

Abstract
Podocyte injury is an early event in diabetic kidney disease and is a hallmark of glomerulopathy. MicroRNA-146a (miR-146a) is highly expressed in many cell types under homeostatic conditions, and plays an important anti-inflammatory role in myeloid cells. However, its role in podocytes is unclear. Here, we show that miR-146a expression levels decrease in the glomeruli of patients with type 2 diabetes (T2D), which correlates with increased albuminuria and glomerular damage. miR-146a levels are also significantly reduced in the glomeruli of albuminuric BTBR ob/ob mice, indicating its significant role in maintaining podocyte health. miR-146a-deficient mice (miR-146a−/−) showed accelerated development of glomerulopathy and albuminuria upon streptozotocin (STZ)-induced hyperglycemia. The miR-146a targets, Notch-1 and ErbB4, were also significantly up-regulated in the glomeruli of diabetic patients and mice, suggesting induction of the downstream TGFβ signaling. Treatment with a pan-ErbB kinase inhibitor erlotinib with nanomolar activity against ErbB4 significantly suppressed diabetic glomerular injury and albuminuria in both WT and miR-146a−/− animals. Treatment of podocytes in vitro with TGF-β1 resulted in increased expression of Notch-1, ErbB4, pErbB4, and pEGFR, the heterodimerization partner of ErbB4, suggesting increased ErbB4/EGFR signaling. TGF-β1 also increased levels of inflammatory cytokine monocyte chemoattractant protein-1 (MCP-1) and MCP-1 induced protein-1 (MCPIP1), a suppressor of miR-146a, suggesting an autocrine loop. Inhibition of ErbB4/EGFR with erlotinib co-treatment of podocytes suppressed this signaling. Our findings suggest a novel role for miR-146a in protecting against diabetic glomerulopathy and podocyte injury. They also point to ErbB4/EGFR as a novel, druggable target for therapeutic intervention, especially because several pan-ErbB inhibitors are clinically available.
Keywords: diabetic nephropathy, kidney, microRNA (miRNA), podocyte, Type 1 diabetes, diabetic glomerulopathy, mir-146a
Introduction
Glomerular injury is a major, progressive complication of diabetes mellitus and the leading cause of end-stage renal disease (ESRD)3 in the United States (1). Its histopathologic changes include mesangial expansion, glomerular basement membrane (GBM) thickening and segmental glomerulosclerosis. Podocytes are insulin-sensitive cells and are injured early in response to diabetic milieu (2, 3), suggesting an important role for these cells in the development and progression of diabetic glomerulopathy. Although recent research has elucidated a number of disease-associated pathways (2, 4–10), how diabetic milieu causes podocyte injury, foot process effacement, and loss is still not completely understood. Clinically useful targets and therapeutics are also sorely needed. Additionally, although histopathologic changes from invasive patient biopsies can clearly distinguish the extent of kidney damage, microalbuminuria is currently the best predictor of progression to ESRD in diabetic patients (11, 12). However, not all patients progress to ESRD at the same rate; some progress faster, whereas others are resistant to further decline in renal function and microalbuminuria is not very helpful in stratifying patients in these groups. Thus, biomarkers for earlier, more sensitive diagnosis of patients who are likely to develop nephropathy or progress faster to ESRD are greatly desired, as it will greatly improve the disease management and patient treatment, and improve clinical trial design.
MicroRNAs (miRNAs) are a family of non-protein-coding RNAs that are ∼22 nucleotides (nt) in length. They sequence-specifically bind the 3′ UTR of target mRNAs, where they promote mRNA degradation or suppress mRNA translation, thus regulating cellular functions. miRNAs are endogenously expressed in the kidney and several have been found to be up- or down-regulated in models of diabetic glomerulopathy (reviewed in Ref. 13) and other renal diseases (14, 15). miR-193a is significantly up-regulated in podocytes in focal segmental glomerulosclerosis, where it directly targets WT1 transcripts (15). miRNAs miR-21, -192, -200b, -200c, -216a, and -217 are induced in the glomerular mesangial cells in animal models of diabetic nephropathy (DN), where a number of them participate in the TGFβ-Smad pathway to mediate glomerular damage (13, 16–19). Similarly, miR-29c is increased in podocytes under high glucose (20), where it promotes apoptosis by activating Rho kinase via suppression of Spry1. Unlike other miRNAs, hyperglycemia down-regulates miR-93 in podocytes and in glomeruli of diabetic animals, thereby increasing VEGF-A expression (21). Podocyte expressed miR-21 is a feedback inhibitor of TGFβ signaling and protects against diabetic glomerulopathy (22). These studies show that diabetic milieu modulates expression of many different miRNAs, and does so differentially in the many cells of the glomerulus. They also suggest that miRNAs play a critical role in the pathophysiology of diabetic glomerular injury.
miR-146a is a key negative regulator of innate immune responses in myeloid cells (23, 24), modulates adaptive immune responses, and has been shown to play central roles in many other cellular functions, including normal hematopoiesis and proliferation of cancer cells (23, 25). miR-146a is also expressed in various endothelial and epithelial cells, although its exact function in these cells is much less clear. Oxidative stress/injury of endothelial cells leads to up-regulation of miR-146a, which is shed into exosomes that are then taken up by cardiomyocytes to mediate peripartum cardiomyopathy (26). In another context, miR-146a induction limits proinflammatory signaling in endothelial cells (27). In the context of diabetic injury, a recent study found that miR-146a is constitutively expressed in the retinal endothelial cells and is down-regulated by high glucose (28). In another study, miR-146a levels were reduced in the retina, kidney, and heart of streptozotocin (STZ)-induced diabetic rats. Recently, Natarajan and co-workers (29) showed that myeloid cell-expressed miR-146a levels increase early in a mouse model of DN, where it plays an anti-inflammatory role by suppressing proinflammatory cytokines in macrophages. It was also recently reported that miR-146a levels increase in the kidney in a model of lupus nephritis (30). Thus, whereas there is a growing body of research showing that miR-146a expression is altered in disease, the role of miR-146a in glomeruli is still unclear and needs further evaluation. Recent unbiased profiling studies reported that miR-146a is highly expressed in podocytes (31, 32), suggesting that it has an important role in maintaining podocyte health. In this study, we examined the role of podocytic miR-146a in diabetic glomerular injury and present our findings below.
Results
Diabetic Patients with Reduced Glomerular miR-146a Levels Show Increased Albuminuria and Glomerular Damage
miR-146a is highly expressed in podocytes (31, 32), suggesting that it might play an important role in maintaining glomerular health. To examine its role, we correlated the expression level of miR-146a in micro-dissected glomeruli of kidney biopsies from patients with T2D and quantified by qRT-PCR clinical and morphometric outcomes (33).
We observed that a subset of patients with low glomerular abundance of miR-146a had high albumin to creatinine ratio (ACR) at the time of biopsy (Fig. 1A) and at a follow-up appointment (Fig. 1B). These data suggest that reduction or loss of miR-146a could be a risk factor for glomerular damage and albuminuria in patients with diabetes. Additionally, we analyzed small RNA sequencing data from microdissected glomeruli (glom) and tubulo-interstitial compartments (TI) of 40 kidney biopsy samples from this cohort and determined that the abundance of the miR-146a cluster miRNAs (34) was slightly, but significantly, enriched in the glomerular as compared with the TI tissue (log2FC [log2(expression in glom) − log2(expression in TI)] = 0.6, p < 0.05). Furthermore, we associated quantitative data of morphometric analyses of glomeruli of these samples (described in Ref. 35) with miR-146a abundance and found a significant correlation between low miR-146a levels and higher mesangial surface area (correlation: 0.351839, p = 0.02) and filtration surface fraction (correlation: −0.309166, p = 0.03), which are parameters associated with increased glomerular damage. Furthermore, patients with low relative expression exhibited relatively higher ACR at follow up compared with subjects with higher miR-146a levels (supplemental Fig. S1). These data support that lower miR-146a expression are associated with increased glomerular damage and albuminuria in diabetic patients.
FIGURE 1.

Down-regulation of miR-146a correlates with increased albuminuria in diabetic patients. A and B, scatter plots showing correlation between relative expression of miR-146a versus urinary ACR in the isolated glomeruli of kidney biopsies from diabetic patients (n = 45). Each dot represents an individual patient. Data are from two time points; one at the time of biopsy (A) and one at the end of observation period (5 ± 1 years post-biopsy, B). Relative miR-146a level of 0.4 is shown to divide the plot into two groups with different ACR values.
To further confirm our findings, we used in situ hybridization (ISH) on human and murine kidney sections. It showed that miR-146a was expressed in the glomeruli of healthy human kidney sections (Fig. 2A) and in the healthy wild-type mouse kidney sections (Fig. 2B). As a control, miR-146a−/− mouse glomeruli showed complete absence of miR-146a staining (Fig. 2B). The glomerular miR-146a levels were dramatically reduced in the kidney sections of diabetic patients. Podocyte loss causes glomerulosclerosis and results in progressive loss of renal function in patients with diabetic nephropathy (7, 36, 37). Kidney sections from diabetic patients also showed reduced staining for podocyte marker synaptopodin (Synpo) (Fig. 2A), suggesting that reduction in miR-146a correlates with reduced podocyte density and that podocyte loss may partially explain the reduced miR-146a expression in the glomeruli of diabetic patients. The glomerular miR-146a levels were also reduced in the kidney sections from the albuminuric BTBR ob/ob mice (Fig. 2B-2C), a genetic model of T2D (38). Quantitative RT-PCR (qRT-PCR) of isolated kidney sections further confirmed that miR-146a levels are significantly reduced in the albuminuric BTBR ob/ob mice (Fig. 2D and supplemental Fig. S2). These data confirm that miR-146a levels are reduced in the glomeruli of diabetic human and mouse kidneys.
FIGURE 2.
