

Abstract
The aim of the study was to investigate the in vitro and in vivo pharmacological profile of cebranopadol, a novel agonist for opioid and nociceptin/orphanin FQ (N/OFQ) receptors (NOP). In vitro cebranopadol was assayed in calcium mobilization studies in cells coexpressing NOP or opioid receptors and chimeric G‐proteins and in a bioluminescence resonance energy transfer (BRET) assay for studying receptor interaction with G‐protein and β‐arrestin 2. The mouse tail withdrawal and formalin tests were used for investigating cebranopadol antinociceptive properties. In calcium mobilization studies cebranopadol showed the following rank order of potency NOP = mu > kappa ≥ delta. In BRET studies, cebranopadol promoted NOP and mu receptors interaction with G‐protein with similar high potency and efficacy. However, cebranopadol did not stimulated NOP–β‐arrestin 2 interactions and displayed reduced potency at mu/β‐arrestin 2. In vivo, cebranopadol exhibits highly potent and extremely long‐lasting antinociceptive effects. The effects of cebranopadol in the tail withdrawal assay were sensitive to both SB‐612111 and naloxone. Collectively the present results confirm and extend previous finding demonstrating that cebranopadol, by acting as mixed NOP/opioid receptor agonist, elicits robust analgesic effects in different pain models.
Keywords: Biased agonism, bioluminescence resonance energy transfer, calcium mobilization, cebranopadol, formalin test, NOP receptor, opioid receptors, pain, tail withdrawal
Abbreviations
- BRET
bioluminescence resonance energy transfer
- CRC
concentration–response curve
- DRC
dose–response curve
- i.v
intravenously
- N/OFQ
nociceptin/orphanin FQ
- NOP
N/OFQ peptide receptor
Introduction
The NOP receptor shows high structural and transductional similarities with classical opioid receptors and share with them the ability to modulate pain responses (Lambert 2008; Lin and Ko 2013; Toll et al. 2016). N/OFQ and selective NOP receptor agonists elicit not only antinociceptive effects at peripheral and spinal levels, but also pronociceptive effects at supraspinal sites (Zeilhofer and Calo 2003; Schroder et al. 2014). NOP agonists are far more effective on inflammatory or neuropathic pain than on acute pain, when injected systemically in rodents (Schroder et al. 2014). However, in nonhuman primates powerful antinociception is observed either after systemic, spinal, or intracisternal administration (Ko and Naughton 2009a; Ko et al. 2009b; Hu et al. 2010; Ding et al. 2015). This suggests that systemic NOP ligands in primates may reduce pain by acting synergistically at both spinal and supraspinal sites (Cremeans et al. 2012; Ding et al. 2015), whereas in rodents there might be opposite effects in brain and spinal cord, which result in a smaller or even absent antinociceptive effect (Zeilhofer and Calo 2003; Schroder et al. 2014).
Several findings suggest that mixed NOP/mu receptor agonists may have innovative analgesic potential. In fact, a synergistic enhancement of antinociception can be achieved by the concurrent activation of NOP and mu receptor with systemically or intrathecally delivered agonists both in rodents (Tian et al. 1997; Courteix et al. 2004) and in monkeys (Ko and Naughton 2009a; Hu et al. 2010; Cremeans et al. 2012).
Novel molecules acting as mixed NOP/opioid receptor agonists (Khroyan et al. 2007; Molinari et al. 2013; Zaveri et al. 2013; Schunk et al. 2014a,b; Sobczak et al. 2014) constitute a unique tool for exploring the potential benefit of NOP and opioid receptor coactivation. One of such ligands, cebranopadol, demonstrated high affinity and efficacy at NOP and mu opioid receptors and displayed robust antinociceptive properties in different rat models of pain (Linz et al. 2014).
The aim of the present study was to further characterize the pharmacological profile of cebranopadol. In vitro cebranopadol was assayed in calcium mobilization studies performed in cells expressing the human recombinant receptors and chimeric G‐proteins and in bioluminescence resonance energy transfer BRET studies investigating its ability to promote NOP and mu receptor interaction with G‐protein and β‐arrestin 2. Moreover, cebranopadol has been evaluated in vivo in mice subjected to the tail withdrawal assay and the formalin test.
Materials and Methods
Calcium mobilization
CHO cells stably coexpressing the human NOP, or kappa, or mu receptor and the C‐terminally modified Gα qi5 and CHO cells coexpressing the delta receptor and the Gα qG66Di5 protein were generated and cultured as described previously (Camarda et al. 2009; Camarda and Calo 2013). The change in fluorescence of the cell‐loaded calcium sensitive dye Fluo‐4 AM was measured with a FlexStation II (Molecular Device, Union City, CA, USA). Agonist responses were quantified as peak change of percent fluorescence intensity over the baseline.
BRET assay
The interaction with G‐protein and β‐arrestin 2 of mu (in SH‐SY5Y neuroblastoma cells) and NOP receptors (in HEK293) were measured using permanently expressing lines cotransfected with each luminescent (Rluc‐tagged) receptor and one of the fluorescent transduction protein (Gβ 1‐RGFP or β‐arrestin 2‐RGFP). Methods for cell culturing, retroviral transduction, and the BRET assay to assess receptor–β‐arrestin interaction (in intact cells) or receptor–G‐protein interaction (in plasma membrane preparations) have been described previously (Molinari et al. 2008; Malfacini et al. 2015). Agonist responses were quantified as stimulated BRET ratio obtained by subtracting the vehicle value to that measured in the presence of ligand.
Animal welfare and ethical statement
All animal care and experimental procedures conformed to the standards of the European Communities Council directives (2010/63/EU) and national regulations (D.L. 26/2014). Studies involving animals are reported in accordance with the ARRIVE guidelines (Kilkenny et al. 2010). The present study was approved by the Ethical Committee for the Use of Laboratory Animals (CEASA) of the University of Ferrara and by the Italian Ministry of Health (authorization number 316/2013‐B). Male CD‐1 mice (25–35 g, total number 300) were from Harlan, Natisone Udine, Italy. They were housed in plexiglas cages (425 × 266 × 155 mm, Techniplast, Buguggiate, Italy), under standard conditions (22°C, 55% humidity, 12‐h light/dark cycle, light on at 07:00 h) with food (standard diet, Mucedola, Settimo Milanese, Italy) and water ad libitum. Each cage was provided with a mouse red house (Tecniplast, Buguggiate, Italy) and nesting materials.
In vivo studies
Drugs were administered intravenously (i.v.) 100 μL/mouse into the vein caudal tail using a 30‐gauge needle attached to a sterile syringe. Each animal was used only once in all in vivo models. Animals were assigned randomly to treatment groups. Different treatment doses and vehicle (saline or 8% DMSO) were tested in a randomized fashion and the operator performing the behavioral tests was blinded with respect to the treatments.
The tail withdrawal test was performed as described previously (Molinari et al. 2013). To reduce animal usage, dose–response curves (DRC) displayed in Figure 8 were obtained by the cumulative addition of doses in the same subject. No signs of distress or overt pain behavior were observed in these mice after repeated i.v. injections. Naloxone (1 mg kg−1) and SB‐612111 (1 mg kg−1) were injected i.v. 5 and 30 min before performing DRC to agonists, respectively.
