

ABSTRACT
Two‐component signaling (TCS) is the primary means by which bacteria, as well as certain plants and fungi, respond to external stimuli. Signal transduction involves stimulus‐dependent autophosphorylation of a sensor histidine kinase and phosphoryl transfer to the receiver domain of a downstream response regulator. Phosphorylation acts as an allosteric switch, inducing structural and functional changes in the pathway's components. Due to their transient nature, phosphorylated receiver domains are challenging to characterize structurally. In this work, we provide a methodology for simulating receiver domain phosphorylation to predict conformations that are nearly identical to experimental structures. Using restrained molecular dynamics, phosphorylated conformations of receiver domains can be reliably sampled on nanosecond timescales. These simulations also provide data on conformational dynamics that can be used to identify regions of functional significance related to phosphorylation. We first validated this approach on several well‐characterized receiver domains and then used it to compare the upstream and downstream components of the fungal Sln1 phosphorelay. Our results demonstrate that this technique provides structural insight, obtained in the absence of crystallographic or NMR information, regarding phosphorylation‐induced conformational changes in receiver domains that regulate the output of their associated signaling pathway. To our knowledge, this is the first time such a protocol has been described that can be broadly applied to TCS proteins for predictive purposes. Proteins 2016; 85:155–176. © 2016 Wiley Periodicals, Inc.
Keywords: response regulator, phosphorelay, post‐translational modifications, molecular dynamics, phosphotransfer, two‐component signaling systems
INTRODUCTION
Two‐component signaling (TCS) systems allow bacteria to respond to external environments using the reversible phosphorylation of conserved histidine and aspartate residues.1, 2 Environmental changes stimulate a histidine kinase (HK) to autophosphorylate on a histidine residue. The phosphoryl group is then passed to an aspartate on the receiver (rec) domain of a downstream response regulator (RR), which modulates a cellular response [Fig. 1(A), top panel]. Some organisms, like plants, fungi and certain bacteria, have developed an expanded system known as a multistep His‐Asp phosphorelay. This uses a hybrid HK that autophosphorylates on a His residue and passes the phosphoryl group to an Asp on an attached rec domain. The phosphoryl group is then transferred to a histidine‐containing phosphotransfer (HPt) protein [Fig. 1(A), bottom panel].2 In plants and fungi, HPt proteins often occupy branch points in the signaling pathway and can transfer phosphoryl groups to multiple downstream RRs. The osmosensing Sln1 pathway in Saccharomyces cerevisiae contains such a system. The HPt protein, Ypd1, acts as the relay point between the upstream hybrid sensor HK, Sln1, and the two downstream RRs, Ssk1 and Skn7.3, 4 To prevent cross‐talk between multiple partners and pathways, protein‐protein interactions within branched systems must be tightly regulated and/or highly specific.5, 6, 7, 8, 9
Figure 1.
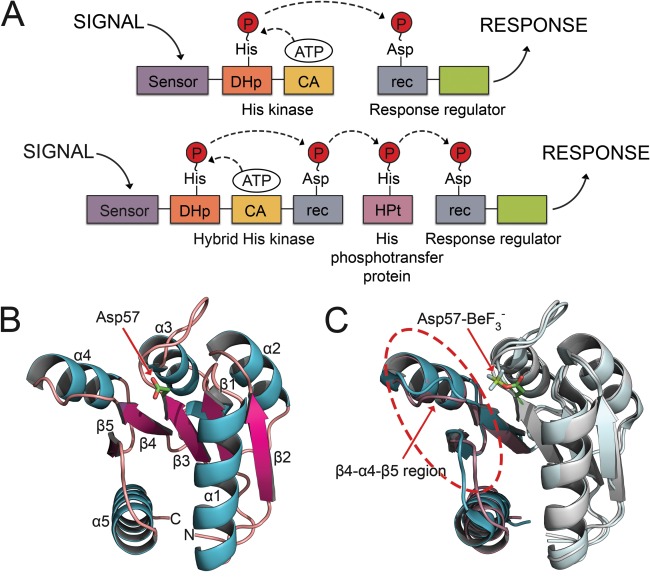
Domain arrangements for TCS systems and topology and induced conformational changes of rec domains. A: Canonical TCS pathway (top) and expanded multistep phosphorelay (bottom). In a typical TCS pathway, the signal is sensed by an HK, which binds ATP in a catalytic domain and autophosphorylates a histidine residue within the DHp. The phosphoryl group is transferred to an aspartate on the receiver domain (rec) of a downstream RR protein. Phosphorylation of the rec domain elicits a cellular response. Expanded phosphorelay systems contain an hybrid HK, with its own receiver domain, and an intermediate known as a HPt. B: The crystal structure of the representative RR, CheY (PDB 3CHY), in its unphosphorylated state shows the common (βα)5 topology. C: Major conformational shifts upon phosphorylation occur within the β4‐α4‐β5 regions (circled). Unphosphorylated CheY (PDB 3CHY) in cyan. bound CheY (PDB 1FQW) in purple. Asp57‐ is shown in stick model.
Nearly all rec domains adopt a common (βα)5 topology [Fig. 1(B)],2 consisting of a five‐stranded, parallel β‐sheet surrounded by five amphipathic α‐helices. Loops located at the C‐terminal ends of β1, β3, and β5 form a docking surface for cognate HKs and HPt proteins. Figure 1(B) shows a representative structure of the chemotaxis RR, CheY.10 Experimental structures show that rec domains use a highly conserved active site geometry that is essential for phosphotransfer. Rec domain active sites contain three conserved acidic residues (Asp or Glu) for coordinating a divalent metal cation, such as Mg2+. The Asp at the C‐terminal end of β3 serves as the site of phosphorylation.2, 11, 12 Two additional residues complete the active site: a highly conserved lysine and a moderately conserved residue (often Gln, Asn, or Lys) located two positions downstream from the phosphorylatable aspartate (D + 2 position).2, 11, 12
The favored model of rec domain switching posits the existence of a dynamic equilibrium between apo and phosphorylated states.13, 14, 15, 16, 17, 18, 19 Phosphorylation is known to induce allosteric and functional changes in rec domains.11, 20, 21, 22, 23, 24 This is thought to cause a population shift by stabilizing the modified state, thereby redistributing the equilibrium.17 This shift is not fully understood, and additional intermediate states have been observed.13, 25, 26, 27, 28 The equilibrium shift model, also known as conformational selection, is in contrast to the more traditional idea of induced fit. In the induced fit scenario, phosphorylation is required to drive the conformational changes in the rec domain, which only occur when the ligand is bound (summarized in Ref. 29, specifically for rec domains in Ref. 13). Despite more recent support for the conformational selection theory, controversy still surrounds the exact mode of rec domain switching.
Structural changes associated with phosphorylation are often subtle and difficult to identify. Structural alignments indicate average RMSDs between apo and phosphorylated conformations range from 1 to 4 Å, but deviations can be significantly greater (>7 Å) in functionally relevant areas. Experimental structures reveal several features common to most known modified rec domain structures.11, 23, 30, 31, 32, 33 Figure 1(C) shows a rec domain alignment of CheY (PDB 3CHY10 in its apo state; cyan) with CheY in its bound state (PDB 1FQW11; purple). In CheY and the majority of other rec domains, large shifts consistently occur within the β4‐α4‐β5 region. Two conserved residues, a Ser/Thr on β4 and a Phe/Tyr/His on β5, adopt characteristically altered orientations in the crystal structures. Upon phosphorylation, the Ser/Thr rotamerizes and/or shifts with the β4α4 loop to form a hydrogen bond with the phosphoryl group, typically at a distance of 2.5–2.8 Å. The aromatic side‐chain of Phe/Tyr/His initially points away from the phosphoryl group, exposed to solvent in the apo state. Upon phosphorylation, it rotamerizes inwards to bury its side‐chain in a hydrophobic pocket recently vacated by the Ser/Thr on β4. These two conserved amino acids are referred to as “switch residues” and are used as traditional indicators of conformational state for rec domains.28 Some studies have also implicated the aromatic residue (Phe/Tyr/His) as a factor in rec domain dimerization, due to its location in the center of a common dimerization interface.34, 35 In CheY, these switch residues are Thr87 and Tyr106. More recently, a quartet of coupled residues have been implicated in directing allosteric changes in CheY.16 The model speculates that Trp58 (D + 1) rotamerizes and shifts upwards, simultaneously increasing contact with Glu89 and decreasing contact with Met85. These changes are thought to be related to the rotamerization of Tyr106 and shifts in the β4α4 region. While these residues are only partially conserved, the relative phosphorylation‐induced changes are visible in many experimental structures.30, 32, 33, 36, 37
Even with highly conserved tertiary structure, genetic diversity between rec domains has led to divergent functional properties, making studies of individual rec domains necessary.11, 20, 21, 23, 24, 38, 39, 40, 41 Depending on their biological role, autodephosphorylation rates of rec domains can span several orders of magnitude, suggesting other structural elements may be affecting the stability of the phosphoaspartate.12, 42, 43, 44 Many rec domains also oligomerize upon phosphorylation, usually through the α4‐β5‐α5 face.30, 34, 45, 46, 47 Non‐canonical interfaces utilizing the α1α5 helices have been observed as well.46, 48, 49 Structural rearrangements associated with phosphorylation can cause changes in surface shape and physicochemical properties, affecting interactions with binding partners.47, 50, 51, 52, 53, 54, 55, 56 While rec structures in their apo states provide basic structural information, they do not reveal the important allosteric changes that occur upon phosphorylation.
Of the >300,000 rec domain sequences found in the NCBI database, ∼150 (non‐redundant) have structures available in the RCSB Protein Data Bank (PDB; http://www.rcsb.org).57, 58, 59 The majority of these structures are unphosphorylated, due to the difficulty of capturing the chemically labile phosphoaspartate species.60, 61, 62, 63, 64 Most “phosphorylated” structures were obtained using phosphoryl analogs, like .11, 23, 30, 31, 32, 33 This approach has met with limited success; fewer than 30 rec domains have been characterized in a conformationally modified state.57, 58 Molecular dynamics (MD) can be used to circumvent the difficulties of characterizing these transient conformations. However, kinetic studies reveal that rec domain phosphoryation and conformational changes occur on high microsecond to low millisecond timescales.20, 21, 65 Simulating these timescales at atomic resolution would be prohibitively expensive and impractical for most research laboratories. To make these studies more accessible, biasing terms can reduce the amount of sampling required.66, 67, 68 The most conserved rec domain feature is the active site geometry required for phosphotransfer.11, 12, 33 Restraints can be used to drive the formation of this active site configuration for any rec domain. This shifts the protein toward a phosphorylated state, leading to allosteric rearrangements throughout the structure. Although these phosphorylated conformations are theoretically observable without restraints, they would require millisecond‐timescale simulations to reliably sample. The active site restraints drastically reduce the conformational search space and allow us to sample phosphorylated conformations in a fraction of the time.69 With this approach, laboratories that lack the capability of millisecond timescale simulations are able to effectively simulate these events.
We have developed a methodology for accessing the phosphorylated conformation of a rec domain protein in a direct and expedient path using active site restraints. To validate this approach, we performed biased, all‐atom simulations on five diverse rec domains with known modified structures. This produced models for the rec domains that are nearly identical to the experimental data, both in global conformation and in key local features. The success of this methodology suggests that while rec domains may utilize some form of conformational selection to achieve allosteric switching, the application of an induced fit model is sufficient to achieve an equivalent effect. Additionally, data on conformational dynamics obtained from each simulation were used to identify areas of major functional significance associated with phosphorylation. Finally, we applied the approach to two rec domains found within the osmoregulatory Sln1 pathway of S. cerevisiae, the upstream Sln1‐rec and the downstream Ssk1‐rec, to compare their structural and functional changes upon activation. The principle of using active site restraints to reduce conformational search space can be applied to other protein families individually, such as kinases and HPt proteins. In addition, this method can be expanded to study the effects of phosphorylation on rec domain interactions with related protein families.
