

Abstract
The reactivities and chemoselectivities of sodium diisopropylamide (NaDA) in N,N-dimethylethylamine (DMEA) are compared with those of lithium diisopropylamide (LDA) in tetrahydrofuran (THF). Metalations of arenes, epoxides, ketones, hydrazones, dienes, and alkyl and vinyl halides are represented. The positive attributes of NaDA–DMEA include high solubility, stability, resistance to solvent decomposition, and ease of preparation. The high reactivities and chemoselectivities often complement those of LDA–THF.
Graphical abstract

Introduction
This paper is the first in a series describing illustrative metalations by sodium diisopropylamine (NaDA) dissolved in N,N-dimethylethylamine (DMEA). The key insight is that NaDA is highly soluble and stable in trialkylamines (eq 1) and can be prepared in 15 min. The utility of NaDA–DMEA is illustrated by showing that its metalations have rates that far exceed those of LDA–THF but give comparable yields and selectivities that are often complementary. To our surprise, we have witnessed no solubility problems with NaDA dissolved in several trialkylamines or with the resulting metalated intermediates. From these results, we argue that synthetic chemists should revisit NaDA.
 |
(1) |
Background
The preparation of lithium diisopropylamide (LDA) by Levine and co-workers1 in 1949 introduced soluble, highly reactive amide bases to the repertoire of synthetic organic chemists. Metalations with LDA began to flourish in the 1960s,2 and LDA became one of the most prominent reagents in organic synthesis.3 Levine4 first synthesized NaDA in 1959 by reacting phenylsodium and diisopropylamine. Improved preparations include those of Lochmann6 (using LDA–t-BuONa5 or n-BuNa–diisopropylamine prepared from n-BuLi–t-BuONa) and Wakefield7 (using a sodium metal–isoprene-based reduction). More recently, Mulvey and co-workers8 have drawn attention to the merits of sodium chemistry by developing new reagents for synthesis and providing an excellent review of these heavy alkali metal bases. Most reports of NaDA focus on exploration for its own sake,9 however, and only a few appear to be consumer-driven applications of NaDA to solve specific problems.10,11
Why has NaDA languished in relative obscurity? Ambiguities about composition—sodium bases versus mixed-metal “superbases”—have been largely resolved,6a and such details certainly would not have deterred synthetic chemists pursuing reactivity and selectivity. The reported thermal instability of solid NaDA7—resolved by refrigeration—also seems insufficient to curb the interest of the synthetic organic community. We suspect the major obstacle to be the scarcity of solvents that afford both high solubility and resistance to base-mediated decomposition.9b,12 This presumption prompted us to examine DMEA and related trialkylamines.
Results
NaDA was prepared by modifying Wakefield’s procedure7 using sodium dispersion in toluene, isoprene, and diisopropylamine, all in DMEA. The sodium dispersion served as both a drying agent for all reagents and a reductant, allowing millimolar-scale synthesis without specialized glassware in 15 min and multiples of that scale in 30 min. (The longer reaction time stems from an exotherm demanding slower addition.) The resulting amide solution can be used directly, stored for later use at −20 °C, or recrystallized to give a white solid. Upgrading purity via recrystallization had a negligible influence on reactivity.
Metalations of representative organic substrates are summarized in Table 1. The yields are of isolated, purified products unless noted otherwise.13 The relative reactivities of NaDA–DMEA and LDA–THF were assessed with either IR or NMR spectroscopies by measuring pseudo-firstorder rate constants or initial rates for 0.10 M solutions of base. The relative rate constants (krel) for NaDA–DMEA versus LDA–THF are often lower limits because LDA is too unreactive or NaDA is too reactive to measure. Large temperature differences demanded by the two reagents prompt us to crudely (but conservatively) use a 2-fold correction for every 10 °C degrees.14
Table 1.