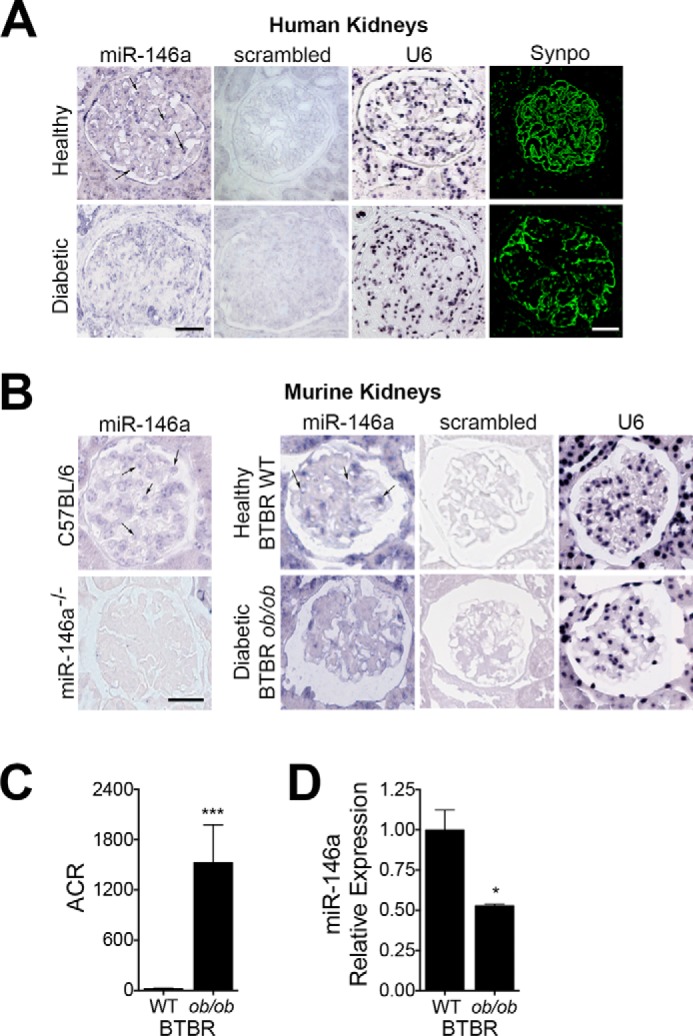
miR-146a levels are reduced in the diabetic human and mouse kidney glomeruli. A and B, representative ISH images of human (A) and mouse (B) kidney sections to detect the expression pattern of miR-146a (indicated with an arrow). Each kidney section was stained with the indicated probe (against miR-146a, a scrambled control or against U6 RNA). Confocal images of immunofluorescently labeled glomeruli (A), stained with the podocyte marker synaptopodin (Synpo, green), show relative podocyte density in the representative healthy and diabetic human kidney sections. Representative ISH images of kidney sections from C57BL/6 WT and miR-146a−/− animals stained with a specific probe against miR-146a are also shown (B). Scale bar, 50 μm (A and B). C, a bar graph showing urinary ACR in 12-week-old BTBR WT and BTBR ob/ob animals. Data shown are mean ± S.E. (n = 5/group). ***, p < 0.001. D, a bar graph showing relative expression level of miR-146a in kidney sections from 12-week-old BTBR WT and BTBR ob/ob mice, as measured by qRT-PCR. Data shown are mean ± S.E. (n = 3). *, p < 0.05.
miR-146a Targets Notch-1 and ErbB4 Are Up-regulated in the Diseased Glomeruli and in Podocytes
Among its various molecular targets, miR-146a directly targets Notch-1 and ErbB4 mRNA (Fig. 3, A and B) (26, 39–41). Notch-1 up-regulation in podocytes correlates with diabetic glomerular injury in experimental models and in patients (39, 41–43). ErbB4 (v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4, also known as HER4) is a member of the epidermal growth factor receptor (EGFR, also known as ErbB) family of tyrosine kinase receptors that often heterodimerizes with EGFR (44). A recent genomewide association study with T1D nephropathy patients showed that an intronic SNP in ErbB4 (rs7588550), which correlates with reduced ErbB4 expression, was significantly associated with protection from T1D (45), suggesting that its up-regulation in the glomeruli may be deleterious. Analysis of publicly available human transcriptomic data in Nephroseq from patients with diabetes mellitus and healthy controls showed that ErbB4 expression was significantly up-regulated in the diseased kidney sections (Fig. 3C) (46). Similarly, isolated kidney sections from miR-146a−/− animals showed no miR-146a expression as compared with the WT (supplemental Fig. S3A), and an increase in basal expression of its targets ErbB4 and Notch-1 in podocytes, but no increase in basal EGFR levels (supplemental Fig. S3B). Western blotting analysis of isolated glomeruli and the primary miR-146a−/− podocytes further confirmed increased expression of ErbB4 over the glomeruli and the podocytes from WT animals (supplemental Fig. S4). Histochemical and immunofluorescence staining of kidney sections from diabetic patients and the diabetic BTBR ob/ob mice also showed up-regulation of miR-146a targets Notch-1 and ErbB4 in the diseased glomeruli versus healthy controls (Fig. 3, D–F). We also observed glomerular up-regulation of the EGFR heterodimerization partner of ErbB4 in these samples, confirming similar findings by others (47–50). Co-staining with podocyte marker synaptopodin showed that the increase in their expression co-localized within podocytes in the glomeruli. Furthermore, expression of miR-146a targets Notch-1 and ErbB4 in the micro-dissected glomeruli of diabetic patients also showed negative association with miR-146a expression (Notch-1, r = −0.273; ErbB4, r = −0.108), providing additional confirmation of the immunofluorescence findings.
FIGURE 3.

miR-146a targets Notch-1 and ErbB4 are up-regulated in the diseased glomeruli. A, primary sequence and predicted secondary structure of pre-miR-146a. miRNA is transcribed in the nucleus as primary miRNA, processed by endonuclease Drosha into pre-miRNA, exported into the cytoplasm, where it is cleaved by RNase Dicer to yield a mature 22-nt duplex miRNA. The sequence of 22-nucleotide (22 nt) mature miR-146a duplex is highlighted in red and blue. Typically, only one strand of the 22-nt duplex gets loaded into an RNA-induced silencing complex (RISC) to suppress target gene expression. In miR-146a, this is typically the 5′-strand (shown in red), also known as miR-146a-5p. B, alignment of miR-146a target sites in the 3′ UTRs of Notch-1 and ErbB4 mRNA (26, 40). C, graph showing relative expression levels of ErbB4 mRNA in various human kidney biopsies, data were derived from Schmid et al. (46). The published microarray data were generated using renal biopsies from patients with histological evidence of DN (n = 13), where the biopsies from cadaveric donors (n = 4), related living donors (n = 3), and from patients with minimal change disease (MCD) (n = 4) without histological or clinical evidence of impaired renal function served as controls. Data shown are mean ± S.E. ****, p < 0.0001. D, representative images showing histochemical analyses of kidney tissue samples from 12-week-old BTBR WT (healthy) and BTBR ob/ob (diabetic) mice using periodic acid-Schiff (PAS) and Masson's trichrome (trichrome) staining showing increased mesangial sclerosis and fibrosis in the diabetic kidneys. Scale bar, 50 μm. E, expression of EGFR, Notch-1, and ErbB4 is up-regulated in the glomeruli of BTBR ob/ob mice. Representative confocal microscopy images of immunofluorescently labeled glomeruli from 12-week-old BTBR WT (healthy) and BTBR ob/ob (diabetic) mice. Kidney sections were imaged after staining with DAPI and antibodies against EGFR, Notch-1, ErbB4, and Synaptopodin (Synpo). Merged DAPI, EGFR and Synpo, DAPI, Notch-1 and Synpo, and DAPI, ErbB4 and Synpo channels are also presented that show podocyte colocalization for these proteins. Scale bar, 50 μm. F, expression of EGFR, Notch-1, and ErbB4 is up-regulated in the glomeruli of diabetic patients. Representative images showing histochemical analyses of kidney tissue samples (left panels) after PAS and trichrome staining showing extensive glomerular expansion, mesangial sclerosis, and fibrosis in the diabetic kidneys. Scale bar, 50 μm. Representative confocal microscopy images of immunofluorescently labeled kidney sections that imaged after staining with DAPI and antibodies against EGFR, Notch-1, ErbB4 and Synpo (right panels). Merged EGFR and Synpo, Notch-1 and Synpo, and ErbB4 and Synpo channels are shown that show podocyte colocalization for these proteins, respectively. Scale bar, 50 μm. Bottom panels present higher magnification views of the boxed regions in the middle panel. G, miR-146a reduces the activity of luciferase linked with 3′ UTRs of ErbB4 and Notch1. Bar graph reporting results from luciferase activity assay from HEK293T cells co-transfected with pre-miR-146a or pre-miR-control and a luciferase reporter plasmid containing either the WT or mutated (mut) sequence of 3′ UTRs of ErbB4 or Notch-1. Luciferase activity was normalized with the activity of β-galactosidase from a co-transfected plasmid. Data shown are mean ± S.E. (n = 4). *, p < 0.05. ***, p < 0.0005.
To confirm direct targeting of Notch-1 and ErbB4 transcripts by miR-146a, we utilized a luciferase reporter vector encoding the 3′ UTR of human ErbB4 or Notch-1 and mutated vectors containing mismatches in the predicted miR-146a binding site in each, respectively (supplemental Fig. S5) (26). Co-transfection of pre-miR-146a and the ErbB4 or the Notch-1 3′ UTR plasmids in HEK293T cells resulted in a significant decrease in the luciferase activity as compared with cells co-transfected with pre-miR-146a and the mutated ErbB4 or Notch-1 3′ UTR plasmids (Fig. 3G). Furthermore, retrovirus-mediated overexpression of miR-146a in WT podocytes also resulted in reduction of ErbB4 expression (supplemental Fig. S6). Together, these data suggest that miR-146a, by directly modulating the expression of its targets ErbB4 and Notch-1, may have an important regulatory role in podocytes and that miR-146a down-regulation may lead to transcriptional up-regulation of these proteins, resulting in glomerular damage in diabetes.
STZ Accelerates Glomerulopathy in miR-146a−/− Animals and Is Attenuated by Erlotinib
To further investigate the role of miR-146a in glomerular function in vivo, we induced hyperglycemia in miR-146a−/− animals using STZ (5). STZ treatment induced similar levels of hyperglycemia (Fig. 4A) and body weight decline (Fig. 4B) in both C57BL/6 wild-type (WT) and the miR-146a−/− animals that persisted throughout the course of this study, confirming recent findings (29). Although the diabetic WT animals showed a slow increase in albuminuria (5, 51, 52), the diabetic miR-146a−/− animals showed a more rapid increase and significantly higher level of albuminuria (Fig. 4C), suggesting that miR-146a deletion greatly accelerates the renal decline in animals, also in line with recent findings (29). Histopathology and morphometric analyses of the kidney sections showed significant mesangial sclerosis in the diabetic WT and miR-146a−/− animals as compared with the healthy controls (Fig. 5, A and B), and an expected increase in leukocyte infiltration with STZ treatment (supplemental Fig. S7) (29). Electron microscopy (EM)-based analyses showed significant foot process effacement (Fig. 5C). The receptor-tyrosine kinase (RTK) EGFR is expressed in the tubules and the glomeruli (53) and promotes TGFβ signaling in diabetes (48–50, 54). Immunofluorescence staining of kidney sections showed increased expression of EGFR along with increased Notch-1 and ErbB4 in the glomeruli of diabetic WT and miR-146a−/− animals, as compared with healthy controls (Fig. 6, A and B). Because the ErbB4 expression increased in both WT and miR-146a−/− diabetic glomeruli, we tested whether treatment with inhibitor erlotinib would offer protection from renal decline. Most small molecule inhibitors target multiple members of the EGFR family and no selective ErbB4 inhibitors are currently available. However, the EGFR inhibitor erlotinib also inhibits ErbB4 with nanomolar affinity (IC50 = 230 nm) (55). Therefore, we tested whether ErbB4 inhibition with this pan-ErbB inhibitor will have efficacy in this model. Administration of erlotinib to the diabetic animals did not change the level of hyperglycemia in either WT or the miR-146a−/− animals (Fig. 4A). However, erlotinib treatment protected both the WT animals, as was recently shown (49, 54), and the miR-146a−/− animals from development of albuminuria (Fig. 4C), and resulted in reduced leukocyte influx in both (supplemental Fig. S7). Furthermore, analysis of kidney sections of erlotinib-treated diabetic mice showed a significant reduction in glomerular mesangial sclerosis and foot process effacement (Fig. 5, A-C), and immunofluorescence analysis showed a significantly reduced expression of EGFR, Notch-1, and ErbB4 in the glomeruli of treated WT and miR-146a−/− animals (Fig. 6, A and B). ISH with a miR-146a probe (Fig. 7A) and immunohistochemical analysis with anti-WT1 antibody (Fig. 7B) showed that expression of miR-146a and WT1 was reduced in the STZ-treated, diabetic WT animals, but that the glomerular miR-146a and WT1 expression was largely preserved in the kidney sections of the animals treated with erlotinib. These data suggest that the ErbB4 pathway is up-regulated in the glomeruli of diabetic animals and that clinically available pan-ErbB inhibitors are potential therapeutics for reducing the glomerular damage.