To investigate potential effects on motor coordination we performed a rotarod test using a constant speed device (Ugo Basile, Varese, Italy). Mice were trained at 15 rpm for 120 sec 1 day before the experiment. Motor performance was calculated as time (sec) spent on rod measured at the antinociceptive peak effect for each drug. A cut‐off time of 120 sec was chosen.
The formalin test procedure and protocol have been described in details (Rizzi et al. 2006). Fentanyl (half‐life <20 min) was applied i.v. (0.01–0.1 mg kg−1) 5 min before and 15 min after formalin injection to assess first‐ and second‐phase effects. Ro 65‐6570 (0.3–1 mg kg−1) and cebranopadol (0.001–1 mg kg−1) were applied i.v. 30 min before formalin injection.
Data analysis
Concentration–response curves (CRC) were fitted with a four‐parameter logistic model using Graph Pad PRISM 5.0 (GraphPad Software In., San Diego, California) to compute the agonist maximal effect (Emax) and its potency (i.e., agonist molar concentration yielding half‐maximal effect expressed as negative logarithm, pEC50). The antagonist potency (pKB) was derived from the rightward shift of agonist CRC using the Gaddum–Schild equation (assuming slope = 1). Data were analyzed using one‐way analysis of variance (ANOVA) followed by a Dunnett's post hoc test or the Student's t‐test. Statistically significant differences are set at P < 0.05.
Materials
All cell culture media and supplements were from Invitrogen (Paisley, UK). All other reagents were from Sigma Chemical Co. (Poole, UK) and were of the highest purity available. DPDPE, endomorphin‐1, naloxone, and SB‐612111 were bought from Tocris Bioscience (Bristol, UK). Fentanyl was bought from SALARAS, Como, Italy) (authorization SP/270, 26/11/2012). Native coelenterazine (CLZN, 5 mmol/L, EtOH) was from Synchem UG & Co. KG (Altenburg, Germany). N/OFQ, dermorphin, dynorphin A, Ro 65‐6570, and cebranopadol were synthesized in house. Stock solutions (1 mmol/L) of peptides and fentanyl were made in distilled water. SB‐612111, Ro 65‐6570, and cebranopadol (10 mmol/L) were solubilized in DMSO. Stock solutions of ligands were stored at −20°C.
Results
Calcium mobilization studies
We compared the potency of cebranopadol to activate the four recombinant human opioid receptor types permanently transfected in CHO cells. In such cells the presence of a transfected Gα q/i hybrid mutant allows measuring Gi/o‐mediated signaling with a common Ca2+ mobilization assay. N/OFQ, fentanyl, DPDPE, and dynorphin A were used as standard ligands for NOP, mu, delta, and kappa receptors. Cebranopadol elicited concentration‐dependent stimulation of calcium release in the four cell lines with maximal effects similar to those of standard ligands, except in kappa cells where it behaved as a partial agonist. The ligand was equipotent in activating NOP and mu receptors, but 10‐fold less potent at delta and kappa receptors (Fig. 1 and Table 1). To verify that the signaling induced by cebranopadol was mediated by occupation of the orthosteric agonist binding site of the receptors, we used receptor‐specific antagonists. In CHONOP cells, the CRC of cebranopadol (as that of N/OFQ) was rightward shifted only by the NOP antagonist SB‐612111 (100 nmol/L), but not by the mu antagonist naloxone (1 μmol/L) (Fig. 2A and C). Conversely, in CHOmu cells cebranopadol and fentanyl were sensitive to naloxone, but not to SB‐612111 (Fig. 2B and D). Surprisingly, SB‐612111 was far more potent in inhibiting cebranopadol‐mediated signaling (pA2>10) than N/OFQ‐mediated signaling (pA2 = 8.85). The potencies of antagonists are reported in Table 2.
Figure 1.
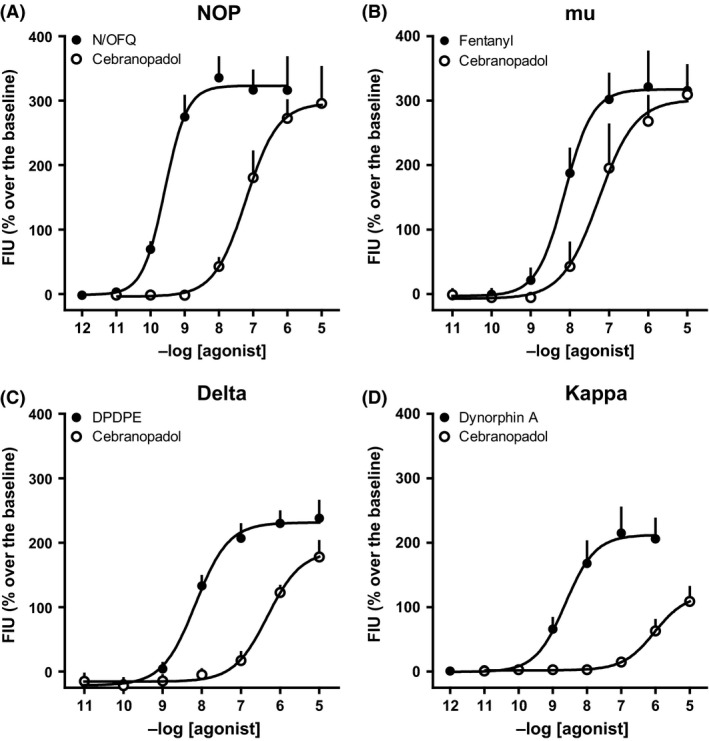
CRC to cebranopadol and standard opioid agonists in calcium mobilization experiments performed on CHO cells coexpressing the NOP (A), mu (B), delta (C), and kappa (D) receptors, and chimeric G‐proteins. Data are mean ± SEM of four experiments performed in duplicate.
Table 1.
Calcium mobilization studies. Potencies and efficacy of N/OFQ, cebranopadol, and standard opioid agonists in CHO cells expressing the human NOP or classical opioid receptors and chimeric proteins
| NOP | mu | delta | kappa | |||||
|---|---|---|---|---|---|---|---|---|
| pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | |
| N/OFQ | 9.59 (9.49–9.68) | 1.00 | Inactive | Inactive | Inactive | |||
| Fentanyl | Inactive | 8.13 (7.90–8.37) | 1.00 | Inactive | Inactive | |||
| DPDPE | Inactive | Inactive | 8.15 (7.88–8.43) | 1.00 | Inactive | |||
| Dynorphin A | Inactive | 6.67 (6.17–7.17) | 0.82 ± 0.10 | 7.73 (7.46–8.00) | 0.99 ± 0.05 | 8.54 (8.18–8.90) | 1.00 | |
| Cebranopadol | 7.28 (6.74–7.81) | 0.89 ± 0.06 | 7.20 (6.51–7.88) | 0.99 ± 0.05 | 6.31 (5.71–6.90) | 0.81 ± 0.06 | 5.98 (5.69–6.27) | 0.55 ± 0.03a |
Inactive means that the compound was inactive up to 1 μmol/L. N/OFQ, fentanyl, DPDPE, and dynorphin A were used as reference agonist for calculating intrinsic activity at NOP, mu, delta, and kappa receptors, respectively.
