

Abstract
Rupture of endosomes and lysosomes is a major cellular stress condition leading to cell death and degeneration. Here, we identified an essential role for the ubiquitin‐directed AAA‐ATPase, p97, in the clearance of damaged lysosomes by autophagy. Upon damage, p97 translocates to lysosomes and there cooperates with a distinct set of cofactors including UBXD1, PLAA, and the deubiquitinating enzyme YOD1, which we term ELDR components for Endo‐Lysosomal Damage Response. Together, they act downstream of K63‐linked ubiquitination and p62 recruitment, and selectively remove K48‐linked ubiquitin conjugates from a subpopulation of damaged lysosomes to promote autophagosome formation. Lysosomal clearance is also compromised in MEFs harboring a p97 mutation that causes inclusion body myopathy and neurodegeneration, and damaged lysosomes accumulate in affected patient tissue carrying the mutation. Moreover, we show that p97 helps clear late endosomes/lysosomes ruptured by endocytosed tau fibrils. Thus, our data reveal an important mechanism of how p97 maintains lysosomal homeostasis, and implicate the pathway as a modulator of degenerative diseases.
Keywords: AAA+‐type ATPase, autophagy, lysosomal membrane permeabilization, multisystem proteinopathy‐1, ubiquitin
Subject Categories: Autophagy & Cell Death; Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The endolysosomal pathway is particularly vulnerable to membrane rupture and lysosomal membrane permeabilization (LMP). LMP is a detrimental consequence of host cell invasion from the endosome to cytosol by virions and bacteria. Moreover, incorporated chemical compounds and mineral crystals can rupture the organelles causing medical conditions such as kidney damage or alveolar inflammation (Salminen et al, 2012). In addition, endogenous factors, such as reactive oxygen species (ROS) or neurotoxic protein aggregates, can induce lysosomal damage. This imposes a severe stress condition that affects cellular homeostasis, and releases cathepsins that in turn can trigger cell death with relevance for neurodegeneration (Boya & Kroemer, 2008; Nixon, 2013; Repnik et al, 2013).
Damaged lysosomes were recently shown to be cleared by selective macroautophagy (called autophagy from here on), which mediates the engulfment of the injured organelles within isolation membranes and ultimately the fusion of the resulting autophagosomes with intact lysosomes for degradation (Dupont et al, 2009; Hung et al, 2013; Maejima et al, 2013). Autophagy of lysosomes (also called lysophagy) involves the established autophagic enzyme machinery that mediates the conversion of the ubiquitin‐like protein LC3 into its lipidated form to decorate the autophagosomal membrane (Mizushima & Levine, 2010; Suzuki & Ohsumi, 2010; Maejima et al, 2013; Klionsky & Schulman, 2014). The association of autophagosomal membranes with the damaged organelle is thought to be largely mediated by extensive ubiquitination of multiple components of the damaged organelle, which recruits the autophagy machinery and LC3‐adapter proteins such as p62/SQSTM1 (Fujita et al, 2013; Shaid et al, 2013; Rogov et al, 2014). Different types of ubiquitin chains have been detected on damaged lysosomes including those with lysine 63 (K63) and lysine 48 (K48) linkages (Fujita et al, 2013). However, the significance of individual chain types for LC3 recruitment and how they are regulated has remained largely elusive (Fujita et al, 2013; Shaid et al, 2013).
Various reports from different organisms have linked the hexameric, ubiquitin‐directed AAA+‐type ATPase p97 (also called VCP or Cdc48) and individual p97 cofactor proteins to the endolysosomal system or autophagy (Ren et al, 2008; Krick et al, 2010; Ossareh‐Nazari et al, 2010; Tanaka et al, 2010; Ramanathan & Ye, 2011; Ritz et al, 2011; Buchan et al, 2013; Johnson et al, 2015). Of note, defective autophagy and compromised lysosome integrity is a prominent cytopathic feature in tissues affected by a VCP/p97‐associated multisystem proteinopathy (MSP) that includes a myopathy and the neurodegenerative components frontotemporal dementia and amyotrophic lateral sclerosis (ALS) (Ju et al, 2009; Tresse et al, 2010; Meyer & Weihl, 2014; Johnson et al, 2015). Commonly, p97 is considered a protein “segregase” that targets ubiquitinated proteins and often facilitates their degradation in the proteasome. To do so, it cooperates with different sets of ubiquitin adapters and additional cofactors. The best‐studied complex is with the heterodimeric Ufd1‐Npl4 ubiquitin adapter that extracts ubiquitinated proteins from cellular structures in a large variety of processes including ER‐associated degradation (Stolz et al, 2011; Meyer et al, 2012; Franz et al, 2014). However, the apparent role of p97 in autophagy and lysosomal homeostasis, and the identity of the relevant p97‐cofactors that explain its molecular mechanism have remained enigmatic.
