

Abstract
Acyl-CoA binding domain-containing 5 (ACBD5) is a peroxisomal protein that carries an acyl-CoA binding domain (ACBD) at its N-terminal region. The recent identification of a mutation in the ACBD5 gene in patients with a syndromic form of retinal dystrophy highlights the physiological importance of ACBD5 in humans. However, the underlying pathogenic mechanisms and the precise function of ACBD5 remain unclear. We herein report that ACBD5 is a peroxisomal tail-anchored membrane protein exposing its ACBD to the cytosol. Using patient-derived fibroblasts and ACBD5 knock-out HeLa cells generated via genome editing, we demonstrate that ACBD5 deficiency causes a moderate but significant defect in peroxisomal β-oxidation of very-long-chain fatty acids (VLCFAs) and elevates the level of cellular phospholipids containing VLCFAs without affecting peroxisome biogenesis, including the import of membrane and matrix proteins. Both the N-terminal ACBD and peroxisomal localization of ACBD5 are prerequisite for efficient VLCFA β-oxidation in peroxisomes. Furthermore, ACBD5 preferentially binds very-long-chain fatty acyl-CoAs (VLC-CoAs). Together, these results suggest a direct role of ACBD5 in peroxisomal VLCFA β-oxidation. Based on our findings, we propose that ACBD5 captures VLC-CoAs on the cytosolic side of the peroxisomal membrane so that the transport of VLC-CoAs into peroxisomes and subsequent β-oxidation thereof can proceed efficiently. Our study reclassifies ACBD5-related phenotype as a novel peroxisomal disorder.
Keywords: genetic disease, lipid metabolism, neurological disease, peroxisome, retinal degeneration, acyl-CoA-binding protein, fatty acid β-oxidation, retinal dystrophy
Introduction
Peroxisomes are ubiquitous single membrane-bounded organelles that play an indispensable role in multiple metabolic pathways, including fatty acid α- and β-oxidation, ether lipid synthesis, docosahexaenoic acid (DHA)2 synthesis, and hydrogen peroxide degradation (1). The functional significance of peroxisomes for human health is manifested by the fact that mutations in human genes encoding peroxisomal proteins lead to a spectrum of inherited peroxisomal disorders (2). Patients with peroxisomal disorders typically develop neurological abnormalities, and the clinical severity ranges from a fatal developmental disorder to a mild degenerative disorder with symptoms such as blindness, hearing impairment, and ataxia, depending on the gene responsible and the nature of the mutation (3).
Peroxisomal disorders can be classified into peroxisome biogenesis disorders (PBDs) and single peroxisomal enzyme deficiencies (SEDs). PBDs, including Zellweger syndrome, are caused by defects in the formation and/or maintenance of functional peroxisomes, and consequently patients with PBDs usually lack the entire peroxisomal metabolic functions (4, 5). The vast majority of PBDs are associated with mutations in PEX genes encoding peroxins, most of which are involved in the import of peroxisomal proteins (5, 6). Conversely, the primary defect in SEDs resides in the peroxisomal matrix enzymes or peroxisomal membrane proteins that mediate metabolite transport. Therefore, specific peroxisome-dependent metabolic pathways are affected in patients with each of the SEDs. Well known SEDs affecting peroxisomal fatty acid β-oxidation pathway include X-linked adrenoleukodystrophy (X-ALD) caused by mutations in the ABCD1 gene (7–9), acyl-CoA oxidase 1 (AOx) deficiency caused by mutations in the AOx gene (10, 11), and D-bifunctional protein deficiency caused by mutations in the HSD17B4 gene (12, 13).
Recently, a homozygous truncating mutation in the ACBD5 gene encoding acyl-CoA binding domain-containing 5 (ACBD5) was reported in a family with retinal dystrophy and severe neurological involvement (Ref. 14; Online Mendelian Inheritance in Man (OMIM) number 616618). ACBD5 is one of the seven members of the mammalian acyl-CoA binding domain-containing protein (ACBP) family characterized by an N-terminal acyl-CoA binding domain (ACBD) (15). Previous proteomic studies using isolated mammalian peroxisomes have identified ACBD5 as a peroxisomal protein (16, 17). Therefore, the newly described ACBD5 deficiency could be considered as a novel peroxisomal disease. Indeed, the three siblings with ACBD5 deficiency also developed white matter disease and spastic paraparesis (14), which are frequently observed in patients with peroxisomal disorders. However, the mechanisms that link ACBD5 deficiency to such a syndromic form of retinal dystrophy remain undefined.
ACBD5 was recently suggested to be involved in basal autophagic degradation of peroxisome, namely pexophagy, in mammalian cell culture (18); however, the precise function of ACBD5 is not fully defined. Because ACBD5 harbors a conserved ACBD at the N terminus, it can be anticipated that ACBD5 exerts its function(s) through binding to acyl-CoAs and that ACBD5 functions in a peroxisome-dependent lipid metabolic pathway. Here, we demonstrate that ACBD5 is a peroxisomal tail-anchored protein exposing its N-terminal ACBD to the cytosol. ACBD5 is required for achieving efficient very-long-chain fatty acid (VLCFA) β-oxidation, and ACBD5 deficiency leads to accumulation of cellular phospholipids containing VLCFAs. Importantly, the N-terminal ACBD and peroxisomal localization of ACBD5 are prerequisite for its function. Moreover, ACBD5 likely binds preferentially very-long-chain fatty acyl-CoAs (VLC-CoAs). Based on our data, we propose that ACBD5 captures VLC-CoAs on the cytosolic side of the peroxisomal membrane to secure efficient transport of VLC-CoAs into peroxisomes for subsequent VLCFA β-oxidation. Our findings provide a mechanistic explanation of how ACBD5 deficiency results in the pathogenesis of retinal dystrophy and associated neurological deficits in patients with ACBD5-related phenotype.
Results
ACBD5 Is a Peroxisomal Tail-anchored Protein Exposing Its Acyl-CoA Binding Domain to the Cytosol
Previous proteomic studies of isolated mammalian peroxisomes identified ACBD5 as a peroxisomal protein (16, 17), and immunofluorescence microscopy of HeLa cells confirmed the peroxisomal localization of endogenous ACBD5 (Fig. 1A and Ref. 18). On the basis of its primary sequence, ACBD5 is predicted to possess a transmembrane domain (TMD) close to the C terminus (Fig. 1B). Therefore, we first investigated whether ACBD5 is an integral membrane protein by subcellular fractionation followed by an alkaline extraction method (19). Consistent with its peroxisomal localization, endogenous ACBD5 was recovered exclusively in the organelle-containing fraction of HeLa cells (Fig. 2A). Upon alkaline extraction, ACBD5 behaved in the same manner as PEX14, a peroxisomal membrane protein, but not AOx, a peroxisomal matrix protein (Fig. 2B), indicating that ACBD5 is a bona fide peroxisomal membrane protein.
FIGURE 1.
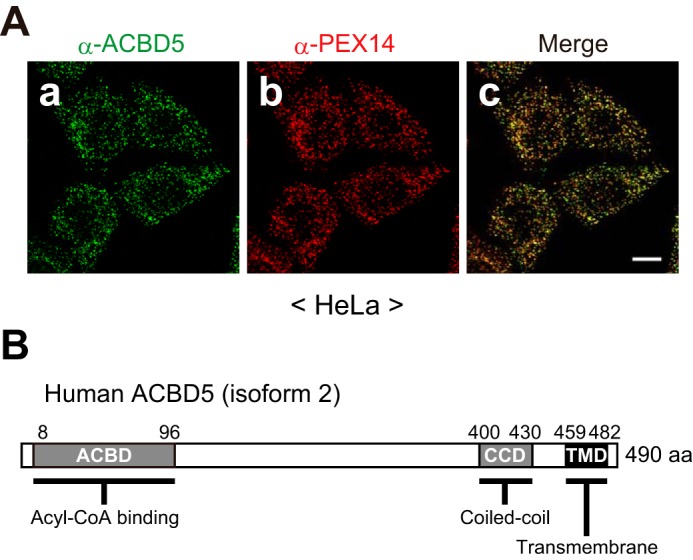
ACBD5 localizes to peroxisomes. A, panels a–c, HeLa cells were immunostained with anti-ACBD5 and anti-PEX14 antibodies. A merged view is also shown. Scale bar, 10 μm. B, schematic representation of the domain structure of ACBD5. Numbers, amino acid positions of human ACBD5 isoform 2 (NCBI reference sequence NP_001035938.1); ACBD, NCBI conserved domain cd00435; CCD, predicted by Paircoil2; TMD, predicted by TMpred.
FIGURE 2.

ACBD5 is a peroxisomal tail-anchored protein. A, HeLa cells were fractionated into total (T; lane 1), cytosolic (C; lane 2), and organelle-containing (O; lane 3) fractions, and equal aliquots of the fractions were subjected to SDS-PAGE and immunoblotting using the indicated antibodies. B, the organelle fraction of HeLa cells (A, lane 3) was treated with 0.1 m Na2CO3 and then separated into soluble (S; lane 2) and membrane (P, pellet; lane 3) fractions. Equal aliquots of each fraction, including total (T; lane 1), were analyzed by immunoblotting with the indicated antibodies. C, HeLa cells expressing FLAG-ACBD5-HA were fixed and immunostained with the indicated antibodies after permeabilization with 1% Triton X-100 (panels a–c) or 50 μg/ml digitonin (panels d–f). D, schematic representation of EGFP-fused human ACBD5 and its variants. Numbers, amino acid positions of human ACBD5 isoform 2 (NCBI reference sequence NP_001035938.1). E, panels a–l, the EGFP-ACBD5 variants shown in D were expressed in HeLa cells and assessed for intracellular localization. EGFP-fused proteins and peroxisomes were detected by EGFP fluorescence and immunostaining with the anti-PTS1 antibody, respectively. Merged views are also shown. F, HeLa cells expressing (lanes 1–3) or not expressing (lanes 4–6) EGFP-ACBD5C40 were fractionated and analyzed as in A. G, the organelle-containing fraction of HeLa cells expressing EGFP-ACBD5C40 (F, lane 3) was subjected to alkaline extraction as in B, and the fractions were likewise verified by immunoblotting. LDH, lactate dehydrogenase, a cytosolic protein; PEX14 and PEX13, peroxisomal membrane proteins; AOx and catalase, peroxisomal matrix enzymes. Of the three chains of AOx (75-kDa A, 52-kDa B, and 23-kDa C), only the AOx-B chain is shown. Scale bars in C and E, 10 μm.