P < 0.05 according to ANOVA followed by the Dunnett's test for multiple comparisons.
Figure 2.
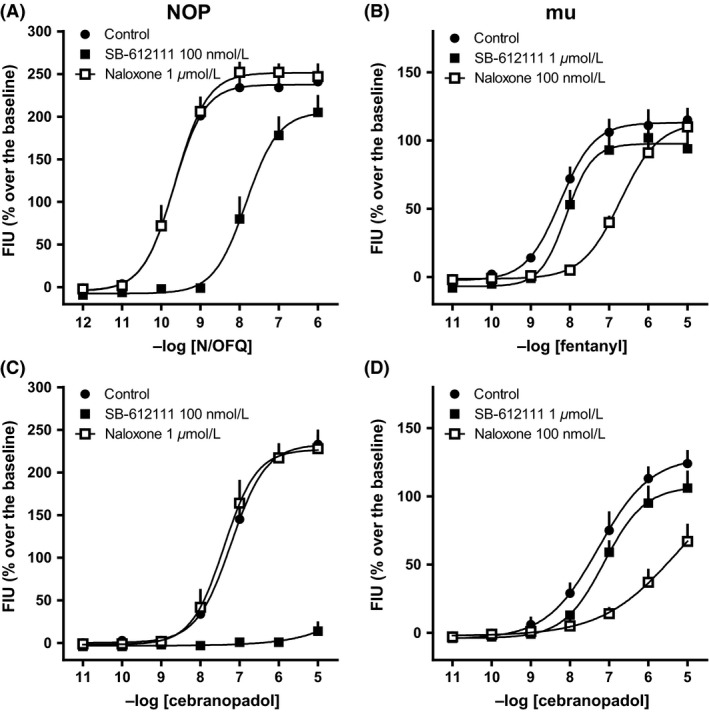
CRC to N/OFQ (A) and cebranopadol (C), in the absence and in the presence of naloxone or SB‐612111 in calcium mobilization experiments performed on CHO cells coexpressing the NOP receptor and the Gα qi5 chimeric protein. CRC to fentanyl (B) and cebranopadol (D), in the absence and in the presence of naloxone or SB‐612111 in calcium mobilization experiments performed on CHO cells coexpressing the mu receptor and the Gα qi5 chimeric protein. Data are mean ± SEM of four experiments performed in duplicate.
Table 2.
Calcium mobilization studies. Potencies (pKB values) of SB‐61211 and naloxone against N/OFQ or cebranopadol in CHO cells expressing the human NOP or mu opioid receptors and chimeric G‐proteins
| NOP | mu | |||
|---|---|---|---|---|
| vs. N/OFQ | vs. cebranopadol | vs. fentanyl | vs. cebranopadol | |
| SB‐612111 | 8.85 (8.30–9.40) | >10 | 5.86 (5.60–6.11) | 6.15 (5.41–6.89) |
| Naloxone | <6 | <6 | 8.18 (7.31–9.05) | 8.61 (8.07–9.15) |
BRET studies
We further examined the receptor specificity of cebranopadol in G‐protein and β‐arrestin coupling assays. In membrane prepared from NOP receptor/G‐protein‐expressing cells the nonpeptide NOP agonist Ro 65‐6570 and cebranopadol were approximately 10‐fold less potent than N/OFQ to enhance receptor–G‐protein interaction (Fig. 3A). However, prolonging the incubation time from 5 to 60 min virtually abolished this difference (Fig. 3B and Table 3). In membranes expressing mu receptor/G‐protein, cebranopadol displayed similar maximal effects, but 10‐fold greater potency than the standard agonists (Fig. 3C and D).
Figure 3.
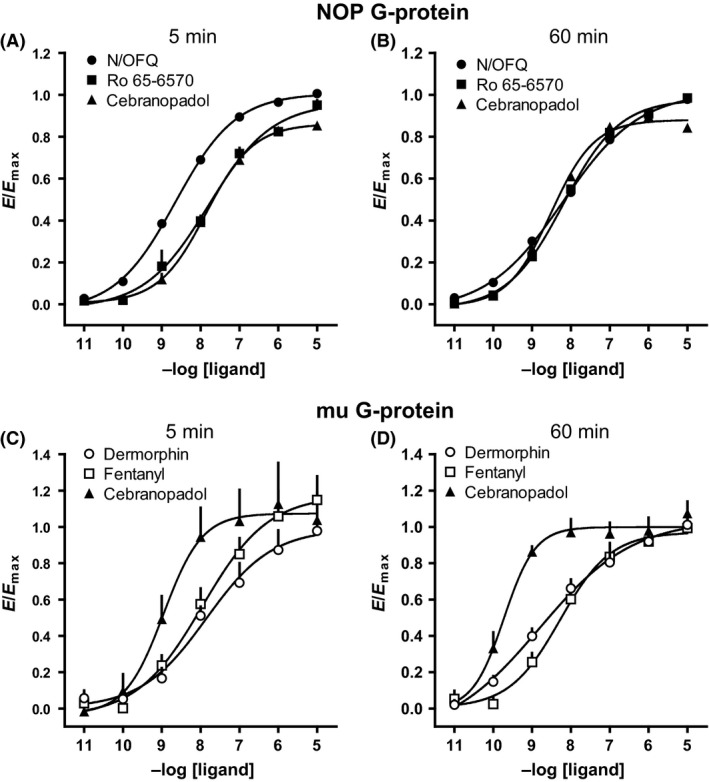
NOP receptor–G‐protein interaction experiments performed on membranes from HEK293 cells: CRC to N/OFQ, Ro 65‐6570, and cebranopadol after 5 (A) or 60 min (B) incubation; mu receptor–G‐protein interaction experiments performed on membranes from SH‐SY5Y cells: CRC to dermophin, fentanyl, and cebranopadol after 5 (C) or 60 min (D) incubation. Data are expressed as mean ± SEM of at least four separate experiments performed in duplicate.
Table 3.
Potencies (pEC50) and efficacies (α) of NOP and mu receptor agonists in promoting receptor interaction with G‐protein and β‐arrestin 2 at two different incubation time periods (5 or 60 min)
| Gβ1 protein | β‐arrestin 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| 5′ | 60′ | 5′ | 60′ | |||||
| pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | |
| NOP receptor | ||||||||
| N/OFQ | 8.60 (8.58–8.63) | 1.00 | 8.16 (7.81–8.51) | 1.00 | 8.21 (7.97–8.45) | 1.00 | 8.18 (7.93–8.44) | 1.00 |
| Ro 65‐6570 | 7.80 (7.45–8.15) | 0.87 ± 0.02 | 8.17 (7.99–8.34) | 0.92 ± 0.01 | 6.62 (6.02–7.23) | 0.65 ± 0.07a | 6.99 (6.47–7.50) | 0.64 ± 0.09a |
| Cebranopadol | 7.87 (7.52–8.21) | 0.83 ± 0.01 | 8.49 (8.43–8.55) | 0.86 ± 0.01 | inactive | Inactive | ||
| mu | ||||||||
| Dermorphin | 7.99 (7.35–8.62) | 1.00 | 8.52 (8.00–9.04) | 1.00 | 7.11 (6.87–7.35) | 1.00 | 8.06 (7.86–8.27) | 1.00 |
| Fentanyl | 7.76 (7.09–8.43) | 1.15 ± 0.12 | 8.28 (7.58–8.98) | 0.93 ± 0.04 | 7.10 (6.83–7.37) | 0.55 ± 0.02a | 7.41 (7.25–7.57) | 0.62 ± 0.03a |
| Cebranopadol | 8.85 (8.51–9.18) | 1.14 ± 0.13 | 9.74 (9.19–10.3) | 1.00 ± 0.04 | 7.30 (7.08–7.52) | 1.21 ± 0.02 | 8.36 (8.07–8.65) | 0.86 ± 0.04 |
Inactive means that the compound was inactive up to 1 μmol/L. N/OFQ and dermorphin were used as reference agonist for calculating intrinsic activity at NOP and mu receptors, respectively.