MATERIALS AND METHODS
Modeling and preparation
Initial structures
Crystal structures of single domain RRs (CheY and Spo0F), rec domains from multi‐domain RRs (PhoP‐rec and FixJ‐rec), and the rec domain of a hybrid HK (Sln1‐rec) were obtained from the PDB.57 Modified rec structures contained both a metal cation and the modified aspartate residue, either with or PO3 2−. For a full list of the proteins used, see Table 1.
Table 1.
Protein Structures used during Simulations
Ssk1‐rec structure prediction
The Protein Homology/analogY Recognition Engine v.2.0 (Phyre2) was used to generate a homology model of apo Ssk1‐rec from S. cerevisiae.70 The Ssk1‐rec domain sequence (residues 505–580:603–651) was used as input for intensive modeling mode. Nonconserved α3β4 loop residues (581–602) were excluded due to poor modeling quality and their location on the surface opposite the active site. The initial model was refined using ∼5 ns of all‐atom MD simulation. The resulting equilbrated structure was extracted and examined using Verify3D and RAMPAGE to assess model quality.71, 72 The Ssk1‐rec model was then prepared and simulated as described in subsequent sections to sample its phosphorylated state.
System preparation
Starting models were prepared from the apo crystal structures. Phosphoaspartate was modeled into the active sites with Coot.73 A Mg2+ atom was also added based on available experimental coordinates. If no cation was present in the experimental structure, coordinates were copied from a homologous structure containing metal. While other metal types have been found in rec domain active sites, Mg2+ is sufficient for function of nearly all rec domains.74, 75, 76 The initial positioning of the metal cation is not criticial in the context of the simulations. Each protein will ultimately adopt its phosphorylated conformation around the cation as long as it is present in the general vicinity of the active site. Initial structures were stripped of all other crystallographic waters, ligands, and additional subunits prior to simulation.
Dowser was used to fill internal water cavities with a default probe radius of 0.2 Å.77, 78 The SOLVATE program was then used to create a contoured solvent shell around each model with a shell thickness of 6.0 Å.67 Systems were loaded into VMD and processed using the AutoPSF plug‐in.79 Finally, structures were immersed in a full orthorhombic water box with 15.0 Å padding on each side using a TIP3P water model with the VMD solvate plug‐in.79 Systems were neutralized and K+/Cl− ions were added to a final concentration of 100 mM using the VMD Autoionize plug‐in.79
Phosphoaspartate
Parameters for dianionic phosphoaspartate (AST) were defined for the CHARMM36 force field based on Damjanović et al. (2009).80, 81, 82 A tetrahedral phosphoryl group exists predominantly in the dianionic form at physiological pH (7.4).63 All simulations were run using the dianionic group instead of the monoanionic form. As described in Ref. 82, charges for the phosphoryl group along with various bond and angle terms were taken from dianionic methylphosphate. Additional dihedral angle terms were taken from phenol phosphate. The torsion angle (X‐CD‐ON2‐X) and partial charges (CG and OD1 atoms) were obtained from previous parameterization studies.82
Molecular dynamics
Simulations
Simulations were prepared and run using VMD and NAMD 2.979, 83 with the CHARMM36 protein force field.80, 81 A 12.0 Å cutoff was used for van der Waals interactions with a switching function distance of 10.0 Å. Long‐range electrostatics were evaluated using the PME (Particle Mesh Ewald) method with a tolerance, interpolation order and grid spacing of 10e−6, 4.0, and 1.0 Å, respectively. A two fs integration step was used for all simulations. The SHAKE algorithm was applied to constrain covalent bonds involving hydrogen atoms throughout. Initial energy minimization was performed on non‐backbone atoms for 100 ps of NVT MD. An all‐atom minimization step was then done for an additional 100 ps of NVT MD with harmonic constraints of 0.5 kcal/mol applied to all Cα atoms. Following this, systems were heated to 310 K by increments of 1 K/ps over ∼310 ps of NVT MD. Equilibration was then performed with Cα atoms restrained for 500 ps of NPT MD. A final, unrestrained equilibration step of 4 ns NPT MD was run to prepare systems for production. Proteins were equilibrated and then simulated in multiple 10 ns production runs to collect 100 ns of effective production data for each rec domain. Trajectories were combined for analysis. Parallel runs were performed to increase conformational sampling.
Simulation restraints
Biasing terms are frequently used to steer systems toward desired states for study. Parameters that regulate this are known as collective variables (colvars).69 A colvar ξ is defined as a differentiable function of the vector of 3N atomic Cartesian coordinates, X
ξ(X) is generally a function with many fewer arguments than 3N, or is a combination of these functions:
with the Cartesian gradient:
where the number of functions z ( α ) is much smaller than the number of atoms. The term z ( α )(X) describes a component and is a function of several atomic coordinates. A colvar is defined by a combination of one or more components. In the simplest scenario, the colvar ξ identifies a single component z, and often relates to properties such as atomic distances or dihedral angles.
Using the colvar module in NAMD, we applied half‐harmonic potential restraints to each rec domain to drive the formation of the appropriate active site geometry.83 Restraints were added during equilibration and maintained for the full 100 ns production data sets. For a list of the restraints used in the CheY representative experiments, see Table 2. While this list was used on every system, minor changes were made if a given protein could not adopt a fully correct active site, often due to initial nonphysiological conformations. A sample colvars list is included for CheY in the Supporting Information (Text S1). It can be adapted for any target rec domain by changing the atomNumbers entries.
Table 2.
Colvars Applied to Drive Active Site formation in CheY
| Type | Group 1 atoms | Group 2 atoms | Boundaries |
|---|---|---|---|
| Distance | Asp57 OD1 | Mg2+ | 1.9–2.2 Å |
| CoordNum | Asp57 PO3 2− | Mg2+ | 1 group |
| Distance | Mg2+ | Initial center of mass | 0.0–0.1 Å |
| Distance | Ala88 N | Asp57 OP2 | 2.0–3.3 Å |
| CoordNum | Asp13 R | Mg2+ | 1 group |
| Distance | Asp57 OP1 | Mg2+ | 0.0–2.2 Å |
| Distance | Asp13 OD1 | Mg2+ | 0.0–2.2 Å |
| Distance | Gln59 O | Mg2+ | 0.0–2.2 Å |
Data analysis
Trajectory analysis
Trajectories were analyzed using VMD and the Bio3D package in R.79, 84, 85 Runs were stripped of all non‐protein atoms using VMD. Partial trajectories were then aligned by Cα atoms using the RMSDTT plug‐in79 and combined to form large ensembles for analysis.
Each simulation produced >20,000 conformations (snapshots) of its protein. Analysis of such high‐dimensionality data is challenging. We performed principal component analysis (PCA) on the full combined trajectories to reduce the complexity of the data and analyze only the dominant conformations. PCA is a multivariate technique to simplify and visualize patterns within structural ensembles, including interconformer relationships.84, 86 Typically, it is performed using a reduced set of atoms, such as an Cα representation. Projection of the conformational distribution onto the subspace defined by the first few principal components (PCs; by eigenvalues) creates a lower‐dimensional representation of the data, making analysis easier and faster.84, 86 We then used a k‐medoids approach to cluster the distribution in PC‐space, grouping conformers based on structural similarity.87, 88 The CLARA (Clustering for Large Applications) algorithm was used to handle the large data sets.88, 89 A range of 4–15 clusters were tested, with the ideal number of clusters estimated using optimum average silhouette width.
We performed dynamical cross‐correlation (DCC) analysis on the combined production trajectories to detect collective motions between Cα atoms during each simulation. Trajectories were first superimposed against the initial frame. A cross‐correlation coefficient, Cij, was calculated for the Cα atoms with the expression:
where Δri and Δrj are the displacements from the mean position of the ith and jth atoms with respect to time.90 Cij values were either positive, representing in‐phase or positively correlated motion, or negative, representing out‐of‐phase or negatively correlated motion. These were collected in matrix form and plotted.
Structural analysis
Structural alignments and analyses were performed using UCSF Chimera.91 Superpositioning to compare per residue deviations was done using the internal β‐sheet, as it showed minimal deviation upon activation in rec domain crystal structures. Graphing was performed in R.85 Electrostatic surface potential was calculated with Adaptive Poisson‐Boltzmann Solver (APBS) package in 0.1M KCl.92 Surface cavities were calculated using the 3D‐Surfer server with the VisGrid algorithm.93–95
Software
MD simulations were performed with NAMD83 on resources provided by the OU Supercomputing Center for Education and Research (OSCER) at the University of Oklahoma. System preparation, visualization and initial trajectory analyses were performed in VMD.79 Models and figures were made in Coot and PyMOL.73, 96 Structural analyses were performed in UCSF Chimera.91 Trajectory analyses and plots were performed with R using the Bio3D, fpc, and cluster packages.84, 88, 97
RESULTS AND DISCUSSION
Restraints drive phosphorylation‐induced conformational changes
Crystal structures of five apo rec domains were obtained from the PDB (see Table 1) for simulations. Phosphorylatable aspartate residues were replaced in the apo models with a custom dianionic phosphoaspartate (AST). A Mg2+ cation was modeled into the center of the active sites to act as a pivot point for each protein. Two options exist and were tested for the positioning of this cation in the apo model(s). The first was to use the same coordinates as the cation found in the modified ( bound) experimental structure for a given rec domain. The second was to align the apo model(s) to a known modified structure of a homologous rec domain containing a bound cation. The metal's coordinates were copied from the homologous structure and inserted into the unphosphorylated system(s). Results from the validation set showed that both approaches are effective, demonstrating how a rec domain with no known modified or metal‐bound structure can be simulated by manually modeling the bound cation. Half‐harmonic potential restraints were applied centered on this metal cation. Any restraints during a simulation can affect the dynamics of the system. Caution should be used when speculating on any mechanism of conformational transition. Nevertheless, we know that the active site is almost always arranged in the previously described geometry, as it is necessary for the chemistry of phosphotransfer. Sampling phosphorylated rec domain conformations without the active site restraints is possible, but would require far longer (millisecond) timescales that are prohibitively expensive and impractical for standard research laboratories. We repeatedly failed to observe complete transitions from apo to phosphorylated states for the rec domains in the validation set without the correct combination of restraints. This suggests that on this timescale, we cannot reliably or adequately sample phosphorylated conformations without the biasing terms.
Biased MD simulations were performed in explicit solvent for each system. Structural comparisons of phosphorylated and apo rec domain crystal structures suggest that the shift toward a phosphorylated conformation is subtle when the entire domain is included (average RMSDs of 1–3 Å).11, 33 Cα deviations observed during the production simulations corroborated this, with most systems stabilizing within the ∼5 ns equilibration period and maintaining RMSDs of 1–2 Å throughout the runs (data not shown). This indicates that the simulated phosphorylation of these rec domains occurs on a similar magnitude as the experimental structures. The subtlety of these perturbations makes overall structural comparisons difficult for both experimental data and predicted models. In contrast, major changes are almost always seen along the β4‐α4‐β5 region, with some deviations occurring on magnitudes of >7 Å in known structures.30, 32, 33, 36, 37 To explore if these changes were due to increased flexibility within this region, or more concerted intramolecular movements, we calculated the per residue RMSF for each protein during the production simulations. Figure 2 shows the plots for the rec domains in the validation set. As expected, the loop regions generally possessed the highest levels of flexibility. The β4‐α4‐β5 region demonstrated no outstanding flexibility relative to the rest of the protein in the majority of the rec domains, suggesting that it is most likely involved in more concerted or specific changes associated with phosphorylation. FixJ‐rec is a possible exception. Comparison of existing crystal structures reveals that the β4‐α4‐β5 region of FixJ‐rec undergoes the most dramatic shift within the validation set. The high RMSF in this region suggests that the protein's intrinsic flexibility may contribute significantly to this deviation, in addition to the allosteric changes induced by phosphorylation.