Next, we analyzed the membrane topology of ACBD5 by means of a differential permeabilization technique. HeLa cells transiently expressing N-terminally FLAG-tagged and C-terminally HA-tagged ACBD5 (FLAG-ACBD5-HA) were immunostained with antibodies against FLAG tag, HA tag, and catalase after full permeabilization of cellular membranes with Triton X-100 or selective permeabilization of the plasma membrane with digitonin. A punctate staining pattern of intraperoxisomal catalase was detected in Triton X-100-permeabilized cells but not in digitonin-permeabilized cells, thereby showing that the peroxisomal membrane remained intact in digitonin-permeabilized cells (Fig. 2C, panels c and f). In Triton X-100-permeabilizaed cells, both the N-terminal FLAG tag and the C-terminal HA tag were detected in a similar punctate pattern that coincided with catalase (Fig. 2C, panels a–c). In contrast, in digitonin-permeabilized cells where antibodies could not access intraperoxisomal epitopes, only the N-terminal FLAG tag was detectable (Fig. 2C, panels d and e). These results strongly suggest that the N-terminal region of ACBD5 is exposed to the cytosol whereas the C-terminal tail is translocated and faces the peroxisomal matrix.
Based on its membrane topology, we predicted that ACBD5 would belong to the tail-anchored protein family, which is characterized by a cytosol-facing N-terminal domain and a single C-terminal TMD (20). In general, the targeting information of tail-anchored proteins resides solely within their C-terminal membrane-anchoring region (21). To test whether the C-terminal region of ACBD5 mediates both peroxisomal targeting and membrane anchoring, we generated several EGFP-fused ACBD5 variants (Fig. 2D) and determined their localization in HeLa cells. As expected, wild-type (WT) EGFP-ACBD5 colocalized with intraperoxisomal proteins harboring peroxisomal targeting signal type 1 (PTS1) (Fig. 2E, panels a–c). EGFP-ACBD5C40 consisting of the C-terminal 40 amino acids (aa) exhibited a similar peroxisomal localization, whereas EGFP-ACBD5ΔC lacking the C-terminal 33 aa did not localize to peroxisomes and rather was detected in cytoplasmic reticular and/or tubular structures (Fig. 2E, panels d–i). The observed localization of EGFP-ACBD5ΔC was unexpected because similar C-terminal deletion mutants of tail-anchored proteins generally show a typical cytosolic localization. Apparently, the localization of EGFP-ACBD5ΔC to cytoplasmic structures was dependent on the coiled coil domain (CCD); a further deletion of the CCD gave rise to a cytosolic localization (ΔCΔCCD; Fig. 2E, panels j–l). Although the physiological significance of this observation was unclear, our data implied that EGFP-ACBD5ΔC failed to reach peroxisomes but associated with cytoplasmic structures via the CCD region. Notably, in subcellular fractionation and alkaline extraction analyses, EGFP-ACBD5C40 showed the same behavior as a peroxisomal membrane protein, PEX13 (Fig. 2, F and G). Therefore, these results demonstrate that the C-terminal TMD-containing region of ACBD5 functions as the signal-anchor sequence for peroxisomal localization. Taken together, we concluded that ACBD5 is a peroxisomal tail-anchored membrane protein and exposes its ACBD to the cytosol.
ACBD5 Is Dispensable for Peroxisome Biogenesis
We next determined whether ACBD5 plays a role in peroxisome biogenesis using human skin fibroblasts derived from a patient with ACBD5 deficiency (ΔACBD5; Ref. 14). Immunofluorescence microscopy revealed that endogenous ACBD5 colocalized with PEX14 in control fibroblasts (derived from a healthy donor), but it was completely absent in ΔACBD5 fibroblasts, thus confirming the complete inactivation of the gene by the previously reported homozygous truncating mutation (Fig. 3A). Remarkably, PEX14-positive punctate structures were observed in ΔACBD5 fibroblasts (Fig. 3A, panel d), implying that the biogenesis of peroxisomal membranes is not affected in ΔACBD5 fibroblasts. To further assess the biogenesis of peroxisomes, the fibroblasts were immunostained for other peroxisomal proteins, including a membrane protein, PMP70; PTS1-harboring matrix enzymes AOx and catalase; and a PTS2-horboring matrix enzyme, alkyldihydroxyacetone phosphate synthase (ADAPS). Here, fibroblasts derived from a PEX10-defective patient with Zellweger syndrome (ΔPEX10) in which peroxisomal matrix protein import is abrogated were also used as an additional control. Results showed that all the peroxisomal proteins tested were imported to punctate structures in ΔACBD5 fibroblasts (Fig. 3B), suggesting that peroxisomal membrane and matrix protein import machineries are not affected in ΔACBD5 fibroblasts. Consistently, the AOx-B chain and the mature form of ADAPS, both of which are proteolytically processed after the import into peroxisomes (22, 23), were detected by immunoblotting analysis in cell lysates of control and ΔACBD5 fibroblasts but not in that of ΔPEX10 fibroblasts (Fig. 3C). Thus, our results indicate that the import of peroxisomal membrane and matrix proteins operates normally in ΔACBD5 fibroblasts and thereby that ACBD5 is dispensable for the biogenesis of peroxisomes.
FIGURE 3.

ACBD5 is dispensable for peroxisome biogenesis. A, fibroblasts derived from a healthy donor (control) and an ACBD5-deficinet patient (ΔACBD5) were immunostained with anti-ACBD5 and anti-PEX14 antibodies. B, fibroblasts from a healthy donor (control; left panels), an ACBD5-deficinet patient (ΔACBD5; middle panels), and a PEX10-deficient patient (ΔPEX10; right panels) were immunostained with antibodies against PMP70 (panels a–c), catalase (panels d–f), AOx (panels g–i), or ADAPS (panels j–l). Cell nuclei were counterstained with Hoechst 33342. C, total cell lysates prepared from control (lane 1), ΔACBD5 (lane 2), and ΔPEX10 (lane 3) fibroblasts were analyzed by SDS-PAGE and immunoblotting using the indicated antibodies. Lamin A/C was used as a loading control. For AOx, only the AOx-B chain is shown. Scale bars in A and B, 20 μm.
Peroxisomal β-Oxidation of Very-long-chain Fatty Acids Is Impaired in ACBD5-deficient Human Skin Fibroblasts
Given that ACBD5 possess a conserved ACBD and that peroxisomes play a prominent role in multiple anabolic and catabolic processes of lipid metabolism, we anticipated that ACBD5 deficiency might influence cellular lipid metabolism. To verify this notion, we performed lipidomic analysis (24) and compared the phospholipid composition of ΔACBD5 fibroblasts with those of fibroblasts derived from a healthy donor (control), a PEX10-deficient patient (ΔPEX10), and patients with two types of peroxisomal β-oxidation deficiency, AOx deficiency (ΔAOx) and X-ALD (ΔABCD1), respectively.
The β-oxidation of VLCFAs is one of the major functions of peroxisomes (2). Notably, laboratory investigations on the ACBD5-deficient patient revealed a slight but distinct perturbation of VLCFA ratios in plasma (Table 1). We therefore focused on the total amount of phosphatidylcholine (PtdCho) species containing VLCFAs with total carbon numbers of 42–48 (VLC-PtdCho). Consistent with our previous report (24), the total amount of VLC-PtdCho was markedly elevated in ΔPEX10 and ΔAOx fibroblasts and was subtly increased in ΔABCD1 fibroblasts (Fig. 4A). Similar to ΔABCD1 fibroblasts, ΔACBD5 fibroblasts exhibited a slight but significant elevation in the total amount of VLC-PtdCho species (Fig. 4A). By contrast, the total amounts of PtdCho species containing medium- and long-chain fatty acids with total carbon numbers of 30–40 were comparable among all the fibroblasts analyzed (Fig. 4B). We have previously shown that almost all VLC-PtdCho species are elevated in peroxisome-deficient and ΔAOx fibroblasts, whereas only the VLC-PtdCho species containing unsaturated, monosaturated, and disaturated VLCFAs accumulate in ΔABCD1 fibroblasts (24). In this context, ΔACBD5 fibroblasts showed accumulation of broad VLC-PtdCho species irrespective of the number of double bonds (Fig. 4, C and D). To understand the mechanism underlying the accumulation of VLC-PtdCho species in ΔACBD5 fibroblasts, we then measured the peroxisomal VLCFA β-oxidation activity and found that the β-oxidation activity toward lignoceric acid (C24:0) in ΔACBD5 fibroblasts was reduced to ∼65% of control fibroblasts (Fig. 4E). Notably, the extent of reduction in the C24:0 β-oxidation activity was milder in ΔACBD5 fibroblasts as compared with that in ΔAOx fibroblasts (Fig. 4E), suggesting that the extent of deficiency in the peroxisomal VLCFA β-oxidation mirrors the level of accumulation of VLC-PtdCho species in these patient-derived fibroblasts.
TABLE 1.