P < 0.05 according to ANOVA followed by the Dunnett's test for multiple comparisons.
As shown in Figure 4A and B, 1 μmol/L naloxone did not affect the stimulatory effects elicited by N/OFQ or cebranopadol in HEK293 cell membranes, while 100 nmol/L SB‐612111 produced a similar rightward shift of the CRC to both agonists showing pKB values of 8.98 and 9.20, respectively. In SH‐SY5Y cell membranes, 1 μmol/L SB‐612111 elicited a slight antagonist effect against both dermorphin and cebranopadol; on the contrary, 100 nM naloxone elicited a similar and robust rightward shift of the CRC to dermorphin and cebranopadol with pKB values of 8.73 and 8.70, respectively (Fig. 4C and D). The pKB values of naloxone and SB‐612111 obtained in these experiments were summarized in Table 4.
Figure 4.
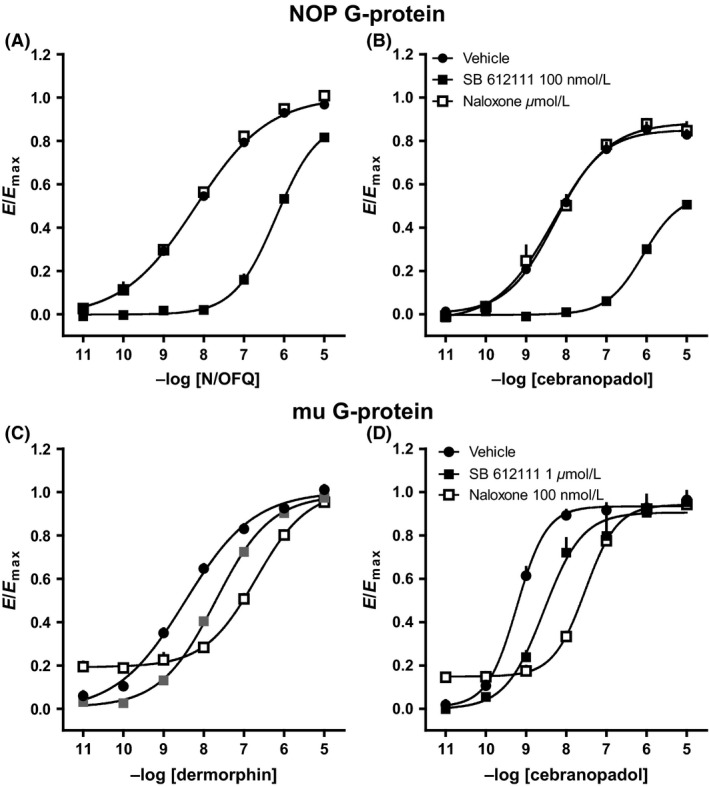
Receptor–G‐protein interaction experiments: effects of SB‐61211 and naloxone against N/OFQ (A) and cebranopadol (B) in membranes from HEK293 cells expressing the NOP receptors and against dermorphin (C) and cebranopadol (D) in membranes from SH‐SY5Y cells expressing mu receptors. Data are expressed as mean ± SEM of four separate experiments performed in duplicate.
Table 4.
Receptor–G‐protein interaction studies. Potencies (pKB values) of SB‐61211 and naloxone against N/OFQ or dermorphine and cebranopadol in membranes from HEK293 cells expressing the NOP or SH‐SY5Y cells expressing mu receptors
| NOP | mu | |||
|---|---|---|---|---|
| vs. N/OFQ | vs. cebranopadol | vs. dermorphin | vs. cebranopadol | |
| SB‐612111 | 8.98 (8.75–9.21) | 9.20 (8.63–9.77) | 6.64 (6.04–7.24) | 6.59 (6.56–6.61) |
| Naloxone | Inactive | Inactive | 8.73 (8.39–9.07) | 8.70 (8.37–9.02) |
With regard to β‐arrestin 2–receptor interactions, we found that cebranopadol was virtually inactive in cells expressing the NOP receptor (Fig. 5A), but maintained full agonist activity in cells expressing the mu receptor (Fig. 5C). Prolonging incubation time only slightly increased cebranopadol potency in mu receptor (Fig. 5D), but did not recover the lack of efficacy (Fig. 5B) in NOP receptor (see summary in Table 3).
Figure 5.
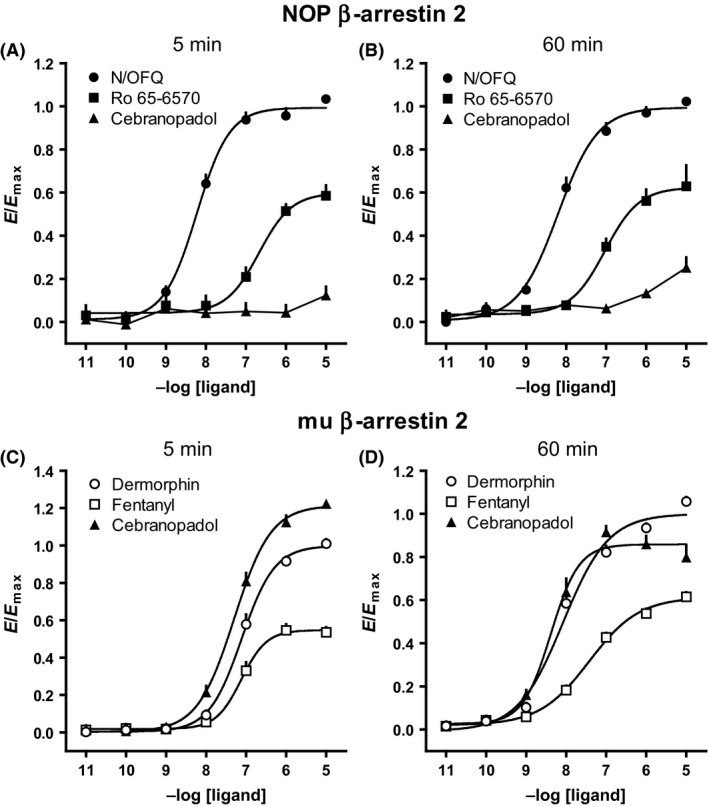
NOP receptor–β‐arrestin 2 interaction experiments performed on HEK293 cells: CRC to N/OFQ, Ro 65‐6570 and cebranopadol after 5 (A) or 60 min (B) incubation; mu receptor–β‐arrestin 2 interaction experiments performed on SH‐SY5Y cells: CRC curves to dermophin, fentanyl and cebranopadol after 5 (C) or 60 min (D) incubation. Data are expressed as mean ± SEM of at least four separate experiments performed in duplicate.