Figure 2.
Residue mobilities during production runs for the five rec domains. Comparison of average root mean square fluctuations (RMSFs) for combined MD trajectories shows regions of relative flexibility in each protein. β‐strands (pink); α‐helices (cyan).
Analysis of collective motions reveals areas of functional relevance
Protein dynamics are intimately involved in enzyme activity and catalysis. Previous work has speculated that areas undergoing collective motions within proteins are likely distinct subdomains of functionally related residues.90, 98, 99, 100, 101, 102, 103, 104 To identify these regions, with the notion that they may prove relevant to the phosphorylated state and protein function, we generated dynamical cross‐correlation maps (DCCM) for all five rec domains.84 Cross‐correlation plots show how various regions of a protein communicate by quantifying their relative motions. Motion can be positively correlated, meaning in the same direction, or negatively correlated (anticorrelated), meaning in the opposite direction. Figure 3 shows maps of correlated and anticorrelated motion within each protein calculated using the combined trajectories. These represent motions with the active site restraints already enforcing the conserved catalytic geometry. Correlation was normalized and scaled between −1.0 and 1.0. Collective motions between −0.25 and 0.25 were not considered significant (Fig. 3 white).100, 105 For CheY, the middle and upper right areas show positively correlated motions between residues 80–89 and 101–110 (Fig. 3, yellow/green). These correspond to the α3‐β4‐α4 and α4‐β5‐α5 regions, suggesting correlated motions between the β4α4 and β5α5 loops and throughout the β4 and β5 strands themselves. In previous studies on activated rec domains, most dramatic shifts were typically observed in these areas, particularly in the loops adjacent to the active site.11, 23, 30, 31, 32, 33 Residues 50–61 and 79–87 (α2‐β3‐α3 and α3‐β4‐α4 regions) also displayed positively correlated motions, again suggesting concerted loop movements and β‐strand interactions. In the lower left, residues 4–14 and 31–38 (β1α1 and α1‐β2‐α2 regions) and residues 4–14 and 50–61 (β1α1 and α2‐β3‐α3 regions) also showed similar positively correlated motions throughout every simulation. The areas of positive correlation suggest high overall stability of the central β‐sheet (as expected) and its interactions with loops, but relatively little correlated motion between it and the flanking helices. Strong negatively correlated motion was found within the CheY molecule between the C‐terminal portion of the α5 helix and the β5 strand and between regions of the α5 helix and most of the α4 helix. This indicates significant concerted movement within the α4‐β5‐α5 face of CheY. Other studies have shown that many rec domains dimerize through this surface.30, 34, 45, 46, 47 While CheY does not form a homodimer, it does binds the downstream flagellar motor switch protein, FliM, through this surface.36 Phosphorylation of CheY is known to increase its binding affinity for FliM 20‐fold.106 The data revealed significant reorientation of key residues (Ala90, Ile95, Tyr106, Lys119, Lys122; relative to the apo conformation) involved in, or potentially blocking, interaction with FliM.36 Each of the five proteins produced remarkably similar patterns of positively correlated motions involving loop regions and the central β‐strands. Due to the high similarity between profiles, this pattern of positively correlated motion can provide a check for the proper simulation of rec domains. Perhaps more important are the unique patterns of negatively correlated motion produced by each protein (Fig. 3, blue/black). These variations offer valuable insights related to differences in rec domain function and effects after allosteric modification. The results suggest that putative regions of functional significance tied to phosphorylation‐induced changes can be identified based on (anti)correlated motions. This provides potential targets for further experimental studies. However, it must be noted that DCCM alone cannot provide conclusive information on conformational transitions. Further study would be valuable for characterizing the extent of these changes, using complementary approaches such as normal mode analysis. The following sections briefly summarize the DCCM results for the four remaining rec domains in the validation set.
Figure 3.
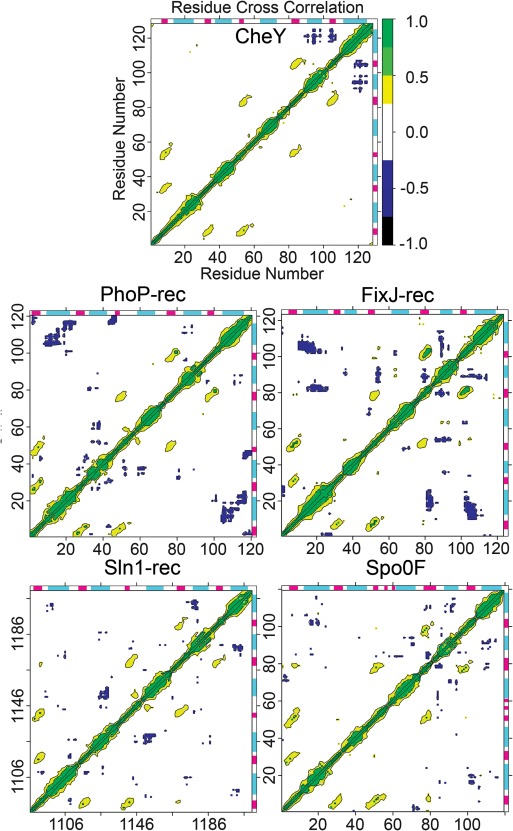
Dynamical cross‐correlation maps (DCCM) reveal collective motions within the phosphorylated rec domains. Positive correlation (yellow/green) suggests movement in phase, or in the same direction. Negative correlation (blue/black) suggests movement out of phase, or along the opposite direction. α‐helix (cyan); β‐strand (magenta).
Major regions of FixJ‐rec that exhibited significant negatively correlated motions were the α4‐β5‐α5 surface, including a large shift in the α4 helix, and the loop regions accessible on the active site surface. Phosphorylation‐dependent FixJ dimerization and further downstream signaling is believed to occur through the α4‐β5‐α5 interface (similar to CheY‐FliM binding).23, 53 Key residues implicated in homodimerization that adopted altered orientations during simulation and were involved in negatively correlated motions include His84, Val87, Val91, Lys95, and the switch residue Phe101.23 Additionally, correlated perturbations seen in the α1α5 region are likely involved in the binding of the upstream partner, FixL. This is supported by the work of Saito, et al. (2003), which suggests that mutations at this surface (particularly on the α1 helix) interfere with phosphotransfer from FixL to FixJ.107 As FixL also exhibits phosphatase activity, this likely affects the stability of phosphorylated FixJ‐rec.
Regions of negatively correlated motion seen in PhoP‐rec correspond to the α2α3 and α1α5 regions, similar to other rec domains. Also present is the characteristic movement of the α4‐β5‐α5 face, though to a much smaller degree than the other rec domains. PhoP dimerizes, even in an unphosphorylated form, through an asymmetric interface, but phosphorylation is required for downstream function (DNA binding).30, 108 Phosphorylation causes a shift in the α4 helix, potentially stabilizing the dimerization interface. Cross‐correlation analysis also revealed significant correlated movements in the loop regions near the active site surface of PhoP‐rec. All known rec domains must bind their upstream donors through this active site to facilitate phosphotransfer, suggesting that the correlated loop motions may be involved in interactions with the upstream partner, PhoQ.
Spo0F regions that demonstrated significant negatively correlated motion correspond to a characteristic rearrangement of the α4‐β5‐α5 face, along with more subtle motions at the α1α5 surface and the loops near the active site. The α4‐β5‐α5 surface of Spo0F is not involved in dimerization and does not directly bind its partner, Spo0B.32 The exact purpose of this area is unknown, and these results suggest a target for additional studies. Collective motion observed between the α1 helix and the α4α5 region is most likely involved in protein‐protein interactions with Spo0B, as phosphorylation is speculated to increase the binding affinity between the two proteins. The co‐crystal structure of the Spo0F•Spo0B complex confirms this, showing direct intermolecular contacts located within this region.32 In addition, previous studies indicated that mutations in α1 helix and the β4α4 region caused defects in binding with KinA.109
Major regions in Sln1‐rec that exhibited significant negatively correlated motion correspond to movements between active site loop regions and between the α2α3, α1α5, and α4‐β5‐α5 surfaces. Sln1 is a fungal hybrid HK believed to dimerize through its dimerization histidine phosphotransfer (DHp) domain.110 The rec domain alone is believed to function as a monomer, though no full‐length structure of the Sln1 hybrid HK exists. Cross‐correlation analysis suggests that the α4‐β5‐α5 surface, and possibly the α1α5 and α2α3 helices, may have significant roles in the signaling pathway. These movements may involve interactions with the downstream partner HPt, Ypd1, or with its own hybrid HK domain. Examination of the existing crystal structures suggest that the α2α3 and α1α5 helix regions are directly involved in binding Ypd1, although the purpose of the α4‐β5‐α5 region remains unknown.4, 33 A more detailed analysis and comparison with another fungal rec domain, Ssk1‐rec, is discussed in later sections.
Trajectory analysis and extraction of representative receiver domain structures
Due to the dynamic nature of proteins, no single “correct” phosphorylated conformation exists for a rec domain. But based on the characteristic changes associated with rec domain phosphorylation found in existing structures, we can identify a representative structure that is most likely to resemble a phosphorylated conformation in a crystal structure. Determining which conformation(s) from within the total ensemble best accomplishes this is indeed challenging. Each simulation produced a substantial amount of conformational data (>20,000 observations). We first had to simplify these complex datasets to identify which conformers were the most significant to this goal. To reduce the dimensionality of the data, we performed PCA on the combined trajectories for each rec domain (data not shown). Performing PCA prior to clustering can save significantly on computational cost and time, as well as filter out high‐frequency conformational variance (noise) from the data.111
Trajectories were clustered in the resulting PC space to quantitatively identify the dominant conformations adopted by the proteins during simulation. Due to the large size of each data set, an implementation of the CLARA algorithm was used.87, 88 The pamk function within the R package fpc was used to estimate the ideal number of clusters based on average silhouette width.89, 97, 112, 113 Data were clustered using the top seven PCs to incorporate ∼50% of the observed data variance. To produce a reasonable number of candidate structures from which to choose a final representative phosphorylated structure, the minimum number of clusters was set to four, and the maximum was set to fifteen. Each cluster was an ensemble of structurally similar conformers. A corresponding medoid was then extracted from each cluster. A medoid is a representative conformer from the cluster that exhibits minimal dissimilarity to the rest of the cluster. This produced a list of candidate structures for the rec domains. For our purposes, the candidates best represent the dominant conformations observed for the phosphorylated rec domains.
Identification and validation of representative predictions using experimental receiver domain structures
Ideally, each candidate (medoid) structure should be analyzed individually for physiological relevance, as all are potentially valid conformations of phosphorylated rec domains. No single scoring method exists to adequately identify the most representative structure of a phosphorylated rec domain. But to fulfill the ultimate goal of this study and for validation purposes, we developed a set of criteria to identify the top 1–2 candidate structures most likely to resemble the phosphorylated crystal structure of each rec domain. All factors must be examined collectively to make an informed decision. The process (depicted in the flowchart in Fig. 4) includes comparisons with the apo conformation to detect characteristic allosteric changes associated with phosphorylation [Fig. 4(A)], quantifying and ranking switch residue and quartet residue reorientations [Fig. 4(B,C)], eliminating inappropriate active site arrangements [Fig. 4(D)] and ranking by conformer cluster population sizes [Fig. 4(E)]. These steps are described in detail for CheY in the text. Results for each rec domain (including CheY) were tabulated and included in the Supporting Information (see Tables S1–S4).
Figure 4.