Reactions of Substrates with NaDAa
| Substrate | Conditions | E+ | Product | krel | Yield | |
|---|---|---|---|---|---|---|
| 1 | n-C8H17Br | 1.2 equiv NaDA 0 °C |
— | 5 | 87% | |
| 2 | 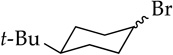 |
1.2 equiv NaDA −78 °C |
— | >500 | 80% | |
| 3 | 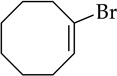 |
1.2 equiv NaDA 0 °C |
— |  |
5 | 87% |
| 4 | 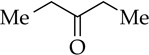 |
1.1 equiv NaDA −78 °C |
TMSCl | 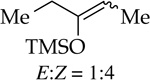 |
N/A | 61% |
| 5 | 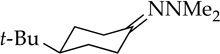 |
1.2 equiv NaDA −78 °C |
CH3I |  |
>300 | 78% |
| 6 | 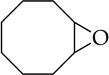 |
1.3 equiv NaDA rt |
H2O | 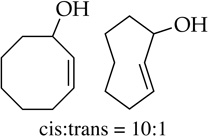 |
>500 | 80% |
| 7 | 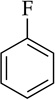 |
1.1 equiv NaDA −78 °C |
CO2 | 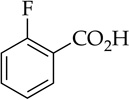 |
N/A | 60% |
| 8 |  |
1.2 equiv NaDA −78 °C |
H2O | 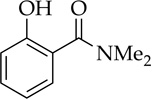 |
>200 | 92% |
| 9 | 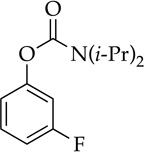 |
1.2 equiv NaDA −78 °C |
CH3OD | 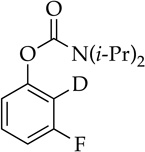 |
>500 | 94% |
| 10 | 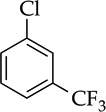 |
1.2 equiv NaDA −78 °C |
CH3OD | 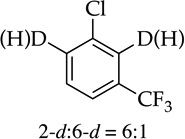 |
1000 | 92% |
| 11 |  |
1.1 equiv NaDA 0 °C |
TIPSCl | 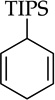 |
>100 | 82% |
| 12 | 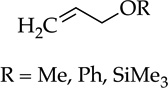 |
1.1 equiv NaDA 0 °C |
— | 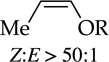 |
>1000 | 83% R = Ph |
krel, relative rate constant; NaDA, sodium diisopropylamide; rt, room temperature; TIPSCl, triisopropylchlorosilane
Discussion
The substrates in Table 1 were chosen to enable a broadly based comparison of NaDA–DMEA and LDA–THF. The dehydrohalogenations in entries 1–3 of Table 1 offer striking contrasts. Whereas n-octyl bromide (entry 1) undergoes clean NaDA-mediated elimination (>100:1), LDA–THF elicits elimination and SN2-like substitution (eq 2).15 Competing substitution–elimination pathways have plagued analogous eliminations of n-alkyl halides.16 Putative trans-diaxial elimination of conformationally anchored cis-4-tert-butylcyclohexyl bromide (entry 2) is highly effective with NaDA. Surprisingly, the equatorial form eliminates with nearly equal efficiency (kaxial/kequatorial = 5), contrasting with the absence of any equatorial elimination by LDA at 25 °C.17 We suspect that a carbenoid mechanism may be involved.18 The comparable rates of NaDA–DMEA and LDA–THF17 for dehydrohalogenations in entries 1 and 3 are outliers in Table 1.
 |
(2) |
Enolization of 3-pentanone (entry 4) is too fast to monitor at standard temperatures. Although the selectivity for NaDA–DMEA is low, it is reversed relative to the 3:1 E/Z selectivity observed for LDA–THF.19 In contrast to ketones, LDA-mediated hydrazone metalations are remarkably slow (entry 5) but are markedly faster with NaDA–DMEA. Trapping the resulting sodium salt with MeI, however, shows a modest 5:1 axial selectivity compared with that for the lithium salt (>15:1).13e
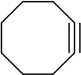
Cyclooctene oxide elimination (entry 6) provided the most unexpected result by giving clean elimination to cyclooctenols in a 10:1 cis:trans selectivity. We can find no precedent for such a base-mediated elimination to give the trans isomer.13f LDA–THF, by contrast, is much slower (requiring reflux) and gives low yields of cis-2-cyclooctenol and bicyclo[3.3.0]octan-1-ol in >13:1 selectivity,20 the latter presumably deriving from carbenoid chemistry.