Given the lack of efficacy for NOP–β‐arrestin 2 interaction, we assessed whether cebranopadol could antagonize the effect of N/OFQ on β‐arrestin coupling. Surprisingly, cebranopadol did not modify the CRC to N/OFQ, whereas SB‐612111 produced a clear shift to the right under the same conditions. Only a small depression of the maximal effect of N/OFQ was detected at 1 μmol/L cebranopadol (Fig. 6A and B).
Figure 6.
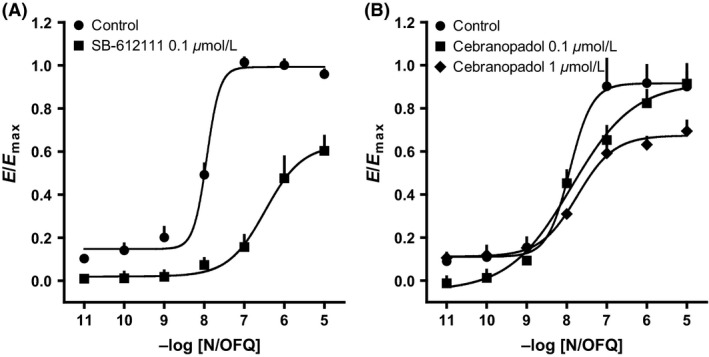
NOP receptor–β‐arrestin 2 interaction experiments performed on HEK293 cells: CRC to N/OFQ in the absence and in the presence of SB‐612111 (A) or cebranopadol (B). Data are expressed as mean ± SEM of four separate experiments performed in duplicate.
Tail withdrawal test
The antinociceptive effect of cebranopadol and fentanyl occurred in the same range of doses (0.01–1 mg kg−1, i.v.), but with very different time courses. Fentanyl analgesia peaked at 5 min postinjection and lasted up to 90 min at the highest dose (Fig. 7A), whereas the effect of cebranopadol had slower onset (peak at 30 min) and lasted at least 2 h before turning toward the baseline (Fig. 7B). In contrast, the NOP selective agonist Ro 65‐6570 tested up to 1 mg kg−1 (i.v.) did not produce statistically significant effects in the mouse tail withdrawal assay (data not shown).
Figure 7.
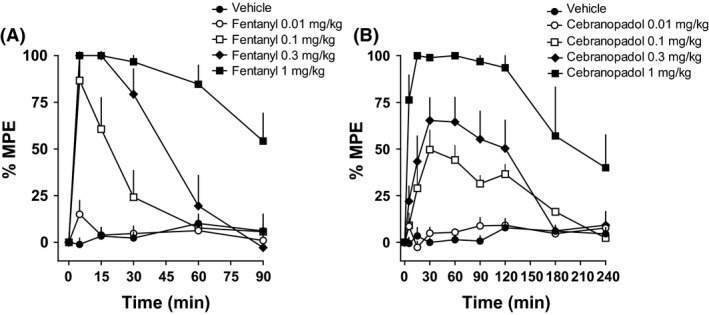
Mouse tail withdrawal assay. DRC to fentanyl (0.01–1 mg kg−1 i.v.) (A) and cebranopadol (0.01–1 mg kg−1 i.v.) (B). Each point represents the mean (eight animals per group) and the vertical bars indicate the SEM.
Mice tested in the rotarod test immediately after the antinociceptive peak effect (induced with 0.3 and 1 mg kg−1 of fentanyl, and 1 mg kg−1 of cebranopadol) showed a typical Straub's tail and hyperactivity, but did not fall down from the apparatus (data not shown). Ro 65‐6570 did not modify animal performance on the rotarod test up to 1 mg kg−1; however at higher doses produced a statistically significant dose‐dependent disruption of locomotor performance (Fig. 8).
Figure 8.
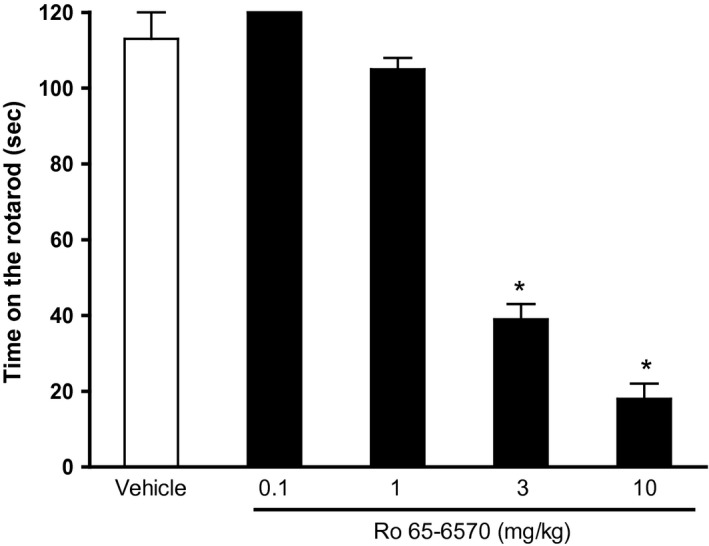
Mouse rotarod test. DRC to Ro 65‐6570 (0.1–10 mg kg−1 i.v.). Histograms indicate the means (eight animals per group) and vertical lines the SEM. *P < 0.05 versus vehicle according to analysis of variance (ANOVA) followed by the Dunnett's test.
We used antagonists (Fig. 9) to investigate the involvement of opioid and NOP receptors in the antinociceptive action of cebranopadol. Neither naloxone nor SB‐612111 (up to 1 mg kg−1) evoked effects on tail withdrawal latency (data not shown). The ED50 of fentanyl (0.03 mg kg−1) was increased by approximately 10‐fold in the presence of naloxone, but was not affected by SB‐612111 (Fig. 9A). In contrast, the ED50 of cebranopadol (0.2 mg kg−1) was increased in a similar manner by both naloxone and SB‐612111 (Fig. 9B).
Figure 9.
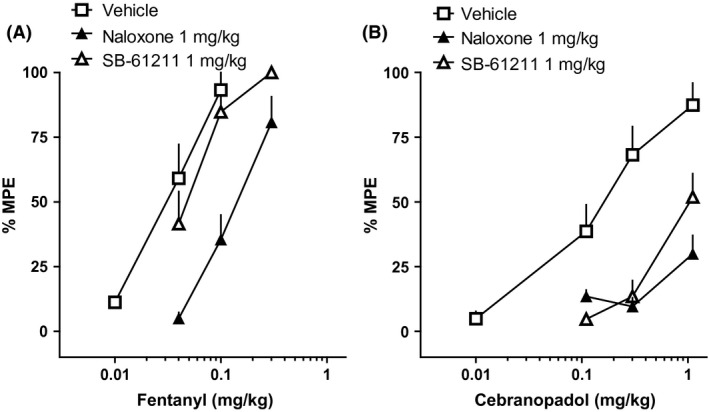
Mouse tail withdrawal assay. Cumulative DRC to fentanyl (A) or to cebranopadol (B) in animal treated with vehicle, naloxone, or SB‐61211. Data are the means (eight animals per group) and vertical lines the SEM.