In this study, we show that p97 translocates to lysosomes and is essential for cell survival after lysosomal damage. We identified three cofactors including a deubiquitinating enzyme (DUB) that interact and cooperate with p97 on the damaged lysosomes. Intriguingly, p97 and its cofactors act downstream of K63 ubiquitination and p62 recruitment. Rather, they specifically remove K48‐linked ubiquitin conjugates to promote autophagosome formation and efficient clearance of the damaged organelles. We further show that disease‐associated mutation of p97 attenuates lysosomal clearance and that damaged lysosomes accumulate in pathological tissue. Moreover, autophagy and the p97 endolysosomal damage complex respond to late endosomes/lysosomes that are damaged by endocytosed tau fibrils, thus implicating the pathway in neurodegenerative disease.
Results
VCP/p97 is a direct and essential component of the lysosomal damage response
To explore whether p97 is involved in the endolysosomal damage response, we used the lysosomotropic reagent L‐leucyl‐L‐leucine methyl ester (LLOMe) in cultured HeLa cells. As previously reported (Maejima et al, 2013), LLOMe specifically and acutely damaged late endosomal and lysosomal compartments as visualized by the translocation of cytosolic galectin‐3 (Gal3) (Fig 1A). Acute recruitment of p62 and LC3 indicated initiation of autophagy of the damaged lysosomes (Figs 1A and EV1A), as expected (Maejima et al, 2013). The damage also triggered a burst of ubiquitination as detected by the pan‐ubiquitin antibody FK2 on the LAMP1‐positive membranes (Fig 1B). Concomitantly, p97 was recruited to lysosomes in the vast majority of cells. This was true for overexpressed GFP‐p97 (Fig 1B) and confirmed for endogenous p97 (Figs 1C and EV1B).
Figure 1. p97 is a direct and essential component of the lysosomal damage response.
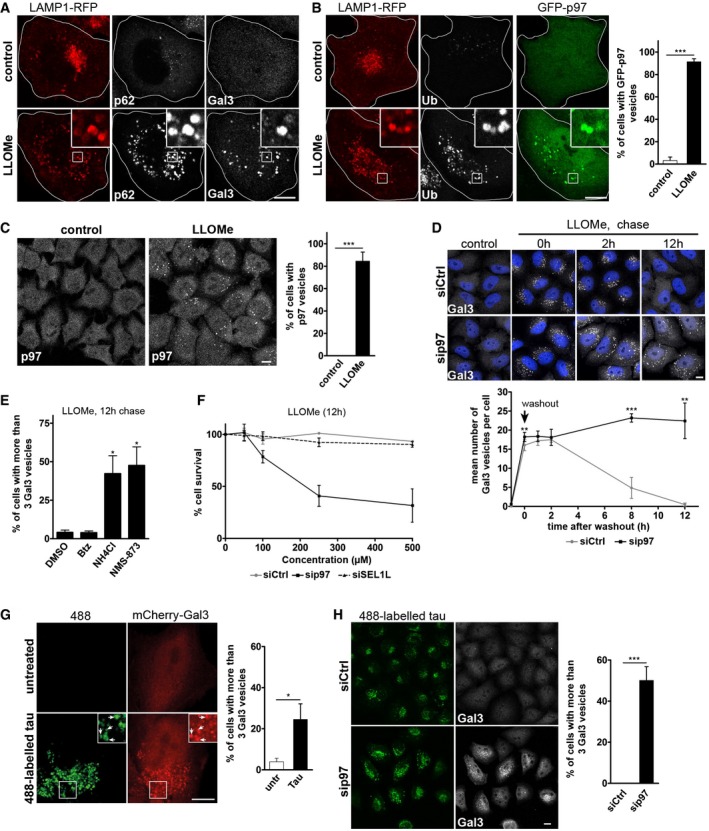
- HeLa cells expressing LAMP1‐RFP were treated with vehicle alone (control) or LLOMe for 1 h to induce lysosome damage. Cells were fixed 2 h after washout, stained with p62 and galectin‐3 (Gal3) antibodies, and analyzed by confocal microscopy.