Orthometalation is the most obvious application of NaDA and has received some attention.9,10 Metalation of fluorobenzene (entry 7) is incomplete at equilibrium, as shown by partial carbonation and by monitoring the reaction with 19F NMR spectroscopy. Precipitation of the arylsodium is observed at approximately 1.0 M at −78 °C. The appearance of benzyne products only at temperatures above −30 °C is notable and surprising.
The rapid orthometalation of the carbamate by NaDA–DMEA (in contrast to LDA–THF) in entry 8 makes the arylsodium observable;13h the Snieckus–Fries rearrangement21 served as an internal quench to illustrate the efficacy of the metalation. The facile haloarene metalations in entries 9 and 10 again underscore a surprising reluctance of the arylsodium to form benzyne at temperatures below −30 °C. Compared with the ~1:1 mixture observed for LDA, the regioselectivity in entry 10 that favors metalation at the 6-position rather than at the doubly activated 2-position is higher,22 but facile equilibration22,23 even at −78 °C affords the 2-sodiated intermediate as the exclusive form, as shown via quenching and 19F NMR spectroscopy.
Metalations of 1,4-cyclohexadiene (entry 11) with subsequent silylation can been carried out, albeit slowly, with LDA–THF, but literature reports all use alkyllithiums.24 Especially notable here is the use of THF as an added ligand after metalation but before silylation; attempts to silylate without adding THF failed with significant loss of yield owing to aromatization and protonation of the sodium salt. Although allyl methyl, phenyl, and trimethylsilyl ethers are not observably metalated by NaDA (entry 12), facile rearrangement to Z-(1-propenyl) ethers as the sole kinetic product compares favorably to a method using t-BuOK–DMSO at >70 °C that gives 10–20:1 Z selectivity and is fast when compared to the recently reported an analogous procedure using LDA by Su and Williard.25
Conclusion
Organosodium chemistry has witnessed brief periods of activity26 followed by long periods of quiescence. We are hoping to regenerate interest. NaDA in DMEA can be prepared in only minutes, shows excellent solubility properties, and is stable as a concentrated stock solution for months with refrigeration. Metalations are breathtakingly fast in many instances, affording surprisingly soluble sodiated products. Importantly, ongoing studies show that DMEA and related trialkylamines are weakly bound to NaDA, which allows facile substitution with stoichiometric quantities of more conventional ligands such as THF, 1,2-dimethoxyethane, and N,N,N′,N′-tetramethylethylenediamine either before or after the metalation. Subsequent reports will show that these ligands often accelerate already rapid metalations by orders of magnitude. The merit of postmetalation ligand substitution is illustrated by the need for THF addition when trapping a dienyl anion with triisopropylchlorosilane. Applications and affiliated reaction mechanisms will be described in due course. Most important, we have yet to uncover any chronic limitations.
Experimental
Reagents and solvents
DMEA, hexane, and THF were distilled from blue or purple solutions containing sodium benzophenone ketyl. All products in Table 1 have been prepared previously13 or are commercially available.
Sodium diisopropylamide
N,N-dimethylethylamine (4.0 mL) and diisopropylamine (0.50 mL, 3.5 mmol) were added to a 15 mL pear-shaped flask. To this flask was added sodium dispersion in toluene (3.0 mL, 35 mmol), which produced effervescence for approximately 10 s. With stirring, isoprene (175 µL, 1.75 mmol) was added over the course of 1 min. Stirring was halted after 5 min, and insoluble materials were allowed to settle, which yielded a yellow supernatant. Variations in the quality (age) of the sodium dispersion can lengthen reaction time. The resulting NaDA solution can be used directly or stored at −20 °C. The reaction was shown to be quantitative by monitoring NaDA formation (δ 3.2–3.3 ppm, septet) and the disappearance of diisopropylamine (δ 2.8–2.9 ppm, octet). Alternatively, the method of Kofron27 was used to titrate the resulting NaDA solution. Diphenylacetic acid (20.0 mg, 0.094 mmol) and 1.0 mL dry THF were added to a vial. Then, the NaDA–DMEA solution was added dropwise at room temperature until yellow coloration persisted, marking the end point of the titration. (The presumed sodium carboxylate can precipitate as a white solid that, interestingly, redissolves with a second equivalent of NaDA, which suggests that the enediolate is soluble.) The results of this titration provide a NaDA titer up to 1.2 times that anticipated if the loss of highly volatile DMEA occurs during NaDA preparation.
