

Abstract
Lithium and valproic acid (VPA) are two primary drugs used to treat bipolar mood disorder and have frequently been used in combination to treat bipolar patients resistant to monotherapy with either drug. Lithium, a glycogen synthase kinase-3 (GSK-3) inhibitor, and VPA, a histone deacetylase (HDAC) inhibitor, have neuroprotective effects. The present study was undertaken to demonstrate synergistic neuroprotective effects when both drugs were coadministered. Pretreatment of aging cerebellar granule cells with lithium or VPA alone provided little or no neuroprotection against glutamate-induced cell death. However, copresence of both drugs resulted in complete blockade of glutamate excitotoxicity. Combined treatment with lithium and VPA potentiated serine phosphorylation of GSK-3 α and β isoforms and inhibition of GSK-3 enzyme activity. Transfection with GSK-3α small interfering RNA (siRNA) and/or GSK-3β siRNA mimicked the ability of lithium to induce synergistic protection with VPA. HDAC1 siRNA or other HDAC inhibitors (phenylbutyrate, sodium butyrate or trichostatin A) also caused synergistic neuroprotection together with lithium. Moreover, combination of lithium and HDAC inhibitors potentiated β-catenin-dependent, Lef/Tcf-mediated transcriptional activity. An additive increase in GSK-3 serine phosphorylation was also observed in mice chronically treated with lithium and VPA. Together, for the first time, our results demonstrate synergistic neuroprotective effects of lithium and HDAC inhibitors and suggest that GSK-3 inhibition is a likely molecular target for the synergistic neuroprotection. Our results may have implications for the combined use of lithium and VPA in treating bipolar disorder. Additionally, combined use of both drugs may be warranted for clinical trials to treat glutamate-related neurodegenerative diseases.
Keywords: lithium, valproic acid, HDAC inhibitors, neuroprotection, GSK-3, bipolar disorder
Introduction
Lithium and valproic acid (VPA) are two first-line treatment drugs for bipolar disorder. The mechanisms underlying their clinical efficacy, however, remain essentially unknown. One of the common effects of lithium and VPA is their ability to protect against apoptotic insults in vitro and in vivo (for review, see Chuang, 2004a). For example, pretreatment with lithium or VPA protects cultured brain neurons from glutamate-induced apoptosis (Nonaka et al., 1998; Hashimoto et al., 2002; Leng and Chuang, 2006). These two drugs have also been shown to display beneficial effects in cellular and animal models of neurodegenerative diseases such as stroke, Alzheimer's disease, Parkinson's disease, Huntington's disease, spinal cord injury, spinal muscular atrophy, retinal degeneration, and human immunodeficiency virus-1 infection (for review, see Tariot et al., 2002; Chuang and Priller, 2006).
Lithium is known to directly inhibit glycogen synthase kinase-3 (GSK-3) activity (Klein and Melton, 1996; Stambolic et al., 1996). GSK-3 is generally considered to have a proapoptotic role, and its inhibition results in cytoprotection (for review, see Bijur and Jope, 2003; Doble and Woodgett, 2003). Lithium also indirectly inhibits GSK-3 by triggering phosphorylation of GSK-3αSer21/βSer9 (Chalecka-Franaszek and Chuang, 1999; De Sarno et al., 2002; Zhang et al., 2003). VPA, also an anticonvulsant, has been reported to inhibit GSK-3β enzymatic activity and induce GSK-3βSer9 phosphorylation in some, but not all, neurally related systems (for review, see Rowe et al., 2007). Conversely, VPA is a direct inhibitor of histone deacetylase (HDAC) (Göttlicher et al., 2001; Phiel et al., 2001). HDAC inhibitors, including phenylbutyrate (PB), sodium butyrate (SB), and trichostatin A (TSA), cause chromatin remodeling through histone hyperacetylation to regulate expression of neuroprotective/neurotrophic proteins and proapoptotic/proinflammatory proteins (for review, see Langley et al., 2005).
Several lines of evidence suggest that neuroprotective/neurotrophic effects of lithium and VPA may be related to their clinical efficacy. Long-term lithium treatment increases total gray matter content (Moore et al., 2000a) and enhances levels of N-acetyl-aspartate, a marker of neuronal viability, in the brain of bipolar patients (Moore et al., 2000b). Moreover, bipolar subjects with past lithium or VPA exposure tend to have greater amygdalar gray volume than control patients without such an exposure (Chang et al., 2005). Interestingly, the loss of the subgenual prefrontal cortex volume found in bipolar patients was essentially suppressed in patients receiving protracted lithium or VPA (Drevets, 2001).
Despite the prominent roles of lithium and VPA in treating bipolar disorder, a significant fraction of patients fails to show an adequate response to either drug. Combined treatment with mood stabilizers has been a frequently used strategy to control bipolar syndromes resistant to monotherapy. One of the most efficacious and safe mood stabilizer combinations appears to be a mixture of lithium and anticonvulsants, notably VPA (for review, see Freeman and Stoll, 1998; Lin et al., 2006). The present study was undertaken to search for an experimental paradigm in which the neuroprotective actions of lithium and VPA or other HDAC inhibitors can be dramatically potentiated and to determine whether GSK-3 is a molecular target for the synergistic neuroprotection elicited by combined drug treatment.
Materials and Methods
Primary cultures of cerebellar granule cells and drug treatment.
Cerebellar granule cells (CGCs) were prepared from 8-d-old Sprague Dawley rats and cultured as described previously (Nonaka et al., 1998), with some modification. Specifically, the dissociated cells were resuspended in serum-free B27/neurobasal medium and plated at a density of 1.2 × 106 cells/ml on 0.01% poly-l-lysine precoated plates. Cytosine arabinofuranoside (10 μm) was added to the cultures 24 h after plating to arrest the growth of non-neuronal cells. Cultures were routinely pretreated with the indicated concentrations of LiCl, sodium VPA, PB, SB, TSA, or a combination of lithium with any of the HDAC inhibitors for indicated times, starting from 1 or 6 d in vitro (DIV), and then exposed to 50 μm glutamate for 24 h to induce neurotoxicity. At the time of experimentation, >92% of cells were CGC neurons.
Measurement of cell viability.
To determine cell survival in a quantitative colorimetric assay, the mitochondrial dehydrogenase activity that reduces 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) was assayed (Nonaka et al., 1998). CGCs cultured on 96-well plates were incubated with MTT (125 μg/ml) added directly to the growth medium for 1 h at 37°C. The medium was then aspirated, and the formazan product was dissolved in dimethylsulfoxide and quantified spectrophotometrically at 540 nm. The results are expressed as a percentage of viability of the control culture.
Lactate dehydrogenase assay.
Cell viability was also quantified with a cytotoxicity detection kit that measures lactate dehydrogenase (LDH) release according to the instructions of the manufacturer (Roche Applied Science, Indianapolis, IN). Briefly, an aliquot of 100 μl of culture medium was taken from the CGC culture grown on a 96-well plate and incubated with the substrate. Total cellular LDH was determined in lysed control cells and compared with LDH levels in treated cell lysates. LDH release into the medium was expressed as a percentage of total LDH.
Analysis of chromatin condensation.
Chromatin condensation was detected by staining of cell nuclei with Hoechst dye 33258. CGCs grown on six-well plates were washed with ice-cold PBS and fixed with 4% formaldehyde in PBS. Cells were then stained with Hoechst 33258 (5 μg/ml) for 5 min at 4°C. Nuclei were visualized under an inverted fluorescence microscope at a wavelength of 360 nm.
Western blotting.
CGC neurons cultured in six-well plates were detached by scraping and then sonicated for 30 s in lysis buffer, as described previously (Leng and Chuang, 2006). Protein concentration was determined with a BCA protein assay kit (Pierce, Rockford, IL). Aliquots containing equal amounts of protein (10 μg) from each sample were mixed with an equal volume of SDS sample buffer, loaded into a 4–12% Nupage Bis-Tris gel, and then subjected to electrophoresis. After separation, proteins were transferred to a polyvinylidene difluoride membrane, which was incubated for 1 h with a primary antibody against p53 (1:1000), GSK-3α/β antibody (1:2000), β-catenin (1:3000) (all from Santa Cruz Biotechnology, Santa Cruz, CA), phospho-GSK-3αSer21/βSer9 (1:1000, Cell Signaling, Beverly, MA), acetylated histone-H3 against both Lys9 and Lys14 acetylation (1:3000), acetylated histone-H3 against Lys9 acetylation (1:2000), acetylated histone-H3 against Lys14 acetylation (1:2000), HDAC1 antibody (1:1000) (all from Millipore, Temecula, CA), phospho-TauSer400, phospho-TauThr205, and total Tau (1:1000; Invitrogen, Carlsbad, CA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000; Advanced Immunochemical, Long Beach, CA), or β-actin (1:5000; Sigma, St. Louis, MO), in 0.01% Tween 20/PBS and then with an HRP-labeled secondary antibody (1:2000; GE Healthcare, Little Chalfont, UK). The reactive bands were visualized by detecting chemiluminescence on the membrane.