Peroxisomal metabolism in this ACBD5-deficient patient
| Value | Ref. value | |
|---|---|---|
| Phytanic acid | 0.34 μm | <12.01 μm |
| Pristanic acid | 0.25 μm | <2.98 μm |
| C22 | 20.5 μm | 40–119 μm |
| C24 | 20 μm | 33–84 μm |
| C26 | 0.65 μm | 0.45–1.32 μm |
| C24/22 | 0.976 | <1.2 |
| C26/22 | 0.032 | <0.028 |
FIGURE 4.

ACBD5-deficient fibroblasts show a mild accumulation of cellular VLC-PtdCho species and a decreased activity in peroxisomal VLCFA β-oxidation. A, cellular levels of VLC-PtdCho in control and the indicated patient-derived fibroblasts were analyzed by LC-ESI-MS/MS. The inset graph shows a detailed comparison among control, ΔACBD5, and ΔABCD1 fibroblasts. B, cellular levels of PtdCho species with carbon numbers from 30 to 40 in the indicated fibroblasts were likewise analyzed by LC-ESI-MS/MS. C and D, VLC-PtdCho species were classified based on the number of double bonds. The amounts of VLC-PtdCho with ≤2 double bonds (C) or ≥3 double bonds (D) are shown for the indicated fibroblasts. E, peroxisomal VLCFA β-oxidation activities in control, ΔACBD5, and ΔAOx fibroblasts were assessed using [14C]lignoceric acid (C24:0) as the substrate. Data shown are the mean of triplicate samples from a representative experiment and are expressed as the percentage of the average activity in control fibroblasts (2.15 nmol/h/mg of protein; set to 100%). F and G, cellular levels of PlsEtn species (F) and DHA phospholipids (DHA-PLs) (G) in the indicated fibroblasts were analyzed by LC-ESI-MS/MS. The DHA phospholipid levels are presented relative to the value in control fibroblasts. Date shown in A–D, F, and G are the mean of four independent experiments. Error bars in A–G represent S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus control fibroblasts; one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test.
Thus, it is most likely that ACBD5 deficiency causes a moderate defect in the peroxisomal β-oxidation of a wide variety of VLCFAs. To examine whether ACBD5 deficiency influences other lipid metabolic processes involving peroxisomal enzymes, we also analyzed the levels of plasmenylethanolamine (PlsEtn), which harbors a vinyl ether-linked long-chain fatty alcohol at its sn-1 position, and phospholipids containing DHA (C22:6n-3). The biosynthesis of PlsEtn is initiated in peroxisomes (25, 26), whereas the biosynthesis of DHA from dietary linolenic acid (C18:3n-3) is completed by peroxisomal β-oxidation (27). Apparently, neither the level of PlsEtn nor that of DHA-containing phospholipids was affected in ΔACBD5 fibroblasts (Fig. 4, F and G). Collectively, our results suggest that ACBD5 deficiency has no impact on either the biosynthesis of PlsEtn or the biosynthesis of DHA and that ACBD5 is involved in the β-oxidation of VLCFAs.
ACBD5 Is Necessary for Efficient Peroxisomal β-Oxidation of VLCFAs
The phenotype of human skin fibroblasts derived from an ACBD5-defective patient suggested a role of ACBD5 as a mediator of peroxisomal VLCFA β-oxidation (Fig. 4). To further assess the importance of ACBD5 in the peroxisomal VLCFA β-oxidation by making use of cells derived from the same genetic background, we next generated ACBD5 knock-out (KO) HeLa cells via the CRISPR/Cas9 gene-editing system. Using two independent guide RNAs (gRNAs; supplemental Fig. S1B), we established two ACBD5-KO clonal cell lines, termed ACBD5-KO1 and ACBD5-KO2, in which ACBD5 protein was not detectable (Fig. 5, A and B; and supplemental Fig. S1B). In both ACBD5-KO1 and ACBD5-KO2 cells, we detected numerous catalase-positive punctate structures (Fig. 5A) and essentially identical amounts of the AOx-B chain as compared with WT cells (Fig. 5, B and C). These results are consistent with our findings obtained with ΔACBD5 fibroblasts and further support the conclusion that ACBD5 is dispensable for peroxisome biogenesis involving peroxisomal import of membrane and matrix proteins.
FIGURE 5.

ACBD5-KO HeLa cells manifest a partial defect in peroxisomal VLCFA β-oxidation. A, WT (panels a–c), ACBD5-KO1 (panels d–f), and ACBD5-KO2 (panels g–i) HeLa cells were immunostained with antibodies against ACBD5 and catalase. Merged views are also shown. Scale bar, 10 μm. B, total cell lysates of WT and ACBD5-KO HeLa cells were analyzed by immunoblotting with the indicated antibodies. For AOx, only the AOx-B chain is shown. C, the relative AOx-B chain levels in B were quantified by densitometry and normalized to tubulin and are shown as the ratio to WT HeLa cells. Data represent the mean of three independent experiments. D, the relative VLCFA β-oxidation activities in WT and ACBD5-KO HeLa cells were assessed as in Fig. 4E. Data shown are the mean of triplicate samples from a representative experiment and are expressed as the percentage relative to the activity in WT HeLa cells. E and F, the cellular levels of total VLC-PtdCho (E) or PtdCho species with carbon numbers of 46 and 48 (VLC-PtdCho with C46 & C48; F) in WT and ACBD5-KO HeLa cells were analyzed by LC-ESI-MS/MS. Data represent the mean of three independent experiments. Error bars in C–F represent S.D. *, p < 0.05; ***, p < 0.001 versus WT HeLa cells; one-way ANOVA with Dunnett's multiple comparison test.
We next evaluated the peroxisomal VLCFA β-oxidation activity in these ACBD5-KO cell lines as well as the parental WT HeLa cells. The activities of C24:0 β-oxidation in ACBD5-KO1 and ACBD5-KO2 cells were significantly decreased as compared with WT HeLa cells (Fig. 5D), thereby suggesting that a reduced level of the peroxisomal VLCFA β-oxidation activity is a general consequence of ACBD5 deficiency. We also analyzed the cellular VLC-PtdCho levels in the ACBD5-KO cell lines. Under our cell culture conditions, the level of VLC-PtdCho species in HeLa cells was lower at about 0.2 nmol/mg of protein (Fig. 5E, WT) than those in human skin fibroblasts showing about 1.3 nmol/mg of protein (Fig. 4A, control). Unlike in ΔACBD5 human skin fibroblasts, the levels of total VLC-PtdCho species in ACBD5-KO1 and ACBD5-KO2 cells were comparable with those in WT HeLa cells (Fig. 5E). Notably, however, a further detailed analysis revealed significant elevation of the level of VLC-PtdCho species containing total carbon numbers of 46 and 48 in the ACBD5-KO cell lines (Fig. 5F).
Given that ACBD5-KO1 and ACBD5-KO2 cells were generated with different gRNAs, the observed effects of ACBD5-KO could be considered to be “on-target,” not “off-target”. Nonetheless, to further rule out potential off-target effects of the CRISPR/Cas9 system-mediated ACBD5-KO, we stably expressed HA-tagged ACBD5 (HA-ACBD5) in ACBD5-KO1 cells and verified the C24:0 β-oxidation activity. We selected two independent clones in which HA-ACBD5 was localized exclusively to peroxisomes, and the HA-ACBD5 level was similar to that of endogenous ACBD5 in WT HeLa cells (Fig. 6, A and B). Unambiguously, the stable expression of HA-ACBD5 indeed restored the C24:0 β-oxidation activity in ACBD5-KO1 cells (Fig. 6C), thereby demonstrating that the effects of ACBD5-KO were on-target. Taken all together, our data strongly suggest that ACBD5 is required for the efficient β-oxidation of VLCFAs in peroxisomes.
FIGURE 6.

ACBD5 is required for efficient peroxisomal VLCFA β-oxidation. A, WT HeLa cells (panels a–c), ACBD5-KO1 cells (panels d–f), and ACBD5-KO1 cells stably expressing HA-ACBD5 (clones 1 and 2; panels g–l) were immunostained with antibodies against HA and catalase. Merged views are also shown. Scale bar, 10 μm. B, total cell lysates of WT HeLa cells (lane 1), ACBD5-KO1 cells (lane 2), and two clones of ACBD5-KO1 cells stably expressing HA-ACBD5 (lanes 3 and 4) were analyzed by immunoblotting as indicated. C, the relative VLCFA β-oxidation activities in the indicated cell lines were assessed as in Fig. 4E. Data are the mean of triplicate samples from a representative experiment and are expressed as the percentage relative to the activity in WT HeLa cells. Error bars represent S.D. **, p < 0.01 versus WT HeLa cells; one-way ANOVA with Dunnett's multiple comparison test.
Acyl-CoA Binding Domain and Peroxisomal Localization of ACBD5 Are Prerequisite for Achieving Efficient Peroxisomal VLCFA β-Oxidation
To understand the molecular basis for the ACBD5-dependent VLCFA β-oxidation, we then asked whether the N-terminal ACBD and peroxisomal localization of ACBD5 are required for such function. To this end, we reconstituted ACBD5-KO1 cells with HA-ACBD5ΔACBD lacking the entire ACBD or HA-ACBD5ΔC devoid of the C-terminal topological signal region (Fig. 7, A and C). In good agreement with the finding that peroxisomal localization of ACBD5 relies specifically on its C-terminal region (Fig. 2, E–G), HA-ACBD5ΔACBD was destined for peroxisomes, whereas HA-ACBD5ΔC was defective in targeting to peroxisomes (Fig. 7B). Similar to transiently expressed EGFP-ACBD5ΔC (Fig. 2E, panel g), HA-ACBD5ΔC stably expressed in ACBD5-KO1 cells was detected in cytoplasmic structures and apparently in the cytosol (Fig. 7B, panel j). Importantly, peroxisomes in ACBD5-KO1 cells expressing HA-ACBD5ΔACBD or HA-ACBD5ΔC remained nearly at the same level of reduced C24:0 β-oxidation activity as in ACBD5-KO1 cells (Fig. 7D), suggesting that both the ACBD and peroxisomal localization of ACBD5 are essential for achieving the efficient β-oxidation of VLCFAs in peroxisomes.