FIGURE 4.

Erlotinib attenuates accelerated development of STZ-induced albuminuria in miR-146a−/− animals. A, both WT and miR-146a−/− animals developed similar and sustained levels of hyperglycemia within 2 weeks of STZ treatment. Treatment with erlotinib starting at 4 weeks after STZ induction did not result in any change in level of hyperglycemia in either strain. Blood glucose levels remained unchanged in the untreated WT and miR-146−/− mice. Data presented on these graphs are mean ± S.E. (n = 3–10/group). B, WT and miR-146a−/− animals displayed equal levels of weight loss upon STZ-induced hyperglycemia that was unaffected by treatment with erlotinib. Data presented on these graphs are mean body weight at the end of the experiment (16 weeks post-STZ) ± S.E. (n = 3–10/group). C, miR-146a−/− animals showed increased albuminuria after ∼8 weeks post-STZ and a significantly increased albuminuria 14 weeks post-STZ, as compared with the WT animals. Treatment with erlotinib significantly reduced the development of albuminuria and both the WT and the miR-146a−/− animals. Data shown are mean ± S.E. (n = 4–10/group). * and #, p < 0.05.
FIGURE 5.

STZ-induced hyperglycemia increases glomerulopathy and foot process effacement in the mouse glomeruli that is suppressed by erlotinib. A, representative images showing PAS-stained kidney tissue samples from WT and miR-146a−/− animals treated with vehicle alone (Control), STZ and vehicle (STZ), or STZ and erlotinib (STZ + Erl) and analyzed 16 weeks post-STZ showing increased mesangial sclerosis in STZ-treated animals that is reduced with erlotinib treatment. Scale bar, 50 μm. B, graphs showing quantification of mesangial matrix expansion from the PAS-stained sections in A. Data shown are normalized to the level of staining in control tissue and are mean ± S.E. (n = 3–5/group). *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, erlotinib protects against STZ-induced podocyte foot process (FP) effacement. Representative electron microscopy images of WT (top panels) and miR-146a−/− (bottom panels) mouse glomeruli treated with vehicle alone (Control), with STZ and vehicle (STZ), or with STZ and erlotinib (STZ + Erl). Scale bar, 2 μm. FP, foot processes; BM, basement membrane. A higher magnification view is presented below every image. Scale bar, 500 nm. A graph on the right shows quantification of the number of FPs per unit glomerular length in each of the samples. Data shown are mean ± S.E. (n = 3/group) and significant difference comparison was performed as compared with the respective controls. *, p < 0.05; **, p < 0.01; ns, not significant.
FIGURE 6.

STZ treatment of WT and miR-146a−/− mice results in increased glomerular injury and induction of miR-146a targets in the mouse glomeruli that is suppressed by erlotinib. A, STZ-induced up-regulation of EGFR, Notch-1, and ErbB4 expression in the mouse glomeruli is reduced by erlotinib. Representative confocal microscopy images of immunofluorescently labeled glomeruli from WT (top panels) and miR-146a−/− (bottom panels) mice treated with vehicle alone (Control), with STZ and vehicle (STZ) or with STZ and erlotinib (STZ + Erl). Kidney sections were imaged after staining with DAPI and antibodies against EGFR, Notch-1, ErbB4, and Synaptopodin (Synpo). Merged DAPI, EGFR and Synpo, DAPI, Notch-1 and Synpo, and DAPI, ErbB4 and Synpo channels are also presented that show podocyte colocalization for these proteins. Scale bar, 50 μm. B, graphs showing the quantification of relative glomerular signal intensity of EGFR, Notch-1, and ErbB4 in samples from A. Data shown are mean ± S.E. (n = 5/group). ns, not significant; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
FIGURE 7.
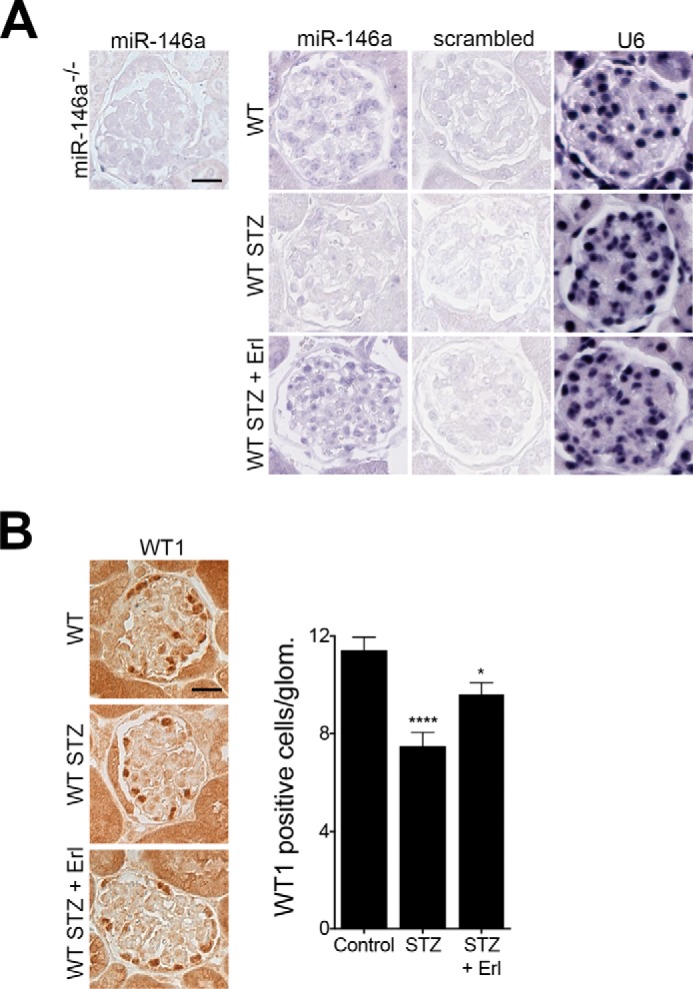
Reduction in glomerular miR-146a levels and podocyte numbers by STZ treatment is suppressed by erlotinib. A, representative ISH images of kidney sections from WT mice treated with vehicle alone (Control), with STZ and vehicle (STZ), or with STZ and erlotinib (STZ + Erl) and stained to detect expression pattern of miR-146a show protection of glomerular miR-146a levels by erlotinib. Each kidney section was stained with the indicated probe (against miR-146a, a scrambled control, or against U6 RNA). Representative ISH image of kidney sections from miR-146a−/− animals stained with a specific probe against miR-146a is also shown. Scale bar, 50 μm. B, erlotinib protects against STZ-induced podocyte loss. Representative immunohistochemical images of glomeruli stained with an antibody against WT1 from WT mice treated with vehicle alone (Control), with STZ and vehicle (STZ), or with STZ and erlotinib (STZ + Erl). Scale bar, 50 μm. Graph on the right shows quantification of WT1 positive cells per glomeruli in these samples. Data shown are mean ± S.E. (n = 3). *, p < 0.05; ****, p < 0.0001.
TGF-β1 Induces ErbB4 Expression and Signaling
Transforming growth factor β1 (TGF-β1) is a pleotropic cytokine and a member of the TGFβ superfamily. It mediates podocyte damage and glomerulosclerosis, and is a key mediator of glomerular injury in diabetes (56, 57). Actin cytoskeleton in podocytes is a critical link between various signaling components and the cellular receptors, whereas playing an essential role in providing stable architectural support to the cell. Immunofluorescence microscopy showed that TGF-β1 induced rearrangement of the actin cytoskeleton resulting in loss of stress fibers in podocytes (58, 59), a hallmark of cellular damage, and that this loss of stress fibers was prevented by co-treatment with erlotinib (Fig. 8A), suggesting a link between TGF-β1 signaling and ErbB4. TGF-β1 imparts its intracellular effects via Smad and MAPK signaling pathways and treatment of podocytes with TGF-β1 showed increased phosphorylation of Smad2/3, Erk, and p38, and expression of cleaved Notch-1, in addition to its autoinduction (Fig. 8B) (56, 57, 60, 61). We also found that TGF-β1 increased expression of ErbB4 and increased ErbB4 phosphorylation (Tyr-984), suggesting that it elicits intracellular signaling. It also enhanced EGFR phosphorylation in the treated cells (at three different sites Tyr-845, Tyr-1068, and Tyr-1173), but not the level of EGFR. Co-immunoprecipitation assays using overexpression vectors in HEK293 cells confirmed that ErbB4 and EGFR heterodimerize on the cell surface (supplemental Fig. S8), suggesting that their observed increase in phosphorylation is likely due to ErbB4/EGFR heterodimerization. miR-146a−/− podocytes showed basally increased expression of ErbB4, cleaved Notch-1 and TGF-β1, and increased phosphorylation of Smad2/3, Erk, p38, ErbB4, and EGFR (Fig. 8B), suggesting that the ErbB4/EGFR pathway is induced in the absence of miR-146a. Co-treatment of WT and miR-146a−/− podocytes with erlotinib to antagonize ErbB4/EGFR signaling showed a marked reduction in the deleterious expression changes induced by TGF-β1. Monocyte chemoattractant protein-1 (MCP-1) is a proinflammatory chemokine that is induced upon RTK-induced STAT3 activation (62). In turn, it increases expression of MCP1-induced protein 1 (MCPIP1), which inhibits miR synthesis and suppresses miR-146a (63, 64). We confirmed that TGF-β1 treatment induced MCP-1 in podocytes (65) and that erlotinib suppressed the TGF-β1-mediated increase in MCP-1 (Fig. 9), suggesting that MCP-1 induction is downstream of EGFR/ErbB4. Similarly, TGF-β1 increased expression of MCPIP1, which was suppressed by erlotinib (Fig. 8, B and C, bottom panel). In sum, these data suggest that diabetic milieu suppresses miR-146a levels in podocytes via induction of MCP1 and MCPIP1, and that reduced miR-146a results in up-regulation of its molecular targets Notch-1 and ErbB4, which also enhances MCP1, thus inducing an autocrine loop (Fig. 10).