Cortical neuronal culture.
Neuron-enriched cerebral cortical cells were prepared from the cerebral cortex of 17-d-old Sprague Dawley rat embryos, as described previously (Leng and Chuang, 2006). Briefly, cortices were dissected from embryonic brain, and the meninges were removed. The cells were dissociated by trypsinization and trituration, followed by DNase treatment. Dissociated cells were resuspended in serum-free B27/neurobasal medium and plated at a density of 3 × 105 cells/cm2 on dishes precoated with 0.01% poly-l-lysine. The cells were maintained at 37°C in the presence of 5% CO2 and 95% air in a humidified incubator. At 6 DIV, cortical neurons were treated with lithium and/or VPA for 24 h for the determination of levels of phospho-GSK-3αSer21/βSer9 and phospho-TauSer400.
GSK-3β kinase activity assay.
GSK-3β activity was measured in a cell-free system, using an immune complex kinase assay. Lysates from CGCs at 7 DIV were prepared in lysis buffer as described previously (Leng and Chuang, 2006). An aliquot of 100 μg of protein extract was incubated at 4°C overnight with 2 μg of the anti-GSK-3β antibody (1:200; BD Bioscience, Palo Alto, CA). The immunocomplex was bound to protein G Sepharose (GE Healthcare) by incubation at 4°C for 2 h and then washed three times with kinase assay buffer (Cell Signaling Technology). Phosphorylation of a typical GSK-3β substrate (Cell Signaling Technology) by the kinase was performed by incubation for 30 min at 37°C in 40 μl of reaction mixture, containing 2 μg of substrate, 30 mm Tris-HCl, pH 7.5, 1 μCi [γ-32P]ATP, 4 mm MgCl2, 2.5 mm β-glycerophosphate, 1 mm DTT, 0.05 mm Na3VO4, 40 μg/ml bovine serum albumin, 100 μm ATP, and GSK-3β immunocomplex in the absence or presence of 3 mm LiCl, 0.8 mm VPA (sodium salt), or a combination of lithium and VPA. The 32P-labeled peptides were recovered on a p81 phosphocellulose paper, washed three times with 0.8% phosphoric acid, and counted with a liquid scintillation counter.
Transfection of small interference RNA specific for GSK-3α, GSK-3β, or HDAC1.
CGCs were transfected with 100 nm GSK-3α, GSK-3β, HDAC1, or scrambled small interference RNA (siRNA) immediately before plating using electroporation with the Nucleofector apparatus in conjunction with a Rat Neuron Nucleofector kit (Amaxa, Gaithersburg, MD) according to the instructions of the manufacturer. The sequence for rat GSK-3α siRNA (αP1269) is CTTCAGTCCTGGTGAACTT, whereas that for rat GSK-3β (βP555) is CCTCTTGCTGGATCCTGAT, as described previously (Liang and Chuang, 2006). Rat HDAC1-targeted siRNA was purchased from Dharmacon (Lafayette, CO). A scrambled siRNA (Dharmacon) was used as an RNA interference control for all siRNA transfection experiments.
Transfection of GSK-3α and GSK-3β plasmid constructs.
Wild-type GSK-3α (hGSK-3α/pMT2) and a dominant-negative mutant of GSK-3α (hGSK-3α/pMT2-KR) were obtained from Dr. Peter S. Klein (University of Pennsylvania School of Medicine, Philadelphia, PA) with the permission of Dr. James R. Woodgett (Ontario Cancer Institute, Toronto, Ontario, Canada). Wild-type GSK-3β (pAdTrack–CMV–GSK-3β) was supplied by Dr. Ratan V. Bhat (AstraZeneca R & D, Södertälje, Sweden). Two dominant-negative mutants (pAdTrack–CMV–GSK-3β–K85R and pAdTrack–CMV–GSK-3β–R96A) for GSK-3β were generated using the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). Transfection of GSK-3 expression vectors (wild-type or dominant-negative mutants) into CGC neurons was conducted at the time of plating using the Nucleofector apparatus (Amaxa), according to the instructions of the manufacturer. The transfection efficiencies were ∼30%. Enhanced green fluorescence protein (eGFP) was cotransfected to ensure that the transfection efficiencies were similar between drug-treated and untreated cultures.
Preparation of nuclear proteins.
CGCs cultured in six-well plates for 7 d were washed once with PBS and then buffer A (20 mm HEPES, pH 7.5, 10 mm KCl, 1 mm MgCl2, 0.02% Triton X-100, 1 mm EGTA, 0.5 mm DTT, 2 mm Na3VO4, 50 mm sodium fluoride, 100 μm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 5 μg/ml pepstatin A, and 1 nm okadaic acid). Cells were then lysed in buffer A with 20 strokes using a Dounce homogenizer. Cell lysates were collected by centrifugation (1000 × g for 10 min) at 4°C in a microcentrifuge tube. The nuclear pellet was washed two times by gently resuspending in 200 μl of wash buffer (10 mm HEPES, pH 7.5, 1 mm MgCl2, 1 mm EGTA, 25 mm NaCl, 2 mm Na3VO4, 50 mm sodium fluoride, 100 μm phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 5 μg/ml pepstatin A) and centrifuging again at 1000 × g. Nuclei were then lysed in high-salt buffer (buffer A containing 5.0 m NaCl), and 20 μg of protein were loaded into each lane for Western blotting analysis, using an antibody against β-catenin, or GAPDH, as described in the preceding section.
Lef/Tcf promoter activity assays.
A luciferase construct containing three wild-type Lef binding sites (Lef-OT) was a kind gift from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). CGCs were transfected with the Lef-OT reporter construct immediately before plating using electroporation with the Nucleofector apparatus (Amaxa), according to the instructions of the manufacturer. The transfection efficiencies were ∼30%. eGFP and secreted alkaline phosphatase were cotransfected to ensure that the transfection efficiencies were similar between drug-treated and untreated cultures. Six days after transfection, cultures were treated with the indicated concentrations of drugs for 24 h. Cells were then harvested, lysed, and assayed for luciferase activity using the luciferase reporter assay system (Promega, Madison, WI). Data were collected with a luminometer (Packard Lumicount; Global Medical Instrumentation, Ramsey, MN). As a control for the luciferase basal expression, an empty vector (promoterless plasmid) was also cotransfected, and the relative activity of luciferase was calculated.
Lef-OT cell culture.
Lef-OT cell line was a kind gift from Dr. Peter S. Klein. The OT cell line was derived by stable transfection of human embryonic kidney 293 (HEK 293) cells with a reporter (OT-luciferase) containing three wild-type Lef binding sites regulating luciferase expression. Cells were selected and maintained in DMEM supplemented with 10% fetal bovine serum and 0.04% G418, an antibiotic used for the selection of eukaryotic cells stably transfected with neomycin resistance genes. After reaching 60–70% confluency, cells were treated with the indicated concentration of drugs and then lysed in reporter lysis buffer for luciferase activity assay (Promega).
Animals and animal treatments.
All procedures were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Two-month-old male CD-1 mice (20–25 g; Charles River, Wilmington, MA) were housed with three to four animals per cage with ad libitum access to water and food under a 12 h light/dark cycle. After a 7 d acclimation period, mice were fed with a chow containing bacon flavor alone, bacon lithium carbonate (3 g/kg), bacon sodium VPA (25 g/kg), or a combination of bacon lithium carbonate and sodium VPA. The control and drug-containing chows were purchased from Bio-Serv (Frenchtown, NJ). These doses of lithium and VPA were chosen because they produced serum drug levels within therapeutic concentrations (Einat et al., 2003). Mice were killed after dietary treatment for 20 d. The brains were removed and dissected, followed by homogenization and sonication for 40 s in lysis buffer as described previously (Leng and Chuang, 2006). An aliquot of 15 μg of total protein extract was used for Western blot detection.
Statistical analyses.
Data are expressed as means ± SEM from at least three independent experiments. Statistical significance was analyzed by one-way ANOVA and the Bonferroni's post hoc test. A p value of ≤0.05 was considered significant.