FIGURE 7.

Acyl-CoA binding domain and peroxisomal localization of ACBD5 are essential for the efficient peroxisomal β-oxidation of VLCFAs. A, schematic representation of HA-ACBD5 and its variants used. ACBD, CCD, and TMD are as in Fig. 2D. B, ACBD5-KO1 cells (panels a–c) and those reconstituted with HA-ACBD5 (WT (clone 2); panels d–f), HA-ACBD5ΔACBD (ΔACBD; panels g–i), or HA-ACBD5ΔC (ΔC; panels j–l) were immunostained with antibodies against HA and catalase. Merged views are also shown. Scale bar, 10 μm. C, total cell lysates of the indicated cell lines were analyzed by immunoblotting with antibodies against HA and tubulin. D, the relative peroxisomal VLCFA β-oxidation activities in the indicated cell lines were assessed as in Fig. 4E. Data are the mean of triplicate samples from a representative experiment and are expressed as the percentage relative to the activity in the HA-ACBD5WT-expressing ACBD5-KO1 cells. Error bars represent S.D. **, p < 0.01; n.s., not significant; one-way ANOVA with Bonferroni's multiple comparison test.
ACBD5 Binds Preferentially to VLC-CoAs
Because reconstitution of ACBD5-KO1 cells with HA-ACBD5ΔACBD failed to restore the impaired C24:0 β-oxidation activity (Fig. 7D), it is most likely that ACBD5 exerts its function by binding to acyl-CoAs. To provide experimental evidence that ACBD5 is indeed potent in binding to acyl-CoAs, we next performed an in vitro acyl-CoA binding assay using a soluble fragment of human ACBD5 that corresponds to the entire cytosolic domain encompassing aa 2–457 (referred to as soluble ACBD5 (sACBD5)). Recombinant sACBD5, sACBD5ΔACBD lacking the ACBD, and GST were purified from Escherichia coli (Fig. 8A) and assessed for binding to [14C]palmitoyl-CoA (C16:0-CoA). Strikingly, sACBD5 showed substantial binding to [14C]C16:0-CoA, whereas sACBD5ΔACBD and GST did not (Fig. 8B), demonstrating that ACBD5 binds to C16:0-CoA practically in a distinct manner dependent on its ACBD.
FIGURE 8.
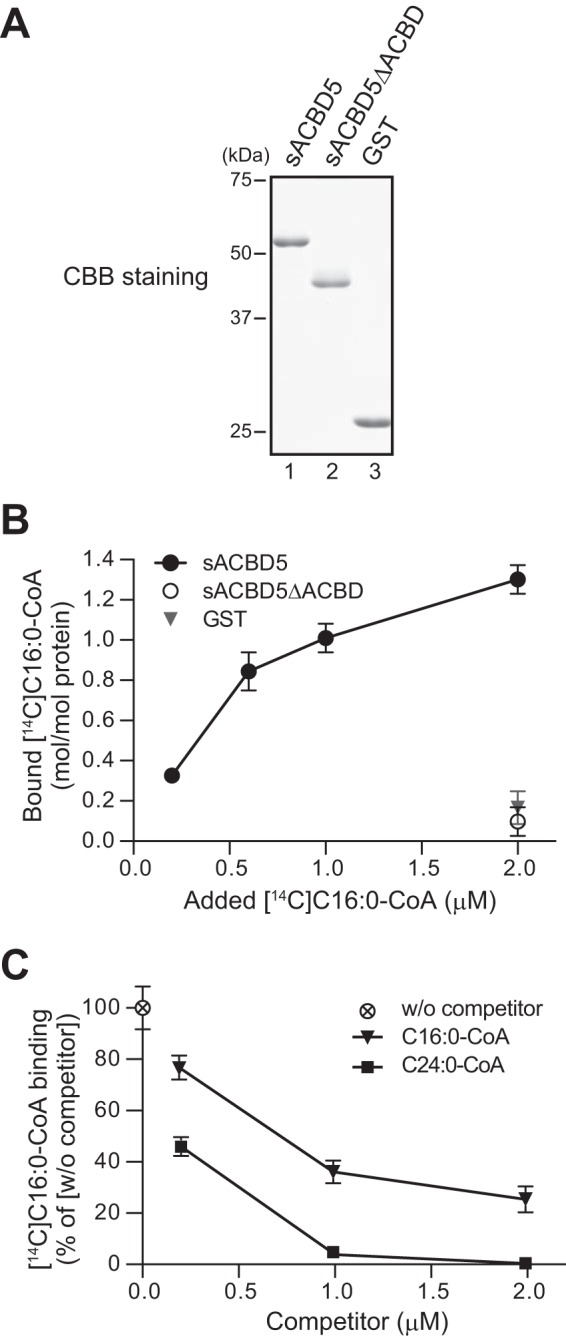
ACBD5 binds preferentially to VLC-CoAs. A, recombinant human ACBD5 cytosolic domain comprising of amino acids 2–457 (sACBD5; lane 1) and its variant lacking the ACBD (sACBD5ΔACBD; lane 2) as well as GST (lane 3) were purified and analyzed by SDS-PAGE. B, recombinant sACBD5, sACBD5ΔACBD, and GST (0.2 μm each), respectively, were incubated with [14C]C16:0-CoA as indicated, and the amounts of [14C]C16:0-CoA bound to each protein were quantified. Data represent the mean ± S.D. (error bars) of triplicate reactions from a representative experiment. C, recombinant sACBD5 (0.2 μm) was incubated with 1.0 μm [14C]C16:0-CoA in the absence (w/o competitor) or presence of increasing amounts of cold C16:0-CoA or C24:0-CoA as a competitor. The amounts of [14C]C16:0-CoA bound by sACBD5 were quantified and expressed as the percentage relative to the binding in the absence of competitor (i.e. w/o competitor). Data shown are the mean ± S.D. (error bars) of triplicate reactions from a representative experiment. CBB, Coomassie Brilliant Blue.
The members of the ACBP family, including the mammalian ACBP family, are thought to bind to medium- and long-chain acyl-CoAs with high affinity (28–30). Furthermore, mammalian ACBD1 was shown to be unable to bind to VLC-CoAs (31). However, in light of the phenotype of ΔACBD5 fibroblasts and ACBD5-KO HeLa cells, we suspected that ACBD5 might have an ability to bind to VLC-CoAs with high affinity. To explore this possibility, we performed the in vitro [14C]C16:0-CoA binding assay with increasing amounts of unlabeled C16:0-CoA or C24:0-CoA. By addition of unlabeled C16:0-CoA and unlabeled C24:0-CoA, the binding of sACBD5 to [14C]C16:0-CoA was dose-dependently decreased (Fig. 8C). Remarkably, the binding to [14C]C16:0-CoA was more effectively reduced by C24:0-CoA than C16:0-CoA (Fig. 8C), hence indicating that ACBD5 binds C24:0-CoA with much higher affinity than C16:0-CoA. Collectively, our findings suggest that ACBD5 is a peroxisomal ACBP possessing a preferential affinity to VLC-CoAs, implying that ACBD5 directly mediates peroxisomal VLCFA β-oxidation via its acyl-CoA binding activity.
Discussion
The discovery of a mutation in the ACBD5 gene in patients with a syndromic form of retinal dystrophy highlights the physiological importance of ACBD5; however, the function of ACBD5 and the pathogenic mechanisms of how ACBD5 deficiency leads to retinal dystrophy remained undefined. In the present study, by characterizing patient-derived human skin fibroblasts, we uncovered that ACBD5 is dispensable for peroxisome biogenesis, including membrane and matrix protein import (Fig. 3), and that ACBD5 deficiency causes a moderate defect in peroxisomal VLCFA β-oxidation and a slight accumulation of cellular VLC-PhoCho species without affecting the cellular levels of PlsEtn species and DHA-containing phospholipids (Fig. 4). Importantly, the phenotype of ΔACBD5 fibroblasts was essentially recapitulated in ACBD5-KO HeLa cells generated via the CRISPR/Cas9 system (Figs. 5 and 6). By taking advantage of ACBD5-KO HeLa cells in facile reconstitution of ACBD5 variants, we showed that both the ACBD and peroxisomal localization of ACBD5 are prerequisite for efficient VLCFA β-oxidation to occur in peroxisomes (Fig. 7). Moreover, ACBD5 was shown to preferentially bind VLC-CoAs (Fig. 8). Based on these findings, we suggest that ACBD5 is directly involved in peroxisomal VLCFA β-oxidation. The precise function of ACBD5 in peroxisomal VLCFA β-oxidation remains to be defined. The loss of ACBD5 results in only a partial but significant reduction in the peroxisomal VLCFA β-oxidation activity. Hence, ACBD5 seems to be required for securing the efficiency of peroxisomal VLCFA β-oxidation. Considering that ACBD5 exposes its ACBD to the cytosol (Fig. 2), we propose that ACBD5 captures VLC-CoAs on the cytosolic side of the peroxisomal membrane so that the uptake of VLC-CoAs into peroxisomes via the peroxisomal ABC transporters (32–34) and subsequent β-oxidation thereof can proceed efficiently. It would be also of interest to investigate whether ACBD5 physically interacts with and/or functions coordinately with the ABC transporters, including ABCD1.