FIGURE 8.

Pan-ErbB inhibitor suppresses inflammation by rescuing podocyte expression of miR-146a and suppressing its targets Notch-1 and ErbB4. A, TGF-β1 treatment reduces F-actin fibers in cultured podocytes and erlotinib protects against this loss. Representative confocal microscopy images of cultured mouse podocytes treated with vehicle alone (Control), TGF-β1 (5 ng/ml), or with TGF-β1 (5 ng/ml) and erlotinib (10 μm) for 24 h and stained with CellMask blue (nuclear stain) and Alexa Fluor 568-labeled phalloidin (to stain F-actin fibers). Scale bar, 20 μm. B and C, TGF-β1 treatment induces expression of Notch-1 and ErbB4 in cultured WT and miR-146a−/− podocytes and erlotinib suppresses it. Immunoblot analysis of various phosphorylated (p-) and total proteins in the lysates from WT (B) and miR-146a−/− (C) podocytes stimulated without or with 5 ng/ml of TGF-β1 (TGF-β1) in the absence or presence of erlotinib (10 μm). GAPDH was used as the loading control. Relative position of the molecular weight markers is shown on the left.
FIGURE 9.
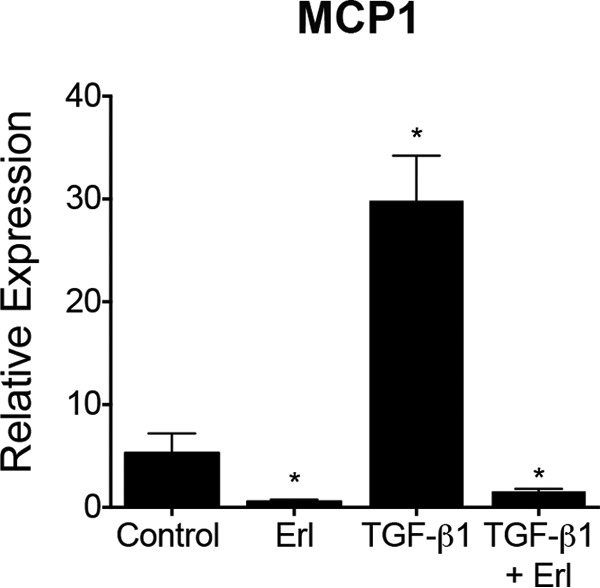
TGF-β1 induced up-regulation of MCP-1 expression in podocytes is suppressed by erlotinib. A bar graph showing the relative expression level of MCP-1 in WT podocytes treated with vehicle DMSO (Control), 10 μm erlotinib (Erl), 5 ng/ml of TGF-β1 (TGF-β1), or 5 ng/ml of TGF-β1 and 10 μm erlotinib (TGF-β1 + Erl) for 24 h, as measured by qRT-PCR. Data were normalized using GAPDH mRNA controls and are mean ± S.E. (n = 3). **, p < 0.005.
FIGURE 10.
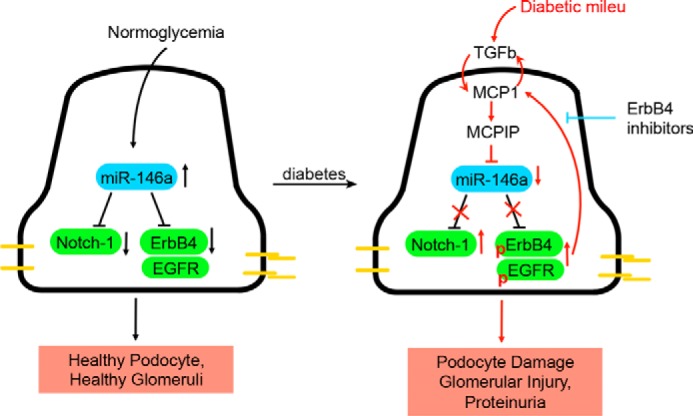
Working model. A diagram showing our working model. Under homeostatic conditions of normoglycemia, podocyte expressed miR-146a suppresses expression of Notch-1 and ErbB4 to maintain healthy cells. Diabetic milieu, including TGF-β1 (and MCP-1), induces MCP1 and MCPIP1 in podocytes, which decreases miR-146a levels. This results in de-repression of Notch-1 and ErbB4, which together with EGFR, induce podocyte injury. Blocking this signaling pathway with ErbB4/EGFR inhibitors suppresses the harmful signaling and decreases podocyte injury in vitro and glomerulopathy in vivo.
Discussion
Podocyte damage and dysfunction is a hallmark of glomerular diseases and is evident in diabetic glomerulopathy. MicroRNAs play a significant role in maintaining podocyte health and in the pathogenesis of podocytopathies. This study provides evidence, for the first time, that miR-146a protects podocytes against diabetic injury and that its loss exacerbates diabetic glomerulopathy. Although miR-146a is most commonly associated with innate immune cells and has primarily been studied in that context, it is also highly expressed in healthy podocytes (31, 32). Our surprising finding that miR-146a expression is significantly reduced in the glomeruli of diabetic patients and diabetic animals, and that its reduction in glomeruli correlates with glomerular damage, support its role in maintaining glomerular health. Furthermore, patients with low glomerular miR-146a levels show significantly faster decline in renal function, as compared with patients with relatively higher levels. Concomitant with changes in miR-146a levels, we also observed up-regulation of direct mRNA targets of miR-146a, namely Notch-1 (39, 40) and ErbB4 (26, 41) (and its binding partner EGFR), in the glomeruli of both diabetic human and mouse kidneys, suggesting a direct molecular link. STZ-induced hyperglycemia significantly accelerated development of albuminuria and glomerular injury in miR-146a−/− animals, as compared with the WT mice, along with an increase in glomerular expression of Notch-1 and ErbB4. Blocking ErbB4 with a known antagonist, erlotinib, protected animals from the development of albuminuria and significantly reduced glomerular damage. This suggests that miR-146a, by directly modulating the expression of Notch-1 and ErbB4, may have an important regulatory role in podocytes. Mechanistic studies with cultured podocytes showed that treatment with TGF-β1, a mediator of diabetic podocyte injury in vivo, increased expression of Notch-1 and ErbB4. TGF-β1 treatment also increased phosphorylation of ErbB4 and its heterodimerization partner EGFR, activating downstream RTK signaling, which has previously been shown to induce proinflammatory pathways via induction of STAT3 (62). Indeed, TGF-β1 increased levels of MCP-1 in podocytes as well as autoinduction of TGFβ. TGF-β1 also increased levels of MCPIP1, a ribonuclease that is up-regulated by MCP-1 and antagonizes miR-146a in cells (63, 64). These data suggest that miR-146a acts as a molecular break on ErbB4 signaling via suppression of ErbB4 levels in podocytes, and that diabetic milieu, via TGF-β1, activates a feed-forward loop, where MCP-1 and MCPIP1 reduce miR-146a levels, thereby activating ErbB4 signaling, which increases autocrine synthesis of MCP-1, further reducing the miR-146a levels (Fig. 10). Data showing that erlotinib treatment counteracts this ErbB4-mediated MCP-1 autocrine loop provides further evidence for this pathway being active in mediating diabetic injury in podocytes. Our findings suggest a novel role for miR-146a in the pathogenesis of diabetic glomerulopathy and as a biomarker for disease progression. They also point to ErbB4 as a novel target for therapeutic intervention.
To date, miR-146a has primarily been defined by its role in an innate immune system, as a negative regulator of proinflammatory NFκB signaling (23, 24). Its currently defined role in modulating kidney injury has also been limited to its function in the immune cells. Indeed, a recent study by Natarajan and co-workers (29) nicely showed that myeloid cell expressed miR-146a levels increase early during DN in mice due to increased influx of miR-146a-expressing myeloid cells, and that miR-146a−/− mice show accelerated DN due to both an increase in macrophage influx in the kidney and an increase in the proinflammatory phenotype of the infiltrated macrophages in the knock-out. However, whether podocyte-expressed miR-146a has any role in podocyte function has never been studied before. The results presented here clearly show that the podocytic miR-146a also has a protective role against diabetic injury, complementing the previous studies. However, because our studies here utilized the global miR-146a−/− (as podocyte-specific miR-146a−/− are not available), one could argue that myeloid cell-expressed miR-146a, due to its role in regulating proinflammatory pathways, is the dominant cell type for the observed effects. However, if macrophage-derived miR-146a was the sole driver of disease pathogenesis, one would expect to find increased miR-146a levels in the tissue from patients with diabetic kidney disease due to increased influx of macrophages. As shown in Figs. 1 and supplemental Fig. S1, we find the exact opposite, in that lower glomerular levels of miR-146a associate with increased proteinuria and glomerular damage. This, in combination with the recent study looking at its role in macrophages (29), suggests both a podocyte intrinsic and extrinsic role for this microRNA in diabetic glomerular injury.
Studies presented here also provide a potential mechanistic understanding behind the previously described role of Notch-1 in diabetic glomerular injury. Notch-1 is a member of a family of four transmembrane proteins that are key developmental proteins (66). Although the Notch pathway is indispensable for renal glomerular and proximal tubule development (67–69), Notch-1 expression is down-regulated in healthy adult kidneys. However, Notch-1 is up-regulated in the glomeruli of diabetic patients and in experimental models of diabetic nephropathy (42, 43). Podocyte-specific overexpression of ICN is sufficient to induce podocyte injury and glomerulosclerosis, suggesting that Notch-1 plays a critical role in the pathogenesis of diabetic glomerulopathy. However, a detailed molecular mechanism behind how Notch-1 is kept in check in healthy podocytes and how it is up-regulated during diabetic injury is not clear. The data presented here suggests that miR-146a may be an important microRNA regulating levels of Notch-1 in podocytes.