Results
Pretreatment of CGCs with lithium and VPA provides neuroprotection against glutamate excitotoxicity in young but not aging cultures
CGCs developed, matured, and aged in cultures. We thus compared the vulnerability of young versus aging CGC cultures to glutamate and their responsiveness to lithium and VPA pretreatment. Young CGCs were pretreated with various concentrations (0.5–3 mm) of LiCl for 6 d (from 1 to 7 DIV) and then exposed to 50 μm glutamate for 24 h (7 to 8 DIV) to induce excitotoxic death. Glutamate exposure induced ∼50% neuronal death as measured by MTT assay, and this excitotoxicity, which was NMDA receptor-mediated, was prevented by lithium pretreatment in a concentration-dependent manner (Fig. 1A). However, pretreatment of aging CGCs with lithium (from 6 to 12 DIV) failed to effectively protect against 50 μm glutamate (24 h, 12–13 DIV) excitotoxicity, which was more severe (∼80%) than that observed in young cultures. Similar stage-dependent in vitro neuroprotection was found with VPA in the concentration range examined (0.1–0.8 mm) (Fig. 1B). Because the maturing cultures (7–8 DIV) were more resistant than the aging cultures (12–13 DIV) to glutamate-induced insult, we examined whether the neuroprotection elicited by mood stabilizers was attributable to the relatively weak excitotoxicity (Fig. 1C). The results show that either lithium or VPA pretreatment remained highly effective in suppressing excitotoxicity elicited by glutamate in a wide concentration range (50–800 μm) in the young cultures.
Figure 1.

Neuroprotection of lithium or VPA in young and aging CGCs. A, CGCs were treated with indicated doses of LiCl (0.5–3 mm) for 6 d beginning from either 1 or 6 DIV and then exposed to 50 μm glutamate for 24 h. Cell viability was quantified by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from six independent cultures. B, The experimental conditions are as described in A except that the treatment drug was VPA (0.1–0.8 mm). C, CGCs were treated with LiCl (3 mm) or VPA (0.4 mm) for 6 d beginning from 1 DIV and then exposed to glutamate (Glut) in a broad concentration range (50–800 μm) for 24 h. Cell viability was quantified by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from four independent cultures. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the untreated control.
Synergistic neuroprotection by cotreatment with lithium and VPA in aging CGC cultures
The presence of 0.8 mm VPA with LiCl during preincubation of CGCs from 6 to 12 DIV caused a concentration-dependent potentiation of the neuroprotection against excitotoxicity, reaching 60, 90, and 100% of the cell viability at 1, 2, and 3 mm lithium, respectively (Fig. 2A). Similarly, the presence of VPA with 3 mm lithium during preincubation resulted in a dose-dependent synergistic neuroprotection that reached a complete rescue at 0.8 mm (Fig. 2B). Measurement of cell death by LDH release confirmed the synergy in neuroprotection elicited by cotreatment with lithium (3 mm) and VPA (0.8 mm) (Fig. 2C). Previously, we showed that glutamate-induced excitotoxicity in CGCs involves p53 upregulation (Chen and Chuang, 1999; Chen et al., 2003). The glutamate-induced increase of p53 protein level was unaffected by treatment with either lithium or VPA alone but was blocked by their cotreatment (Fig. 2D).
Figure 2.

Synergistic neuroprotective effects of lithium and VPA in aging CGC cultures. A, B, CGCs at 6 DIV were continuously treated with 0.8 mm VPA in the presence of 0.5–3.0 mm LiCl (A) or 3 mm LiCl in the presence of 0.2–1.0 mm VPA (B). Cells were stimulated with 50 μm glutamate at 12 DIV and assayed for cell viability by MTT assay 24 h later. Data are means ± SEM from five independent cultures. Note that maximal synergism was observed with 0.8 mm VPA and 3 mm LiCl. C, CGCs at 6 DIV were pretreated with LiCl (3 mm), VPA (0.8 mm), or in combination for 6 d before glutamate (Glut; 50 μm) treatment for 24 h. Data of LDH released into the medium are expressed as means ± SEM of percentage of total LDH for three independent cultures. ***p < 0.001. D, Combined lithium and VPA treatment blocked glutamate-induced p53 upregulation. CGCs at 6 DIV were pretreated with LiCl (3 mm), VPA (0.8 mm), or both for 6 d before glutamate (50 μm) treatment for 24 h. Cells were then harvested for Western blotting of p53. Shown are results from a typical experiment that was repeated three times. Arrows, Note that glutamate-induced p53 upregulation was unaffected by lithium or VPA alone but was blocked by their cotreatment.
Morphological evidence for synergistic neuroprotection by lithium and VPA
CGCs were pretreated with lithium (3 mm) or VPA (0.8 mm) either alone or in combination from 6 to 12 DIV, followed by glutamate (50 μm) exposure for 24 h. When viable cells were detected by incubation with calcein-AM, little or no neuroprotection was elicited by either lithium or VPA; however, we observed synergistic neuroprotection when both drugs were present (Fig. 3A). Similar synergy in neuroprotection was observed when CGC cultures were stained with MTT, which labels viable neuronal cell bodies and processes (Fig. 3B). When Hoechst 33258 dye was used to identify neurons undergoing chromatin condensation, a hallmark of apoptosis, synergistic neuroprotection against glutamate-induced apoptosis was also evident (Fig. 3C).
Figure 3.
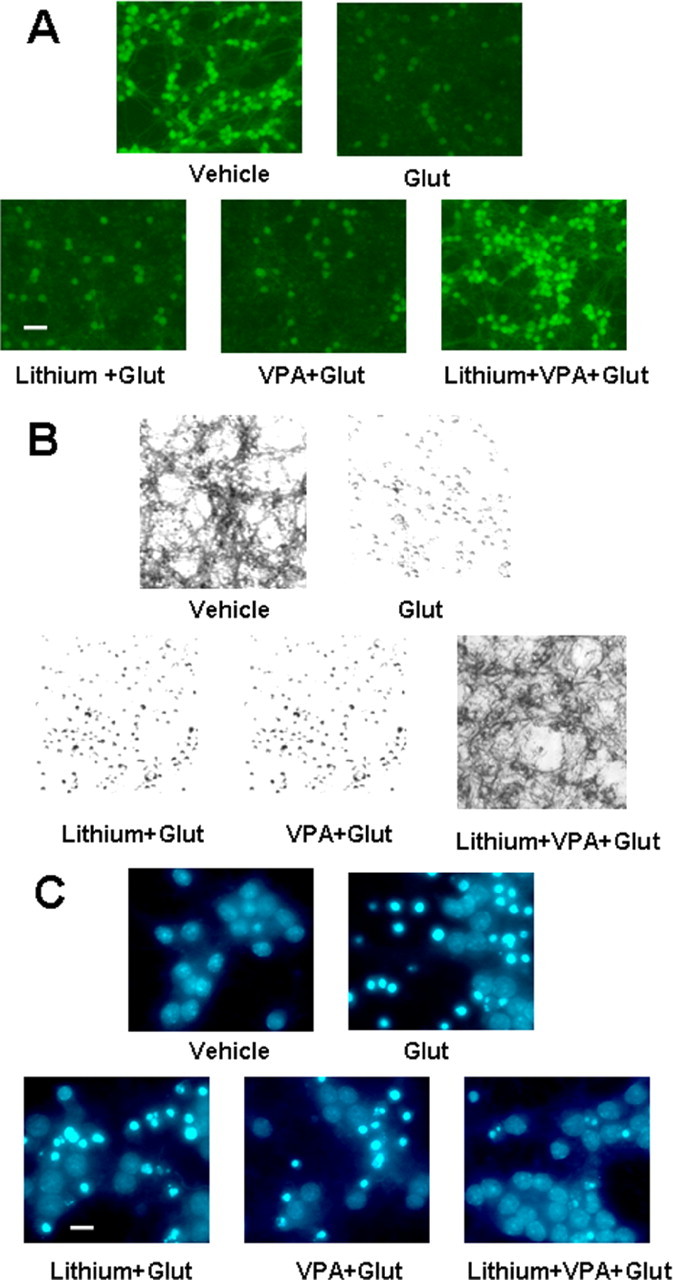
Morphological assessments of lithium–VPA synergism. A, Calcein-AM staining. CGCs at 6 DIV were pretreated with 3 mm LiCl, 0.8 mm VPA, or both in combination for 6 d before glutamate (Glut) exposure for 24 h. Cells were stained with calcein-AM and examined under an inverted fluorescence microscope. Scale bar, 30 μm. B, MTT staining. Experimental conditions are as described above, except that cells were stained with MTT. Photomicrographs were taken using an inverted light microscope. C, Hoechst dye nuclear staining. CGCs were treated as described above. After exposure to glutamate for 24 h, cells were fixed with 4% formaldehyde and then stained with Hoechst dye 33258 (5 μg/ml in PBS) for 5 min at 4°C. Photomicrographs were taken using an inverted UV illumination microscope. Scale bar, 10 μm.