Although we propose such a direct role of ACBD5 in peroxisomal VLCFA β-oxidation, in light of the recently proposed role of ACBD5 in basal pexophagy, it remains possible that ACBD5 might indirectly maintain the integrity of peroxisomal VLCFA β-oxidation pathway by removing defective peroxisomes. In the yeast Pichia pastoris, Atg37 has been shown to be an ACBP that resides in peroxisomal membranes and regulates phagosome formation during pexophagy through its binding activity toward Atg37 and C16:0-CoA (18, 35). Nazarko et al. (18) identified ACBD5 as the human orthologue of Atg37 and suggested by means of RNA interference that ACBD5 is required for pexophagy, although the molecular basis of the ACBD5-mediated pexophagy in mammals is obscure. Accordingly, ACBD5 deficiency might potentially lead to the accumulation of defective peroxisomes, culminating in a global defect in the peroxisomal functions. However, the effect of ACBD5 deficiency appears to be restricted to the peroxisomal β-oxidation pathway; our lipidomic analysis suggests that the biosynthesis of PlsEtn and that of DHA, which rely on peroxisomal enzymes (25, 27), are not affected in ΔACBD5 fibroblasts (Fig. 4, F and G). Furthermore, at least under our cell culture conditions, the level of AOx-B chain in ACBD5-KO HeLa cells was similar to that in WT HeLa cells (Fig. 5), implying that a marked accumulation of peroxisomes is unlikely in ACBD5-KO HeLa cells. Further studies are required to clarify the molecular function of ACBD5 in peroxisomal VLCFA β-oxidation and/or mammalian pexophagy and to better explain how ACBD5 deficiency affects the peroxisomal VLCFA β-oxidation pathway.
ACBD5 belongs to the ACBP family, members of which are suggested to be involved in diverse biological processes, including lipid metabolism, cellular differentiation, and apoptosis (15, 28, 29, 36, 37). In general, ACBPs are thought to bind medium- to long-chain acyl-CoAs. For instance, mammalian ACBD1 (also known as ACBP/DBI) and its yeast homologue Acb1p bind C14–C22 acyl-CoAs with high affinity but are unable to bind VLC-CoAs (31, 38, 39). Intriguingly, ACBD5 showed a greater affinity for C26:0-CoA than for C16:0-CoA (Fig. 8), consistent with its potential role in peroxisomal VLCFA β-oxidation. Thus, ACBD5 may differ from other ACBPs in terms of substrate specificity. However, the binding property of ACBD5 apparently needs to be determined in more detail to further establish the physiological role of ACBD5 as peroxisomal fatty acid oxidation pathways in humans can potentially metabolize various substrates such as saturated VLCFAs, unsaturated VLCFAs, and branched-chain fatty acids (see also below and Ref. 1). Conversely, given that ACBPs are suggested to function via interaction with a multitude of proteins (29, 37), it would also be of importance and interest to search for protein interactors of ACBD5. Furthermore, several ACBPs have been suggested to form a dimer (30, 37, 40). Intriguingly, we have detected self-association, including a dimer formation of ACBD5.3 The oligomeric status of ACBD5 and its physiological relevance should be investigated in the future.
In the ACBD5 gene, a mutation predicted to disrupt a consensus splice donor site was identified in three siblings suffering from a syndromic form of retinal dystrophy (14). As reported (14), the aberrant transcript expected to encode a truncated ACBD5 protein retaining most of its cytosolic region was detected in ΔACBD5 fibroblasts.4 However, even though the anti-ACBD5 polyclonal antibody was raised to the cytosolic region of ACBD5, immunoblotting analysis with the antibody failed to detect the truncated ACBD5 protein in ΔACBD5 fibroblasts (Fig. 3C), consistent with the earlier data (14), hence implying that the truncated ACBD5 protein was degraded. Besides vision impairment, the ACBD5-deficient patients also manifested in infancy clinical symptoms such as severe white matter disease, spastic paraparesis, and cognitive impairment (14). These clinical features are commonly observed in patients with PBDs and in patients with SEDs, including X-ALD, AOx deficiency, and D-bifunctional protein deficiency (3, 4). Because morphological and biochemical studies on ΔACBD5 fibroblasts suggested that peroxisome biogenesis is normal in the absence of ACBD5 (Fig. 3), the ACBD5 deficiency can be categorized as an SED with typical neurological deficits.
The levels of VLCFAs and branched-chain fatty acids are widely utilized for the initial screening of peroxisomal disorders (41). Laboratory investigations on one of the three ACBD5-deficient patients revealed that the plasma C26/C22 ratio was slightly elevated, whereas the concentrations of branched-chain fatty acids phytanic acid and pristanic acid were normal (Table 1), implying that the peroxisomal VLCFA β-oxidation, but not the peroxisomal branched-chain fatty acid oxidation, was affected in the patient. Consistently, lipidomic profiling of fibroblasts from the ACBD5-deficient patient suggests that ACBD5 deficiency impairs the peroxisomal β-oxidation of VLCFAs (Fig. 4A). By contrast, as mentioned above, the peroxisome-dependent syntheses of PlsEtn and DHA in ΔACBD5 fibroblasts appear to be normal (Fig. 4, F and G). Remarkably, the extent of VLC-PtdCho accumulation in ΔACBD5 fibroblasts is similar to that in ΔABCD1 fibroblasts and is much lower than that in ΔAOx fibroblasts (Fig. 4A), which probably mirrors the slightly elevated plasma C26/C22 ratio in the ACBD5-deficient patient. Of note, in contrast to ΔABCD1 fibroblasts showing no accumulation of PtdCho species that contain polyunsaturated VLCFAs, ΔACBD5 fibroblasts exhibit an overall accumulation of VLC-PtdCho species, including those with polyunsaturated VLCFAs (Fig. 4, C and D). With respect to this finding, it is noteworthy that in addition to accumulation of saturated and monounsaturated VLCFAs accumulation of polyunsaturated VLCFAs has been implicated in the pathogenesis of peroxisomal β-oxidation disorders (3, 11, 24, 42). Collectively, we suggest that ACBD5 deficiency is a novel peroxisomal β-oxidation disorder and that the mild but global accumulation of VLCFAs in plasma and in cellular phospholipids contributes to the pathogenesis of brain and retinal phenotypes caused by ACBD5 deficiency. Nonetheless, although the levels of phytanic acid and pristanic acid were normal at least in plasma from an ACBD5-deficient patient, it may still be possible that ACBD5 deficiency affects the cellular homeostasis of branched-chain fatty acids. Future cellular and biochemical studies could address this issue.
An important issue remains as to how ACBD5 deficiency and its apparently accompanying accumulation of VLCFAs cause the ACBD5-linked phenotype, especially the retinal degeneration. Photoreceptor cells are highly enriched in PtdCho species with polyunsaturated long-chain fatty acids and/or polyunsaturated VLCFAs (43–47), and it is well established that DHA is essential for normal retinal development and function (43, 48–50). Notably, in patients with PBDs, the DHA level in retina is markedly reduced, which is thought to give rise to the pigmentary retinal disorder in these patients (51, 52). Meanwhile, growing evidence also suggests the roles of polyunsaturated VLCFAs in maintaining the integrity of photoreceptor cells (53). The physiological consequence of these fatty acids is particularly highlighted by autosomal dominant Stargardt-like macular dysdrophy-3 that is caused by mutations in the gene encoding ELOVL4 (ELOVL fatty acid elongase 4) that catalyzes the elongation of VLCFAs; this fatty acid elongase is required for the synthesis of retinal polyunsaturated VLCFAs (53–55). In the case of ACBD5 deficiency, the accumulation of polyunsaturated VLCFAs (Fig. 4, C and D) seems to be responsible for the retinal degeneration because the loss of ACBD5 is unlikely to affect the peroxisome-dependent synthesis of DHA (Fig. 4G). In regard to this issue, it would be interesting to note that patients with ACBD5 deficiency and those with X-ALD both develop severe white matter disease, whereas only the former patients manifest retinopathy. This might be explained by the aforementioned fact that loss of ACBD5 causes the accumulation of a broad range of VLCFAs, including polyunsaturated species, whereas ABCD1 deficiency causes the accumulation of only saturated and monounsaturated VLCFAs (Fig. 4, C and D and Ref. 24). Thereby, we suggest that ACBD5 deficiency and its associated perturbation of peroxisomal β-oxidation of VLCFAs, especially polyunsaturated VLCFAs, may cause dysregulated fatty acid homeostasis in the retina, giving rise to the severe photoreceptor degeneration. Nonetheless, future investigations should determine the affected retinal cell types as well as the retinal lipid profiles in the patients with ACBD5 deficiency and/or in animal models of ACBD5 deficiency to better understand the mechanistic link between ACBD5 deficiency and retinal dystrophy.
In summary, the present work demonstrates that ACBD5 is a peroxisomal tail-anchored protein involved in peroxisomal VLCFA β-oxidation via preferential binding to VLC-CoAs and that ACBD5 deficiency causes defective peroxisomal VLCFA β-oxidation, resulting in VLCFA accumulation. Our findings shed light on the function of ACBD5 and the potential pathogenic mechanism by which ACBD5 deficiency leads to a syndromic form of retinal dystrophy.
Experimental Procedures
Cell Culture and DNA Transfection
Human skin fibroblasts from a normal control (control; identification number TIG120; Ref. 56), a patient with ACBD5 deficiency (ΔACBD5; identification number 10DG1872; Ref. 14), a PEX10-defective patient with Zellweger syndrome (ΔPEX10; Ref. 57), a patient with AOx deficiency (ΔAOx; identification number PDL30092; Refs. 10 and 11), and a patient with X-ALD (ΔABCD1; identification number PDL6886; Ref.56) were used. Human skin fibroblasts and HeLa cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum at 37 °C under 5% CO2. DNA transfection was done using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Antibodies
Rabbit antibodies to ADAPS (23), AOx (22), catalase (58), PTS1 peptide (59), PEX13 (60), PEX14 (61), PMP70 (22, 62), and HA epitope tag (63) and guinea pig antibodies raised to human catalase (catalase from human erythrocytes; Sigma-Aldrich) and PEX14 (64) were used. Rabbit anti-ACBD5 antibody was raised against recombinant sACBD5 protein (aa 2–457 of human ACBD5 isoform 2; see below). The following antibodies were purchased: rabbit anti-FLAG antibody (Sigma-Aldrich); mouse monoclonal antibodies against HA epitope tag (16B12; Covance), GFP (B-2; Santa Cruz Biotechnology), and α-tubulin (DM1A; Abcam); and goat antibodies against lactate dehydrogenase antibody (Rockland) and lamin A/C (Santa Cruz Biotechnology).