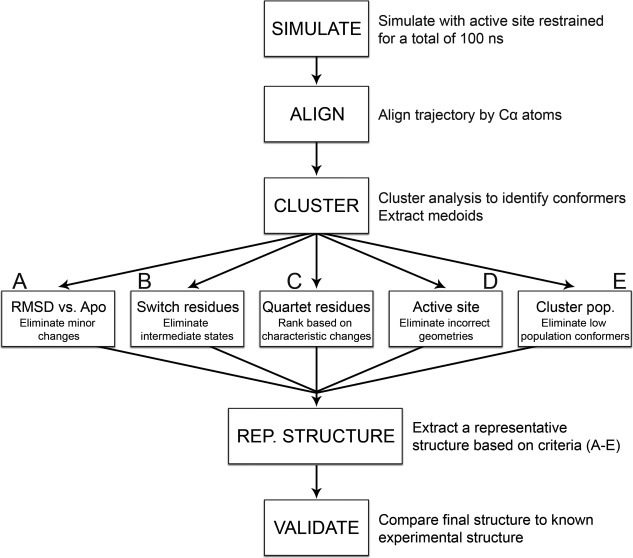
Flowchart summarizing the methodology. Initial apo structure is phosphorylated and simulated with active site restraints. The resulting combined ensemble is aligned to remove translational and rotational fluctuation. Conformers are then clustered together based on similarity using k‐medoids based algorithm. Candidate structures (medoids) are extracted for each cluster. Each medoid is analyzed to identify the candidate most likely to resemble a phosphorylated crystal structure using the following criteria: (A) Candidates are aligned to their apo structure to eliminate medoids showing little to no conformational changes (∼3.5 Å cutoff). B: Switch residue positions are analyzed. Medoids lacking fully buried aromatic side chains and hydrogen‐bonded Thr/Ser residues (2.9 Å cutoff) are ignored. C: Quartet residue positions are measured and compared between medoids. D: Active site geometry is checked. Medoids lacking fully conserved active site arrangements are ignored. E: Medoids from transient clusters are eliminated (∼1500 conformer cutoff). Using these criteria, a final representative structure is identified. This structure is then validated against existing structural data.
Structural alignments were performed with each set of candidate structures and their corresponding apo crystal structure [Fig. 4(A)]. Figure 5(A) shows a superposition of all seven candidate structures extracted from the CheY trajectories aligned to the apo CheY structure (PDB 3CHY; black ribbon) using the invariant β‐sheet core.10 Most of the conformational variance we observed between clusters was small (∼1.5 Å) and unsurprisingly located in the flexible loop regions. Qualitatively, this can be seen in the CheY alignment in Figure 5(A), where the majority of the conformers show little deviation, especially throughout the helix and strand elements. Other rec domains (Spo0F; see Supporting Information Fig. S4) exhibited far more diversity among the medoid conformers. The largest changes induced by phosphorylation occur in the β4‐α4‐β5 region of CheY. This area has consistently shown high deviations in nearly all modified rec structures.11, 23, 31, 32, 33 The shift from apo to phosphorylated state is quantified in Figure 5(B) for CheY, which shows the per residue all‐atom RMSD after alignment between the apo (PDB 3CHY) and bound crystal (PDB 1FQW) in black trace. The grey traces in Figure 5(B) shows the same for each medoid of CheY aligned to the apo crystal. The colored trace shows the RMSD between the apo crystal structure and a sample medoid structure, demonstrating a significant conformational shift characteristic of phosphorylation. By examining the deviations in the β4‐α4‐β5 region [Fig. 5(B), shaded area; Supporting Information Figs. S1–S4), we were able to eliminate any candidate structures that showed unusually low relative conformational changes associated with phosphorylation, indicating a closer resemblance to the apo conformation. For CheY, typical max deviations observed among the medoids were ∼4.5–5 Å within this area. A single medoid was found [the conformer that best represents cluster 7 (see Supporting Information Table S1), which contains ∼5% of the total ensemble] with significantly smaller changes (∼2.5–3 Å). This medoid was assigned lower priority based on these RMSD calculations. The remaining candidates displayed more characteristic changes when aligned to the apo structure, and comparison between the colored and black traces suggest a phosphorylation‐induced conformational shift on a similar magnitude. Alignments and RMSD calculations for the other protein systems are available in the Supporting Information (Figs. S1–S4). A threshold of ≤ 3.5 Å was used for most of the rec domains. However, a lower cutoff of 3.0 Å was applied to Sln1‐rec, which exhibited consistently smaller changes overall (see Supporting Information Table S4).
Figure 5.
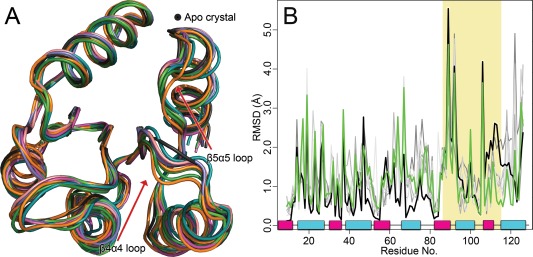
Alignments of candidate structures to the unphosphorylated crystal structure of CheY. A: Superposition of apo crystal structure (PDB 3CHY, black) with candidate (medoid) structures identified during cluster analysis. B: Per residue, all‐atom RMSD for phosphorylated structures versus the apo crystal structure. Shaded area corresponds to β4‐α4‐β5 regions. Trace color corresponds to chosen structures in alignment: example candidate structure versus apo crystal (green); crystal vs. apo crystal (black). Traces for additional candidate structures appear in gray. α‐helix (cyan); β‐strand (magenta).
Switch residue orientation was also examined in each of the extracted candidate structures [Fig. 4(B)]. Figure 6 shows a close‐up comparison of the switch residues found in CheY. In Figure 6(A), the apo crystal structure of CheY shows the unmodified positions of the switch residues. The side‐chain of Tyr109 adopts a torsion angle (χ 1) of −25° and Thr87 is positioned 6.1 Å away from the phosphorylatable Asp57. Figure 6(B) shows how Thr87 shifts to within 2.5 Å of Asp57, and Tyr109 adopts a torsion angle (χ 1) of 76° in the bound crystal structure. Figure 6(C) demonstrates the same “switched” orientations in a sample medoid structure predicted for CheY. A similar shift of Thr87 was observed in ∼100% of the structural ensemble for CheY, indicating an almost complete and instantaneous switch upon adoption of the catalytic active site geometry. However, medoids extracted from clusters 2, 5, and 7 (which contain ∼32% of the total structural ensemble) exhibited a smaller rotamerization of Tyr109 (torsion angle χ 1 of ∼60°). These medoids were assigned lower priority. Medoids extracted from clusters 1, 3, 4, and 6 (which contain the other ∼68% of the total structural ensemble) adopted rotamers corresponding to more buried orientations characteristic of phosphorylated conformations (torsion angle χ 1 of ∼75–80°). In CheY, these differences were subtle, but in other rec domains (Sln1‐rec, Spo0F) the switch residues showed far greater deviations. Candidates that did not exhibit proper switch residue reorientations were eliminated as possible representative structures. A cutoff of 2.9 Å was applied between the Thr/Ser switch residue side‐chain and the phosphoryl group. Aromatic switch residue torsions angles are typically unique and were considered relative only to other medoids of the same protein. Minor or incomplete rotamerizations (partially buried orientations) were assigned lower priority. Each of the five rec domains in the validation set produced at least one candidate structure with switch residue orientations matching the modified crystal structures, except for PhoP‐rec, where Tyr98 remained completely in a solvent‐exposed orientation in 100% of the total ensemble. Further examination of the PhoP‐rec structure reveals that the rec domain crystallized as a homodimer, with Tyr98 located in the center of the dimerization interface.30 Previous studies have implicated the aromatic switch residue in certain dimeric RR interactions.34, 35 Comparison of the modified crystal structure and the predicted model reveals similar environments around the Tyr98 side‐chain, with adequate volume to allow rotamerization in both. In E. coli PhoP, the pocket created by the shift in the β4α4 loop is less hydrophobic in nature than the other rec domains in the validation set, likely making rotamerization less favorable. We speculate that by using only monomeric PhoP‐rec during the simulations, the rotamerization was not inducible. Also of significant note is the aromatic switch residue His101 in Spo0F. In the crystal structures of Spo0F, the side‐chain of His101 adopts multiple conformations, but remains solvent‐exposed regardless of phosphorylation state.32 This residue and region have no known role in dimerization or partner binding for Spo0F, which may be why it fails to rotamerize.15, 32, 114, 115 Most of the extracted Spo0F candidate structures exhibited various degrees of rotamerization at His101. The majority adopted a partially buried orientation (medoids extracted from clusters 1, 3–12, which contain ∼91% of the total structural ensemble), likely resembling an intermediate conformation. A single medoid (extracted from cluster 2, which contains ∼9% of the total structural ensemble) was identified that remained fully solvent‐exposed. No candidates were observed to adopt a completely buried orientation. This supports the existence of an intermediate state(s) in solution, although medoids with partially exposed rotamers were eliminated as possible representative structures. More importantly, it indicates that this computational approach can capture unique allosteric changes associated with phosphorylation that may differ between rec domain proteins. Figures of switch residues for the remaining rec domains can be found in the Supporting Information along with tabulated results (Tables S1–S5; Figs. S5–S8).
Figure 6.
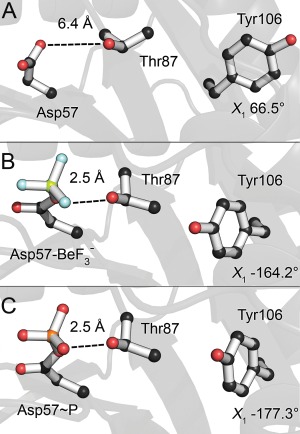
Characteristic switch residues shift upon phosphorylation in CheY. A: Apo orientation (PDB 3CHY). B: bound orientation (PDB 1FQW). Thr87 has shifted to form a hydrogen bond with Asp57. Tyr106 has rotamerized inwards into the hydrophobic pocket. C: A sample candidate structure of phosphorylated CheY demonstrates near‐identical orientations to the bound crystal.
Quartet residue positions were also analyzed within the medoid structures [Fig. 4(C)]. These residues have been hypothesized to form an allosteric network, able to switch between apo and phosphorylated‐like conformational states.16 Comparisons rely on the relative changes in interatomic distances between residues upon transitioning from apo to phosphorylated conformation. While most quartet residues share moderate to low sequence conservation, we found that their relationships are relatively maintained in the rec domains used in the study and likely in most rec domains. In the CheY candidates, we examined the distance between Trp58 (D + 1 position) and the switch Thr87, which are consistently found in closer proximity in modified crystal structures. All medoids showed a decrease in the atomic distance between these side chains, although only the most dramatic shifts were considered for potential representative structures. We also examined the distance between Trp58 and Met85, which is believed to increase upon phosphorylation. Again, all medoids showed an increase in relative distance upon phosphorylation, but higher priority was placed on the medoids that exhibited the largest shifts. Interaction between Trp58 and Glu89 is also thought to increase as a result of phosphorylation. Each medoid showed various interatomic distances, but only two exhibited strong interaction between these residues (extracted from clusters 4 and 6, which contain ∼29% of the total structural ensemble). Also analyzed were the rotamerization of Trp58, related to the interaction with Glu89, and the rotamerization of Met85. Only two medoids (extracted from clusters 4 and 5, which contain ∼40% of the total structural ensemble) showed sufficiently large rotamerization at Trp58, going from 75° to ∼110° upon phosphorylation. The χ 1 torsion angle of Met85 shifts from 70° (apo crystal) to −178° ( crystal). A similar rotamerization is observed in four different CheY medoids (extracted from clusters 2 and 4–6, which contain ∼61% of the total structural ensemble). Interestingly, the near‐complete “phosphorylated” organization of quartet residues is observed in only two candidates (medoids extracted from clusters 4 and 6, which contain ∼31% of the total structural ensemble). Analysis of the final quartet residue, Tyr109, can be found in the previous section. By ranking the changes in interatomic distance between the quartet residues in relation to the apo state, we were able to narrow down which medoids were more likely to resemble the phosphorylated crystal structures for each rec domain. Tabulated results for the other systems (M55, I80, E89 in Spo0F; V1145, A1171, F1175 in Sln1‐rec; L55, V80, H84 in FixJ‐rec; L52, V77, R81 in PhoP‐rec) can be found in the Supporting Information (Tables S1–S5).