Lithium and VPA cotreatment potentiates GSK-3 activity inhibition
GSK-3 is an evolutionary conserved kinase consisting of α and β isoforms and involved in the phosphorylation of an array of proteins, notably transcription factors (for review, see Grimes and Jope, 2001). Lithium inhibits GSK-3 activity by binding to the magnesium-sensitive active site of the enzyme (Klein and Melton, 1996; Stambolic et al., 1996) and also indirectly through inducing phosphorylation of GSK-3αSer21 and GSK-3βSer9 (Chalecka-Franaszek and Chuang, 1999; De Sarno et al., 2002; Zhang et al., 2003). Emerging evidence supports the idea that GSK-3 inhibition is involved in the neuroprotective effects of lithium (Bhat et al., 2000; De Sarno et al., 2002; Li et al., 2002; Phiel et al., 2003; Liang and Chuang, 2007). Therefore, we first examined the effects of VPA on levels of lithium-induced GSK-3α/β serine phosphorylation (Fig. 4). Levels of phospho-GSK-3βSer9, the predominant phosphorylated GSK-3 isoform present in CGCs, were markedly enhanced by lithium treatment for 30 min, 1 d, or 3 d (Fig. 4A–C). VPA treatment failed to change basal GSK-3βSer9 phosphorylation levels but did potentiate lithium-induced increase in GSK-3βSer9 phosphorylation at all three time points. Relatively weak levels of phospho-GSK-3αSer21 were also detected in the untreated conditions, which were enhanced by lithium treatment. Similar to the effects on GSK-3βSer9 phosphorylation, the copresence of VPA potentiated the effects of lithium on phospho-GSK-3αSer21, notably after treatment for 3 d (Fig. 4C).
Figure 4.

VPA potentiates lithium-induced serine phosphorylation of GSK-3α and β isoforms. CGCs at 6 DIV were treated with 3 mm LiCl and/or 0.8 mm VPA for 30 min (A), 24 h (B), or 72 h (C). Cells were harvested for Western blotting analysis of levels of phospho-GSK-3αSer21and phospho-GSK-3βSer9. β-Actin was used as a loading control. Typical Western blots are shown at the panels, and quantified results (means ± SEM of percentage of control from four independent experiments) are at the right. *p < 0.05, **p < 0.01, ***p < 0.001 between the indicated groups.
Because GSK-3 is a major kinase that phosphorylates the cytoskeletal protein Tau (Hong et al., 1997), the effects of lithium and/or VPA on Ser/Thr phosphorylation levels of Tau were also examined. Treatment with lithium or VPA decreased levels of p-TauSer400 by 50 or 40%, respectively, whereas cotreatment with both drugs decreased the phosphorylation levels by almost 90% (Fig. 5A,B). Cotreatment with lithium and VPA also reduced phospho-TauThr205 levels more than either drug alone. In rat cortical neurons, cotreatment with lithium and VPA for 1 d starting from 6 DIV also increased phospo-GSK-3αSer21/βSer9 and decreased phospho-TauSer400 more than either drug alone (Fig. 5C). Finally, effects of lithium and VPA on the enzyme activity of GSK-3β were examined in a cell-free system. GSK-3β protein was immunoprecipitated from CGC lysates using an anti-GSK-3β antibody, and the immunocomplex was assayed for the ability of GSK-3β to phosphorylate its protein substrate. The GSK-3β enzymatic activity was inhibited >40% by lithium and >20% by VPA, whereas the copresence of lithium and VPA inhibited the activity by ∼70% (Fig. 5D).
Figure 5.

Combined treatment with lithium and VPA potentiates inhibition of GSK-3 activity. CGCs at 6 DIV were treated with 3 mm LiCl, 0.8 mm VPA, or a combination of the two for 24 h. Cells were harvested for Western blotting of levels of p-TauSer400, p-TauThr205, total Tau, and GAPDH (as a loading control). Typical blots are shown in A, and quantified results (means ± SEM from three independent experiments) are in B. *p < 0.05, **p < 0.01 between the indicated groups. Note that p-TauSer400, p-TauThr205, and total Tau appeared as doublets in the Western blots, which likely reflects differential degrees of glycosylation (Liu et al., 2002). C, Cortical neurons at 6 DIV were treated with 3 mm LiCl, 0.8 mm VPA, or their combination for 24 h. Cells were harvested for Western blotting analysis of p-GSK-3αSer21, p-GSK-3βSer9, and p-TauSer400. D, Cell lysates prepared from CGCs at 7 DIV were immunoprecipitated with anti-GSK-3β antibody. The immunocomplex was then assayed for GSK-3β enzymatic activity in the absence or presence of LiCl (3 mm), VPA (0.8 mm), or a combination of both. The data are means ± SEM of percentage of control from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 between the indicated groups.
Silencing GSK-3 mimics lithium-induced potentiation of neuroprotection in the presence of VPA
Because lithium alone failed to elicit neuroprotection against glutamate excitotoxicity in aging CGCs, we first examined whether this inability is related to the ineffectiveness of lithium to inhibit GSK-3. CGCs at 3, 6, or 13 DIV were treated with 3 mm LiCl for 30 min, and then levels of phospho-GSK-3βSer9 were measured. Basal phospho-GSK-3βSer9 levels were found to be decreased in a culture-time-dependent manner (Fig. 6A,B). These results are reminiscent of age-induced dephosphorylation of phospho-GSK-3βSer9 in rat cortical neurons (Liang and Chuang, 2007) and suggest an increase in GSK-3β activity. Moreover, lithium-induced elevation of phospho-GSK-3βSer9 also diminished with the age of the culture, notably from 6 to 13 DIV. The dose responses of lithium-induced increase in phospho-GSK-3βSer9 and neuroprotection in aging CGCs were also studied. The optimal dose of lithium to increase phospho-GSK-3βSer9 and to rescue neurons from glutamate excitotoxicity remained at 3 mm (Fig. 6C–E), thus indicating that the decreased response of GSK-3 to lithium cannot be overcome by increasing the concentration of this drug.
Figure 6.

Effects of lithium treatment on levels of phospho-GSK-3βSer9 and glutamate excitotoxicity in aging CGCs. CGCs at 3, 6, or 13 DIV were treated with vehicle or 3 mm LiCl for 30 min and then harvested for Western blotting of phospho-GSK-3βSer9, using β-actin as the loading control. Typical blots are shown in A, and quantified results (means ± SEM from 4 independent cultures) are shown in B. **p < 0.01 between the indicated groups. Note that basal and lithium-stimulated levels of phospho-GSK-3βSer9 were diminished with successive days in culture. Cells at 6 DIV were also treated with 1–9 mm LiCl for 30 min followed by determination of phospho-GSK-3βSer9 levels using β-actin as the loading control. Typical blots are shown in C, and quantified results (means ± SEM from four independent cultures) are shown in D. *p < 0.05, **p < 0.01, ***p < 0.001 compared with the vehicle-treated control. E, For examining the effects of lithium on glutamate-induced excitotoxicity, cells at 6 DIV were treated with 1–9 mm LiCl for 6 d followed by exposure to 50 μm glutamate for 24 h. Cell viability was then determined by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from three independent cultures. *p < 0.05 compared with the vehicle-treated control. Note that the optimal concentration of lithium for inducing an increase in phospho-GSK-3βSer9 and reducing glutamate-induced cell death remained at 3 mm.
Although GSK-3 is likely the major target of lithium to elicit neuroprotection, other targets of this drug have also been suggested for other actions (for review, see Gould et al., 2004; Chuang and Priller, 2006; Rowe et al., 2007). To provide additional evidence that synergistic neuroprotection provided by lithium and VPA is related to GSK-3 inhibition, CGCs were transfected with GSK-3α siRNA and/or GSK-3β siRNA before plating, followed by treatment with 0.8 mm VPA for 6 d starting at 6 DIV. Our previous study has shown that these siRNAs for GSK-3α and GSK-3β are isoform specific in silencing their expression in rat cortical neurons (Liang and Chuang, 2007). Consistent with these results, transfection of CGCs with a mixture of GSK-3α siRNA and GSK-3β siRNA caused a marked reduction in protein levels of both GSK-3 isoforms measured 6 d later (Fig. 7A). Treatment with either GSK-3α siRNA or GSK-3β siRNA alone failed to show neuroprotection against glutamate excitotoxicity (Fig. 7B). However, exposure to GSK-3α siRNA or GSK-3β siRNA followed by VPA treatment caused a significant increase in cell viability from ∼20 to 50% in glutamate-treated cells, whereas the scrambled siRNA, used as a control, was ineffective. In CGC cultures transfected with a mixture of GSK-3α siRNA and GSK-3β siRNA in conjunction with VPA treatment, an additional increase in cell viability to nearly 80% was observed. Thus, silencing the expression of GSK-3α and/or GSK-3β together with VPA treatment mimics the synergistic effects of lithium and VPA in neuroprotection. Similar results were observed when a dominant-negative mutant of GSK-3α, pMT2-KR, was used to replace GSK-3α siRNA and a dominant-negative mutant of GSK-3β, K85R, was used to replace GSK-3β siRNA (data not shown). In another experiment, the synergistic neuroprotective effects of lithium and VPA were found to be reduced by overexpressing wild-type GSK-3α or GSK-3β but, as expected, unaffected by overexpression of a dominant-negative mutant of GSK-3α, pMT2-KR, or two dominant-negative mutants of GSK-3β, K85R and R96A (Fig. 7C). Neither wild-type nor dominant-negative mutants of GSK-3α and GSK-3β alone significantly affected glutamate-induced neuronal death. Together, these results lend additional support to the view that GSK-3 is a molecular target for lithium and VPA to induce synergy in their neuroprotective actions.