- p97 translocates to ruptured lysosomes. Cells co‐expressing LAMP1‐RFP and GFP‐p97 were treated as in (A) and stained with the pan‐ubiquitin antibody FK2 (Ub). Percentage of cells showing recruitment of p97 in three independent experiments (mean ± SD). Student's unpaired t‐test.
- Immunodetection of endogenous p97 after LLOMe treatment as in (A). Percentage of cells showing recruitment of p97 in three independent experiments (mean ± SD). Student's unpaired t‐test.
- Clearance of damaged lysosomes depends on p97. Cells were transfected with control (Ctrl) or p97 siRNA oligos, fixed at indicated time points after LLOMe washout, and stained for endogenous Gal3. Quantification of the number of Gal3‐positive vesicles per cell. Data represent mean ± SD of four independent experiments with ≥ 30 cells quantified per condition (one‐sided Welch's unequal variance t‐tests comparing the siCtrl with sip97).
- Cells were chased after LLOMe treatment for 12 h with vehicle alone (DMSO), bortezomib (Btz, 10 nM), NH4Cl (20 mM), or the p97 inhibitor NMS‐873 (5 μM), and Gal3 vesicles quantified. Data represent mean ± SEM of three independent experiments with ≥ 30 cells quantified per condition (one‐sided Welch's t‐tests).
- p97 is essential for cell survival after lysosome rupture. Cells were transfected with control (Ctrl), p97, or SEL1L siRNA oligos and treated with indicated concentrations of LLOMe for 12 h. Cell viability was measured using the MTS assay. Data represent mean ± SD of three independent experiments.
- Tau fibrils are endocytosed and induce lysosomal damage. Tau fibrils were generated in vitro, fluorescently labeled by Dylight 488, and fragmented by sonication (488‐labeled tau). HeLa cells expressing mCherry‐Gal3 were incubated with 300 nM tau fibrils for 24 h. Arrows indicate compartments that contain 488‐labeled tau and are positive for mCherry‐Gal3 indicating lysosome rupture. Percentage of cells with Gal3‐positive vesicles was quantified from three independent experiments (mean ± SD). Student's unpaired t‐test.
- Accumulation of tau‐induced lysosomal damage upon p97 depletion. Cells transfected with control (Ctrl) or p97 siRNA oligos were treated as in (G). Percentage of cells with Gal3‐positive vesicles was quantified from three independent experiments (mean ± SD). Student's unpaired t‐test.
Figure EV1. p97 is a direct component of the lysosome damage response (related to Fig 1).
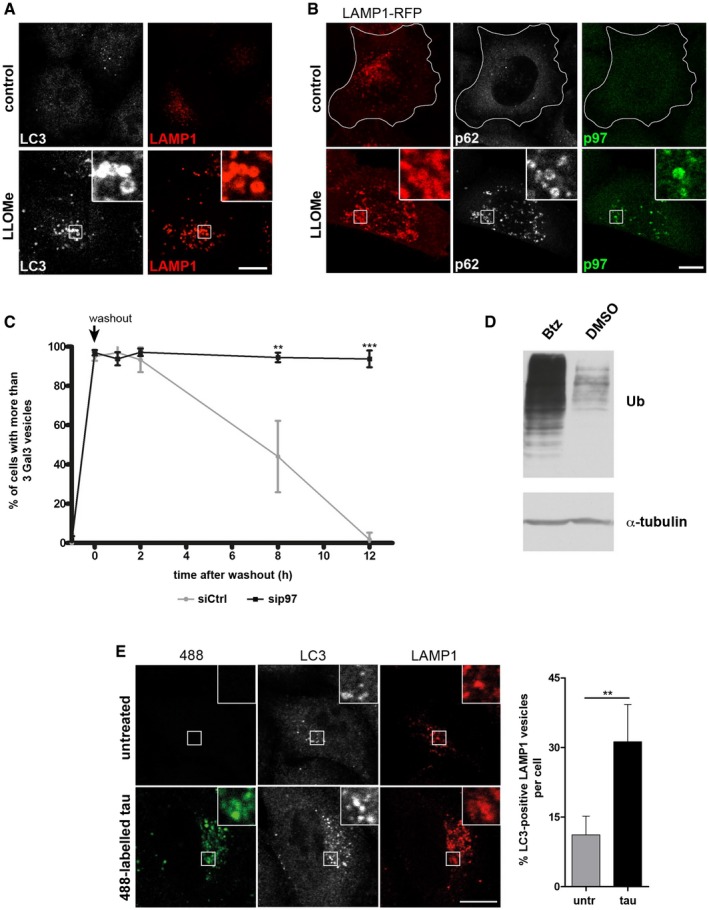
- LLOMe ruptures lysosomes and induces an autophagic response. HeLa cells were treated with vehicle alone (control) or LLOMe for 1 h to induce lysosome damage. Cells were fixed 2 h after washout, stained with LAMP1 and LC3 antibodies, and analyzed by confocal immunofluorescence microscopy.