Prior studies have also shown that EGFR phosphorylation and activation in podocytes promotes glomerular injury in response to inflammatory and diabetic stimuli, and that its pharmacologic blockade or podocyte-specific deletion is protective (47–50). EGFR phosphorylation leads to induction of TGFβ-Smad-2/3 signaling, resulting in podocyte apoptosis. However, whether induction and activation of the ErbB4 heterodimerization partner of EGFR in podocytes plays a role in disease pathogenesis and what that role might be has not been described before. The results presented here suggest that both proteins are induced in the diabetic glomeruli. Similarly, the pan-ErbB kinase inhibitor erlotinib binds to EGFR with a significantly higher affinity (2 nm) than ErbB4 (55) and has been shown to protect against DN (47–50), which might suggest that the efficacy of erlotinib observed here is likely due to its inhibition of EGFR over ErbB4, suggesting that EGFR is the main culprit in disease pathogenesis. However, here we find that, at least with the TGF-β1 treatment of podocytes in vitro, ErbB4 levels increased in cells without affecting levels of EGFR, suggesting that ErbB4 up-regulation has pathogenic relevance. TGF-β1 treatment also increased phosphorylated forms of both EGFR and ErbB4, suggesting that the heterodimeric receptor signals together for the induction of downstream signaling pathways and that erlotinib inhibits this signaling. Still, the studies presented here cannot rule out that the protective effects of erlotinib are solely due to it inhibiting EGFR. Future studies with tissue-specific ErbB4 knockouts and knockins, as well as any compounds that selectively target ErbB4 over EGFR will help clarify this better.
Finally, ErbB4, like Notch-1, is a key developmental protein, which is expressed in developing nephrons and plays a role in epithelial cell proliferation and tubulogenesis, and is present in podocytes (70–73). It shows minimal expression in the adult kidney. Although ErbB4 is highly up-regulated in the kidneys with polycystic kidney disease, its genetic deletion protected animals from polycystic kidney disease, suggesting a complex and unclear role for ErbB4 in development and disease pathogenesis (74). Here, our results show that ErbB4 is up-regulated in diabetic glomeruli and in podocytes, suggesting that it plays a role in diabetic podocyte injury. Additionally, we find that ErbB4 shows higher expression at basal level in miR-146a−/− podocytes, suggesting regulation by miR-146a. These results also provide a potential explanation for the recent findings from a genomewide association study with >11,000 type 1 diabetes (T1D) patients aiming to look at an association for DN as a primary phenotype, which reported that an intronic SNP in ERBB4 (rs7588550, p = 2.1 × 10−7) that results in reduced expression of ErbB4 provides the strongest protective effect (45).
In summary, our findings suggest a novel role for miR-146a in the pathogenesis of diabetic glomerulopathy and as a biomarker for disease progression. They also point to ErbB4 as a novel target for therapeutic intervention early in the disease process. Given that several ErbB kinase inhibitors, which also target ErbB4, are in the clinic as potent anti-cancer agents, our findings also suggest that such clinically available agents are potential therapeutics for diabetic kidney disease.
Experimental Procedures
Materials
The cell culture reagents were purchased from Life Technologies, except for the fetal bovine serum (FBS), which was from HyClone (Logan, UT). Alexa Fluor 568-labeled phalloidin and HCS CellMask blue were obtained from Life Technologies. Rat tail collagen I, was from Sigma and mouse recombinant interferon-γ was from Cell Sciences (Canton, MA). Recombinant TGF-β1 was from R&D Systems (Minneapolis, MN) and erlotinib was from LC Laboratories (Woburn, MA). The list of primary antibodies used in Western blotting is as follows. The polyclonal goat anti-synaptopodin antibody (P-19) and rabbit anti-ErbB4 antibody (C-18) were from Santa Cruz (Dallas, TX); rabbit anti-EGFR was from Millipore (number 06-847, Darmstadt, Germany); polyclonal rabbit anti-Notch1 was from Rockland (number 100-401-407, Limerick, PA); rabbit anti-podocin antibody was from Sigma. Antibodies against cleaved Notch1 (Val-1744) and anti-GAPDH (6C5) were obtained from Abcam (Cambridge, MA). The anti-EGFR (D38B1), anti-p-EGFR (Tyr-845), anti-p-EGFR (Tyr-1068) (D7A5), anti-p-EGFR (Tyr-1173) (53A5), anti-p-ErbB4 (Tyr-984), anti-ERK (137F5), anti-p-ERK (Thr-202/Tyr-Y204), anti-p38, anti-phospho-p38 (Thr-180/Tyr-182) (28B10), anti-Smad2/Smad3, anti-p-Smad2 (Ser-465/467)/Smad3 (Ser-423/425) (D27F4), and anti-TGF-β1 antibodies were from Cell Signaling (Danvers, MA). Retroviral vector for ectopic miR-146a expression has been described previously (25). Control vector LV015-G encoding a scrambled sequence was from Applied Biological Materials (Richmond, BC, Canada).
Animals
Animal care and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with institutional guidelines. The C57BL/6J wild-type (WT) and miR-146a−/− (catalog number 016239) (75) mice were purchased from the Jackson Laboratory (Bar Harbor, ME).
Blood Glucose and Urinary Albumin and Creatinine Measurements
Blood glucose was measured from blood obtained from the tail vein of mice by using a FreeStyle Freedom lite glucometer (Abbott, Abbott Park, IL). For urinary albumin and creatinine measurements, spot urine samples were collected non-invasively from mice. Urinary albumin and creatinine concentrations were measured using a mouse albumin ELISA (Bethyl Laboratories, Montgomery, TX) and a creatinine assay (Exocell, Philadelphia, PA), respectively. Urine albumin:creatinine ratios were then calculated.
Primary Podocyte Isolation
Primary mouse podocytes were isolated according to published protocols (76). Briefly, kidneys were collected from 8- to 16-week-old mice. Freshly collected kidneys were mashed with cold PBS through test sieves (Retsch, Newtown, PA) whose pore diameters are 180, 75, and 52 μm, sequentially. Glomeruli in the sieve of 52-μm pores were collected in PBS. After spinning down, they were resuspended with RPMI 1640 medium containing 10% FBS and plated on collagen I-coated plates for 14 days. Cells were trypsinized and filtered with a 40-μm strainer. Filtered cells were spun down and seeded on collagen I-coated plates for culturing.
Cell Culture and Immunofluorescence Staining
Immortalized murine podocytes have been described and were cultured according to published protocols (77, 78). Cells were transduced with miR-146a or scrambled sequence containing vectors as previously described (25). For immunofluorescence staining of cultured cells, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% bovine or donkey serum (Sigma). After incubating with primary antibodies, secondary antibodies and/or Alexa 568-conjugated phalloidin (Life Technologies) were incubated with cells. Stained cells were imaged with a Zeiss 700 LSM confocal microscope (Zeiss, Hartford, CT) and Opera Imaging System (PerkinElmer Life Sciences), as described (58).
For tyramide signal amplification-mediated immunofluorescence co-staining of synaptopodin with the markers Notch-1, EGFR, or ErbB4 in formalin-fixed tissue sections, human or mouse kidney tissue was fixed in formalin and embedded in paraffin for further processing. Tissue sections (3 μm) were deparaffinized and hydrated through xylenes and graded alcohol series before acidic antigen retrieval (number H-3300, Vector Laboratories, Burlingame, CA). Sections were incubated in 0.1% Triton X-100 for 10 min at room temperature, washed, and incubated with blocking buffer for 1 h at room temperature. Sections were then incubated with primary antibodies in blocking buffer at 4 °C overnight. Tissues were washed and incubated with 0.3% hydrogen peroxide for 30 min at room temperature to block endogenous peroxidase activity. Sections were then incubated with blocking buffer containing HRP-labeled secondary antibody polymer (number MP-7401, Vector Laboratories) and donkey anti-goat AF488 (number A-11055, Thermo Fisher) for 30 min at room temperature. Sections were washed, incubated with tyramide signal amplification reagent for 10 min, washed, and stained with DAPI for 3 min at room temperature. Stained tissues were imaged with a Zeiss 700 LSM confocal microscope and the images were quantified and analyzed using ImageJ software (NIH, Bethesda, MD).
Electron Microscopy Imaging and Quantification
For transmission electron microscopy, tissues were fixed, embedded, sectioned, and stained as previously described (79). Briefly, kidney sections from 25-week-old miR-146a−/− mice treated with vehicle (sodium citrate), STZ, or STZ and erlotinib were sectioned and imaged as recently described (58). Images were acquired using Zeiss Sigma HD VP electron microscope (Zeiss). Transmission electron microscopy micrographs were analyzed using SmartTiff software (Zeiss). To reduce potential bias, the analyses were performed in a blinded fashion, where the tissues were sectioned, stained, imaged, and analyzed by an independent observer that was blinded to the tissue identity. Furthermore, micrographs selected at random from three different glomeruli per condition were used for analysis. Length of GBM was measured using the point-to-point tool on images acquired at ×5000 magnification. Each secondary foot process was tallied manually and divided into total GBM length to calculate number of foot processes per micron of GBM. This ratio was used as an indicator of foot process effacement.
Luciferase Plasmids
For the luciferase assay, 3′ UTR of wild type or mutated ErbB4 or Notch-1 were cloned into luciferase reporter plasmid pmirGLO plasmid (Promega, Madison, WI) following the manufacturer's instructions. The inserted 3′ UTR sequences were (also in supplemental Fig. S5): wild type ErbB4 3′ UTR: AAGAAATGTCCACATAACTTCGTGGTAGATTCCAGTTCTTGTGTACGAGCCTGCCCTAGT, mutated ErbB4 3′ UTR: AAGAAATGTCCACATAACTTGCTAAAAGATCCCAATTCTTATGTACGAGCCTGCCCTAGT; wild type Notch1 3′ UTR: GGAAAAACATATCTGTTCCAAGAAAATAAACTAGTTCTCAGAGCCTTGATTTTCCTGG, mutated Notch1 3′ UTR: GGAAAAACATATCTGTTCGATCTACTTACACTGGATCTCAGAGCCTTGATTTTCCTGG.
Luciferase Assay
MicroRNA precursors pre-miR 146a-5p (hsa-miR-146a-5p, catalog number AM17100, Life Technologies) or the control precursor pre-miR Negative Control #1 (AM17110, Life Technologies) were transfected into HEK293T cells in 96-well cell culture plates using Lipofectamine 3000 (Life Technologies) and following the manufacturer's protocols. After 24 h, the cells were further transfected with reporter luciferase expression plasmids (pmirGLO, Promega) containing sequence from wild type or mutated ErbB4 3′ UTR or Notch1 3′ UTR as above (supplemental Fig. S5), along with the β-galactosidase expression plasmid (80). To quantify luciferase activity in each well, cells were washed with PBS and lysed using supplied lysis buffer 48 h after plasmid transfection. Cellular lysates were used in a luciferase assay and a β-galactosidase assay. The luminescence activity in the lysate was measured 15 min after mixing the cell lysate with a luciferase substrate (Promega). The β-galactosidase activity in the lysate was measured by incubating the cell lysate with assay buffer containing o-nitrophenyl-β-galactoside (Sigma) for 30 min and, subsequently, using sodium carbonate to stop the reaction and measuring absorbance at 420 nm. Luminescence values were normalized to each absorbance readings at 420 nm of the lysate (β-galactosidase activity).