Figure 7.
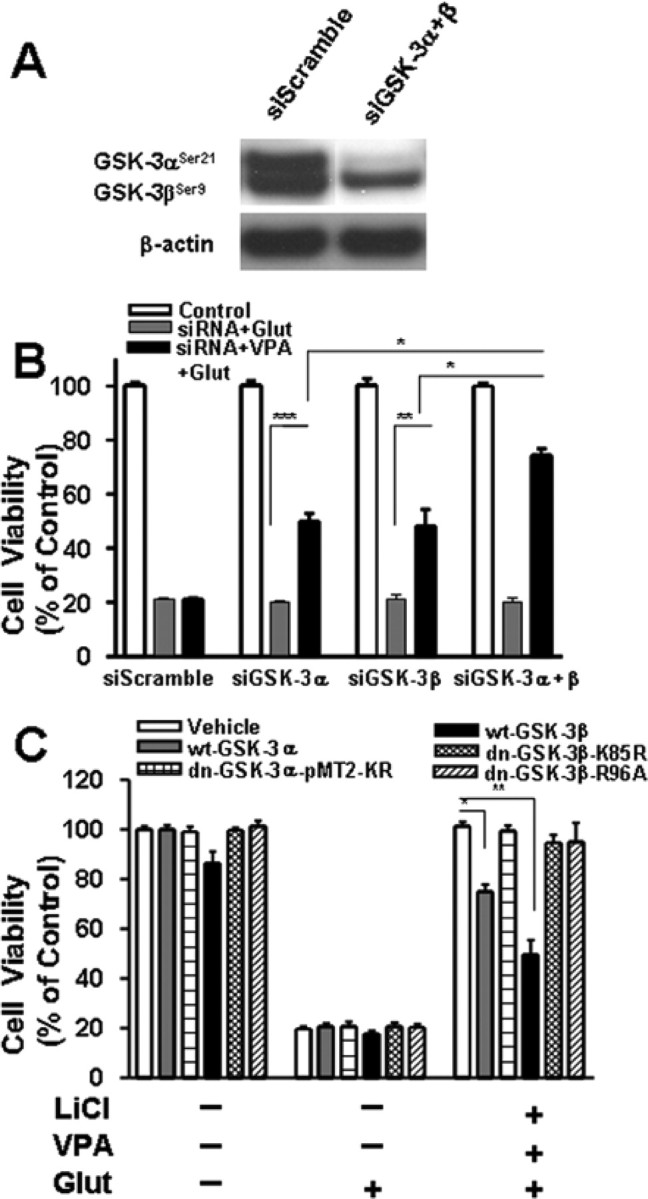
Effects of rat GSK-3 isoform-specific siRNAs and GSK-3β plasmids on glutamate-induced excitotoxicity. Cells were transfected with 100 nm siRNA specific to either GSK-3α (siGSK-3α; αP1269) or GSK-3β (siGSK-3β; βP555) before plating by electroporation. Scrambled siRNA (siScramble) at a corresponding concentration was used as the transfection control. Western blotting revealed that protein levels of both GSK-3α and GSK-3β were markedly decreased 6 d after transfection with a mixture of siRNA for GSK-3α and siRNA for GSK-3β (siGSK-3α + β) (A). Transfected cells were also treated with 0.8 mm VPA or its vehicle from 6 to 12 DIV and then exposed to 50 μm glutamate (Glut) for 24 h (B). Cell viability was measured by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 between the indicated groups. Note that transfection with siGSK-3α and/or siGSK-3β was neuroprotective only in the VPA-treated groups. In another experiment (C), CGCs were transfected at the time of plating with a plasmid of wild-type GSK-3α (wt-GSK-3α), GSK-3α dominant-negative mutant (dn-GSK3α-pMT2-KR), wild-type GSK-3β (wt-GSK-3β), or GSK-3β dominant-negative mutant (dn-GSK-3β-K85R or dn-GSK-3β-R96A). Cells were then treated with a combination of LiCl (3 mm) and VPA (0.8 mm) or vehicle from 6 to 12 DIV, followed by 24 h exposure to glutamate (50 μm). Cell viability determined by MTT assay was expressed as means ± SEM from three independent experiments. Note that neuroprotection elicited by lithium and VPA was attenuated by wild-type GSK-3α or GSK-3β but not their dominant-negative mutants.
Pretreatment with lithium and HDAC1 siRNA or other HDAC inhibitors also induces synergies in neuroprotection
Because VPA is an inhibitor of HDAC, we attempted to investigate whether the synergy in neuroprotection is related to this ability. Our recent study showed that silencing the expression of HDAC1 isoform with its specific siRNA mimics the ability of VPA to activate BDNF promoter IV in rat cortical neurons (Yasuda et al., 2008). Therefore, we first tested the effects of HDAC1 siRNA on neuroprotection against glutamate excitotoxicity. Transfection of CGCs with HDAC1 siRNA before plating resulted in a marked decrease in HDAC1 protein level measured 6 d later (Fig. 8A). Treatment with HDAC1 siRNA alone failed to protect against glutamate excitotoxicity in aging CGCs (Fig. 8B). However, HDAC1 siRNA in conjunction with subsequent lithium treatment caused robust neuroprotection. In contrast, treatment with the scrambled control siRNA had no effect. Next, we examined the effects of PB and SB, which are fatty acid derivatives that are structurally similar to VPA. Reminiscent to the effects of VPA, preincubation with PB or SB (from 6 to 12 DIV) induced limited protection of CGCs against glutamate excitotoxicity (Fig. 8C,D). More importantly, preincubation with lithium in conjunction with PB or SB completely protected CGCs from excitotoxicity. Like VPA, PB and SB did not change GSK-3αSer21/βSer9 phosphorylation levels but potentiated lithium-induced serine phosphorylation levels of GSK-3α/β, after a 1 d treatment (Fig. 8E–H). Similar synergism in neuroprotection and potentiation in lithium-induced GSK-3αSer21/βSer9 phosphorylation was observed when TSA, a hydroxamate, was used as an HDAC inhibitor during pretreatment (Fig. 9A–C).
Figure 8.

HDAC1-specific siRNA and HDAC inhibitors PB and SB mimic VPA-induced synergy in neuroprotection. Cells were transfected with 100 nm siRNA specific to rat HDAC1 isoform (siHDAC1) before plating by electroporation. Scrambled siRNA (siScramble) was used as the transfection control. Western blotting revealed that protein levels of HDAC1 were attenuated 6 d after transfection with siHDAC1, whereas the levels of β-actin were unchanged (A). Transfected cells were also treated with 3 mm LiCl or its vehicle from 6 to 12 DIV and then exposed to 50 μm glutamate (Glut) for 24 h (B). Cell viability was measured by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from six independent cultures. Note that siHDAC1 was neuroprotective only in the group treated with lithium. CGCs at 6 DIV were pretreated for 6 d with 3 mm LiCl in the absence or presence of 1 mm PB (C) or 1 mm SB (D) before treatment with 50 μm glutamate for 24 h. Cell viability was quantified by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from four independent cultures. Cells at 6 DIV were also treated with lithium in conjunction with PB in (E) or SB (F) for 24 h and then harvested for Western blotting analysis of pGSK-3αSer21/βSer9 and β-actin (used as a loading control). G, Quantified results of GSK-3α and GSK-3β serine phosphorylation in CGCs treated with lithium and/or PB. H, Quantified results of GSK-3α and GSK-3β serine phosphorylation in CGCs treated with lithium and/or SB. Data are means ± SEM of percentage of vehicle control from three independent experiments.
Figure 9.