IR spectroscopic analyses
IR spectra were recorded by using an in situ IR spectrometer fitted with a 30-bounce, silicon-tipped probe. The spectra were acquired in 16 scans at a gain of 1 and a resolution of 4 cm−1. A representative reaction was carried out as follows: The IR probe was inserted through a nylon adapter and O-ring seal into an oven-dried, cylindrical flask fitted with a magnetic stir bar and a T-joint. The T-joint was capped with a septum for injections and a nitrogen line. After evacuation under full vacuum, heating, and flushing with nitrogen, the flask was charged with NaDA (62 mg, 0.50 mmol) in 4.9 mL DMEA and cooled in a dry ice–acetone bath prepared with fresh acetone. After recording a background spectrum, we added substrate stock (100 µL, 0.50 mmol) with stirring. For the most rapid reactions, IR spectra were recorded every 6 s.
NMR spectroscopic analyses
NMR samples for monitoring reactions were prepared by using stock solutions and sealed with partial vacuum or under ambient argon pressure with two natural rubber septa. Standard 1H and 19F NMR spectra were recorded at 500 and 470 MHz, respectively. In the case of 1H NMR spectroscopic analysis, the loss of NaDA and formation of diisopropylamine can be monitored (vide supra) in addition to characteristic changes corresponding to substrate.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM039764) for support.
Footnotes
The authors declare no competing financial interests.
References and Footnotes
- 1.Hammell M, Levine R. J. Org. Chem. 1950;15:162. [Google Scholar]
- 2.Creger PL. J. Am. Chem. Soc. 1967;89:2500. [Google Scholar]
- 3.(a) Bakker WII, Wong PL, Snieckus V. Lithium Diisopropylamide. In: Paquette LA, editor. e-EROS. New York: Wiley; 2001. [Google Scholar]; (b) Clayden J. In: Organolithiums: Selectivity for Synthesis. Baldwin JE, Williams RM, editors. New York: Pergamon Press; 2002. [Google Scholar]; (c) Hartung CG, Snieckus V. In: Modern Arene Chemistry. Astruc D, editor. Chapter 10. Weinheim: Wiley- VCH; 2002. [Google Scholar]; (d) http://chem.wisc.edu//areas/reich/group/index.htm. [Google Scholar]
- 4.Raynolds S, Levine R. J. Am. Chem. Soc. 1959;82:472. [Google Scholar]
- 5.(a) Lochmann L, Trekoval J. J. Organomet. Chem. 1979;179:123. [Google Scholar]; (b) Lochmann L, Janata M. Eur. J. Chem. 2014;12:537. [Google Scholar]
- 6.(a) Lochmann L, Pospíšil J, Lím D. Tetrahedron Lett. 1966;2:257. [Google Scholar]; (b) Lochmann L. Eur. J. Inorg. Chem. 2000:1115. [Google Scholar]
- 7.Barr D, Dawson AJ, Wakefield BJ. J. Chem. Soc., Chem. Commun. 1992:204. [Google Scholar]
- 8.Mulvey RE, Robertson SD. Angew. Chem., Int. Ed. 2013;52:11470. doi: 10.1002/anie.201301837. [DOI] [PubMed] [Google Scholar]