Western Blotting
Podocytes were cultured in 100-mm dishes under non-permissive conditions at 37 °C without γ-interferon for 10–14 days to promote differentiation. Differentiated podocytes were maintained in serum-free medium overnight before treating with recombinant mouse TGFβ1 (5 ng/ml) and erlotinib (10 μm) for 24 h. Subsequently, cells were washed with ice-cold PBS and lysed using cold lysis buffer (RIPA containing EDTA and EGTA, Boston Bioproducts, Ashland, MA) supplemented with protease inhibitor (Roche Life Science, Indianapolis, IN) and phosphatase inhibitor (Sigma). Cell lysates were incubated on ice for 30 min (with intermittent vortexing) and centrifuged in a tabletop microcentrifuge at 13,000 rpm for 15 min at 4 °C. Supernatants were carefully transferred to new microcentrifuge tubes and protein concentrations were determined by Bradford assay (Bio-Rad). Equal amounts of protein from each sample were loaded to NuPAGE Novex 4–12% BisTris gels (Life Technologies) and transferred to an Immobilon-P PVDF membrane (EMD Millipore, Billerica, MA). The membrane was blocked with 5% BSA (ThermoFisher Scientific) in TBS/Tween-20 (0.05%) (Boston Bioproducts, Ashland, MA), followed by incubation with primary antibodies at 4 °C overnight. After washing with TBS containing Tween 20, the membrane was incubated with a secondary antibody conjugated to horseradish peroxidase (Promega) for 1 h at room temperature. Blots were developed using SuperSignal West Pico chemiluminescent substrate (ThermoFisher Scientific) using X-ray films (Kodak, Rochester, NY) and using an AX-700LE film processor (Alphatek, Houston, TX).
Co-immunoprecipitation Assay
HEK293T cells were transfected with mammalian expression plasmids coding for 20 μg of EGFR (11011, Addgene), or 20 μg of ErbB4 (29536, Addgene) or both using Lipofectamine 3000 and Opti-MEM (Life Technologies) in 100-mm cell culture dishes following the manufacturer's protocols. After 48 h, cells were lysed with Nonidet P-40 lysis buffer (Boston Bioproducts) supplemented with protease inhibitor (Roche Life Science) and phosphatase inhibitor (Sigma). Total cell lysates containing 800 μg of protein were incubated with primary antibodies (either rabbit isotope control antibody or anti-ErbB4 antibody (Santa Cruz Biotechnology)) overnight at 4 °C. Subsequently, protein G beads (Life Technologies) pre-blocked with BSA were added to the lysate/antibody mixture and incubated for 4 h at 4 °C. Next, the beads were thoroughly washed and bound protein was eluted using Laemmli buffer (Bio-Rad) and boiled for 5 min. Eluted proteins were used for Western blotting, as described above.
miRNA Analysis by qRT-PCR
MicroRNA analysis was performed using qRT-PCR protocol as previously described (22). Briefly, total RNAs or miRNA fractions were isolated from mouse kidney tissue or from mouse podocytes using miRNeasy Mini Kit (Qiagen, Valencia, CA) according to the manufacturer provided protocol and quantified using NanoDrop (ThermoFisher). The isolated RNAs (0.5–2.0 μg) were used as template for cDNA synthesis using a High Capacity cDNA Archive Kit (Life Technologies). qRT-PCR was performed using CFX96TM Real-time System (Bio-Rad) and the following TaqMan® Gene expression assays (Life Technologies) were used: miR-146a (A25576, ID: 478399_mir), miR361 (A25576, ID: 478056_mir), and Gapdh (4331182, ID: Mm99999915_g1). For analysis, the fold-change in mRNA levels between various groups was determined after normalizing each mRNA expression level with Gapdh (2−ΔΔCt method). The fold-change in miR-146a levels between various groups was determined after normalizing the miR-146a expression level in each group with levels of miR361.
STZ-induced Hyperglycemia
Hyperglycemia was induced in the wild type C57B/L6 and the miR-146a−/− mice according to published protocols (5). Briefly, 8–10-week-old male mice were administered two doses of STZ (Sigma) (125 mg/kg body weight) in 50 mm sodium citrate buffer, pH 4.5, intraperitoneally on days 1 and 4. Glucose levels from tail blood were measured with an Accu-Check glucometer (Roche Life Science). Animals with glucose levels >400 mg/dl on two consecutive measurements were regarded as hyperglycemic. The mice received no insulin during the study period. Urinary albumin and creatinine were analyzed before injection and 2, 4, 6, 8, 10, 12, 14, and 16 weeks after the STZ injection. Erlotinib (37 mg/kg), in saline containing 1% Tween 20, 25% kolliphor, and 2.5% DMSO, was administered intraperitoneally every other day starting at week 4 post-STZ to a group of mice, according to literature protocols. Kidneys were harvested and processed for histological and ultrastructural analyses after 16 weeks post-STZ.
Tissue Histochemical and Immunofluorescence Staining
Mouse kidneys were harvested after perfusion with PBS. One section of the removed kidney was fixed in 10% formalin and embedded in paraffin and another part was immediately snap frozen in OCT embedding compound on liquid nitrogen and stored at −80 °C. Paraffin-embedded sections (4 μm) were stained with hemotoxylin-eosin (H&E), periodic acid-Schiff (PAS), or Masson's trichrome. Stained slides were blindly evaluated by an experienced pathologist and scanned using Aperio software (Leica, Buffalo Grove, IL). Fibrosis was indicated as a percent of tissue area stained blue with Masson's trichrome. Quantification for glomerular volume and mesangial expansion was performed according to published methods using ImageJ software (National Institutes of Health, Bethesda, MD) (29). Glomerular podocyte density was determined for using anti-WT1 (sc-15421, Santa Cruz) immunohistochemical staining and manual counting of WT1-positive cells from at least 5 glomeruli per sample (n = 3) in a blinded fashion. CD11b-positive cells were analyzed by immunohistochemical staining using anti-CD11b antibody (ab75476, Abcam). For immunohistochemical staining, tissue sections were deparaffinized in xylene and rehydrated through descending concentrations of ethanol and subjected to antigen retrieval by steam heating in an acidic pH solution (Citrate-based, Vector Laboratories). Subsequently, sections were incubated in 0.3% hydrogen peroxidase in water for 30 min followed by blocking (4% FBS, 4% BSA, 0.4% fish gelatin) at room temperature for 1 h. Thereafter, sections were incubated with primary antibodies at 4 °C overnight, followed by washing and incubation with appropriate secondary HRP-labeled secondary antibody polymer (Vector Labs) for 30 min at room temperature. Signal development was accomplished by using the DAB substrate kit (Vector Laboratories). CD11b quantification was done by counting marker positive cells in four independent, randomly chosen areas analyzed at ×40 using a light microscope for each kidney tissue (n = 3–5/treatment group).
For immunofluorescence analyses, frozen tissue sections were prepared and fixed in −20 °C acetone before immunofluorescence staining and analyses. Sections were blocked at room temperature for 1 h and incubated with the primary antibodies against total Notch-1 (polyclonal rabbit anti-Notch1, (catalog number 100-401-407, Rockland, Limerick, PA), ErbB4 (polyclonal rabbit anti-ErBB4, catalog number sc-283, C-18, Santa Cruz, Dallas, TX), EGFR (polyclonal rabbit anti-EGFR catalog number 06-847, Millipore, Darmstadt, Germany), and synaptopodin (polyclonal goat, SC-21537, Santa Cruz) at 4 °C overnight. Slides were incubated with appropriate fluorescently labeled secondary antibodies (Life Technologies) and mounted with DAPI containing mounting solution (Vector Laboratories). Fluorescence images were acquired using a Zeiss 700 LSM confocal microscope with a ×20 objective and analyzed using the Zen software (Zeiss, Hartford, CT). Quantification was performed using ImageJ Software (NIH).
Human Samples
Human specimens and clinical data were from recently described studies (22, 33). Briefly, kidney biopsy samples were collected from Southwestern American Indians enrolled in a randomized, placebo-controlled clinical trial to test the renoprotective efficacy of Losartan in early type 2 diabetic kidney disease (ClinicalTrials.gov no. NCT00340678). Glomeruli and tubulointerstitial fractions were isolated by microdissection, and the small RNA fraction was isolated using AllPrep kit (Qiagen). For expression analysis of miR-146a and the mRNAs, TaqMan primers (Applied Biosystems) and the 7900HT Fast Real-time PCR System (Applied Biosystems) were used according to the manufacturer's protocols as described. Patients with ACR>1g/mg were excluded. The small RNA fraction of the isolated RNA was used for construction of small RNA libraries. cDNA library preparation and multiplexed RNA sequencing was performed as previously described (22). This study was approved by the Review Board of the National Institutes of Health, NIDDK, and each participant gave informed consent.
miRNA Expression Profiling on Human Samples
miRNA profiling by quantitative real-time PCR on human specimens was performed as recently described using small RNA fractions (<200 nucleotides) was isolated from microdissected glomeruli and TaqMan miRNA assays (Applied Biosystems) (22). miRNA expression values and threshold cycle (CT) were normalized by U6 small nuclear RNA (snRNA), and RNU44 and RNU48 small nucleolar RNA (snoRNA). ΔCycle time (ΔCT) was calculated by subtracting the CT of the miRNAs from the CT geometric mean of snRNA and snoRNA.
For miRNA expression profiling by small RNA sequencing, the isolated small RNA fraction, as above, was used for construction of small RNA libraries. cDNA library preparation and multiplexed RNA sequencing was performed as previously described (22, 81, 82). Data were analyzed as described (22), where the miRNA annotation was adopted from literature definitions and the reads from miRNAs that were at the same precursor cistronic transcription unit in the genome were collapsed together into one miRNA cluster annotation. For normalization, the miRNA sequence reads were normalized by the total reads of each sample, as described (22).
Statistical Analysis
Data were analyzed using Excel (Microsoft, Redmond, WA) and Prism (GraphPad, San Diego, CA) softwares and were compared with using the Student's t test, where appropriate. p < 0.05 was considered statistically significant.