- Endogenous p97 translocates to ruptured lysosomes. Cells transiently expressing LAMP1‐RFP were treated as in (A) and stained for endogenous p97 and p62.
- Quantification of the percentage of cells with more than three Gal3 vesicles per cell for each condition from Fig 1D.
- Cells were treated with 10 nM bortezomib (Btz) or with vehicle alone (DMSO) for 12 h and ubiquitin conjugates (Ub) in whole‐cell lysates were detected by immunoblotting.
- Tau fibrils are endocytosed and induce autophagy. Tau fibrils were generated in vitro, fluorescently labeled by Dylight 488, and fragmented by sonication (488‐labeled tau). HeLa cells were incubated with 300 nM tau fibrils for 24 h, fixed, and stained with LC3 and LAMP1 antibodies. Untreated cells served as a control. Quantification of the fraction of LC3‐positive LAMP1 vesicles per cell.
To evaluate the functional significance of p97 recruitment, we monitored the autophagic clearance of Gal3‐labeled damaged lysosomes over time after LLOMe washout (Fig 1D). In control cells, the mean number of Gal3‐positive lysosomes per cell reached a maximum during the treatment and began to diminish 2 h after washout until they fully disappeared during the course of 12 h in agreement with previous reports (Maejima et al, 2013). In cells depleted of p97 by siRNA, clearance of Gal3 vesicles was dramatically inhibited and Gal3 vesicles persisted for at least 12 h as shown by the mean number per cell (Fig 1D), and also the percentage of cells with persisting Gal3 (Fig EV1C).
Consistent with a role of p97 and autophagy in lysosomal clearance, treatment with the allosteric p97 ATPase inhibitor NMS‐873 (Magnaghi et al, 2013), or autophagic inhibition with ammonium chloride in the chase media affected Gal3 clearance (Fig 1E). In contrast, proteasome inhibition with bortezomib had no effect on Gal3 clearance (Fig 1E), although accumulation of polyubiquitinated proteins in whole‐cell extracts was detected (Fig EV1D). In control‐depleted cells, LLOMe treatment alone even up to 500 μM had only little effect on cell survival (Fig 1F). However, p97 depletion resulted in quantitative cell death when combined with LLOMe treatment in a dose‐dependent manner (Fig 1F), showing that p97 is essential for restoration of cellular homeostasis after lysosomal damage. In contrast, depletion of SEL1L, which like p97 is involved in the ER stress response, had no effect on cell survival.
To examine whether p97 and autophagy respond also to lysosomal damage caused by agents other than LLOMe, we used neurotoxic aggregates, which can damage lysosomes when endocytosed (Freeman et al, 2013). Tau fibrils are released from cells in a number of neurodegenerative pathologies. We generated fluorescently labeled fragments of tau fibrils in vitro that are capable of inducing aggregation of endogenous tau in cells when added to the culture media (Poepsel et al, 2015). Consistent with a previous report (Wu et al, 2013), tau fibrils were endocytosed and transported to LAMP1‐positive compartments (Fig EV1E). Crucially, the endocytosed tau fibrils induced translocation of Gal3 and colocalized with LC3 indicating lysosome rupture and initiation of autophagy (Figs 1G and EV1E). Consistent with a role for p97 in lysophagy, cellular depletion of p97 led to an accumulation of Gal3 vesicles during the incubation with tau fibrils (Fig 1H).
Disease‐associated mutations of p97 compromise clearance of damaged lysosomes
Mutations in p97 cause a degenerative disorder marked by the accumulation of aberrant autophagosomes and endolysosomes in affected tissues. Consistent with this, mouse embryonic fibroblasts (MEFs) generated from p97R155H/wt knockin mice showed impaired clearance of lysosomes after LLOMe treatment as compared with p97wt/wt control MEFs (Fig 2A). This was also true for other disease‐associated mutations including L198W and A232E when overexpressed in stable inducible cell lines (Figs 2B and EV2A). Western blot analysis of LC3 lipidation in these cells revealed an increase and persistence of LC3‐II after LLOMe washout suggesting impairment in autophagy flux and clearance during lysophagy (Figs 2C and EV2B for quantification). Moreover, the affected skeletal muscle tissue from patients carrying p97 disease mutations accumulated Gal3‐positive vesicles in the sarcoplasm, whereas this was not observed in control muscle (Fig 2D). The Gal3 signal often colocalized with caveolin‐3 (Cav3) (Fig 2D), which we showed earlier accumulates at the limiting membrane of late endosomes and lysosomes as a consequence of the disease mutation (Ritz et al, 2011). Moreover, Gal3 localized with LAMP2 at sites of lysosomal membrane accumulation typical for the disease (Fig EV2C), indicating that p97 disease mutations interfere with the ability to efficiently clear damaged late endosomes and lysosomes from affected tissues.