Mammalian Expression Plasmids
The cDNA encoding human ACBD5 isoform 2 (NCBI reference sequence NM_001042473.3) was obtained by reverse transcription-PCR using total RNA isolated from human control skin fibroblasts, and the internal BamHI site was removed by introducing a silent mutation. To generate FLAG-ACBD5-HA, a BamHI-XbaI fragment encoding ACBD5 (aa 2–490) C-terminally fused with HA epitope tag was PCR-amplified and ligated into the BamHI-XbaI sites of pcDNAZeo/FLAG (65). To construct EGFP-ACBD5, EGFP-ACBD5ΔC, and EGFP-ACBD5ΔCCDΔC, BamHI-SalI fragments encoding ACBD5 (aa 2–490), ACBD5ΔC (aa 2–457), and ACBD5ΔCCDΔC (aa 2–399 plus 432–457) were amplified by standard or overlap extension PCR, and each was cloned into the BamHI-XhoI sites of pcDNAZeo/EGFP (65). For EGFP-ACBD5C40, a HindIII-BamHI fragment encoding EGFP followed by a flexible linker (GGGGSGGGGS) and a BamHI-SalI fragment encoding the C-terminal 40 aa of ACBD5 were PCR-amplified and ligated together into the HindIII-XhoI sites of pcDNA3.1/Zeo(+) vector (Invitrogen).
To facilitate stable expression of HA-ACBD5 and its variants, BamHI-SalI fragments encoding ACBD5, ACBD5ΔC, and ACBD5ΔACBD (aa 2–7 plus 97–490) were PCR-amplified, and each was cloned into the BamHI-XhoI sites of pcDNAZeo/HA (65). Using the resultant plasmids as a template, the HA-ACBD5, HA-ACBD5ΔC, and HA-ACBD5ΔACBD sequences were amplified as NheI-NotI fragments and separately ligated into the corresponding sites of a modified pIRESpuro3 vector (Clontech), termed pEF-IRESpuro3, where the original CMV promoter had been replaced by the EF1α promoter from pEF1/myc-His A (Invitrogen).
Subcellular Fractionation and Alkaline Extraction
Cells were collected and resuspended in ice-cold homogenizing buffer (0.25 m sucrose, 20 mm Hepes-KOH, pH 7.4, 1 mm EDTA, and protease inhibitor mixture (5 μg/ml aprotinin and 10 μg/ml each antipain, chymostatin, E-64, leupeptin, and pepstatin)) containing 100 μg/ml digitonin. Samples were kept on ice for 10 min to permeabilize the plasma membrane and then centrifuged at 20,000 × g for 10 min to yield cytosolic and organelle-containing fractions. For alkaline extraction (19), organelle-containing fractions were treated with 0.1 m Na2CO3 on ice for 30 min. Soluble and membrane fractions were separated by ultracentrifugation at 100,000 × g for 30 min.
Lipidomic Analysis
Lipidomic analysis was performed as described (24). Briefly, total lipids were extracted from 50 μg of cellular protein by the method of Bligh and Dyer (66). Fifty picomoles of 1-heptadecanoyl-sn-glycero-3-phosphocholine, 1,2-didodecanoyl-sn-glycero-3-phosphocholine, and 1,2-didodecanoyl-sn-glycero-3-phosphoethanolamine were added as internal standards. The organic phase was evaporated under the nitrogen stream and suspended in methanol. Liquid chromatography coupled with electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) was performed using a 4000 Q-TRAP quadrupole linear ion trap hybrid mass spectrometer (AB Sciex) equipped with an ACQUITY UPLC System (Waters) and an ACQUITY UPLC BEH C18 column (1.7 μm; 1.0 × 150 mm; Waters). The data were analyzed and quantified using Analyst software (AB Sciex).
Measurement of Fatty Acid β-Oxidation Activity
Fatty acid β-oxidation activity was determined essentially as described (67). Cells grown in 12-well plates were preincubated at 37 °C for 1 h in serum-free DMEM. After the medium was changed to fresh serum-free DMEM, 2 nmol of [14C]C24:0 (American Radiolabeled Chemicals) dissolved in 0.1 m Tris-HCl, pH 8.0, containing 10 mm α-cyclodextrin was added to the culture medium, and cells were further incubated at 37 °C for 2–2.5 h. The radioactivity of acid-soluble metabolites was measured using a Beckman LS6500 scintillation counter. Fatty acid β-oxidation activity was calculated as nmol/h/mg of protein and expressed as indicated in the figure legends.
Generation of ACBD5-KO HeLa Cell Lines Using the CRISPR/Cas9 System
To generate ACBD5-KO HeLa cell lines, we used two independent gRNAs specific for human ACBD5 gene (ACBD5 gRNA1 and ACBD5 gRNA2). The target sequences of these gRNAs were selected using an online CRISPR design tool (Massachusetts Institute of Technology) as depicted in supplemental Fig. S1. To construct the plasmid coexpressing Cas9 and each gRNA, a pair of oligonucleotides containing the gRNA target sequence was annealed and ligated into the BbsI-linearized pX330 vector (Addgene 42230; a gift from Feng Zhang; Ref. 68). Oligonucleotide pairs used were: 5′-CACCGCTCAAACCTAGTCTCGTGCA-3′ and 5′-AAACTGCACGAGACTAGGTTTGAGC-3′ for ACBD5 gRNA1 and 5′-CACCGCTCAAACCTAGTCTCGTGCA-3′ and 5′-AAACTGCACGAGACTAGGTTTGAGC-3′ for ACBD5 gRNA2. The resultant plasmids, pX330/ACBD5gRNA1 and pX330/ACBD5gRNA2, were separately transfected into HeLa cells. At 3 days after transfection, cells were diluted and seeded into 96-well plates to isolate clonal cell lines, which were screened for ACBD5 deficiency by immunofluorescence microscopy and immunoblotting. To confirm the presence of frameshifting insertions/deletions, genomic DNA was extracted from ACBD5-KO clonal cell lines, and the genomic region encompassing the gRNA target sequences was PCR-amplified using the following primers: forward, 5′-TCTCGGTGAGTTGTTTTGCTGTCGT-3′; and reverse, 5′-TCCCCTTCCACTACATGGCTCCTAC-3′. The PCR product was subcloned into T-Vector pMD19 (Takara Bio Inc.) and sequenced (supplemental Fig. S1B). Two clonal ACBD5-KO cell lines generated through each gRNA, termed ACBD5-KO1 and ACBD5-KO2, were used in this study.
To establish clonal ACBD5-KO1 cell lines stably expressing WT or variant HA-ACBD5, ACBD5-KO1 cells were transfected with the above described pEF-IRESpuro3-based plasmids and selected with 1 μg/ml puromycin. Single clones were likewise isolated by limiting dilution and subsequently assessed for the expression of HA-ACBD5 variants by immunofluorescence microscopy as well as immunoblotting.
Expression and Purification of Recombinant sACBD5 and sACBD5ΔACBD
For bacterial expression and purification of recombinant sACBD5 (aa 2–457) and sACBD5ΔACBD (aa 2–7 plus 97–457), the corresponding cDNA fragments were PCR-amplified and respectively cloned into pGEX-6P-1 (GE Healthcare) via the BamHI/SalI sites. Using the resultant plasmids, sACBD5 and sACBD5ΔACBD were each expressed as a GST fusion protein in E. coli Rosetta 2(DE3)/pLysS cells (Novagen). Protein expression was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside at 30 °C for 4 h. Cells expressing the GST fusion proteins were harvested, resuspended in lysis buffer (PBS containing 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, and 5 mm dithiothreitol), and disrupted by sonication. Lysates were cleared by centrifugation and incubated with glutathione-Sepharose beads (GE Healthcare) at 4 °C for 1.5 h on a rotating wheel to purify the GST fusion proteins. After extensive washing, sACBD5 and sACBD5ΔACBD were released from the beads by treatment with PreScission protease (GE Healthcare), which cleaved off the GST tag, according to the manufacturer's instructions.
In Vitro Acyl-CoA Binding Assay
Binding of the purified recombinant proteins to [14C]C16:0-CoA (Moravek Biochemicals, Inc.) was determined essentially as described (69). In brief, binding reactions were prepared by mixing 0.2 μm recombinant protein (sACBD5, sACBD5ΔACBD, or GST) and [14C]C16:0-CoA in binding buffer (10 mm potassium phosphate, pH 7.4). Where indicated, unlabeled C16:0-CoA or C24:0-CoA (Avanti Polar Lipids) was also added to the reaction mixtures. The concentrations of radiolabeled and unlabeled acyl-CoAs were as indicated in the figure legends. The reaction mixtures were incubated at 37 °C for 30 min and subsequently chilled on ice. After removing the free [14C]C16:0-CoA by Lipidx-1000 (PerkinElmer Life Sciences), the radioactivity in the reactions was measured using a Beckman LS6500 scintillation counter.
Other Methods
Immunoblotting and immunofluorescence microscopy were done as described (65). Unless otherwise indicated, fixed cells were permeabilized with 1% Triton X-100 in PBS prior to immunostaining. Statistical analyses were performed as indicated in the figure legends using GraphPad Prism v6.0h (GraphPad Software, Inc.).