Formalin test
The intraplantar injection of 20 μL of 1.5% formalin solution into the dorsal surface of the right hind paw produced a biphasic nociceptive response. The I° phase started immediately after formalin injection and lasted for 10 min, while the II° phase was prolonged, starting approximately 15–20 min after the injection and lasting for about 40 min. Neither vehicles (saline or 8% DMSO) produced any pain‐related behavior (data not shown). Fentanyl dose‐dependently inhibited both the I° and the II° phase of the assay at 0.03 and 0.1 mg kg−1, with estimated ED50 of 0.03 and 0.04 mg kg−1 for the I° and the II° phase, respectively (Fig. 10A and B). Ro 65‐6570 also produced dose‐dependent antinociceptive effects, but a complete DRC of this compound could not be obtained (Fig. 10C and D) due to its disrupting locomotor performance at high doses. Cebranopadol was also very effective in inhibiting the nociceptive effect of formalin, and produced a statistically significant effect starting at 0.01 mg kg−1. Cebranopadol ED50 values for the I° and the II° phase were 0.04 and 0.03 mg kg−1, respectively (Fig. 10E and F).
Figure 10.
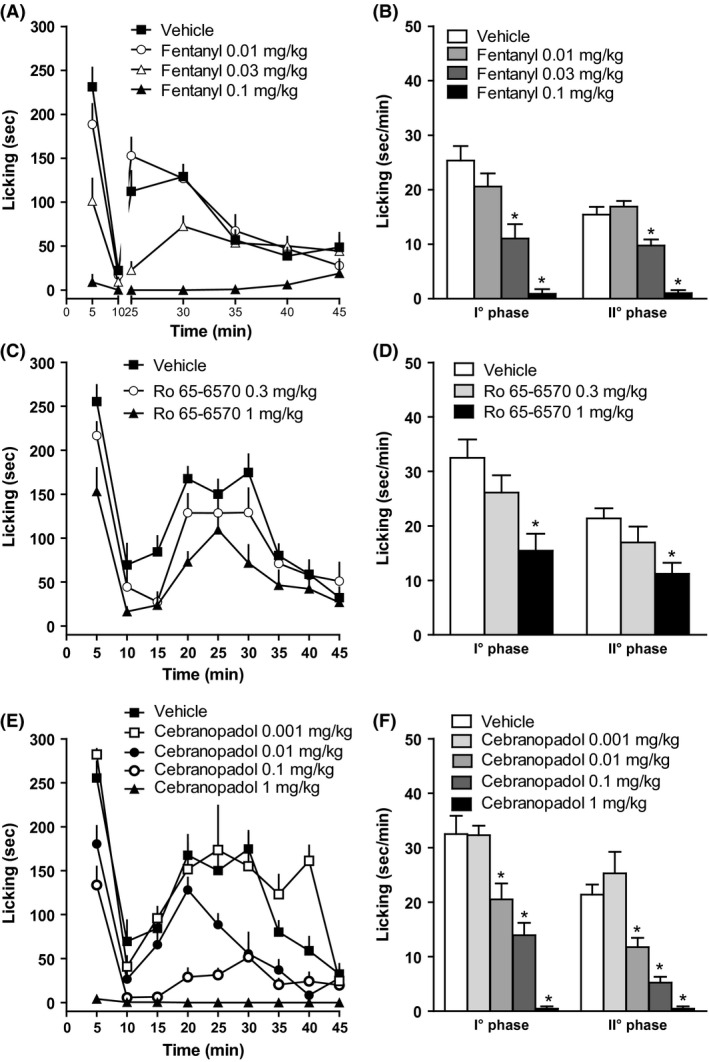
Time course of formalin‐induced pain behavior in mice treated i.v. with fentanyl (0.01–0.1 mg kg−1, A) or Ro 65‐6570 (0.3–1 mg kg−1, C), or cebranopadol (0.001–1 mg kg−1, E). (B), (D), and (F): Formalin‐induced pain behavior during the I° and II° phases. Each point represents the mean (9–11 animals per group) and the vertical bars indicate the SEM. *P < 0.05 versus vehicle according to analysis of variance (ANOVA) followed by the Dunnett's test.
Discussion
The present study confirm and extend previous findings (Linz et al. 2014) by demonstrating that cebranopadol acts as a mixed NOP/opioid receptor agonist. In addition, results obtained in the BRET assay suggest that cebranopadol behaves as G‐protein biased agonist at mu and particularly at NOP receptors. In vivo in mice cebranopadol promoted brilliant antinociceptive effects via simultaneous activation of NOP and opioid receptors. Interestingly cebranopadol displayed higher analgesic potency against inflammatory than nociceptive pain. Altogether this study support the proposal that cebranopadol represents the prototype of a novel class of analgesics, that is, mixed NOP/opioid agonists characterized by high effectiveness particularly for inflammatory/neuropathic pain associated with better tolerability compared to classical opioids (Lin and Ko 2013; Toll 2013; Schroder et al. 2014).
The relative potencies of cebranopadol for activating Gq/i‐mediated calcium signaling through the four opioid receptors (NOP ≈ mu > delta ≥ kappa) agree with the profile of affinities measured in receptor binding assays (Linz et al. 2014). Moreover, this ligand displayed full agonism at NOP, mu, and delta receptors and partial agonism at the kappa receptor, which is also consistent with data obtained in [35S]GTPγS binding assays (Linz et al. 2014).
However, we found that cebranopadol displayed ~10‐fold greater potency for mu than NOP receptors when the agonism of this ligand was compared in a BRET assay that can directly measure receptor–G‐protein coupling. In such assays we also noted that incubation time had a significant effect on increasing the apparent potency of ligands, and this enhancement of potency was far larger for cebranopadol than for other agonists. This suggests that cebranopadol may have unusually slow kinetics of receptor activation. Consistent with this conclusion is also the observation that the absolute potency values measured in the calcium assay are significantly lower than those determined here in BRET assays and in [35S]GTPγS binding assays reported by others (Linz et al. 2014). Our previous studies suggest that the calcium assay tends to underestimate the potency of agonists with slow kinetics of action (Camarda et al. 2009; Rizzi et al. 2014; Ruzza et al. 2014). Thus, the relatively low potency displayed by cebranopadol in the calcium assay may result from the nonequilibrium condition (Charlton and Vauquelin 2010) existing between the slow receptor activation induced by this compound and the transient nature of the calcium response.
The anomalous kinetics of cebranopadol might also be responsible for the surprising behavior of the NOP antagonist SB‐612111 in the calcium assay. This ligand exhibited the expected competitive antagonism toward N/OFQ, but unexpectedly excessive potency and apparent insurmountable behavior in inhibiting Ca2+ signaling induced by cebranopadol. The combination of slow activation kinetics by cebranopadol and slow receptor dissociation of SB‐612111 (Spagnolo et al. 2007) could make the condition for steady‐state measurement of response unreachable in the calcium assay, thus producing noncompetitive antagonism behavior (Vauquelin et al., 2006). This interpretation is supported by the results obtained by testing SB‐612111 in the BRET G‐protein assay where the compound displayed similar antagonist potency versus N/OFQ and cebranopadol.