We next examined the phosphorylated active sites of each candidate structure [Fig. 4(D)]. Proper active site geometry is essential for the phosphotransfer reaction to occur in all rec domains, and a divalent metal cation is required for catalysis. Figure 7 shows this geometry for CheY. In Figure 7(A), the active site lacks the metal cation, leading to a disordered side‐chain arrangement. With the addition of a Mg2+ cation and the phosphoryl analog , the active site side chains arrange in an octahedral configuration centered around the metal ion [Fig. 7(B)]. This is also demonstrated in a sample medoid for CheY [Fig. 7(C)]. Despite the restraints, we frequently observed incomplete or incorrect active sites in the extracted medoids. If a candidate structure failed to fully adopt the conserved active site arrangement, we eliminated it as a possible representative structure. One of the most frequently seen deviations was a failure to recruit the two water molecules needed to complete the octahedral coordination around the cation. This was usually due to one of the conserved acidic residues forming multiple points of coordination between their side chains and the metal, rather than a single point of coordination from each. Those candidate structures with a warped phosphoryl group geometry were placed assigned lower priority as well. Among the CheY candidates, a single medoid structure adopted the ideal catalytic active site, extracted from cluster 4 (which contains ∼14% of the total ensemble). This medoid also had the closest to ideal linear phosphoryl geometry as well (measured by dihedral angle of the phosphoaspartate bond). The remaining candidates failed to properly coordinate the metal cation and/or the phosphoryl group. Active site results for the other rec domains used in this study were analyzed and tabulated (see Supporting Information, Tables S1–S5; Figs. S9–S12).
Figure 7.
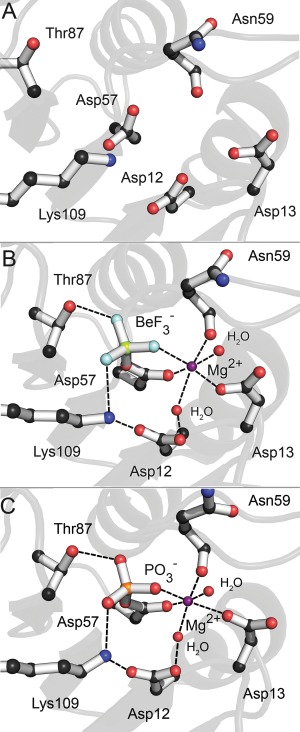
Highly conserved active site geometry of active CheY. A: Disordered active site side chains of apo CheY (PDB 3CHY). B: Active site side chains of CheY bound to (PDB 1FQW). The addition of Mg2+ orders the active site, generating an octahedral coordination geometry with the recruitment of two additional water molecules. Switch residue Thr87 has shifted to hydrogen bond with Asp57. C: Active site side chains in a sample candidate structure of phosphorylated CheY demonstrates near‐identical geometry to the bound crystal.
Finally, the population sizes of the clusters were calculated [Fig. 4(E)]. When performing geometrical clustering on MD simulations, we assume that conformations grouped into the same cluster are structurally similar, and therefore lie within the same basin on the free‐energy landscape of the protein.116 Clusters are typically different sizes, due to conformational sampling progressing according to a Boltzmann distribution during simulation. Sparsely populated clusters are more likely to represent transient and/or higher energy states. When trying to identify which conformations will best resemble a crystal structure, which we normally assume is at an energetic minimum, neither of these effects is desirable. For this reason, medoids extracted from minor clusters (<1500 conformers) were typically eliminated, unless otherwise described. Additionally, if multiple medoids were identified as likely representative structures, the candidate from the larger cluster was retained.
Taking all of these criteria into consideration, a process of elimination allowed for the convenient identification of the top representative structure most likely to resemble a phosphorylated structure for each rec domain. This is apparent in the tabulated results seen in the Supporting Information (Tables S1–S5). Table cells retained for analysis are shown in grey. Medoids with solid grey in every column were found to be most representative of phosphorylated conformation. Each of these representative structures was then compared to their modified crystal structure counterpart to validate the predictions. We first performed overall structural alignments to compare global protein structure. Figure 8(A) shows the final CheY representative model (the medoid extracted from cluster 4, which contains ∼14% of the total structural ensemble) superimposed to the bound crystal structure (PDB 1FQW, dark blue ribbon). Structural alignments were also performed on each of the other rec domains (see Supporting Information, Figs. S13–S16). Overall average RMSDs for the predicted models ranged from 0.5 to 1.0 Å when compared to the existing modified crystal structures obtained from the PDB. While these measurements suggest that the models are nearly identical to their experimental counterparts, RMSD averages are often misleading, especially considering that the majority of the important conformational changes occur in flexible loop areas. Global RMSDs comparing apo and modified crystal structures are typically higher, but only moderately so. In addition, both our simulations and structures of identical proteins obtained from different experimental sources (crystallography, NMR) suggest that the same rec domain in a common phosphorylated state may deviate by up to 2 Å, presumably through the protein's inherent flexibility. This likely generates considerable “noise,” making it challenging to identify significant conformational differences. A more prudent analysis should focus on those areas known to change significantly and directly in response to phosphorylation. To this end, we generated per residue Cα RMSD traces for the β4‐α4‐β5 region, to compare the deviations between both the apo versus bound crystal structures and the predicted models vs. bound crystal structures. Structures were aligned by the invariant β‐sheet core, as this was found to exhibit the least amount of deviation upon phosphorylation. Figure 8(B) contains two RMSD traces for CheY. The black trace represents the deviation between the apo crystal (PDB 3CHY) and bound crystal (PDB 1FQW) structures, showing the degree to which the experimental conformations change. The colored trace represents the deviation between the top predicted model and the bound crystal (PDB 1FQW) structure, showing how closely the prediction matches the experimental conformation. While natural fluctuations were present in most, the top representative models were still consistently and significantly closer to the experimental modified structures in every example, except for PhoP‐rec, which showed only moderate improvement (see Supporting Information, Fig. S13). In experimental structures, PhoP‐rec experiences only minor changes upon phosphorylation, suggesting that visualizing the significant differences using simple RMSD measurements may be ineffective. Additional comparisons for the rest of the proteins are available in the Supporting Information (Figs. S13–S16).
Figure 8.
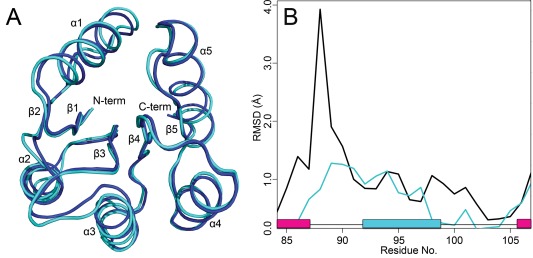
Global structural alignment of the top representative model to the BeF3 − bound crystal structure of CheY and comparisons of the β4‐α4‐β5 region. A: Final predicted model (light cyan). Crystal structure of CheY with bound (PDB 1FQW, dark blue). B: Per residue Cα RMSD for apo vs. crystals (black), and for predicted model versus crystal (cyan). The black trace shows the differences between the phosphorylated and unphosphorylated conformations. The cyan trace shows how closely the prediction matches the true crystal structure. α‐helix (cyan); β‐strand (magenta).
To further analyze the conformational changes in the β4‐α4‐β5 region, we calculated the pseudodihedral angle formed by consecutive Cα atoms in the β4α4 loop for each of the rec domains. This loop is related to the reorientation of the conserved switch Thr/Ser residue and is intimately involved in the transition between apo and phosphorylated conformations. Representing flexible regions in proteins using pseudodihedral angles is an effective way of comparing local conformations without relying on structural alignment methods and RMSD calculations.117 Table 3 contains the angles observed in the modified crystal structures, the top predicted models and the apo crystal structures for each of the proteins within the validation set. All models match their corresponding modified crystal structure closely, except for the seemingly incorrect Spo0F. Futher investigation revealed the existence of multiple conformations for the β4α4 loop in the bound crystal structure of Spo0F (PDB 2FTK), suggesting that the phosphorylated rec domain can adopt at least two separate forms. In addition, this bound Spo0F structure was crystallized bound to its cognate phosphotransferase, Spo0B, which interacts extensively with the β4α4 loop. In the absence of its binding partner, the β4α4 loop likely adopts an altered conformation. Analysis of the NMR structural ensemble of bound Spo0F (PDB 1PUX) shows pseudodihedral angles for the β4α4 loop ranging from 70–95°. These results suggest that our predicted structure likely resembles a phosphorylated crystal structure of monomeric Spo0F.
Table 3.
Pseudodihedral Angles Defined by Consecutive Cα Atoms in the β4α4 Loop
| Protein | Modified crystal structure | Predicted structure | Apo crystal structure | Residues included |
|---|---|---|---|---|
| CheY | 160.36 | 161.12 | −125.10 | Ala88‐Lys91 |
| PhoP‐rec | 172.45 | 167.78 | −168.23 | Ala80‐Ser83 |
| Sln1‐rec | −169.75 | −177.81 | 160.29 | Ala1174‐Asp1177 |
| FixJ‐rec | 132.20 | 127.22 | −7.51 | Gly83‐Asp86 |
| Spo0F | 52.56, 109.61 | 65.09 | 56.46 | Ala83‐Glu86 |
As a final validation step, we compared the physicochemical characteristics of the existing crystal structures with the predicted representative models. Properties such as electrostatic potential, hydrophobicity and geometric surface features provide insight into the biological functions of proteins. By identifying differences between the phosphorylation states of the rec domain crystal structures, and determining whether those differences are also present in the representative models, we can more accurately determine the quality of the models as predictive tools. Any visible changes that occur upon phosphorylation can be used to identify functionally relevant areas and targets for future experimental studies. Figure 9 shows the most significant changes found in the CheY structures. Structures were aligned and then arranged side‐by‐side to compare common regions. We first calculated the electrostatic surface potential for each of the structures (apo crystal, bound crystal, predicted model) using the APBS package.92 Figure 9(A) shows the results for CheY when viewed along the β4‐α4‐β5 surface. Upon phosphorylation, characteristic shifts facilitated by the movement of the β4α4 loop in CheY lead to a 20‐fold increase in binding affinity for its partner, FliM.106 This is accompanied by a change in the electrostatic potential of the α4‐β5‐α5 face, caused by residue shifts near the α4 and α5 helices and the β4α4 loop [Fig. 9(A)]. The structure of CheY bound to FliM (PDB 1F4V) reveals that the areas of altered potential, particularly the β4α4 loop, interact directly with the N‐terminal region of FliM [Fig. 9(A)].36 These changes are nearly identical in the bound crystal structure and the predicted model, but are absent in the apo structure. An implementation of the VisGrid algorithm on the 3D‐SURFER server was then used to characterize the local geometric features of the proteins.93, 95 VisGrid classifies the shape of a molecule's surface based on a visibility criterion, essentially the fraction of open or visible directions from a given target position. Cavities and protrusions are defined as clusters of positions based on the level of their visibility. VisGrid provides a simple and robust method of comparing the relative geometric shapes and regions of potential functional significance for different protein structures. Figure 9(B) shows the first pocket detected in CheY. The bound structure and model both show a cavity near the center of the active site surface, but this cavity is largely absent in the apo structure. Figure 9(C) shows a second pocket found on the β4‐α4‐β5 surface. While the pocket is detected in all three structures, it has a significantly altered shape and location in the apo structure. Figure 9(D) shows a protrusion detected on the active site surface. This protrusion is formed by the loops adjacent to the active site. No large or continuous protrusion was detected at this location in the apo crystal structure. Similar comparisons were performed for each of the other rec domains from this study (data not shown). The predicted models closely matched the modified crystal structures for all members of the validation set. Nearly every unique structural difference identified between the modified and apo crystal structures was accounted for in the models, including regions of known functional significance.