Author Contributions
Y. Y. and Y. F. designed the research. Y. Y., K. S., K. N., and M. A.-O. performed the experiments. Y. A. performed the lipid analysis by LC-ESI-MS/MS. Y. Y. and Y. F. interpreted the data. Y. Y., F. S. A., and Y. F. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank K. Shimizu for preparing figures and the other members of our laboratory for discussions.
This work was supported in part by Grants-in-aid for Scientific Research 24247038, 25112518, 25116717, 26116007, 15K14511, and 15K21743 (to Y. F.); Kyushu University Interdisciplinary Programs in Education and Projects in Research Development (to Y. Y.); a grant from the King Salman Center for Disability Research grant (to F. S. A.); and grants from the Takeda Science Foundation, the Naito Foundation, and Japan Foundation for Applied Enzymology (to Y. F.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Fig. S1.
K. Nakagawa and Y. Fujiki, unpublished data.
Y. Yagita and Y. Fujiki, unpublished data.
- DHA
- docosahexaenoic acid
- aa
- amino acid
- ABC
- ATP-binding cassette
- ACBD5
- acyl-CoA binding domain-containing 5
- ACBP
- acyl-CoA binding domain-containing protein
- ANOVA
- analysis of variance
- AOx
- acyl-CoA oxidase 1
- CCD
- coiled-coil domain
- CRISPR
- clustered regularly interspaced short palindromic repeats
- EGFP
- enhanced GFP
- ELOVL
- elongation of very-long-chain fatty acids
- ESI
- electrospray ionization
- gRNA
- guide RNA
- PBD
- peroxisome biogenesis disorder
- PtdCho
- phosphatidylcholine
- PlsEtn
- plasmenylethanolamine
- PTS1
- peroxisomal targeting signal type 1
- sACBD5
- soluble ACBD5
- SED
- single peroxisomal enzyme deficiency
- TMD
- transmembrane domain
- VLC-CoA
- very-long-chain fatty acyl-CoA
- VLCFA
- very-long-chain fatty acid
- X-ALD
- X-linked adrenoleukodystrophy.
References
- 1. Wanders R. J., and Waterham H. R. (2006) Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 75, 295–332 [DOI] [PubMed] [Google Scholar]
- 2. Wanders R. J. (2014) Metabolic functions of peroxisomes in health and disease. Biochimie 98, 36–44 [DOI] [PubMed] [Google Scholar]
- 3. Berger J., Dorninger F., Forss-Petter S., and Kunze M. (2016) Peroxisomes in brain development and function. Biochim. Biophys. Acta 1863, 934–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waterham H. R., Ferdinandusse S., and Wanders R. J. (2016) Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 1863, 922–933 [DOI] [PubMed] [Google Scholar]
- 5. Weller S., Gould S. J., and Valle D. (2003) Peroxisome biogenesis disorders. Annu. Rev. Genomics Hum. Genet. 4, 165–211 [DOI] [PubMed] [Google Scholar]
- 6. Fujiki Y., Okumoto K., Mukai S., Honsho M., and Tamura S. (2014) Peroxisome biogenesis in mammalian cells. Front. Physiol. 5, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kemp S., Pujol A., Waterham H. R., van Geel B. M., Boehm C. D., Raymond G. V., Cutting G. R., Wanders R. J., and Moser H. W. (2001) ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum. Mutat. 18, 499–515 [DOI] [PubMed] [Google Scholar]
- 8. Mosser J., Douar A.-M., Sarde C.-O., Kioschis P., Feil R., Moser H., Poustka A.-M., Mandel J.-L., and Aubourg P. (1993) Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361, 726–730 [DOI] [PubMed] [Google Scholar]
- 9. Smith K. D., Kemp S., Braiterman L. T., Lu J. F., Wei H. M., Geraghty M., Stetten G., Bergin J. S., Pevsner J., and Watkins P. A. (1999) X-linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem. Res. 24, 521–535 [DOI] [PubMed] [Google Scholar]
- 10. Ferdinandusse S., Denis S., Hogenhout E. M., Koster J., van Roermund C. W., IJlst L., Moser A. B., Wanders R. J., and Waterham H. R. (2007) Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum. Mutat. 28, 904–912 [DOI] [PubMed] [Google Scholar]
- 11. Poll-The B. T., Roels F., Ogier H., Scotto J., Vamecq J., Schutgens R. B., Wanders R. J., van Roermund C. W., van Wijland M. J., Schram A. W., Tager J. M., and Saudubray J. M. (1988) A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy). Am. J. Hum. Genet. 42, 422–434 [PMC free article] [PubMed] [Google Scholar]
- 12. Ferdinandusse S., Denis S., Mooyer P. A., Dekker C., Duran M., Soorani-Lunsing R. J., Boltshauser E., Macaya A., Gärtner J., Majoie C. B., Barth P. G., Wanders R. J., and Poll-The B. T. (2006) Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann. Neurol. 59, 92–104 [DOI] [PubMed] [Google Scholar]
- 13. Suzuki Y., Jiang L. L., Souri M., Miyazawa S., Fukuda S., Zhang Z., Une M., Shimozawa N., Kondo N., Orii T., and Hashimoto T. (1997) D-3-Hydroxyacyl-CoA dehydratase/D-3-hydroxyacyl-CoA dehydrogenase bifunctional protein deficiency: a newly identified peroxisomal disorder. Am. J. Hum. Genet. 61, 1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abu-Safieh L., Alrashed M., Anazi S., Alkuraya H., Khan A. O., Al-Owain M., Al-Zahrani J., Al-Abdi L., Hashem M., Al-Tarimi S., Sebai M.-A., Shamia A., Ray-Zack M. D., Nassan M., Al-Hassnan Z. N., et al. (2013) Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23, 236–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan J., Liu J., Culty M., and Papadopoulos V. (2010) Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog. Lipid Res. 49, 218–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kikuchi M., Hatano N., Yokota S., Shimozawa N., Imanaka T., and Taniguchi H. (2004) Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem. 279, 421–428 [DOI] [PubMed] [Google Scholar]
- 17. Wiese S., Gronemeyer T., Ofman R., Kunze M., Grou C. P., Almeida J. A., Eisenacher M., Stephan C., Hayen H., Schollenberger L., Korosec T., Waterham H. R., Schliebs W., Erdmann R., Berger J., et al. (2007) Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell. Proteomics 6, 2045–2057 [DOI] [PubMed] [Google Scholar]
- 18. Nazarko T. Y., Ozeki K., Till A., Ramakrishnan G., Lotfi P., Yan M., and Subramani S. (2014) Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J. Cell Biol. 204, 541–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kutay U., Hartmann E., and Rapoport T. A. (1993) A class of membrane proteins with a C-terminal anchor. Trends Cell Biol. 3, 72–75 [DOI] [PubMed] [Google Scholar]
- 21. Borgese N., Brambillasca S., and Colombo S. (2007) How tails guide tail-anchored proteins to their destinations. Curr. Opin. Cell Biol. 19, 368–375 [DOI] [PubMed] [Google Scholar]
- 22. Tsukamoto T., Yokota S., and Fujiki Y. (1990) Isolation and characterization of Chinese hamster ovary cell mutants defective in assembly of peroxisomes. J. Cell Biol. 110, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Honsho M., Yagita Y., Kinoshita N., and Fujiki Y. (2008) Isolation and characterization of mutant animal cell line defective in alkyl-dihydroxyacetonephosphate synthase: localization and transport of plasmalogens to post-Golgi compartments. Biochim. Biophys. Acta 1783, 1857–1865 [DOI] [PubMed] [Google Scholar]
- 24. Abe Y., Honsho M., Nakanishi H., Taguchi R., and Fujiki Y. (2014) Very-long-chain polyunsaturated fatty acids accumulate in phosphatidylcholine of fibroblasts from patients with Zellweger syndrome and acyl-CoA oxidase1 deficiency. Biochim. Biophys. Acta 1841, 610–619 [DOI] [PubMed] [Google Scholar]
- 25. Braverman N. E., and Moser A. B. (2012) Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 1822, 1442–1452 [DOI] [PubMed] [Google Scholar]
- 26. Wanders R. J., Schumacher H., Heikoop J., Schutgens R. B., and Tager J. M. (1992) Human dihydroxyacetonephosphate acyltransferase deficiency: a new peroxisomal disorder. J. Inher. Metab. Dis. 15, 389–391 [DOI] [PubMed] [Google Scholar]
- 27. Ferdinandusse S., Denis S., Mooijer P. A., Zhang Z., Reddy J. K., Spector A. A., and Wanders R. J. (2001) Identification of the peroxisomal β-oxidation enzymes involved in the biosynthesis of docosahexaenoic acid. J. Lipid Res. 42, 1987–1995 [PubMed] [Google Scholar]
- 28. Faergeman N. J., Wadum M., Feddersen S., Burton M., Kragelund B. B., and Knudsen J. (2007) Acyl-CoA binding proteins; structural and functional conservation over 2000 MYA. Mol. Cell. Biochem. 299, 55–65 [DOI] [PubMed] [Google Scholar]
- 29. Xiao S., and Chye M.-L. (2011) New roles for acyl-CoA-binding proteins (ACBPs) in plant development, stress responses and lipid metabolism. Prog. Lipid Res. 50, 141–151 [DOI] [PubMed] [Google Scholar]
- 30. Soupene E., and Kuypers F. A. (2015) Ligand binding to the ACBD6 protein regulates the acyl-CoA transferase reactions in membranes. J. Lipid Res. 56, 1961–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosendal J., Ertbjerg P., and Knudsen J. (1993) Characterization of ligand binding to acyl-CoA-binding protein. Biochem. J. 290, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Netik A., Forss-Petter S., Holzinger A., Molzer B., Unterrainer G., and Berger J. (1999) Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): implications for therapy. Hum. Mol. Genet. 8, 907–913 [DOI] [PubMed] [Google Scholar]