The ability of cebranopadol to activate both NOP and mu receptors is confirmed by the pharmacological profile emerging from our analgesiometric tests in vivo. Both in the tail withdrawal and in the formalin assay cebranopadol displayed robust analgesic effects, which were equivalent in intensity to those of fentanyl, although slower in onset and much longer lasting. Unlike fentanyl, however, cebranopadol was blocked by both mu and NOP receptor antagonists. This indicates that both receptor types contribute to the antinociceptive response of this compound.
Also, consistent with the coinvolvement of mu and NOP receptors in cebranopadol analgesia is the observation that its antinociceptive potency was sevenfold greater in the formalin test. This model is deemed more predictive of efficacy for inflammatory and neuropathic pain than the tail withdrawal assay. Accordingly, the NOP‐selective standard agonist Ro 65‐6570 produced significant antinociception only in the former but not in the latter assay. Such data perfectly agree with previous studies reported in rats (Linz et al. 2014) and mice (Schunk et al. 2014b).
Another interesting feature of cebranopadol is the lack of ability to elicit sedation, a common response to high doses of NOP agonists (Lambert 2008; Toll et al. 2016) that can limit their therapeutic usefulness (Woodcock et al. 2010). The absence of sedative effects of cebranopadol may derive from its ability to simultaneously activate both NOP and opioid (particularly mu) receptors. Alternatively the NOP G‐protein biased agonism (see below) behavior of cebranopadol might be relevant to this issue. Clearly further studies are needed to deeply investigate the signaling mechanisms involved into the sedative effects of NOP agonists.
Collectively, our findings support the notion that cebranopadol is the prototype of a novel class of analgesics, that is, mixed NOP/opioid agonists, which are characterized by high effectiveness against inflammatory/neuropathic pain and better tolerability than classical opioids (Lin and Ko 2013; Toll 2013; Schroder et al. 2014).
However, the effects of cebranopadol in promoting β‐arrestin coupling of mu and NOP receptor deserve further comments and raise novel questions on the molecular mechanisms underlying the pharmacological profile of this ligand.
One question regards the variation of efficacy between G‐protein and arrestin interactions that cebranopadol differentially displayed in mu and NOP receptors. In the mu receptor, cebranopadol maintained full agonism for both G‐protein and arrestin interactions, unlike the standard ligand fentanyl, which displayed full agonism for G‐protein and partial agonism for arrestin. Conversely, in the NOP receptor, cebranopadol exhibited total loss of efficacy for arrestin coupling, while the NOP agonist Ro 65‐6570 changed from full G‐protein agonism to partial arrestin agonism, just like fentanyl did in the mu receptor. The phenomenon that synthetic agonists, such as fentanyl and Ro 65‐6570, tend to show more efficacy at G‐proteins than arrestin, whereas naturally occurring peptides maintain full efficacy at both transduction proteins, was extensively documented in opioid receptors (Molinari et al. 2010; Chang et al. 2015; Malfacini et al. 2015). Although the use of different cell lines is a limitation of our findings, the comparison of the action of cebranopadol with those elicited by peptide agonists clearly suggest that cebranopadol has a significant level of biased G‐protein efficacy selectively on the NOP receptor. This may explain the gap between in vitro and in vivo data that our study reveals to exist for the different efficacies of cebranopadol at mu and NOP receptors. In vitro, cebranopadol shows 10‐fold greater potency at mu than at NOP receptors for G‐protein coupling and a dramatic turn from full mu agonism to lack of NOP agonism for arrestin coupling. Yet, the reported binding affinities of this ligand for the two receptors are virtually identical (Linz et al. 2014). This clearly suggests that cebranopadol is a more efficacious agonist in activating mu than NOP receptors. In vivo, however, cebranopadol appears to exert strong and comparable analgesic effects originating from both mu and NOP receptors. A plausible explanation for this discrepancy lies in the strong G‐protein bias that this agonist shows at NOP receptors. In fact, the absence of the restraining effect of arrestin‐dependent desensitization in NOP but not in mu‐mediated signaling may compensate for the difference in efficacy, thus resulting in a balanced analgesia from both receptors.
A second question concerns the noticeable failure of cebranopadol to interfere with N/OFQ‐mediated stimulation of NOP receptor–arrestin interaction. In such BRET experiments, the ligand did not shift the EC50 but only slightly reduced the Emax of the N/OFQ CRC at a concentration that exceeds by almost a 1000‐fold the binding constant of cebranopadol for NOP receptor. These results stand in contrast with the behavior of NOP partial agonists such as [F/G]N/OFQ(1‐13)‐NH2 or UFP‐113, which we have reported earlier (Malfacini et al. 2015). These ligands like cebranopadol had no efficacy on receptor–arrestin coupling, but unlike cebranopadol were potent and competitive antagonists of the N/OFQ effect in the BRET arrestin assay. It is not possible to rationalize such findings without suggesting an unexpected complexity in the way cebranopadol regulate NOP receptor–arrestin interactions. We do not want to propose merely speculative hypotheses without the support of experiments specifically focused to elucidate the molecular mechanism by which cebranopadol interacts with the NOP receptor–arrestin complex.
In conclusion, the present study together with positive results from phase II clinical studies (Lambert et al. 2015) strongly support the notion that dual mu/NOP receptor agonists such as cebranopadol constitute an innovative class of analgesics worth of further development. Additional efforts to elucidate the exact molecular mechanism of action of these molecules are also necessary in order to improve future rational ligand design and drug discovery in this novel class of drugs.
Author contributions
A. R., C. R., and G. C. participated in research design. A. R., M. C. C., C. R., D. M., and F. F. conducted experiments and performed data analysis. C. T., S. B., and R. G. designed, synthesized, and purified peptides, Ro 65‐6570, and cebranopadol. A. R., T. C., and G. C. wrote the first draft of the manuscript. The final version of the article was revised by T. C. and G. C.
Disclosure
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by the funds from the Italian Ministry of Health (FIRB Grant RBFR109SBM to C. T.) and the University of Ferrara (FAR grant to G. C.).