Figure 9.
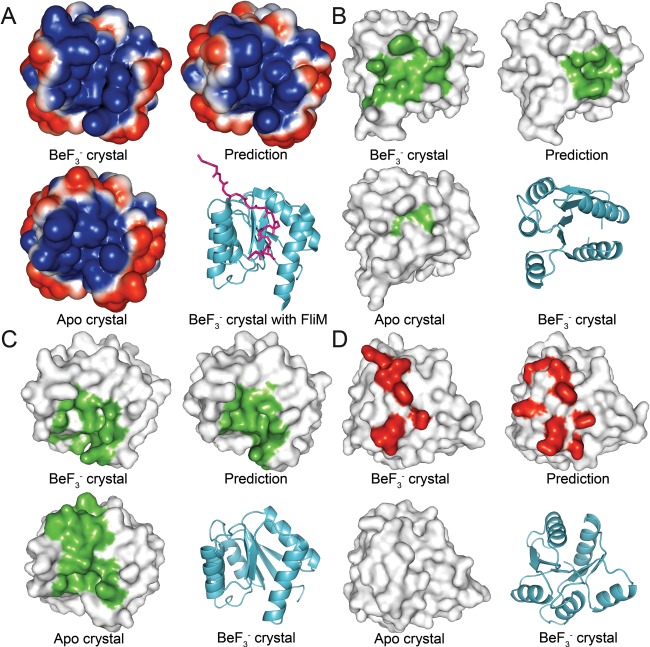
Changes in physicochemical surface properties upon phosphorylation of CheY. Structures were aligned and partitioned into quadrants. View is of the same region on every structure. A: Electrostatic surface potential calculated for the CheY crystal structures and the representative model. Shown is the β4‐α4‐β5 face. A ribbon representation of the structure of CheY bound to FliM (PDB 1F4V) is shown for reference. Negative potential is blue; positive potential is red; FliM is magenta. B: Detected molecular cavity located near the active site of CheY. C: Detected molecular cavity located on the β4‐α4‐β5 surface. D: Detected molecular protrusion located near the active site of CheY.
By combining comparisons of global structure, functionally relevant areas, physicochemical characteristics and local geometric features, we have demonstrated that the models predicted using the previously described approach provide accurate representations of phosphorylated rec domain structures. Although changes are challenging to detect, the predictions are able to capture the key structural features associated with the phosphorylation of rec domains. The fact that these features are not detectable in the apo crystal structures shows the value that such models hold for guiding experimental studies.
Phosphorylation of Ssk1‐rec and comparison with the upstream Sln1‐rec
Having validated the new methodology, we then compared the rec domains of the upstream hybrid HK, Sln1, with the downstream RR, Ssk1, from the S. cerevisiae osmotic stress response pathway. Ssk1‐rec is slightly unusual in that the phosphorylated conformation is the resting state, and activation occurs only upon dephosphorylation. While crystal structures of apo and bound Sln1‐rec exist,4, 33 no structure is currently available for the Ssk1‐rec domain. A homology model for apo Ssk1‐rec was generated using the Phyre2 server as an alternative.70 Though they share little overall sequence similarity, Ssk1‐rec possesses a canonical rec domain at residues 506–651 that aligns well with Sln1‐rec, except for a short, 22‐residue loop (residues 580–603) between α3 and β4 on the surface distal to the active site. This loop consistently modeled poorly and was excluded from the final structure prediction.
We subjected the apo Ssk1‐rec model to energy minimization and equilibration to alleviate any non‐physiological structural features or model bias produced by homology modeling. The resulting structure was validated with the Verify3D server.71 Ramachandran plots generated with RAMPAGE indicated that ∼99% of residues fell within favored or allowed regions.72 This “relaxed” model was phosphorylated and used for restrained MD. Trajectories were analyzed and clustered to extract candidate structures for phosphorylated Ssk1‐rec. A final representative medoid was chosen based on the previously described criteria. We then compared this model and the dynamic information obtained from the simulations with the Sln1‐rec data, to provide insight into the components of the fungal signaling pathway.
The characteristic pattern of positively correlated motion found in all rec domains can be seen in the DCCM for Ssk1‐rec (Supporting Information Fig. S17). Additional patches were also observed between the β1 strand with the α2 helix, and the β3 strand with the α4β5 region. None of the other rec domains in this study displayed positively correlated motion in these areas. Ssk1‐rec also demonstrated a large network of negatively correlated movements, far more extensive than in Sln1‐rec. These areas of negative correlation correspond to the β1 strand with the α5 helix, the β1 strand with the α3 helix/loop, the α1 helix/loop with the β3 strand, the α1 helix/loop with the β4 loop/strand, the α1 helix/loop with the β5 strand, the entire β2‐α2‐β3 region with the α3β4 region, the entire β4‐α4‐β5 region with the α5 helix, and the α5 helix with the β1, β2 and β3 strands. The results suggest that Ssk1‐rec undergoes a multitude of significant collective movements in its phosphorylated state, in addition to the characteristic perturbations at the β4‐α4‐β5 surface. Ssk1‐rec and Sln1‐rec appear to share negatively correlated motions between their α2 and α3 regions and their α4 and α5 regions, though in both cases the affected areas are larger in Ssk1‐rec.
The differences seen in the collective motions of Ssk1‐rec and Sln1‐rec are likely attributable to the distinct functional roles of each protein. Sln1‐rec, as part of a hybrid HK, interacts with its upstream donor (its own catalytic kinase domain) and downstream acceptor (the HPt protein, Ypd1) through a common binding surface formed by the loops adjacent to the active site and portions of the α1 and α5 helices. The interface between the rec and HK domains of Sln1 is likely similar, though no structure currently exists for the complex. The concentrated regions of negatively correlated motion seen in Sln1‐rec are localized to these relatively small areas. We speculate that this is due to the presence of only the single binding surface. In contrast, Ssk1‐rec must bind the upstream Ypd1, the downstream Ssk2 and possibly its own extensive N‐terminal domain. The Ypd1 binding interface is known to be homologous to Sln1‐rec, formed at the active site surface by adjacent loop regions.4 However, mutational studies by Horie et al. (2008) suggest that the Ssk2 binding site is located elsewhere on the rec domain.118 The same researchers also determined that Ssk1 forms a homodimer in solution, though whether the dimerization interface is a part of the rec domain or the N‐terminal region is unknown. These multiple regions of functional significance likely correspond to the far more complex network of collective motions detected in phosphorylated Ssk1‐rec.
Most significant structural changes induced by phosphorylation in Ssk1‐rec were centered around the β4‐α4‐β5 region, with the β4α4 loop and N‐terminal portion of the α4 helix undergoing a dramatic shift away from the α4‐β5‐α5 face. Large changes also occurred in the α1, α2 and C‐terminal half of α5 helices. Smaller shifts were observed in the N‐terminal portion of the α3 helix and the β1α1, β3α3, and β5α5 loops. Supporting Information Fig. S18 shows a structural alignment of the top representative structure and the apo Ssk1‐rec model. Visually, the shift in the α4 helix and loop is far more dramatic than in Sln1‐rec. The α1, α2, and α5 helices of Ssk1‐rec may also be functionally important, as their shifts correspond to strong, broad collective motions not seen in Sln1‐rec. Surface analysis shows that phosphorylated Ssk1‐rec forms additional protrusions on the β4α4 loop and helix, the β5α5 loop and down the α5 helix (data not shown). Additionally, the α4‐β5‐α5 surface cavity undergoes significant shape changes (data not shown).
Horie et al. (2008) discovered a mutation (D628G) on Ssk1‐rec that abolished binding with Ssk2 and inhibited the activation of Ssk2 by wild‐type Ssk1‐rec through dimerization.118 This suggests that the likely binding surface for Ssk2 is located on the α4‐β5‐α5 surface of Ssk1‐rec. The D628G mutation occurs next to the aromatic switch residue, Tyr629. The authors hypothesized that replacing this aspartate with a glycine locks the protein into a conformation that is unable to bind Ssk2. They also speculated that phosphorylation induces a similar shift, possibly through the characteristic rotamerization of the aromatic switch residue. Our results may shed further light on this phenomenon.
Surprisingly, Tyr629 did not rotamerize in any of the phosphorylated simulations, remaining completely solvent‐exposed in both phosphorylated and apo conformations. We speculate that this is caused by the severe shift of the β4α4 loop and α4 helix upon phosphorylation. Unlike most of the other rec domains, the pocket created by the shift of the Thr/Ser switch residue in Ssk1‐rec is significantly less hydrophobic due to Lys618. In the apo Ssk1‐rec structure, the side chains Lys618 and Tyr629 are hydrogen‐bonded and solvent‐exposed, pointing outwards. Upon phosphorylation, the conformational change causes Lys618 to shift up and in, partially occupying the pocket that would normally be filled by the rotamerized Tyr629 and forming hydrogen bonds with residues on the α4 helixand β4α4 loop. PhoP‐rec contains a similar organization with residues Lys87 and Tyr98, although it lacks the dramatic shift in the α4 helix upon phosphorylation. Because of this, Lys87 never enters the pocket, and Tyr98 is able to rotamerize and satisfy an additional hydrogen bond with Arg81. These results suggest that unlike the majority of rec domains, Tyr629 is incapable of rotamerizing while Ssk1‐rec is phosphorylated. This may be related to Ssk1‐rec requiring dephosphorylation to activate further downstream binding.
Although Asp628 does not appear to affect the rotamerization of Tyr629, it may play a role in the overall conformational change that occurs in the α4‐β5‐α5 surface. Comparison of the Ssk1‐rec models reveals that phosphorylation causes the α4 and α5 helices to shift away from the β5 strand in an opening motion. The DCCM also shows negatively correlated motion between several areas on the α5 helix and the β5 strand (Supporting Information Fig. S17), including Asp628. In the representative phosphorylated model, the side‐chain of Asp628 forms an hydrogen bond with Trp646 on the α5 helix. Lys642 is also near, potentially creating an additional salt bridge. We compared brief trajectories of phosphorylated and unphosphorylated Ssk1‐rec to investigate the role of Asp628 (data not shown) . When phosphorylated, Asp628 forms a salt bridge with Lys642 in ∼67% of frames and a hydrogen bond with Trp646 in ∼32% of frames. When unphosphorylated, Asp628 forms a salt bridge with Lys642 in ∼85% of frames, and a hydrogen bond with Trp646 in ∼84% of frames. These preliminary numbers suggest a significant change in interaction stability corresponding to the α5 helix shifting closer to the β5 strand upon dephosphorylation. We speculate that mutating Asp628 to glycine abolishes this effect, preventing binding to Ssk2 through the α4‐β4‐α5 surface.