Author Contributions
H. W. L., S. Q. K., S. J. K., and M. H. F. designed and performed in vitro and in vivo experiments with erlotinib and analyzed data; H. W. L., M. M. A., T. G., and K. H. K. performed Western blotting analysis, PCR, and qPCR and analyzed data; S. Q. K., S. J. K., F. G., K. S., P-L. T., and T. B. H. designed, performed, and analyzed immunofluorescence studies; N. J. T. performed TEM studies; D. J. C. helped with histopathology; M. H. F., S. J. K., J. L. Z., L. F. M., D. B., and T. B. H. helped design in vivo assays using miR-146a KO animals; M. K., M. B., J. R., and V. G. analyzed human data; V. G. designed and supervised the studies and H. W. L., S. Q. K., and V. G. co-wrote the paper. All authors reviewed and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Hatem Elshabrawy, Saravana Kanagavelu, Steve Mangos, Andrew Armstrong, Isabel Fernandez, Prachal Bhargava, Dony Maiguel, Tristan Hays, and Alex Braley for generous technical help with the podocyte cell-based assays, animal husbandry, and helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK084195 and R01HL109582 (to V. G.) and R01DK106512 and R01DK107984 (to V. G. and J. R.), the Nephcure Foundation and resources from the Rush University Medical Center. V. G. and J. R. are inventors on pending patent applications related to this study. These authors and the Rush University Medical Center have the potential for financial benefit from their future commercialization. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S8.
- ESRD
- end-stage renal disease
- GBM
- glomerular basement membrane
- miRNA
- microRNA
- DN
- diabetic nephropathy
- STZ
- streptozotocin
- qRT
- quantitative RT
- T2D
- type 2 diabetes
- ACR
- albumin to creatinine ratio
- TI
- tubulo-interstitial
- ISH
- in situ hybridization
- EGFR
- epidermal growth factor receptor
- RTK
- receptor-tyrosine kinase
- MCP-1
- monocyte chemoattractant protein-1
- nt
- nucleotide
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. USRDS: the United States Renal Data System (2003) Am. J. Kidney Dis. 42, 1–230 [PubMed] [Google Scholar]
- 2. Wolf G., Chen S., and Ziyadeh F. N. (2005) From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 54, 1626–1634 [DOI] [PubMed] [Google Scholar]
- 3. Coward R. J., Welsh G. I., Yang J., Tasman C., Lennon R., Koziell A., Satchell S., Holman G. D., Kerjaschki D., Tavaré J. M., Mathieson P. W., and Saleem M. A. (2005) The human glomerular podocyte is a novel target for insulin action. Diabetes 54, 3095–3102 [DOI] [PubMed] [Google Scholar]
- 4. Remuzzi G., Benigni A., and Remuzzi A. (2006) Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J. Clin. Investig. 116, 288–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gödel M., Hartleben B., Herbach N., Liu S., Zschiedrich S., Lu S., Debreczeni-Mór A., Lindenmeyer M. T., Rastaldi M. P., Hartleben G., Wiech T., Fornoni A., Nelson R. G., Kretzler M., Wanke R., et al. (2011) Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 121, 2197–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartleben B., Gödel M., Meyer-Schwesinger C., Liu S., Ulrich T., Köbler S., Wiech T., Grahammer F., Arnold S. J., Lindenmeyer M. T., Cohen C. D., Pavenstädt H., Kerjaschki D., Mizushima N., Shaw A. S., Walz G., and Huber T. B. (2010) Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 120, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spurney R. F., and Coffman T. M. (2008) Stressed-out podocytes in diabetes? J. Am. Soc. Nephrol. 19, 2035–2037 [DOI] [PubMed] [Google Scholar]
- 8. Pagtalunan M. E., Miller P. L., Jumping-Eagle S., Nelson R. G., Myers B. D., Rennke H. G., Coplon N. S., Sun L., and Meyer T. W. (1997) Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 99, 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shankland S. J. (2006) The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 69, 2131–2147 [DOI] [PubMed] [Google Scholar]
- 10. Wiggins R. C. (2007) The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 71, 1205–1214 [DOI] [PubMed] [Google Scholar]
- 11. Mogensen C. E., Chachati A., Christensen C. K., Close C. F., Deckert T., Hommel E., Kastrup J., Lefebvre P., Mathiesen E. R., and Feldt-Rasmussen B. (1985) Microalbuminuria: an early marker of renal involvement in diabetes. Uremia Invest. 9, 85–95 [DOI] [PubMed] [Google Scholar]
- 12. Remuzzi G., Macia M., and Ruggenenti P. (2006) Prevention and treatment of diabetic renal disease in type 2 diabetes: the BENEDICT study. J. Am. Soc. Nephrol. 17, S90–97 [DOI] [PubMed] [Google Scholar]
- 13. Kato M., Park J. T., and Natarajan R. (2012) MicroRNAs and the glomerulus. Exp. Cell Res. 318, 993–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chau B. N., Xin C., Hartner J., Ren S., Castano A. P., Linn G., Li J., Tran P. T., Kaimal V., Huang X., Chang A. N., Li S., Kalra A., Grafals M., Portilla D., et al. (2012) MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4, 121ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gebeshuber C. A., Kornauth C., Dong L., Sierig R., Seibler J., Reiss M., Tauber S., Bilban M., Wang S., Kain R., Böhmig G. A., Moeller M. J., Gröne H. J., Englert C., Martinez J., and Kerjaschki D. (2013) Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat. Med. 19, 481–487 [DOI] [PubMed] [Google Scholar]
- 16. Zhang Z., Peng H., Chen J., Chen X., Han F., Xu X., He X., and Yan N. (2009) MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Lett. 583, 2009–2014 [DOI] [PubMed] [Google Scholar]
- 17. Chandrasekaran K., Karolina D. S., Sepramaniam S., Armugam A., Wintour E. M., Bertram J. F., and Jeyaseelan K. (2012) Role of microRNAs in kidney homeostasis and disease. Kidney Int. 81, 617–627 [DOI] [PubMed] [Google Scholar]
- 18. Ho J., and Kreidberg J. A. (2012) The long and short of microRNAs in the kidney. J. Am. Soc. Nephrol. 23, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J. Y., Yong T. Y., Michael M. Z., and Gleadle J. M. (2010) Review: the role of microRNAs in kidney disease. Nephrology 15, 599–608 [DOI] [PubMed] [Google Scholar]
- 20. Long J., Wang Y., Wang W., Chang B. H., and Danesh F. R. (2011) MicroRNA-29c is a signature microRNA under high glucose conditions that targets Sprouty homolog 1, and its in vivo knockdown prevents progression of diabetic nephropathy. J. Biol. Chem. 286, 11837–11848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Long J., Wang Y., Wang W., Chang B. H., and Danesh F. R. (2010) Identification of microRNA-93 as a novel regulator of vascular endothelial growth factor in hyperglycemic conditions. J. Biol. Chem. 285, 23457–23465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lai J. Y., Luo J., O'Connor C., Jing X., Nair V., Ju W., Randolph A., Ben-Dov I. Z., Matar R. N., Briskin D., Zavadil J., Nelson R. G., Tuschl T., Brosius F. C. 3rd, Kretzler M., and Bitzer M. (2015) MicroRNA-21 in glomerular injury. J. Am. Soc. Nephrol. 26, 805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao J. L., Rao D. S., Boldin M. P., Taganov K. D., O'Connell R. M., and Baltimore D. (2011) NF-κB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. U.S.A. 108, 9184–9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taganov K. D., Boldin M. P., Chang K. J., and Baltimore D. (2006) NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boldin M. P., Taganov K. D., Rao D. S., Yang L., Zhao J. L., Kalwani M., Garcia-Flores Y., Luong M., Devrekanli A., Xu J., Sun G., Tay J., Linsley P. S., and Baltimore D. (2011) miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 208, 1189–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halkein J., Tabruyn S. P., Ricke-Hoch M., Haghikia A., Nguyen N. Q., Scherr M., Castermans K., Malvaux L., Lambert V., Thiry M., Sliwa K., Noel A., Martial J. A., Hilfiker-Kleiner D., and Struman I. (2013) MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J. Clin. Investig. 123, 2143–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng H. S., Sivachandran N., Lau A., Boudreau E., Zhao J. L., Baltimore D., Delgado-Olguin P., Cybulsky M. I., and Fish J. E. (2013) MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol. Med. 5, 1017–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feng B., Chen S., McArthur K., Wu Y., Sen S., Ding Q., Feldman R. D., and Chakrabarti S. (2011) miR-146a-mediated extracellular matrix protein production in chronic diabetes complications. Diabetes 60, 2975–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhatt K., Lanting L. L., Jia Y., Yadav S., Reddy M. A., Magilnick N., Boldin M., and Natarajan R. (2016) Anti-inflammatory role of microRNA-146a in the pathogenesis of diabetic nephropathy. J. Am. Soc. Nephrol. 27, 2277–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ichii O., Otsuka S., Sasaki N., Namiki Y., Hashimoto Y., and Kon Y. (2012) Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney Int. 81, 280–292 [DOI] [PubMed] [Google Scholar]
- 31. Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A. O., Landthaler M., Lin C., Socci N. D., Hermida L., Fulci V., Chiaretti S., et al. (2007) A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boerries M., Grahammer F., Eiselein S., Buck M., Meyer C., Goedel M., Bechtel W., Zschiedrich S., Pfeifer D., Laloë D., Arrondel C., Gonçalves S., Krüger M., Harvey S. J., Busch H., Dengjel J., and Huber T. B. (2013) Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 83, 1052–1064 [DOI] [PubMed] [Google Scholar]
- 33. Weil E. J., Fufaa G., Jones L. I., Lovato T., Lemley K. V., Hanson R. L., Knowler W. C., Bennett P. H., Yee B., Myers B. D., and Nelson R. G. (2013) Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes 62, 3224–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farazi T. A., Horlings H. M., Ten Hoeve J. J., Mihailovic A., Halfwerk H., Morozov P., Brown M., Hafner M., Reyal F., van Kouwenhove M., Kreike B., Sie D., Hovestadt V., Wessels L. F., van de Vijver M. J., and Tuschl T. (2011) MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 71, 4443–4453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weil E. J., Lemley K. V., Mason C. C., Yee B., Jones L. I., Blouch K., Lovato T., Richardson M., Myers B. D., and Nelson R. G. (2012) Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 82, 1010–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wharram B. L., Goyal M., Wiggins J. E., Sanden S. K., Hussain S., Filipiak W. E., Saunders T. L., Dysko R. C., Kohno K., Holzman L. B., and Wiggins R. C. (2005) Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 16, 2941–2952 [DOI] [PubMed] [Google Scholar]
- 37. Meyer T. W., Bennett P. H., and Nelson R. G. (1999) Podocyte number predicts long-term urinary albumin excretion in Pima indians with type II diabetes and microalbuminuria. Diabetologia 42, 1341–1344 [DOI] [PubMed] [Google Scholar]