- 9.(a) Munguia T, Bakir ZA, Cervantes-Lee F, Metta-Magana A, Pannell KH. Organometallics. 2009;28:5777. [Google Scholar]; (b) Andrews PC, Barnett NDR, Mulvey RE, Clegg W, O'Neil PA, Barr D, Cowton L, Dawson AJ, Wakefield BJ. J. Organomet. Chem. 1996;518:85. [Google Scholar]
- 10.(a) Boeckman RK, Boehmler DJ, Musselman RA. Org. Lett. 2001;3:3777. doi: 10.1021/ol016738i. [DOI] [PubMed] [Google Scholar]; (b) LaMontagne MP, Ao MS, Markovac A, Menke JR. J. Med. Chem. 1976;19:363. doi: 10.1021/jm00225a003. [DOI] [PubMed] [Google Scholar]; (c) Bond JL, Krottinger D, Schumacher RM, Sund EH, Weaver TJ. J. Chem. Eng. Data. 1973;18:349. [Google Scholar]; (d) Levine R, Raynolds S. J. Org. Chem. 1960;25:530. [Google Scholar]; (e) Mamane V, Louërat F, Iehl J, Abboud M, Fort Y. Tetrahedron. 2008;64:10699. [Google Scholar]; (f) Baum BM, Levine R. J. Heterocyclic Chem. 1966;3:272. [Google Scholar]; (g) Tsuruta T. Makromol. Chem. 1985;13:33. [Google Scholar]; (h) Andrikopoulos PC, Armstrong DR, Clegg W, Gilfillan CJ, Hevia E, Kennedy AR, Mulvey RE, O’Hara CT, Parkinson JA, Tooke DM. J. Am. Chem. Soc. 2004;126:11612. doi: 10.1021/ja0472230. [DOI] [PubMed] [Google Scholar]
- 11.Inorganic chemists have found NaDA useful for installing the diisopropylamido moiety into transition metal coordination spheres: Spallek T, Heß O, Meermann-Zimmermann M, Meermann C, Klimpel MG, Estler F, Schneider D, Scherer W, Tafipolsky M, Törnroos KW, Maichle-Mössmer C, Sirsch P, Anwander R. Dalton Trans. 2016;45:13750. doi: 10.1039/c6dt01568a.
- 12.To circumvent solubility issues, Mioskowski and co-workers generated n-BuNa in the presence of arenes to achieve orthometalation: Gissot A, Becht JM, Desmurs JR, Pévère V, Wagner A, Mioskowski C. Angew. Chem., Int. Ed. Engl. 2002;41:340. doi: 10.1002/1521-3773(20020118)41:2<340::aid-anie340>3.0.co;2-d.
- 13.The products in Table 1 that are not commercially available but have been prepared previously are listed according to table entry number as follows: (a) entry 2: Scott WJ, Stille JK. J. Am. Chem. Soc. 1986;108:3033. doi: 10.1021/ja00263a015. (b) entry 3: Chen W, Wang D, Dai C, Hamelberg D, Wang B. Chem. Commun. 2012;48:1736. doi: 10.1039/c2cc16716f. (c) entry 4: Tirpak RE, Rathke MW. J. Org. Chem. 1982;47:5099. (d) entry 5: Corey EJ, Enders D. Tetrahedron Lett. 1976;17:3. (e) entry 6: Whitham GH, Wright M. J. Chem. Soc. C. 1971:883. (f) entry 8: García F, McPartlin M, Morey JV, Nobuto D, Kondo Y, Naka H, Uchiyama M, Wheatley AEH. Eur. J. Org. Chem. 2008:644. (g) entry 9: Singh KJ, Collum DB. J. Am. Chem. Soc. 2006;128:13753. doi: 10.1021/ja064655x. (h) entry 11: Landais Y, Zekri E. Eur. J. Org. Chem. 2002:4037. (i) entry 12: Taskinen E. Tetrahedron. 1993;49:11389.
- 14.We often find a 3-fold rate change with each 10 °C to be a better approximation.