CONCLUSIONS
We have presented a computational methodology that utilizes biased MD to predict the phosphorylated conformations of RR rec domains. This approach uses multiple half‐harmonic restraints to drive formation of the conserved rec domain active site. This rearrangement pulls the rest of the protein into its phosphorylated state, allowing sampling of modified conformations on a nanosecond timescale. The biasing terms make these studies accessible to research laboratories that may lack the specialized equipment and experience required to simulate this process on the millisecond timescale on which it normally occurs. We validated this technique on five diverse, well‐characterized rec domains: CheY, PhoP‐rec, FixJ‐rec, Sln1‐rec, and Spo0F. Cross‐correlation analysis revealed areas of correlated motion that have been implicated in phosphorylation‐mediated behavior for the five rec domains. This information can be used to guide further experimental studies toward areas of functional interest. We extracted representative models for each rec domain and compared them to the existing crystal structures using global structure, switch residue and quartet residue orientations, active site geometry, loop conformations, and physicochemical surface properties to demonstrate the accuracy of the predictions. The models closely matched the experimentally determined structures of the conformationally modified rec domains in all aspects, validating the approach and suggesting that an induced fit model of conformational switching is sufficient to sample these transient phosphorylated conformations. Researchers studying TCS pathways can use this approach to gain structural insight into specific rec domain characteristics, such as changes in surface properties and binding surface accessibility. We demonstrated this by comparing the known and predicted structures of the S. cerevisiae signaling proteins, Sln1‐rec and Ssk1‐rec, respectively. This information allows for more complete analysis of the functional differences in these two rec domains in the absence of experimental structural data. We predict that this approach can also be applied to signaling complexes, to study changes in protein‐protein interactions within TCS systems as rec domains transition from apo to phosphorylated conformations.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the OU Supercomputing Center for Education and Research (OSCER) at the University of Oklahoma for providing the computer resources for this study. They also thank Katie Branscum, Emily Kennedy, Skyler Hebdon, Jamie Sykes, Smita Menon, Krutik Soni, and Paul Sims for intellectual input. Finally, they thank the anonymous reviewers for their helpful comments.
All work in this study was performed at the University of Oklahoma.
REFERENCES
- 1. Stock AM, Robinson VL, Goudreau PN. Two‐component signal transduction. Annu Rev Biochem 2000;69:183–215. [DOI] [PubMed] [Google Scholar]
- 2. West AH, Stock AM. Histidine kinases and response regulator proteins in two‐component signaling systems. Trends Biochem Sci 2001;26:369–376. [DOI] [PubMed] [Google Scholar]
- 3. Posas F, Wurgler‐Murphy SM, Maeda T, Witten EA, Thai TC, Saito H. Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1‐YPD1‐SSK1 “two‐component” osmosensor. Cell 1996;86:865–875. [DOI] [PubMed] [Google Scholar]
- 4. Xu Q, Porter SW, West AH. The yeast YPD1/SLN1 complex: insights into molecular recognition in two‐component signaling systems. Structure 2003;11:1569–1581. [DOI] [PubMed] [Google Scholar]
- 5. Fisher SL, Jiang W, Wanner BL, Walsh CT. Cross‐talk between the histidine protein kinase VanS and the response regulator PhoB. Characterization and identification of a VanS domain that inhibits activation of PhoB. J Biol Chem 1995;270:23143–23149. [DOI] [PubMed] [Google Scholar]
- 6. Laub MT, Goulian M. Specificity in two‐component signal transduction pathways. Annu Rev Genet 2007;41 :121–145. [DOI] [PubMed] [Google Scholar]
- 7. Rowland MA, Deeds EJ. Crosstalk and the evolution of specificity in two‐component signaling. Proc Natl Acad Sci USA 2014;111:5550–5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silva JC, Haldimann A, Prahalad MK, Walsh CT, Wanner BL. In vivo characterization of the type A and B vancomycin‐resistant enterococci (VRE) VanRS two‐component systems in Escherichia coli: a nonpathogenic model for studying the VRE signal transduction pathways. Proc Natl Acad Sci USA 1998;95:11951–11956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willett JW, Tiwari N, Müller S, Hummels KR, Houtman JCD, Fuentes EJ, Kirby JR. Specificity residues determine binding affinity for two‐component signal transduction systems. MBio 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7‐A resolution. J Biol Chem 1991;266:15511–15519. [DOI] [PubMed] [Google Scholar]
- 11. Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem 2001;276:16425–16431. [DOI] [PubMed] [Google Scholar]
- 12. Thomas SA, Brewster JA, Bourret RB. Two variable active site residues modulate response regulator phosphoryl group stability. Mol Microbiol 2008;69:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dyer CM, Dahlquist FW. Switched or not: the structure of unphosphorylated CheY bound to the N terminus of FliM. J Bacteriol 2006;188:7354–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao R, Mukhopadhyay A, Fang F, Lynn DG. Constitutive activation of two‐component response regulators: characterization of VirG activation in Agrobacterium tumefaciens . J Bacteriol 2006;188:5204–5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gardino AK, Volkman BF, Cho HS, Lee SY, Wemmer DE, Kern D. The NMR solution structure of ‐activated Spo0F reveals the conformational switch in a phosphorelay system. J Mol Biol 2003;331:245–254. [DOI] [PubMed] [Google Scholar]
- 16. McDonald LR, Boyer JA, Lee AL. Segmental notions, not a two‐state concerted switch, underlie allostery in CheY. Structure 2012;20:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Volkman BF, Lipson D, Wemmer DE, Kern D. Two‐state allosteric behavior in a single‐domain signaling protein. Science 2001;291:2429–2433. [DOI] [PubMed] [Google Scholar]
- 18. Gardino AK, Villali J, Kivenson A, Lei M, Liu CF, Steindel P, Eisenmesser EZ, Labeikovsky W, Wolf‐Watz M, Clarkson MW, Kern D. Transient non‐native hydrogen bonds promote activation of a signaling protein. Cell 2009;139:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao R, Mack TR, Stock AM. Bacterial response regulators: Versatile regulatory strategies from common domains. Trends Biochem Sci 2007;32:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bourret RB, Thomas SA, Page SC, Creager‐Allen RL, Moore AM, Silversmith RE. Measurement of response regulator autodephosphorylation rates spanning six orders of magnitude. Methods Enzymol 2010;471:89–114. [DOI] [PubMed] [Google Scholar]
- 21. Bobay BG, Hoch JA, Cavanagh J. Dynamics and activation in response regulators: The β4‐α4 loop. Biomol Concepts 2012;3:175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Groban ES, Narayanan A, Jacobson MP. Conformational changes in protein loops and helices induced by post‐translational phosphorylation. PLoS Comput Biol 2006;2:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama J‐P. Conformational changes induced by phosphorylation of the FixJ receiver domain. Structure 1999;7:1505–1515. [DOI] [PubMed] [Google Scholar]
- 24. Kaserer AO, Andi B, Cook PF, West AH. Effects of osmolytes on the SLN1‐YPD1‐SSK1 phosphorelay system from Saccharomyces cerevisiae . Biochemistry 2009;48:8044–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Re SD, Tolstykh T, Wolanin PM, Stock JB. Genetic analysis of response regulator activation in bacterial chemotaxis suggests an intermolecular mechanism. Protein Sci 2002;11:2644–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guhaniyogi J, Robinson VL, Stock AM. Crystal structures of beryllium fluoride‐free and beryllium fluoride‐bound CheY in complex with the conserved C‐terminal peptide of CheZ reveal dual binding modes specific to CheY conformation. J Mol Biol 2006;359:624–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simonovic M, Volz K. A distinct meta‐active conformation in the 1.1‐A resolution structure of wild‐type apo CheY. J Biol Chem 2001;276:28637–28640. [DOI] [PubMed] [Google Scholar]
- 28. Stock AM, Guhaniyogi J. A new perspective on response regulator activation. J Bacteriol 2006;188:7328–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci 2010;35:539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bachhawat P, Stock AM. Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J Bacteriol 2007;189:5987–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cho H, Wang W, Kim R, Yokota H, Damo S, Kim SH, Wemmer D, Kustu S, Yan D. acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a complex with phosphoserine phosphatase. Proc Natl Acad Sci USA 2001;98:8525–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Varughese KI, Tsigelny I, Zhao H. The crystal structure of beryllofluoride Spo0F in complex with the phosphotransferase Spo0B represents a phosphotransfer pretransition state. J Bacteriol 2006;188:4970–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao X, Copeland DM, Soares AS, West AH. Crystal structure of a complex between the phosphorelay protein YPD1 and the response regulator domain of SLN1 bound to a phosphoryl analog. J Mol Biol 2008;375:1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Toro‐Roman A, Wu T, Stock AM. A common dimerization interface in bacterial response regulators KdpE and TorR. Protein Sci 2005;14:3077–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hickey JM, Lovell S, Battaile KP, Hu L, Middaugh CR, Hefty PS. The atypical response regulator protein ChxR has structural characteristics and dimer interface interactions that are unique within the OmpR/PhoB subfamily. J Biol Chem 2011;286:32606–32616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SY, Cho HS, Pelton JG, Yan D, Henderson RK, King DS, Huang L, Kustu S, Berry EA, Wemmer DE. Crystal structure of an activated response regulator bound to its target. Nat Struct Biol 2001;8:52–56. [DOI] [PubMed] [Google Scholar]
- 37. Gouet P, Fabry B, Guillet V, Birck C, Mourey L, Kahn D, Samama JP. Structural transitions in the FixJ receiver domain. Structure 1999;7:1517–1526. [DOI] [PubMed] [Google Scholar]
- 38. Bourret RB. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol 2010;13:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hess JF, Oosawa K, Kaplan N, Simon MI. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell 1988;53:79–87. [DOI] [PubMed] [Google Scholar]
- 40. Janiak‐Spens F, Sparling JM, Gurfinkel M, West AH. Differential stabilities of phosphorylated response regulator domains reflect functional roles of the yeast osmoregulatory SLN1 and SSK1 proteins. J Bacteriol 1999;181:411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weiss V, Magasanik B. Phosphorylation of nitrogen regulator I (NRI) of Escherichia coli. Proc Natl Acad Sci USA 1988;85:8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pazy Y, Wollish AC, Thomas SA, Miller PJ, Collins EJ, Bourret RB, Silversmith RE. Matching biochemical reaction kinetics to the timescales of life: structural determinants that influence the autodephosphorylation rate of response regulator proteins. J Mol Biol 2009;392:1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goudreau PN, Lee PJ, Stock AM. Stabilization of the phospho‐aspartyl residue in a two‐component signal transduction system in Thermotoga maritima . Biochemistry 1998;37:14575–14584. [DOI] [PubMed] [Google Scholar]
- 44. Jagadeesan S, Mann P, Schink CW, Higgs PI. A novel “four‐component” two‐component signal transduction mechanism regulates developmental progression in Myxococcus xanthus . J Biol Chem 2009;284:21435–21445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Menon S, Wang S. Structure of the response regulator PhoP from Mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry 2011;50:5948–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bachhawat P, Swapna GV, Montelione GT, Stock AM. Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure 2005;13:1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fiedler U, Weiss V. A common switch in activation of the response regulators NtrC and PhoB: phosphorylation induces dimerization of the receiver modules. embo J 1995;14:3696–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sola M, Gomis‐Ruth FX, Serrano L, Gonzalez A, Coll M. Three‐dimensional crystal structure of the transcription factor PhoB receiver domain. J Mol Biol 1999;285:675–687. [DOI] [PubMed] [Google Scholar]