Rizzi A., Cerlesi M. C., Ruzza C., Malfacini D., Ferrari F., Bianco S., Costa T., Guerrini R., Trapella C., Calo' G.. Pharmacological characterization of cebranopadol a novel analgesic acting as mixed nociceptin/orphanin FQ and opioid receptor agonist, Pharma Res Per, 4(4), 2016, e00247, doi: 10.1002/prp2.247
References
- Camarda V, Calo G (2013). Chimeric G proteins in fluorimetric calcium assays: experience with opioid receptors. Methods Mol Biol 937: 293–306. [DOI] [PubMed] [Google Scholar]
- Camarda V, Fischetti C, Anzellotti N, Molinari P, Ambrosio C, Kostenis E, et al. (2009). Pharmacological profile of NOP receptors coupled with calcium signaling via the chimeric protein G alpha qi5. Naunyn Schmiedebergs Arch Pharmacol 379: 599–607. [DOI] [PubMed] [Google Scholar]
- Chang SD, Mascarella SD, Spangler SM, Gurevich VV, Navarro HA, Carroll FI, et al. (2015). Quantitative Signaling and Structure‐Activity Analyses Demonstrate Functional Selectivity at the Nociceptin/Orphanin FQ Opioid Receptor. Mol Pharmacol 88: 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton SJ, Vauquelin J (2010). Elusive equilibrium: the challenge of interpreting receptor pharmacology using calcium assays. Br J Pharmacol 161: 1250–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courteix C, Coudore‐Civiale MA, Privat AM, Pelissier T, Eschalier A, Fialip J (2004). Evidence for an exclusive antinociceptive effect of nociceptin/orphanin FQ, an endogenous ligand for the ORL1 receptor, in two animal models of neuropathic pain. Pain 110: 236–245. [DOI] [PubMed] [Google Scholar]
- Cremeans CM, Gruley E, Kyle DJ, Ko MC (2012). Roles of mu‐opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine‐induced physiological responses in primates. J Pharmacol Exp Ther 343: 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Hayashida K, Suto T, Sukhtankar DD, Kimura M, Mendenhall V, et al. (2015). Supraspinal actions of N/OFQ, morphine and substance P in regulating pain and itch in nonhuman primates. Br J Pharmacol 172: 3302–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Calo G, Guerrini R, Ko MC (2010). Long‐lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP‐112. Pain 148: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, et al. (2007). SR 16435 [1‐(1‐(bicyclo[3.3.1]nonan‐9‐yl)piperidin‐4‐yl)indolin‐2‐one], a novel mixed nociceptin/orphanin FQ/mu‐opioid receptor partial agonist: analgesic and rewarding properties in mice. J Pharmacol Exp Ther 320: 934–943. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Naughton NN (2009a). Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J Pain 10: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP (2009b). Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology 34: 2088–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DG (2008). The nociceptin/orphanin FQ receptor: a target with broad therapeutic potential. Nat Rev Drug Discov. 7: 694–710. [DOI] [PubMed] [Google Scholar]
- Lambert DG, Bird MF, Rowbotham DJ. (2015). Cebranopadol: a first in‐class example of a nociceptin/orphanin FQ receptor and opioid receptor agonist. Br J Anaesth 114: 364–366. [DOI] [PubMed] [Google Scholar]
- Lin AP, Ko MC (2013). The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem Neurosci 4: 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, et al. (2014). Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 349: 535–548. [DOI] [PubMed] [Google Scholar]
- Malfacini D, Ambrosio C, Gro' MC, Sbraccia M, Trapella C, Guerrini R, et al. (2015). Pharmacological profile of nociceptin/orphanin FQ receptors interacting with G‐proteins and β‐arrestins 2. PLOSONE 10: e0132865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari P, Casella I, Costa T (2008). Functional complementation of high‐efficiency resonance energy transfer: a new tool for the study of protein binding interactions in living cells. Biochem J 409: 251–261. [DOI] [PubMed] [Google Scholar]
- Molinari P, Vezzi V, Sbraccia M, Grò C, Riitano D, Ambrosio C, et al. (2010). Morphine‐like opiates selectively antagonize receptor‐arrestin interactions. J Biol Chem 285: 12522–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari S, Camarda V, Rizzi A, Marzola G, Salvadori S, Marzola E, et al. (2013). [Dmt1]N/OFQ(1‐13)‐NH2: a potent nociceptin/orphanin FQ and opioid receptor universal agonist. Br J Pharmacol 168: 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi A, Nazzaro C, Marzola GG, Zucchini S, Trapella C, Guerrini R, et al. (2006). Endogenous nociceptin/orphanin FQ signalling produces opposite spinal antinociceptive and supraspinal pronociceptive effects in the mouse formalin test: pharmacological and genetic evidences. Pain 124: 100–108. [DOI] [PubMed] [Google Scholar]
- Rizzi A, Malfacini D, Cerlesi MC, Ruzza C, Marzola E, Bird MF, et al. (2014). In vitro and in vivo pharmacological characterization of nociceptin/orphanin FQ tetrabranched derivatives. Br J Pharmacol 171: 4138–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzza C, Rizzi A, Malfacini D, Pulga A, Pacifico S, Salvadori S, et al. (2014). Pharmacological characterization of tachykinin tetrabranched derivatives. Br J Pharmacol 171: 4125–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Lambert DG, Ko MC, Koch T (2014). Functional plasticity of the N/OFQ‐NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol 171: 3777–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunk S, Linz K, Frormann S, Hinze C, Oberborsch S, Sundermann B, et al. (2014a). Discovery of Spiro[cyclohexane‐dihydropyrano[3,4‐b]indole]‐amines as Potent NOP and Opioid Receptor Agonists. ACS Med Chem Lett 5: 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunk S, Linz K, Hinze C, Frormann S, Oberborsch S, Sundermann B, et al. (2014b). Discovery of a Potent Analgesic NOP and Opioid Receptor Agonist: Cebranopadol. ACS Med Chem Lett 5: 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak M, Cami‐Kobeci G, Salaga M, Husbands SM, Fichna J (2014). Novel mixed NOP/MOP agonist BU08070 alleviates pain and inhibits gastrointestinal motility in mouse models mimicking diarrhea‐predominant irritable bowel syndrome symptoms. Eur J Pharmacol 736: 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo B, Carra G, Fantin M, Fischetti C, Hebbes C, McDonald J, et al. (2007). Pharmacological characterization of the nociceptin/orphanin FQ receptor antagonist SB‐612111 [(‐)‐cis‐1‐methyl‐7‐[[4‐(2,6‐dichlorophenyl)piperidin‐1‐yl]methyl]‐6,7,8,9‐tetrah ydro‐5H‐benzocyclohepten‐5‐ol]: in vitro studies. J Pharmacol Exp Ther 321: 961–967. [DOI] [PubMed] [Google Scholar]
- Tian JH, Xu W, Fang Y, Mogil JS, Grisel JE, Grandy DK, et al. (1997). Bidirectional modulatory effect of orphanin FQ on morphine‐induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br J Pharmacol 120: 676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L (2013). The use of bifunctional NOP/mu and NOP receptor selective compounds for the treatment of pain, drug abuse, and psychiatric disorders. Curr Pharm Des 19: 7451–7460. [PubMed] [Google Scholar]
- Toll L, Bruchas MR, Calo' G, Cox BM, Zaveri NT (2016). Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid sistems. Pharmacol Reviews 68: 419–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauquelin G, Liefde I (2006). Slow antagonist dissociation and long‐lasting in vivo receptor protection. Trends Pharmacol Sci 27: 355–359. [DOI] [PubMed] [Google Scholar]
- Woodcock A, McLeod RL, Sadeh J, Smith JA (2010). The efficacy of a NOP1 agonist (SCH486757) in subacute cough. Lung 188(Suppl 1): S47–S52. [DOI] [PubMed] [Google Scholar]
- Zaveri NT, Jiang F, Olsen C, Polgar WE, Toll L (2013). Designing bifunctional NOP receptor‐mu opioid receptor ligands from NOP receptor‐selective scaffolds. Part I. Bioorg Med Chem Lett 23: 3308–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Calo G (2003). Nociceptin/orphanin FQ and its receptor–potential targets for pain therapy? J Pharmacol Exp Ther 306: 423–429. [DOI] [PubMed] [Google Scholar]