- 15.Contrary to our perception, LDA-mediated dehydrohalogenations are conspicuously underrepresented in the published literature for vague reasons. Nikitin KV, Andryukhova NP. Can. J. Chem. 2004;82:571. Vinick FJ, Desai MC, Jung S, Thadeio P. Tetrahedron Lett. 1989;30:787. Altman J, Wilchek M. J. Heterocyclic Chem. 1988;25:915. (d) For references to amide-mediated dehydrohalogenations and reviews on eliminations, see: Kopka IE, Nowak MA, Rathke MW. Synth. Commun. 1986;16:27. (e) Also, see ref 17
- 16.(a) Smith M, March J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 6th. Chapters 10. New York: Wiley-Interscience; 2007. p. 17. [Google Scholar]; (b) Carey FA, Sundberg RJ. Advanced Organic Chemistry. 5th. Chapters 4. New York: Springer; 2007. p. 5. [Google Scholar]; (c) Saunders WH, Jr, Cockerill AF. Mechanisms of Elimination Reactions. II. New York: John Wiley & Sons; 1973. pp. 60–68. [Google Scholar]; (d) Baciocchi Ein. In: The Chemistry of Halides, Pseudo Halides and Azides, Supplement D. Patai S, Rappoport Z, editors. Chichester, U.K.: Wiley; 1983. [Google Scholar]
- 17.Ma Y, Ramírez A, Singh KJ, Keresztes I, Collum DB. J. Am. Chem. Soc. 2006;128:15399. doi: 10.1021/ja060964b. [DOI] [PubMed] [Google Scholar]
- 18.Cristol SJ, Whittemore CA. J. Org. Chem. 1969;34:705. [Google Scholar]
- 19.(a) Ireland RE, Mueller RH, Willard AK. J. Am. Chem. Soc. 1976;98:2868. [Google Scholar]; (b) Miller DJ, Saunders WH., Jr J. Org. Chem. 1982;47:5039. [Google Scholar]; (c) Xie LF, Saunders WH. J. Am. Chem. Soc. 1991;113:3123. [Google Scholar]
- 20.(a) Boeckman RK. Tetrahedron Lett. 1977;49:4281. [Google Scholar]; (b) Apparu M, Barrelle M. Tetrahedron. 1978;34:1541. [Google Scholar]; Whitesell JK, White PD. Synthesis. 1975:602. [Google Scholar]
- 21.(a) Sibi MP, Snieckus V. J. Org. Chem. 1983;48:1935. [Google Scholar]; (b) Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]; (c) Hartung CG, Snieckus V. In: Modern Arene Chemistry. Astruc D, editor. Chapter 10. Weinheim: Wiley-VCH; 2002. [Google Scholar]; (d) Zhao Y, Snieckus V. J. Am. Chem. Soc. 2014;136:11224. doi: 10.1021/ja503819x. [DOI] [PubMed] [Google Scholar]
- 22.Hoepker AC, Gupta L, Ma Y, Faggin MF, Collum DB. J. Am. Chem. Soc. 2011;133:7135. doi: 10.1021/ja200906z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Cottet F, Schlosser M. Eur. J. Org. Chem. 2004:3793. [Google Scholar]; (b) Trecourt F, Mallet M, Marsais F, Quéguiner G. J. Org. Chem. 1988;53:1367. [Google Scholar]; (c) Gros PC, Fort Y. Eur. J. Org. Chem. 2009:4199. [Google Scholar]; (d) Fukuda T, Ohta T, Sudo E, Iwao M. Org. Lett. 2010;12:2734. doi: 10.1021/ol100810c. [DOI] [PubMed] [Google Scholar]; (e) Viciu M, Gupta L, Collum DB. J. Am. Chem. Soc. 2010;132:6361. doi: 10.1021/ja910834b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landais Y, Zekri E. Tetrahedron Lett. 2001;42:6547. [Google Scholar]
- 25.(a) Taskinen E. Tetrahedron. 1993;48:11389. [Google Scholar]; (b) Su C, Williard PG. Org. Lett. 2010;12:5378. doi: 10.1021/ol102029u. [DOI] [PubMed] [Google Scholar]
- 26.For an interesting historical perspective on organoalkali metal chemistry, see: Seyferth D. Organometallics. 2006;25:2. Seyferth D. Organometallics. 2009;28:2.
- 27.Kofron WG, Baclawski LM. J. Org. Chem. 1976;41:1879. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.