- 49. Trajtenberg F, Albanesi D, Ruétalo N, Botti H, Mechaly AE, Nieves M, Aguilar PS, Cybulski L, Larrieux N, de Mendoza D, Buschiazzo A. Allosteric activation of bacterial response regulators: The role of the cognate histidine kinase beyond phosphorylation. MBio 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McCleary WR. The activation of PhoB by acetylphosphate. Mol Microbiol 1996;20:1155–1163. [DOI] [PubMed] [Google Scholar]
- 51. Lee SY, De La Torre A, Yan D, Kustu S, Nixon BT, Wemmer DE. Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev 2003;17:2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wassmann P, Chan C, Paul R, Beck A, Heerklotz H, Jenal U, Schirmer T. Structure of ‐modified response regulator PleD: implications for diguanylate cyclase activation, catalysis, and feedback inhibition. Structure 2007;15:915–927. [DOI] [PubMed] [Google Scholar]
- 53. Da Re S, Schumacher J, Rousseau P, Fourment J, Ebel C, Kahn D. Phosphorylation‐induced dimerization of the FixJ receiver domain. Mol Microbiol 1999;34:504–511. [DOI] [PubMed] [Google Scholar]
- 54. Park S, Meyer M, Jones AD, Yennawar HP, Yennawar NH, Nixon BT. Two‐component signaling in the AAA + ATPase DctD: binding Mg2+ and selects between alternate dimeric states of the receiver domain. faseb J 2002;16:1964–1966. [DOI] [PubMed] [Google Scholar]
- 55. Sheftic SR, White E, Gage DJ, Alexandrescu AT. NMR structure of the HWE kinase associated response regulator Sma0114 in its activated state. Biochemistry 2014;53:311–322. [DOI] [PubMed] [Google Scholar]
- 56. Park AK, Moon JH, Lee KS, Chi YM. Crystal structure of receiver domain of putative NarL family response regulator spr1814 from Streptococcus pneumoniae in the absence and presence of the phosphoryl analog beryllofluoride. Biochem Biophys Res Commun 2012;421:403–407. [DOI] [PubMed] [Google Scholar]
- 57. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy Sean R, Griffiths‐Jones S, Howe KL, Marshall M, Sonnhammer ELL. The Pfam Protein Families Database. Nucleic Acids 2002;30:276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tatusova T, Ciufo S, Fedorov B, O'Neill K, Tolstoy I. RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res 2014;42:(Database issue):D553–D559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Attwood PV, Besant PG, Piggott MJ. Focus on phosphoaspartate and phosphoglutamate. Amino Acids 2011;40:1035–1051. [DOI] [PubMed] [Google Scholar]
- 61. Carpenter FH. The free energy change in hydrolytic reactions: the non‐ionized compound convention. J Am Chem Soc 1960;82:1111–1122. [Google Scholar]
- 62. Hayes DM, Kenyon GL, Kollman PA. Theoretical calculations of the hydrolysis energies of some “high‐energy” molecules. A survey of some biologically important hydrolytic reactions. J Am Chem Soc 1978;100:4331–4340. [Google Scholar]
- 63. Koshland DE. Effect of catalysts on the hydrolysis of acetyl phosphate. Nucleophilic displacement mechanisms in enzymatic reactions. J Am Chem Soc 1952;74:2286–2292. [Google Scholar]
- 64. Ruben EA, Plumley JA, Chapman MS, Evanseck JD. Anomeric effect in “high energy” phosphate bonds. Selective destabilization of the scissile bond and modulation of the exothermicity of hydrolysis. J Am Chem Soc 2008;130:3349–3358. [DOI] [PubMed] [Google Scholar]
- 65. Feher VA, Cavanagh J. Millisecond‐timescale motions contribute to the function of the bacterial response regulator protein Spo0F. Nature 1999;400:289–293. [DOI] [PubMed] [Google Scholar]
- 66. Carter EA, Ciccotti G, Hynes JT, Kapral R. Constrained reaction coordinate dynamics for the simulation of rare events. Chem Phys Lett 1989;156:472–477. [Google Scholar]
- 67. Grubmüller H. Predicting slow structural transitions in macromolecular systems: Conformational flooding. Phys Rev E 1995;52:2893–2906. [DOI] [PubMed] [Google Scholar]
- 68. Huber T, Torda A, van Gunsteren W. Local elevation: a method for improving the searching properties of molecular dynamics simulation. J Comput‐Aid Mol Des 1994;8:695–708. [DOI] [PubMed] [Google Scholar]
- 69. Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Mol Phys 2013;111:3345–3362. [Google Scholar]
- 70. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protocols 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three‐dimensional profiles. Methods Enzymol 1997;277:396–404. [DOI] [PubMed] [Google Scholar]
- 72. Lovell SC, Davis IW, Arendall WB, III , de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins 2003;50:437–450. [DOI] [PubMed] [Google Scholar]
- 73. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Cryst D 2010;66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kojetin DJ, Thompson RJ, Benson LM, Naylor S, Waterman J, Davies KG, Opperman CH, Stephenson K, Hoch JA, Cavanagh J. Structural analysis of divalent metals binding to the Bacillus subtilis response regulator Spo0F: the possibility for in vitro metalloregulation in the initiation of sporulation. Biometals 2005;18:449–466. [DOI] [PubMed] [Google Scholar]
- 75. Lukat GS, Stock AM, Stock JB. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry 1990;29:5436–5442. [DOI] [PubMed] [Google Scholar]
- 76. Stock AM, Martinez‐Hackert E, Rasmussen BF, West AH, Stock JB, Ringe D, Petsko GA. Structure of the Mg2+‐bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry 1993;32:13375–13380. [DOI] [PubMed] [Google Scholar]
- 77. Gumbart J, Trabuco LG, Schreiner E, Villa E, Schulten K. Regulation of the protein‐conducting channel by a bound ribosome. Structure 2009;17:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins 1996;24:433–438. [DOI] [PubMed] [Google Scholar]
- 79. Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph 1996;14:33–38. [DOI] [PubMed] [Google Scholar]
- 80. Brooks BR, Brooks CL, III , Mackerell AD, Jr. , Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem 2009;30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph‐McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz‐Kuczera J, Yin D, Karplus M. All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 1998;102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 82. Damjanović A, García‐Moreno E B, Brooks BR. Self‐guided Langevin dynamics study of regulatory interactions in NtrC. Proteins 2009;76:1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006;22:2695–2696. [DOI] [PubMed] [Google Scholar]
- 85. Team RDC . R: A language and environment for statistical computing. R Found 2013. [Google Scholar]
- 86. Skjaerven L, Martinez A, Reuter N. Principal component and normal mode analysis of proteins; a quantitative comparison using the GroEL subunit. Proteins 2011;79:232–243. [DOI] [PubMed] [Google Scholar]
- 87. Kaufman L, Rousseeuw P. Clustering by means of Medoids. Reports of the faculty of Mathematics and Informatics; 1987.
- 88. Maechler MR,P, Struyf A, Hubert M, Hornik K. Cluster analysis basics and extensions. R Package Version 1.14.4; 2013.
- 89. Kaufman L, Rousseeuw P. Clustering Large Applications (Program CLARA). Finding Groups in Data: An Introduction to Cluster Analysis. Hoboken, NJ, USA: Wiley; 2008. p. 126–163.
- 90. Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 1991;11:205–217. [DOI] [PubMed] [Google Scholar]
- 91. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 92. Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 2007;35:(Web Server issue):W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. La D, Esquivel‐Rodriguez J, Venkatraman V, Li B, Sael L, Ueng S, Ahrendt S, Kihara D. 3D‐SURFER: software for high‐throughput protein surface comparison and analysis. Bioinformatics 2009;25:2843–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sael L, Li B, La D, Fang Y, Ramani K, Rustamov R, Kihara D. Fast protein tertiary structure retrieval based on global surface shape similarity. Proteins 2008;72:1259–1273. [DOI] [PubMed] [Google Scholar]
- 95. Li B, Turuvekere S, Agrawal M, La D, Ramani K, Kihara D. Characterization of local geometry of protein surfaces with the visibility criterion. Proteins 2008;71:670–683. [DOI] [PubMed] [Google Scholar]
- 96. Schrodinger L. The PyMOL Molecular Graphics System version 1.8.; 2015.
- 97. Hennig C. Fpc: Flexible Procedures for Clustering. R Package Version 2.1‐10; 2015.
- 98. Swaminathan S, Harte WE, Beveridge DL. Investigation of domain structure in proteins via molecular dynamics simulation: application to HIV‐1 protease dimer. J Am Chem Soc 1991;113:2717–2721. [Google Scholar]
- 99. Pieniazek SN, Hingorani MM, Beveridge DL. Dynamical allosterism in the mechanism of action of DNA mismatch repair protein MutS. Biophys J 2011;101:1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. VanSchouwen B, Akimoto M, Sayadi M, Fogolari F, Melacini G. Role of Dynamics in the Autoinhibition and Activation of the Hyperpolarization‐activated Cyclic Nucleotide‐modulated (HCN) Ion Channels. J Biol Chem 2015;290:17642–17654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kormos BL, Baranger AM, Beveridge DL. A study of collective atomic fluctuations and cooperativity in the U1A‐RNA complex based on molecular dynamics simulations. J Struct Biol 2007;157:500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wan H, Hu JP, Tian XH, Chang S. Molecular dynamics simulations of wild type and mutants of human complement receptor 2 complexed with C3d. Phys Chem Chem Phys 2013;15:1241–1251. [DOI] [PubMed] [Google Scholar]
- 103. Xu L, Kong R, Zhu J, Sun H, Chang S. Unraveling the conformational determinants of LARP7 and 7SK small nuclear RNA by theoretical approaches. Mol Biosyst 2016;12:2613–2621. [DOI] [PubMed] [Google Scholar]
- 104. Bhattacharyya M, Vishveshwara S. Elucidation of the conformational free energy landscape in H.pylori LuxS and its implications to catalysis. BMC Struct Biol 2010;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lukman S, Grant BJ, Gorfe AA, Grant GH, McCammon JA. The Distinct Conformational Dynamics of K‐Ras and H‐Ras A59G. PLoS Comput Biol 2010;6:e1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bren A, Eisenbach M. The N terminus of the flagellar switch protein, FliM, is the binding domain for the chemotactic response regulator, CheY. J Mol Biol 1998;278:507–514. [DOI] [PubMed] [Google Scholar]
- 107. Saito K, Ito E, Hosono K, Nakamura K, Imai K, Iizuka T, Shiro Y, Nakamura H. The uncoupling of oxygen sensing, phosphorylation signalling and transcriptional activation in oxygen sensor FixL and FixJ mutants. Mol Microbiol 2003;48:373–383. [DOI] [PubMed] [Google Scholar]
- 108. Kato A, Tanabe H, Utsumi R. Molecular characterization of the PhoP‐PhoQ two‐component system in Escherichia coli K‐12: identification of extracellular Mg2+‐responsive promoters. J Bacteriol 1999;181:5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tzeng YL, Hoch JA. Molecular recognition in signal transduction: the interaction surfaces of the Spo0F response regulator with its cognate phosphorelay proteins revealed by alanine scanning mutagenesis. J Mol Biol 1997;272:200–212. [DOI] [PubMed] [Google Scholar]
- 110. Maeda T, Wurgler‐Murphy SM, Saito H. A two‐component system that regulates an osmosensing MAP kinase cascade in yeast. Nature 1994;369:242–245. [DOI] [PubMed] [Google Scholar]
- 111. Wolf A, Kirschner KN. Principal component and clustering analysis on molecular dynamics data of the ribosomal L11.23S subdomain. J Mol Model 2013;19:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Duda RO, Hart PE. Pattern classification and scene analysis. New York, NY, USA: Wiley; 1973.
- 113. Caliński T, Harabasz J. A dendrite method for cluster analysis. Commun Stat 1974;3:1–27. [Google Scholar]
- 114. Varughese KI. Conformational Changes of Spo0F along the Phosphotransfer Pathway. J Bacteriol 2005;187:8221–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zapf J, Sen U, Madhusudan Hoch JA, Varughese KI. A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure 2000;8:851–862. [DOI] [PubMed] [Google Scholar]
- 116. Fraccalvieri D, Pandini A, Stella F, Bonati L. Conformational and functional analysis of molecular dynamics trajectories by Self‐Organising Maps. BMC Bioinformatics 2011;12:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. DeWitte RS, Shakhnovich EI. Pseudodihedrals: simplified protein backbone representation with knowledge‐based energy. Protein Sci 1994;3:1570–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Horie T, Tatebayashi K, Yamada R, Saito H. Phosphorylated Ssk1 prevents unphosphorylated Ssk1 from activating the Ssk2 mitogen‐activated protein kinase kinase kinase in the yeast high‐osmolarity glycerol osmoregulatory pathway. Mol Cell Biol 2008;28:5172–5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information