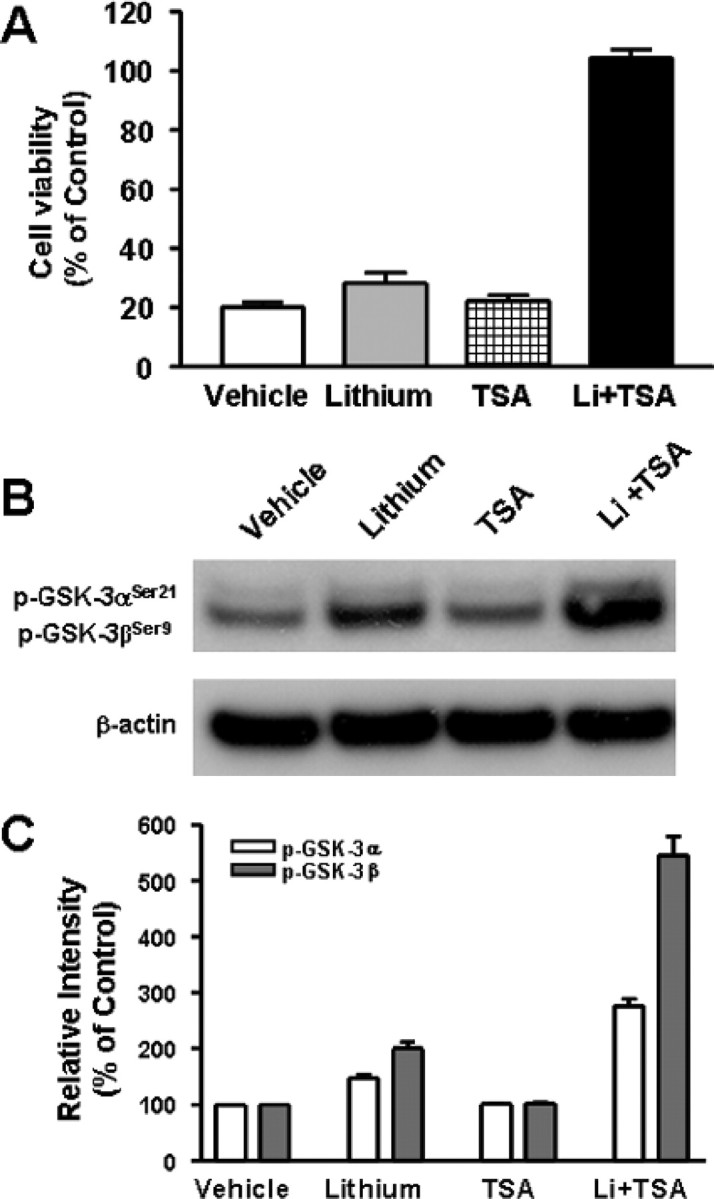
Combined treatment with TSA and lithium also induces synergy in neuroprotection and GSK-3 serine phosphorylation. A, CGCs at 6 DIV were pretreated with 3 mm LiCl and/or 20 nm TSA for 6 d and then exposed to 50 μm glutamate for 24 h. Cell viability was quantified by MTT assay and expressed as means ± SEM of percentage of vehicle-treated control from five independent cultures. B, C, CGCs at 6 DIV were treated with 3 mm LiCl and/or 20 nm TSA for 24 h and then harvested for Western blotting of GSK-3αSer21/βSer9 and β-actin (used as a loading control). Quantified results of GSK-3 serine phosphorylation are means ± SEM of percentage of vehicle control from three independent experiments.
HDAC inhibitors potentiate lithium-induced activation of Lef/Tcf-dependent transcription
Inhibition of GSK-3 activity results in a decrease in the phosphorylation of its substrate β-catenin and hence stabilization of this transcription factor (Hedgepeth et al., 1997). β-Catenin then accumulates in the cytoplasm and translocates to the nucleus with another transcription factor Lef/Tcf to activate transcription (Barker et al., 2000). Therefore, we investigated the effects of lithium and/or VPA on Lef/Tcf-dependent transcription. CGCs were transfected by electroporation with a firefly luciferase reporter containing three wild-type Lef binding sites (Lef-OT) before plating and then treated with lithium (3 mm) and/or VPA (1–4 mm) for 24 h (6 to 7 DIV). Treatment with either lithium or VPA alone resulted in a fivefold to sevenfold increase in Lef-luciferase activity (Fig. 10A). Cotreatment with lithium and VPA elicited a more than additive increase in the activation of Lef-luciferase activity at 2, 3, and 4 mm. We next determined levels of β-catenin proteins in nuclei isolated from CGCs after drug treatment. As expected, incubation with lithium (3 mm) for 1 d enhanced nuclear β-catenin levels, and this effect was enhanced by cotreatment with VPA (Fig. 10B).
Figure 10.

Combined treatment with LiCl and VPA potentiates Lef/Tcf-dependent transcriptional activity and nuclear β-catenin protein level in CGCs. A, CGCs were transfected with Lef-OT reporter construct before plating, using an Amaxa Nucleofector. At 6 DIV, CGCs were treated with 3 mm LiCl and/or the indicated concentrations of VPA for 24 h. Cells were then lysed for assay of Lef/Tcf-dependent luciferase activity. Data are means ± SEM of fold increase relative to the empty vector control from three independent experiments. B, CGCs at 6 DIV were treated with 3 mm LiCl, 0.8 mm VPA, or their combination for 24 h. Cells were then harvested, and nuclear proteins were prepared for Western blotting analysis of β-catenin. GAPDH was used as a loading control.
It has been reported that VPA at relatively high concentrations activates Lef/Tcf-dependent transcription and induces synergism with lithium when equal concentrations of both drugs are present in HEK 293 cells (Phiel et al., 2001). We thus studied activation of Lef-dependent transcription by lithium and VPA or other HDAC inhibitors. Treatment of HEK 293 cells with lithium for 24 h caused a concentration-dependent increase in Lef-luciferase activity with a >30-fold increase at 20 mm (Fig. 11A). Incubation with VPA also induced a similar dose-dependent increase in Lef-luciferase activity, and the presence of both VPA and lithium together at equal concentrations triggered a synergy (up to 170-fold) in Lef-dependent transcription (Fig. 11B), reminiscent of the previously reported results (Phiel et al., 2001). We further expanded the above results to show that similar synergism with lithium occurred when other HDAC inhibitors (PB and TSA) were used in combination with lithium during incubation (Fig. 11C,D). Together, these results further suggest that the GSK-3-mediated β-catenin/Lef signaling cascade contributes to the synergism in neuroprotection elicited by cotreatment with lithium and an HDAC inhibitor.
Figure 11.

Cotreatment with lithium and HDAC inhibitors induces synergy in Lef/Tcf-dependent transcriptional activation in HEK 293 cells. The OT cell line was derived from HEK 293 cells and stably transfected with a reporter containing three wild-type Lef binding sites regulating the luciferase expression. A, OT cells were incubated with the indicated concentrations of LiCl in the range of 2–20 mm for 24 h. B–D, OT cells were incubated with VPA, PB, or TSA at the indicated concentrations in the absence or presence of LiCl at doses shown in A. After 24 h incubation, cells were harvested for luciferase activity assay. Data are means ± SEM of fold increase relative to untreated control from three independent experiments.
Effects of lithium on HDAC inhibitor-induced histone acetylation
Given that HDAC is a target of VPA, we also examined whether the presence of lithium causes a synergy of HDAC inhibition with VPA or other HDAC inhibitors. Treatment of CGCs with VPA for 24 h (from 6 to 7 DIV) induced a marked increase in levels of histone-H3 Lys9 and Lys14 acetylation, suggesting HDAC inhibition (Fig. 12A). Although lithium by itself failed to change histone Lys9 and Lys14 acetylation levels, it slightly enhanced VPA-induced histone-H3 acetylation, when used in combination. Qualitatively similar results were obtained when levels of histone acetylation at either Lys9 or Lys14 were separately measured using antibodies that specifically recognize histone acetylation at either residue. Similar slight potentiation of histone acetylation was observed when other HDAC inhibitors (SB, PB, and TSA) were used in conjunction with lithium (Fig. 12B–D).
Figure 12.

Effects of HDAC inhibitors in the absence or presence of lithium on histone acetylation. CGCs at 6 DIV were treated with 3 mm LiCl in the absence or presence of 0.8 mm VPA (A), 1 mm SB (B), 1 mm PB (C), or 20 nm TSA (D) for 24 h. Cells were harvested for Western blotting analysis of acetylated histone-H3 (AH3) using antibodies directed against both Lys9 and Lys14 acetylation or only Lys9 or Lys14 acetylation for A and B, and only Lys9 and Lys14 coacetylations were measured for C and D. β-Actin protein levels were used as the loading control.
Effects of chronic treatment with lithium and/or VPA on levels of phospho-GSK-3βSer9
To assess the effects of mood stabilizers in vivo, mice were subjected to dietary treatment with lithium carbonate (3 g/kg) and/or VPA (25 g/kg) for 20 d. Western blotting analysis revealed that lithium markedly increased the levels of phospho-GSK-3βSer9 in the lysate of the frontal cortex (Fig. 13A,C). VPA treatment also significantly increased cortical phospho-GSK-3βSer9 levels but to a lesser extent, whereas cotreatment with lithium and VPA elicited a more than additive effect. Similar potentiation of GSK-3βSer9 phosphorylation was observed in the cerebellum of these mice treated with lithium and/or VPA (Fig. 13B,D).
Figure 13.

Effects of chronic treatment of mice with lithium and/or VPA on levels of phospho-GSK-3βSer9 in brains. CD-1 mice were treated with chow containing lithium carbonate (3 g/kg) and/or VPA (25 g/kg) for 20 d. Animals were killed, and brains were removed for Western blotting analyses of levels of p-GSK-3βSer9 and β-actin (used as the loading control) in the frontal cortex (A) and cerebellum (B). Each lane represents the result of an individual animal. C, D, Quantification of results of p-GSK-3βSer9 levels in the frontal cortex and cerebellum. Data are means ± SEM of six animals in each group. *p < 0.05, **p < 0.01, ***p < 0.001 between the indicated groups.
