

The mammalian target of rapamycin (mTOR) is a central regulator of cell growth, proliferation and metabolism by forming at least 2 functionally distinct complexes, mTOR complex-1 (mTORC1) and mTOR complex-2 (mTORC2).1 To exert their biological functions, mTORC1 phosphorylates various substrates including S6K, 4E-BP1 and SREBP to promote protein synthesis, while mTORC2 phosphorylates multiple AGC kinases including Akt, SGK and PKC to facilitate cell proliferation and survival.2 mTORC2 can release mTORC1 inhibition in large through Akt-mediated phosphorylation of TSC2 or PRAS40, whereas mTORC1/S6K can negatively regulate the mTORC2/Akt signaling through phosphorylation of IRS-1, GRB10 and Sin1.3 Hyper-activation of the mTOR signaling pathway has been frequently observed in many types of human cancers, thereby attracting intensive research attention as an ideal drug target for cancer therapies. To this end, the anti-tumor effects of rapamycin or its analogs (rapalogs), the most well-studied mTOR inhibitors, have been thoroughly evaluated in clinics. However, rapamycin treatment has displayed poor outcomes for most cancer types and severe side-effects in patients, which may be in part due to the insensitivity of mTORC2 to rapamycin, as well as the reactivation of the PI3K/Akt signaling upon mTORC1 inhibition to unleash the negative feedback loop.4 Notably, rapamycin also leads to a G1-phase cell cycle arrest mediated in part by TGF-β signaling to promote cell survival.5 Therefore, it is a burning question to further improve the anti-cancer efficacy of rapamycin, given that fully understanding molecular details of mTOR signaling circuits might allow us to overcome these defects.
In this issue of Cell Cycle, an elegant study by Saqcena et al. shed new light on resolving these problems.6 Specifically, when MDA-MB-231 breast cancer cells and Calu-1 lung cancer cells are synchronized in S-phase, high doses of rapamycin alone or in combination with the PI3K inhibitor, LY2940002, markedly induced cellular apoptosis (Fig. 1). More importantly, in K-Ras mutant cancer cells such as MDA-MB-231 and Calu-1, but not K-Ras-WT cell lines including MCF7, deprivation of glutamine (Gln) could bypass a Gln-dependent G1 cell cycle checkpoint and subsequently arrested cells in S-phase, instead of G1-phase. As a result, Gln depletion greatly enhanced rapamycin-induced cellular apoptosis in mutant K-Ras-driven cancer cells (Fig. 1).
Figure 1.
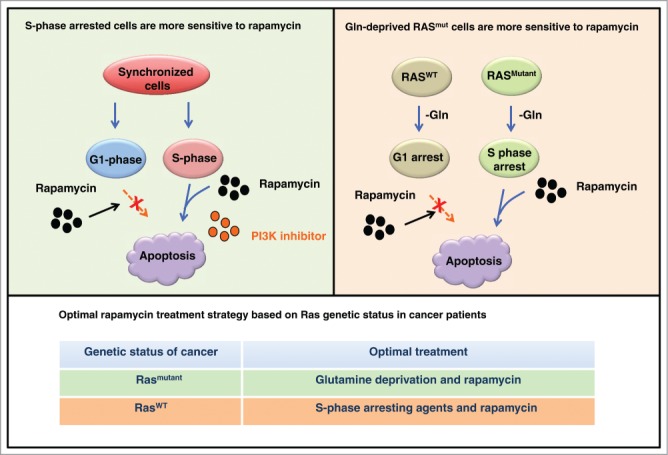
The cell cycle status controls the anti-cancer effects of rapamycin treatments. Rapamycin alone or in combination with the PI3K inhibitors promotes cellular apoptosis in cells synchronized in S-phase, but not in G1-phase of the cell cycle. On the other hand, blockade of glutamine (Gln) utilization leads to a S-phase arrest and induces rapamycin-mediated cellular apoptosis in Ras-mutant cancer cells. These results could provide the rationale to guide the optimization strategy for the clinical usage of rapamycin, based on Ras genetic status in cancer patients.
Thus, Saqcena et al. reveal that rapamycin exerts an augmented capacity to eliminate cancer cells arrested in the S-phase, rather than in the G1 phase, in large through promoting cellular apoptosis. More importantly, this study also suggests Gln deprivation-induced S-phase arrest as a possible option to enhance rapamycin-induced cellular apoptosis, at least in K-Ras mutant driven cancers. This study further indicates that for the remaining 70% of human cancers with wild-type K-Ras, a prior S-phase arrest by agents such as hydroxyurea (HU),7 may also benefit rapamycin treatment via elevating cellular apoptosis (Fig. 1). However, the detailed molecular mechanism(s) underlying the specific role for the pro-apoptotic effects of rapamycin in a cell cycle-dependent manner warrants further investigation. For example, does inhibiting mTOR by rapamycin in S-phase lead to replication stress, which in turn results in apoptosis? If so, better clinical outcomes could be achieved using a combination of rapamycin with DNA replication inhibitors. In addition, it seems that the effects of rapamycin are cell type or tissue context-dependent, as rapamycin treatment exhibits a better clinical efficacy in metastatic renal cell carcinomas than other types of human cancers.4 Therefore, further investigation is required to uncover the molecular mechanisms underlying how rapamycin functions in different cancer types, which will provide the rationale and facilitate the optimization for the clinical application of rapamycin as an anti-cancer drug, alone or in combination with other agents to benefit more cancer patients.
References
- 1.Wullschleger S, et al.. Cell 2006; 124:471–84; PMID:16469695; http://dx.doi.org/ 10.1016/j.cell.2006.01.016 [DOI] [PubMed] [Google Scholar]
- 2.Hung CM, et al.. Cold Spring Harb Perspect Biol 2012; 4(12); pii: a008771; PMID: 23124837; doi: 10.1101/cshperspect.a008771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fruman DA, et al.. Nat Rev Drug Discov 2014; 13:140–56; PMID:24481312; http://dx.doi.org/ 10.1038/nrd4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun SY. Cancer Lett 2013; 340:1–8; PMID:23792225; http://dx.doi.org/ 10.1016/j.canlet.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee A, et al.. Cancer Lett 2015; 360:134–40; PMID:25659819; http://dx.doi.org/ 10.1016/j.canlet.2015.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saqcena M, et al.. Cell Cycle 2015:14(14):2285-92; PMID: 25945415; PMID:25945415; http://dx.doi.org/ 10.1080/15384101.2015.1046653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koc A, et al.. J Biol Chem 2004; 279:223–30; PMID:14573610; http://dx.doi.org/ 10.1074/jbc.M303952200 [DOI] [PubMed] [Google Scholar]