshet Y., Karp C. L., and Karin N. (2014) CXCL11-dependent induction of FOXP3-negative regulatory T cells suppresses autoimmune encephalomyelitis. J. Clin. Invest. 124, 2009–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simmons G., Clapham P. R., Picard L., Offord R. E., Rosenkilde M. M., Schwartz T. W., Buser R., Wells T. N., and Proudfoot A. E. (1997) Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276, 276–279 [DOI] [PubMed] [Google Scholar]
- 18. Mack M., Luckow B., Nelson P. J., Cihak J., Simmons G., Clapham P. R., Signoret N., Marsh M., Stangassinger M., Borlat F., Wells T. N., Schlöndorff D., and Proudfoot A. E. (1998) Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J. Exp. Med. 187, 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaertner H., Lebeau O., Borlat I., Cerini F., Dufour B., Kuenzi G., Melotti A., Fish R. J., Offord R., Springael J. Y., Parmentier M., and Hartley O. (2008) Highly potent HIV inhibition: engineering a key anti-HIV structure from PSC-RANTES into MIP-1 β/CCL4. Protein Eng. Des. Sel. 21, 65–72 [DOI] [PubMed] [Google Scholar]
- 20. Saita Y., Kodama E., Orita M., Kondo M., Miyazaki T., Sudo K., Kajiwara K., Matsuoka M., and Shimizu Y. (2006) Structural basis for the interaction of CCR5 with a small molecule, functionally selective CCR5 agonist. J. Immunol. 177, 3116–3122 [DOI] [PubMed] [Google Scholar]
- 21. Ferain T., Hoveyda H., Ooms F., Schols D., Bernard J., and Fraser G. (2011) Agonist-induced internalization of CC chemokine receptor 5 as a mechanism to inhibit HIV replication. J. Pharmacol. Exp. Ther. 337, 655–662 [DOI] [PubMed] [Google Scholar]
- 22. Sabroe I., Peck M. J., Van Keulen B. J., Jorritsma A., Simmons G., Clapham P. R., Williams T. J., and Pease J. E. (2000) A small molecule antagonist of chemokine receptors CCR1 and CCR3: Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J. Biol. Chem. 275, 25985–25992 [DOI] [PubMed] [Google Scholar]
- 23. de Mendonça F. L., da Fonseca P. C., Phillips R. M., Saldanha J. W., Williams T. J., and Pease J. E. (2005) Site-directed mutagenesis of CC chemokine receptor 1 reveals the mechanism of action of UCB 35625, a small molecule chemokine receptor antagonist. J. Biol. Chem. 280, 4808–4816 [DOI] [PubMed] [Google Scholar]
- 24. Wise E. L., Duchesnes C., da Fonseca P. C., Allen R. A., Williams T. J., and Pease J. E. (2007) Small molecule receptor agonists and antagonists of CCR3 provide insight into mechanisms of chemokine receptor activation. J. Biol. Chem. 282, 27935–27943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amat M., Benjamim C. F., Williams L. M., Prats N., Terricabras E., Beleta J., Kunkel S. L., and Godessart N. (2006) Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br. J. Pharmacol. 149, 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Black J. W., and Leff P. (1983) Operational models of pharmacological agonism. Proc. R Soc. Lond B Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 27. Govaerts C., Bondue A., Springael J. Y., Olivella M., Deupi X., Le Poul E., Wodak S. J., Parmentier M., Pardo L., and Blanpain C. (2003) Activation of CCR5 by chemokines involves an aromatic cluster between transmembrane helices 2 and 3. J. Biol. Chem. 278, 1892–1903 [DOI] [PubMed] [Google Scholar]
- 28. Cooper M. J., and Anders M. W. (1974) Metabolic and pharmacodynamic interactions of enantiomers of propoxyphene and methorphan. Life Sci. 15, 1665–1672 [DOI] [PubMed] [Google Scholar]
- 29. Nichols A. J., Sulpizio A. C., Ashton D. J., Hieble J. P., and Ruffolo R. R. Jr. (1989) The interaction of the enantiomers of carvedilol with α1- and β1-adrenoceptors. Chirality 1, 265–270 [DOI] [PubMed] [Google Scholar]
- 30. Bartsch W., Sponer G., Strein K., Müller-Beckmann B., Kling L., Böhm E., Martin U., and Borbe H. O. (1990) Pharmacological characteristics of the stereoisomers of carvedilol. Eur. J. Clin. Pharmacol. 38, S104–S107 [DOI] [PubMed] [Google Scholar]
- 31. Mehvar R., and Brocks D. R. (2001) Stereospecific pharmacokinetics and pharmacodynamics of β-adrenergic blockers in humans. J. Pharm. Pharm. Sci. 4, 185–200 [PubMed] [Google Scholar]
- 32. Kim I. M., Tilley D. G., Chen J., Salazar N. C., Whalen E. J., Violin J. D., and Rockman H. A. (2008) β-blockers alprenolol and carvedilol stimulate β-arrestin-mediated EGFR transactivation. Proc. Natl. Acad. Sci. U.S.A. 105, 14555–14560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rentsch K. M. (2002) The importance of stereoselective determination of drugs in the clinical laboratory. J. Biochem. Biophys. Methods 54, 1–9 [DOI] [PubMed] [Google Scholar]
- 34. Steen A., Thiele S., Guo D., Hansen L. S., Frimurer T. M., and Rosenkilde M. M. (2013) Biased and constitutive signaling in the CC-chemokine receptor CCR5 by manipulating the interface between transmembrane helices 6 and 7. J. Biol. Chem. 288, 12511–12521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ballesteros J. A., and Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations of G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 36. El-Asmar L., Springael J. Y., Ballet S., Andrieu E. U., Vassart G., and Parmentier M. (2005) Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol. Pharmacol. 67, 460–469 [DOI] [PubMed] [Google Scholar]
- 37. van der Westhuizen E. T., Breton B., Christopoulos A., and Bouvier M. (2014) Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509 [DOI] [PubMed] [Google Scholar]