Discussion
In this study, we showed for the first time that cotreatment of CGCs with lithium and VPA induced synergistic neuroprotective effects in a time-dependent manner. Thus, pretreatment with either drug alone provided little or no neuroprotection against glutamate-induced excitotoxicity in aging CGCs, whereas their copresence produced a complete blockade of glutamate-induced neuronal death, as assessed biochemically and morphologically. Our results demonstrated that potentiation of GSK-3 inhibition is closely associated with the synergy of neuroprotection. For example, the presence of VPA enhanced lithium-induced increase in GSK-3αSer21/βSer9 phosphorylation in both CGCs and cortical neurons. Lithium and VPA cotreatment caused an additive decrease in GSK-3 activity in CGCs, as assayed by TauSer400 and TauThr205 phosphorylation levels in intact cells and GSK-3β enzymatic activity in a cell-free system. Furthermore, chronic treatment of mice with therapeutic doses of lithium and VPA also resulted in an additive increase in phospho-GSK-3αSer21/βSer9 levels in the frontal cortex and cerebellum. It is generally believed that GSK-3 inhibition has a neuroprotective role and is most likely a major mechanism responsible for the neuroprotective effects of lithium (De Sarno et al., 2002; Li et al., 2002; Chen et al., 2004; Liang and Chuang, 2007). Thus, potentiation of GSK-3 inhibition by the combination of lithium and VPA treatment appears to be an important target involved in the synergism of neuroprotection. This notion is further supported by our observation that transfection with siRNAs or dominant-negative mutants for GSK-3α and/or GSK-3β mimicked the synergistic neuroprotection of lithium with VPA and that overexpression of wild-type GSK-3β suppressed the synergistic effects of these mood stabilizers.
Research on the effects of VPA on GSK-3 activity has generated inconsistent results. VPA inhibits GSK-3 through an increase in GSK-3 serine phosphorylation (Chen et al., 1999; Kim et al., 2005; Kozlovsky et al., 2006) and suppresses GSK-3 activity in neuroblastoma cells overexpressing GSK-3β, as assessed by Tau phosphorylation (Grimes and Jope, 2001). In contrast, other studies suggested that this drug fails to affect GSK-3 activity (Phiel et al., 2001; Hall et al., 2002; Eickholt et al., 2005; Jin et al., 2005). In CGCs, VPA alone did inhibit GSK-3 activity, although the drug did not seem to affect the levels of GSK-3α and GSK-3β serine phosphorylation, in contrast to an increase in the phosphorylation found in the mouse brain after VPA dietary treatment. Together, these results suggest that the effects of VPA on the GSK-3 activity and the regulatory mechanisms, such as its serine phosphorylation, depend on the experimental paradigms and conditions used.
Transfection of CGCs with HDAC1 siRNA mimicked the ability of HDAC inhibitors to synergistically protect neurons in the presence of lithium, suggesting that at least this HDAC subtype is involved. The potentiation of lithium-induced GSK-3 serine phosphorylation and neuroprotection by VPA is also mimicked by lithium combination with structurally similar HDAC inhibitors PB and SB and structurally dissimilar TSA, a hydroxamate. These results suggest that the potentiation is a common feature for all HDAC inhibitors examined and that HDAC inhibition is one of the initial actions of VPA and other HDAC inhibitors. Cotreatment of HDAC inhibitors with lithium appeared to have only slight effects on HDAC inhibitor-induced histone hyperacetylation. However, given that Western blotting measures genome-wide levels of histone acetylation in bulk chromatin and that HDAC inhibitors are likely to affect a small fraction of nucleosomes to regulate gene transcription, additional experiments are necessary to address the issue regarding the roles of HDAC-regulated genes in mediating the neuroprotective synergy. Both the nuclear levels of β-catenin protein and β-catenin-Lef/Tcf-mediated transcriptional activation were enhanced by the combined treatment with lithium and VPA. Moreover, cotreatment with lithium and VPA or other HDAC inhibitors, both at relatively high concentrations, caused a robust synergism in Lef/Tcf-dependent transcription in HEK 293 cells. It requires additional investigation as to whether the potentiation by VPA of lithium-induced elevation of GSK-3 serine phosphorylation, reduction of GSK-3 enzymatic activity, and Lef/Tcf transcriptional activity requires the intrinsic activity of VPA to inhibit HDAC.
A number of reports support the notion that GSK-3 inhibition is related to the antidepressant-like and antimania-like effects in rodent models. For example, treatment with a GSK-3 peptide inhibitor or heterozygous gene knock-out of GSK-3β in mice shows antidepressant-like effects in a forced swim test (Kaidanovich-Beilin et al., 2004; O'Brien et al., 2004). A novel GSK-3 inhibitor acting as an ATP competitor also produces antidepressant-like behavior in rats (Gould et al., 2004). In addition, the same GSK-3 inhibitor reduces amphetamine-induced hyperactivity in rats, a mania-like behavior (Gould et al., 2004). Reducing GSK-3 activity by pharmacological inhibition or genetic deletion has also been reported to suppress dopamine-dependent locomotor hyperactivity (Beaulieu et al., 2004).
Postmortem studies reported a reduction in cortical volume, a decrease in the density of glial cells and neurons, as well as neuronal atrophy in the prefrontal, orbital, and cingulate cortices, amygdala, and other brain areas in patients with bipolar disorder (for review, see Manji et al., 2003). Structural imaging studies showed a reduction in gray matter volume in orbital and medial prefrontal cortices, ventral striatum, and hippocampus as well as enlargement of the third ventricle in bipolar patients (for review, see Manji et al., 2003; Chuang and Manji, 2007). Protracted treatments of bipolar patients with lithium or VPA have been found to suppress the loss of gray matter volume in the prefrontal cortex and amygdala compared with patients receiving no such treatment (Drevets, 2001; Chang et al., 2005). These and other studies suggest that neuroprotective and neurotrophic effects of mood stabilizers, most likely through GSK-3 inhibition, enhance cellular resilience and plasticity and, in turn, contribute to their clinical efficacy (for review, see Manji et al., 2001; Bachmann et al., 2005), although additional longitudinal clinical studies are required to firmly establish this linkage.
Combined treatment with lithium and VPA has been used frequently to treat bipolar disorder resistant to monotherapy with either drug (for review, see Lin et al., 2006). It is recognized that lithium and VPA combination is the first-line treatment for some bipolar subtypes such as those characterized by rapid cycling or mixed episodes and substance abuse comorbidity (for review, see Lin et al., 2006). Bipolar patients receiving a lithium-VPA cotherapy also seem to be less likely to develop a relapse compared with patients receiving lithium monotherapy (for review, see Solomon et al., 1998). Additionally, glutamate overflow and its receptor hyperactivity have been suggested to be a critical factor involved in the pathogenesis of stress-related depression (for review, see Zarate et al., 2003). Thus, synergistic neuroprotective effects against glutamate excitotoxicity elicited by lithium and VPA cotreatment may have implications in their therapeutic mechanisms for bipolar patients. In this context, it is noteworthy that the combination of lithium and VPA reportedly causes an additive suppression of brain levels of myristoylated alanine-rich C kinase substrate, a potential molecular target of mood stabilizers (Lenox et al., 1996). Several atypical antipsychotic drugs have also been used in conjunction with lithium or VPA to maximize the clinical efficacy for bipolar disorder (for review, see Freeman and Stoll, 1998; Zarate and Quiroz, 2003; Bachmann et al., 2005; Lin et al., 2006). It would thus be of interest to investigate whether the synergy in neuroprotection can be elicited by antipsychotic drug cotreatment with lithium/VPA.
Glutamate-induced excitotoxicity has been implicated in ischemic stroke and many neurodegenerative diseases, including Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis, spinal cord injury, head trauma, status epilepticus, Parkinson's disease, and cerebellar degeneration (for review, see Chuang, 2004b; Chuang and Priller, 2006). Pre- or post-insult treatment with lithium or VPA reduces brain infarct volume after cerebral ischemia and improves neurological performance in rats (Nonaka and Chuang, 1998; Ren et al., 2003, 2004; Xu et al., 2003; Kim et al., 2007). These two drugs also exhibit neuroprotective effects in animal models of a variety of neurodegenerative diseases from several independent studies (for review, see Tariot et al., 2002; Chuang and Priller, 2006). Notably, lithium, by acting on GSK-3, decreases the production of β-amyloid peptide from amyloid precursor protein (APP) (Phiel et al., 2003), lowers the phosphorylation and aggregation of Tau protein (Noble et al., 2005), and provides neuroprotective effects in APP transgenic mice (Rockenstein et al., 2007). HDAC inhibitors have also been shown to enhance learning, memory, and synaptic plasticity through chromatin remodeling and cAMP response element-binding protein (CREB):CREB binding protein-dependent transcriptional activation (Fischer et al., 2007; Vecsey et al., 2007). In light of the novel findings in the present study, it may be suggested that combined use of lithium and VPA or other HDAC inhibitors may be a rational strategy in clinical trials for neurodegenerative diseases, particularly those linked to glutamate excitotoxicity.