- 33. Morita M., and Imanaka T. (2012) Peroxisomal ABC transporters: Structure, function and role in disease. Biochim. Biophys. Acta 1822, 1387–1396 [DOI] [PubMed] [Google Scholar]
- 34. Wiesinger C., Kunze M., Regelsberger G., Forss-Petter S., and Berger J. (2013) Impaired very long-chain acyl-CoA β-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J. Biol. Chem. 288, 19269–19279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nazarko T. Y. (2014) Atg37 regulates the assembly of the pexophagic receptor protein complex. Autophagy 10, 1348–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burton M., Rose T. M., Faergeman N. J., and Knudsen J. (2005) Evolution of the acyl-CoA binding protein (ACBP). Biochem. J. 392, 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neess D., Bek S., Engelsby H., Gallego S. F., and Færgeman N. J. (2015) Long-chain acyl-CoA esters in metabolism and signaling: role of acyl-CoA binding proteins. Prog. Lipid Res. 59, 1–25 [DOI] [PubMed] [Google Scholar]
- 38. Faergeman N. J., Sigurskjold B. W., Kragelund B. B., Andersen K. V., and Knudsen J. (1996) Thermodynamics of ligand binding to acyl-coenzyme A binding protein studied by titration calorimetry. Biochemistry 35, 14118–14126 [DOI] [PubMed] [Google Scholar]
- 39. Gaigg B., Neergaard T. B., Schneiter R., Hansen J. K., Faergeman N. J., Jensen N. A., Andersen J. R., Friis J., Sandhoff R., Schrøder H. D., and Knudsen J. (2001) Depletion of acyl-coenzyme A-binding protein affects sphingolipid synthesis and causes vesicle accumulation and membrane defects in Saccharomyces cerevisiae. Mol. Biol. Cell 12, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Augoff K., Kolondra A., Chorzalska A., Lach A., Grabowski K., and Sikorski A. F. (2010) Expression, purification and functional characterization of recombinant human acyl-CoA-binding protein (ACBP) from erythroid cells. Acta Biochim. Pol. 57, 533–540 [PubMed] [Google Scholar]
- 41. Shimozawa N. (2011) Molecular and clinical findings and diagnostic flowchart of peroxisomal diseases. Brain Dev. 33, 770–776 [DOI] [PubMed] [Google Scholar]
- 42. Wanders R. J. (2004) Metabolic and molecular basis of peroxisomal disorders: A review. Am. J. Med. Genet. A 126A, 355–375 [DOI] [PubMed] [Google Scholar]
- 43. Fliesler S. J., and Anderson R. E. (1983) Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 22, 79–131 [DOI] [PubMed] [Google Scholar]
- 44. Aveldaño M. I., and Bazán N. G. (1983) Molecular species of phosphatidylcholine, -ethanolamine, -serine, and -inositol in microsomal and photoreceptor membranes of bovine retina. J. Lipid Res. 24, 620–627 [PubMed] [Google Scholar]
- 45. Aveldaño M. I. (1987) A novel group of very long chain polyenoic fatty acids in dipolyunsaturated phosphatidylcholines from vertebrate retina. J. Biol. Chem. 262, 1172–1179 [PubMed] [Google Scholar]
- 46. Aveldaño M. I., and Sprecher H. (1987) Very long chain (C24 to C36) polyenoic fatty acids of the n-3 and n-6 series in dipolyunsaturated phosphatidylcholines from bovine retina. J. Biol. Chem. 262, 1180–1186 [PubMed] [Google Scholar]
- 47. Harkewicz R., Du H., Tong Z., Alkuraya H., Bedell M., Sun W., Wang X., Hsu Y.-H., Esteve-Rudd J., Hughes G., Su Z., Zhang M., Lopes V. S., Molday R. S., Williams D. S., et al. (2012) Essential role of ELOVL4 protein in very long chain fatty acid synthesis and retinal function. J. Biol. Chem. 287, 11469–11480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. SanGiovanni J. P., and Chew E. Y. (2005) The role of ω-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog. Retin. Eye Res. 24, 87–138 [DOI] [PubMed] [Google Scholar]
- 49. Neuringer M., and Jeffrey B. G. (2003) Visual development: neural basis and new assessment methods. J. Pediatr. 143, S87–95 [DOI] [PubMed] [Google Scholar]
- 50. Bazan N. G. (2007) Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 48, 4866–4881 [DOI] [PubMed] [Google Scholar]
- 51. Martinez M. (1989) Polyunsaturated fatty acid changes suggesting a new enzymatic defect in Zellweger syndrome. Lipids 24, 261–265 [DOI] [PubMed] [Google Scholar]
- 52. Martinez M. (1992) Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney and retina of patients with peroxisomal disorders. Brain Res. 583, 171–182 [DOI] [PubMed] [Google Scholar]
- 53. Agbaga M.-P., Mandal M. N., and Anderson R. E. (2010) Retinal very long-chain PUFAs: new insights from studies on ELOVL4 protein. J. Lipid Res. 51, 1624–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bernstein P. S., Tammur J., Singh N., Hutchinson A., Dixon M., Pappas C. M., Zabriskie N. A., Zhang K., Petrukhin K., Leppert M., and Allikmets R. (2001) Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest. Ophthalmol. Vis. Sci. 42, 3331–3336 [PubMed] [Google Scholar]
- 55. Zhang K., Kniazeva M., Han M., Li W., Yu Z., Yang Z., Li Y., Metzker M. L., Allikmets R., Zack D. J., Kakuk L. E., Lagali P. S., Wong P. W., MacDonald I. M., Sieving P. A., et al. (2001) A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 27, 89–93 [DOI] [PubMed] [Google Scholar]
- 56. Itoyama A., Honsho M., Abe Y., Moser A., Yoshida Y., and Fujiki Y. (2012) Docosahexaenoic acid mediates peroxisomal elongation, a prerequisite for peroxisome division. J. Cell Sci. 125, 589–602 [DOI] [PubMed] [Google Scholar]
- 57. Okumoto K., Itoh R., Shimozawa N., Suzuki Y., Tamura S., Kondo N., and Fujiki Y. (1998) Mutation in PEX10 is the cause of Zellweger peroxisome deficiency syndrome of complementation group B. Hum. Mol. Genet. 7, 1399–1405 [DOI] [PubMed] [Google Scholar]
- 58. Shimozawa N., Tsukamoto T., Suzuki Y., Orii T., and Fujiki Y. (1992) Animal cell mutants represent two complementation groups of peroxisome-defective Zellweger syndrome. J. Clin. Investig. 90, 1864–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Otera H., Okumoto K., Tateishi K., Ikoma Y., Matsuda E., Nishimura M., Tsukamoto T., Osumi T., Ohashi K., Higuchi O., and Fujiki Y. (1998) Peroxisome targeting signal type 1 (PTS1) receptor is involved in import of both PTS1 and PTS2: studies with PEX5-defective CHO cell mutants. Mol. Cell. Biol. 18, 388–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mukai S., and Fujiki Y. (2006) Molecular mechanisms of import of peroxisome-targeting signal type 2 (PTS2) proteins by PTS2 receptor Pex7p and PTS1 receptor Pex5pL. J. Biol. Chem. 281, 37311–37320 [DOI] [PubMed] [Google Scholar]
- 61. Shimizu N., Itoh R., Hirono Y., Otera H., Ghaedi K., Tateishi K., Tamura S., Okumoto K., Harano T., Mukai S., and Fujiki Y. (1999) The peroxin Pex14p: cDNA cloning by functional complementation on a Chinese hamster ovary cell mutant, characterization, and functional analysis. J. Biol. Chem. 274, 12593–12604 [DOI] [PubMed] [Google Scholar]
- 62. Kinoshita N., Ghaedi K., Shimozawa N., Wanders R. J., Matsuzono Y., Imanaka T., Okumoto K., Suzuki Y., Kondo N., and Fujiki Y. (1998) Newly identified Chinese hamster ovary cell mutants are defective in biogenesis of peroxisomal membrane vesicles (peroxisomal ghosts), representing a novel complementation group in mammals. J. Biol. Chem. 273, 24122–24130 [DOI] [PubMed] [Google Scholar]
- 63. Otera H., Harano T., Honsho M., Ghaedi K., Mukai S., Tanaka A., Kawai A., Shimizu N., and Fujiki Y. (2000) The mammalian peroxin Pex5pL, the longer isoform of the mobile peroxisome targeting signal (PTS) type 1 transporter, translocates Pex7p-PTS2 protein complex into peroxisomes via its initial docking site, Pex14p. J. Biol. Chem. 275, 21703–21714 [DOI] [PubMed] [Google Scholar]
- 64. Mukai S., Ghaedi K., and Fujiki Y. (2002) Intracellular localization, function, and dysfunction of the peroxisome-targeting signal type 2 receptor, Pex7p, in mammalian cells. J. Biol. Chem. 277, 9548–9561 [DOI] [PubMed] [Google Scholar]
- 65. Yagita Y., Hiromasa T., and Fujiki Y. (2013) Tail-anchored PEX26 targets peroxisomes via a PEX19-dependent and TRC40-independent class I pathway. J. Cell Biol. 200, 651–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 67. Suzuki Y., Shimozawa N., Yajima S., Yamaguchi S., Orii T., and Hashimoto T. (1991) Effects of sodium 2-[5-(4-chlorophenyl)pentyl]-oxirane-2-carboxylate (POCA) on fatty acid oxidation in fibroblasts from patients with peroxisomal diseases. Biochem. Pharmacol. 41, 453–456 [DOI] [PubMed] [Google Scholar]
- 68. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rasmussen J. T., Börchers T., and Knudsen J. (1990) Comparison of the binding affinities of acyl-CoA-binding protein and fatty-acid-binding protein for long-chain acyl-CoA esters. Biochem. J. 265, 849–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.