- 38. Hudkins K. L., Pichaiwong W., Wietecha T., Kowalewska J., Banas M. C., Spencer M. W., Mühlfeld A., Koelling M., Pippin J. W., Shankland S. J., Askari B., Rabaglia M. E., Keller M. P., Attie A. D., and Alpers C. E. (2010) BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. 21, 1533–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mei J., Bachoo R., and Zhang C. L. (2011) MicroRNA-146a inhibits glioma development by targeting Notch1. Mol. Cell. Biol. 31, 3584–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bai Y., Qian C., Qian L., Ma F., Hou J., Chen Y., Wang Q., and Cao X. (2012) Integrin CD11b negatively regulates TLR9-triggered dendritic cell cross-priming by upregulating microRNA-146a. J. Immunol. 188, 5293–5302 [DOI] [PubMed] [Google Scholar]
- 41. Horie T., Ono K., Nishi H., Nagao K., Kinoshita M., Watanabe S., Kuwabara Y., Nakashima Y., Takanabe-Mori R., Nishi E., Hasegawa K., Kita T., and Kimura T. (2010) Acute doxorubicin cardiotoxicity is associated with miR-146a-induced inhibition of the neuregulin-ErbB pathway. Cardiovasc. Res. 87, 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Niranjan T., Bielesz B., Gruenwald A., Ponda M. P., Kopp J. B., Thomas D. B., and Susztak K. (2008) The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 14, 290–298 [DOI] [PubMed] [Google Scholar]
- 43. Waters A. M., Wu M. Y., Onay T., Scutaru J., Liu J., Lobe C. G., Quaggin S. E., and Piscione T. D. (2008) Ectopic notch activation in developing podocytes causes glomerulosclerosis. J. Am. Soc. Nephrol. 19, 1139–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tao R. H., and Maruyama I. N. (2008) All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 121, 3207–3217 [DOI] [PubMed] [Google Scholar]
- 45. Sandholm N., Salem R. M., McKnight A. J., Brennan E. P., Forsblom C., Isakova T., McKay G. J., Williams W. W., Sadlier D. M., Makinen V. P., Swan E. J., Palmer C., Boright A. P., Ahlqvist E., Deshmukh H. A., et al. (2012) New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 8, e1002921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmid H., Boucherot A., Yasuda Y., Henger A., Brunner B., Eichinger F., Nitsche A., Kiss E., Bleich M., Gröne H. J., Nelson P. J., Schlöndorff D., Cohen C. D., Kretzler M., and European Renal cDNA Bank (ERCB) Consortium. (2006) Modular activation of nuclear factor-κB transcriptional programs in human diabetic nephropathy. Diabetes 55, 2993–3003 [DOI] [PubMed] [Google Scholar]
- 47. Bollée G., Flamant M., Schordan S., Fligny C., Rumpel E., Milon M., Schordan E., Sabaa N., Vandermeersch S., Galaup A., Rodenas A., Casal I., Sunnarborg S. W., Salant D. J., Kopp J. B., et al. (2011) Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 17, 1242–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen J., Chen J. K., and Harris R. C. (2015) EGF receptor deletion in podocytes attenuates diabetic nephropathy. J. Am. Soc. Nephrol. 26, 1115–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang M. Z., Wang Y., Paueksakon P., and Harris R. C. (2014) Epidermal growth factor receptor inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes 63, 2063–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Advani A., Wiggins K. J., Cox A. J., Zhang Y., Gilbert R. E., and Kelly D. J. (2011) Inhibition of the epidermal growth factor receptor preserves podocytes and attenuates albuminuria in experimental diabetic nephropathy. Nephrology 16, 573–581 [DOI] [PubMed] [Google Scholar]
- 51. Alpers C. E., and Hudkins K. L. (2011) Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 20, 278–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brosius F. C. 3rd, Alpers C. E., Bottinger E. P., Breyer M. D., Coffman T. M., Gurley S. B., Harris R. C., Kakoki M., Kretzler M., Leiter E. H., Levi M., McIndoe R. A., Sharma K., Smithies O., Susztak K., et al. (2009) Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 20, 2503–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Breyer M. D., Redha R., and Breyer J. A. (1990) Segmental distribution of epidermal growth factor binding sites in rabbit nephron. Am. J. Physiol. 259, F553–558 [DOI] [PubMed] [Google Scholar]
- 54. Chen J., Chen J. K., Nagai K., Plieth D., Tan M., Lee T. C., Threadgill D. W., Neilson E. G., and Harris R. C. (2012) EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 23, 215–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., and Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 56. Böttinger E. P., and Bitzer M. (2002) TGF-β signaling in renal disease. J. Am. Soc. Nephrol. 13, 2600–2610 [DOI] [PubMed] [Google Scholar]
- 57. Schiffer M., Bitzer M., Roberts I. S., Kopp J. B., ten Dijke P., Mundel P., and Böttinger E. P. (2001) Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Investig. 108, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee H. W., Khan S. Q., Faridi M. H., Wei C., Tardi N. J., Altintas M. M., Elshabrawy H. A., Mangos S., Quick K. L., Sever S., Reiser J., and Gupta V. (2015) A podocyte-based automated screening assay identifies protective small molecules. J. Am. Soc. Nephrol. 26, 2741–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu D. T., Bitzer M., Ju W., Mundel P., and Böttinger E. P. (2005) TGF-β concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. J. Am. Soc. Nephrol. 16, 3211–3221 [DOI] [PubMed] [Google Scholar]
- 60. Xavier S., Niranjan T., Krick S., Zhang T., Ju W., Shaw A. S., Schiffer M., and Böttinger E. P. (2009) TβRI independently activates Smad- and CD2AP-dependent pathways in podocytes. J. Am. Soc. Nephrol. 20, 2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Susztak K., Raff A. C., Schiffer M., and Böttinger E. P. (2006) Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55, 225–233 [PubMed] [Google Scholar]
- 62. Liu N., Guo J. K., Pang M., Tolbert E., Ponnusamy M., Gong R., Bayliss G., Dworkin L. D., Yan H., and Zhuang S. (2012) Genetic or pharmacologic blockade of EGFR inhibits renal fibrosis. J. Am. Soc. Nephrol. 23, 854–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suzuki H. I., Arase M., Matsuyama H., Choi Y. L., Ueno T., Mano H., Sugimoto K., and Miyazono K. (2011) MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell 44, 424–436 [DOI] [PubMed] [Google Scholar]
- 64. Qu B., Cao J., Zhang F., Cui H., Teng J., Li J., Liu Z., Morehouse C., Jallal B., Tang Y., Guo Q., Yao Y., and Shen N. (2015) Type I interferon inhibition of microRNA-146a maturation through up-regulation of monocyte chemotactic protein-induced protein 1 in systemic lupus erythematosus. Arthritis Rheumatol. 67, 3209–3218 [DOI] [PubMed] [Google Scholar]
- 65. Lee E. Y., Chung C. H., Khoury C. C., Yeo T. K., Pyagay P. E., Wang A., and Chen S. (2009) The monocyte chemoattractant protein-1/CCR2 loop, inducible by TGF-β, increases podocyte motility and albumin permeability. Am. J. Physiol. Renal Physiol. 297, F85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Artavanis-Tsakonas S., Rand M. D., and Lake R. J. (1999) Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 [DOI] [PubMed] [Google Scholar]
- 67. Cheng H. T., Kim M., Valerius M. T., Surendran K., Schuster-Gossler K., Gossler A., McMahon A. P., and Kopan R. (2007) Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134, 801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barak H., Surendran K., and Boyle S. C. (2012) The role of Notch signaling in kidney development and disease. Adv. Exp. Med. Biol. 727, 99–113 [DOI] [PubMed] [Google Scholar]
- 69. Sirin Y., and Susztak K. (2012) Notch in the kidney: development and disease. J. Pathol. 226, 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Carpenter G. (2003) ErbB-4: mechanism of action and biology. Exp. Cell Res. 284, 66–77 [DOI] [PubMed] [Google Scholar]
- 71. Veikkolainen V., Naillat F., Railo A., Chi L., Manninen A., Hohenstein P., Hastie N., Vainio S., and Elenius K. (2012) ErbB4 modulates tubular cell polarity and lumen diameter during kidney development. J. Am. Soc. Nephrol. 23, 112–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zeng F., Zhang M. Z., Singh A. B., Zent R., and Harris R. C. (2007) ErbB4 isoforms selectively regulate growth factor induced Madin-Darby canine kidney cell tubulogenesis. Mol. Biol. Cell 18, 4446–4456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Coaxum S. D., Garnovskaya M. N., Gooz M., Baldys A., and Raymond J. R. (2009) Epidermal growth factor activates Na+/H+ exchanger in podocytes through a mechanism that involves Janus kinase and calmodulin. Biochim. Biophys. Acta 1793, 1174–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zeng F., Miyazawa T., Kloepfer L. A., and Harris R. C. (2014) Deletion of ErbB4 accelerates polycystic kidney disease progression in cpk mice. Kidney Int. 86, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lu L. F., Boldin M. P., Chaudhry A., Lin L. L., Taganov K. D., Hanada T., Yoshimura A., Baltimore D., and Rudensky A. Y. (2010) Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 142, 914–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shankland S. J., Pippin J. W., Reiser J., and Mundel P. (2007) Podocytes in culture: past, present, and future. Kidney Int. 72, 26–36 [DOI] [PubMed] [Google Scholar]
- 77. Mundel P., Reiser J., Zúñiga Mejía Borja A., Pavenstädt H., Davidson G. R., Kriz W., and Zeller R. (1997) Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 236, 248–258 [DOI] [PubMed] [Google Scholar]
- 78. Saleem M. A., O'Hare M. J., Reiser J., Coward R. J., Inward C. D., Farren T., Xing C. Y., Ni L., Mathieson P. W., and Mundel P. (2002) A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J. Am. Soc. Nephrol. 13, 630–638 [DOI] [PubMed] [Google Scholar]
- 79. Wei C., Möller C. C., Altintas M. M., Li J., Schwarz K., Zacchigna S., Xie L., Henger A., Schmid H., Rastaldi M. P., Cowan P., Kretzler M., Parrilla R., Bendayan M., Gupta V., et al. (2008) Modification of kidney barrier function by the urokinase receptor. Nat. Med. 14, 55–63 [DOI] [PubMed] [Google Scholar]
- 80. Chen H., Yang K., Choi S., Fischer J. H., and Jeong H. (2009) Up-regulation of UDP-glucuronosyltransferase (UGT) 1A4 by 17β-estradiol: a potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab. Dispos. 37, 1841–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ben-Dov I. Z., Muthukumar T., Morozov P., Mueller F. B., Tuschl T., and Suthanthiran M. (2012) MicroRNA sequence profiles of human kidney allografts with or without tubulointerstitial fibrosis. Transplantation 94, 1086–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Farazi T. A., Brown M., Morozov P., Ten Hoeve J. J., Ben-Dov I. Z., Hovestadt V., Hafner M., Renwick N., Mihailović A., Wessels L. F., and Tuschl T. (2012) Bioinformatic analysis of barcoded cDNA libraries for small RNA profiling by next-generation sequencing. Methods 58, 171–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.