In conclusion, our results demonstrated robust synergistic neuroprotective effects against glutamate excitotoxicity when the mood stabilizers lithium and VPA were used in combination. We also provided evidence that GSK-3 inhibition was correlated with the synergy of neuroprotection elicited by these two drugs. Our findings have implications in the combined use of both drugs in treating bipolar disorder and further suggest potential utility of a combination of lithium and HDAC inhibitors in intervening glutamate-related neurodegeneration.
Footnotes
This work was supported by the Intramural Research Program of National Institute of Mental Health, National Institutes of Health (NIH). We sincerely thank Dr. Weihan Wang of Uniformed Services University of the Health Sciences (Bethesda, MD) for his valuable assistance in the course of this study. We also thank the NIH Fellows Editorial Board for critical readings and revisions of this manuscript.
References
- Bachmann RF, Schloesser RJ, Gould TD, Manji HK. Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol. 2005;32:173–202. doi: 10.1385/MN:32:2:173. [DOI] [PubMed] [Google Scholar]
- Barker N, Morin PJ, Clevers H. The yin-yang of TCF/β-catenin signaling. Adv Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. NeuroReport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Barnea-Goraly N, Karchemskiy A, Simeonova DI, Barnes P, Ketter T, Reiss AL. Cortical magnetic resonance imaging findings in familial pediatric bipolar disorder. Biol Psychiatry. 2005;58:197–203. doi: 10.1016/j.biopsych.2005.03.039. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Bower KA, Ma CL, Fang SY, Thiele CJ, Luo J. Glycogen synthase kinase 3β (GSK3β) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- Chen RW, Chuang DM. Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression: a prominent role in neuroprotection against excitotoxicity. J Biol Chem. 1999;274:6039–6042. doi: 10.1074/jbc.274.10.6039. [DOI] [PubMed] [Google Scholar]
- Chen RW, Qin ZH, Ren M, Kanai H, Chalecka-Franaszek E, Leeds P, Chuang DM. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: roles in glutamate excitotoxicity and lithium neuroprotection. J Neurochem. 2003;84:566–575. doi: 10.1046/j.1471-4159.2003.01548.x. [DOI] [PubMed] [Google Scholar]
- Chuang DM. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol. 2004a;16:83–90. doi: 10.1615/critrevneurobiol.v16.i12.90. [DOI] [PubMed] [Google Scholar]
- Chuang DM. Lithium neuroprotection from glutamate excitotoxicity. Lithium and mood stabilizers: mechanism of action. Clinical Neurosci Res. 2004b;4:243–252. [Google Scholar]
- Chuang DM, Manji HK. In search of the holy grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked? Biol Psychiatry. 2007;62:4–6. doi: 10.1016/j.biopsych.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Priller J. Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Müller-Oerlingausen B, editors. Lithium in neuropsychiatry: the comprehensive guide. Abington, UK: Taylor and Francis; 2006. pp. 381–397. [Google Scholar]
- De Sarno P, Li XH, Richard JS. Regulation of Akt and glycogen synthase kinase-3 phosphorylation by sodium valproate and lithium. Neuropharmacology. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- Eickholt BJ, Towers GJ, Ryves WJ, Eikel D, Adley K, Ylinen LM, Chadborn NH, Harwood AJ, Nau H, Williams RS. Effects of valproic acid derivatives on inositol trisphosphate depletion, teratogenicity, glycogen synthase kinase-3β inhibition, and viral replication: a screening approach for new bipolar disorder drugs derived from the valproic acid core structure. Mol Pharmacol. 2005;67:1426–1433. doi: 10.1124/mol.104.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Freeman MP, Stoll AL. Mood stabilizer combinations: a review of safety and efficacy. Am J Psychiatry. 1998;155:12–21. doi: 10.1176/ajp.155.1.12. [DOI] [PubMed] [Google Scholar]
- Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TD, Quiroz JA, Singh J, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry. 2004;9:734–755. doi: 10.1038/sj.mp.4001518. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hall AC, Brennan A, Goold RG, Cleverley K, Lucas FR, Gordon-Weeks PR, Salinas PC. Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol Cell Neurosci. 2002;20:257–270. doi: 10.1006/mcne.2002.1117. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80:589–597. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS. Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol. 1997;185:82–91. doi: 10.1006/dbio.1997.8552. [DOI] [PubMed] [Google Scholar]
- Hong M, Chen DCR, Klein PS, Lee VMY. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- Jin N, Kovacs AD, Sui Z, Dewhurst S, Maggirwar SB. Opposite effects of lithium and valproic acid on trophic factor deprivation-induced glycogen synthase kinase-3 activation, c-Jun expression and neuronal cell death. Neuropharmacology. 2005;48:576–583. doi: 10.1016/j.neuropharm.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. HDAC inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. Molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky N, Amar S, Belmaker RH, Agam G. Psychotropic drugs affect Ser9-phosphorylated GSK-3β protein levels in rodent frontal cortex. Int J Neuropsychopharmacol. 2006;9:337–342. doi: 10.1017/S1461145705006097. [DOI] [PubMed] [Google Scholar]
- Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord. 2005;4:41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- Leng Y, Chuang DM. Endogenous α-synuclein is potently induced by valproate and participates in the neuroprotection against glutamate excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenox RH, McNamara RK, Watterson JM, Watson DG. Myristoylated alanine-rich C kinase substrate (MARCKS): a molecular target for the therapeutic action of mood stabilizers in the brain? J Clin Psychiatry. 1996;57:23–31. [PubMed] [Google Scholar]
- Li X, Bijur GN, Jope RS. Glycogen synthase kinase-3β, mood stabilizers, and neuroprotection. Bipolar Disord. 2002;4:137–144. doi: 10.1034/j.1399-5618.2002.40201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem. 2006;281:30479–30484. doi: 10.1074/jbc.M607468200. [DOI] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- Lin D, Mok H, Yatham LN. Polytherapy in bipolar disorder. CNS Drugs. 2006;20:29–42. doi: 10.2165/00023210-200620010-00003. [DOI] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3β. FEBS Lett. 2002;530:209–214. doi: 10.1016/s0014-5793(02)03487-7. [DOI] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, Gray NA, Zarate CA, Charney DS. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry. 2003;53:707–742. doi: 10.1016/s0006-3223(03)00117-3. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK. Lithium-induced increase in human brain grey matter. Lancet. 2000a;356:1241–1242. doi: 10.1016/s0140-6736(00)02793-8. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB, Faulk MW, Koch S, Glitz DA, Jolkovsky L, Manji HK. Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2's neurotrophic effects? Biol Psychiatry. 2000b;48:1–8. doi: 10.1016/s0006-3223(00)00252-3. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Chuang DM. Neuroprotective effects of chronic lithium on focal cerebral ischemia in rats. NeuroReport. 1998;9:2081–2084. doi: 10.1097/00001756-199806220-00031. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-d-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci USA. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VMY, Klein PS. GSK-3α regulates production of Alzheimer's disease amyloid-β peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Ren M, Senatorov VV, Chen RW, Chuang DM. Post-insult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci USA. 2003;100:6210–6215. doi: 10.1073/pnas.0937423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong MR, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB, Crews L, Masliah E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci. 2007;27:1981–1991. doi: 10.1523/JNEUROSCI.4321-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31:920–931. doi: 10.1016/j.neubiorev.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Keitner GI, Ryan CE, Miller IW. Lithium plus valproate as maintenance polypharmacy for patients with bipolar I disorder. J Clin Psychopharmacol. 1998;18:38–49. doi: 10.1097/00004714-199802000-00007. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Tariot PN, Loy R, Ryan JM, Porsteinsson A, Ismail S. Mood stabilizers in Alzheimer's disease: symptomatic and neuroprotective rationales. Adv Drug Deliv Rev. 2002;54:1567–1577. doi: 10.1016/s0169-409x(02)00153-9. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JH, Culman J, Blume A, Brecht S, Gohlke P. Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke. 2003;34:1287–1292. doi: 10.1161/01.STR.0000066308.25088.64. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2008 doi: 10.1038/sj.mp.4002099. in press. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Quiroz JA. Combination treatment in bipolar disorder: a review of controlled trials. Bipolar Disord. 2003;5:217–225. doi: 10.1034/j.1399-5618.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Du J, Quiroz J, Gray NA, Denicoff KD, Singh J, Charney DS, Manji HK. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann NY Acad Sci. 2003;1003:273–291. doi: 10.1196/annals.1300.017. [DOI] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium: evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]