Figure 2. Disease‐associated mutations of p97 compromise clearance of damaged lysosomes.
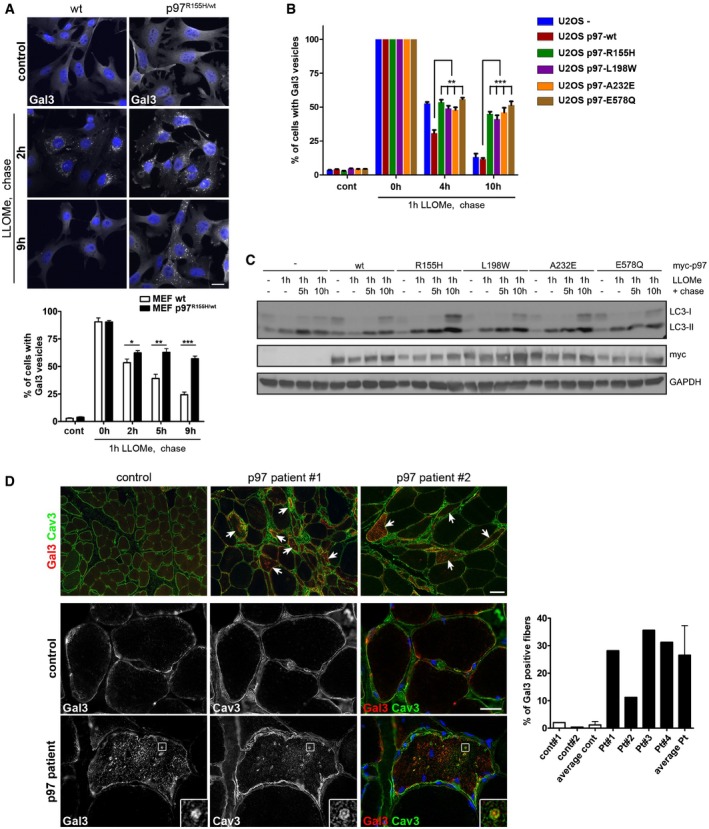
- Impaired clearance of damaged lysosomes in cells expressing a p97 disease mutant. Wild‐type and p97R155H/+ MEFs were LLOMe‐treated for 1 h and released for indicated times. Percentage of cells with Gal3‐positive vesicles was quantified from three independent experiments (mean ± SD, Student's unpaired t‐test). *P < 0.05; **P < 0.01; ***P < 0.001. Scale bar, 25 μm.
- Stable U2OS cell lines were doxycycline‐induced to express myc‐tagged p97 wild‐type or disease mutants as indicated. Induced parental cells served as control (U2OS‐). Cells were treated with LLOMe for 1 h and chased for indicated time before fixation. Percentage of cells with Gal3‐positive vesicles was quantified from three independent experiments. Comparable expression levels were verified (see Fig EV2A). **P < 0.01; ***P < 0.001. Student's unpaired t‐test.
- Stable U2OS cell lines described in (B) were treated with LLOMe for 1 h and chased for indicated time. LC3 and GAPDH levels were analyzed by immunoblotting. Quantification is shown in Fig EV2B.
- Accumulation of damaged lysosomes in affected p97 patient skeletal muscle. Patient biopsies immunostained for Gal3 and caveolin‐3 (Cav3). Quantification from 4 random 10× fields for each biopsy and means for controls and patients, respectively. Patients carry p97 mutations R155H (#1‐3) or R93C for #4. Patients #2 and #3 are cousins. Scale bar, 100 μm. Arrows indicate single myofibers with increased Gal3 staining.
Source data are available online for this figure.
Figure EV2. Compromised clearance of damaged lysosomes in p97 mutant‐expressing cells, and vacuolated muscle fibers from a p97 patient are positive for Gal3 and LAMP2 (related to Fig 2).
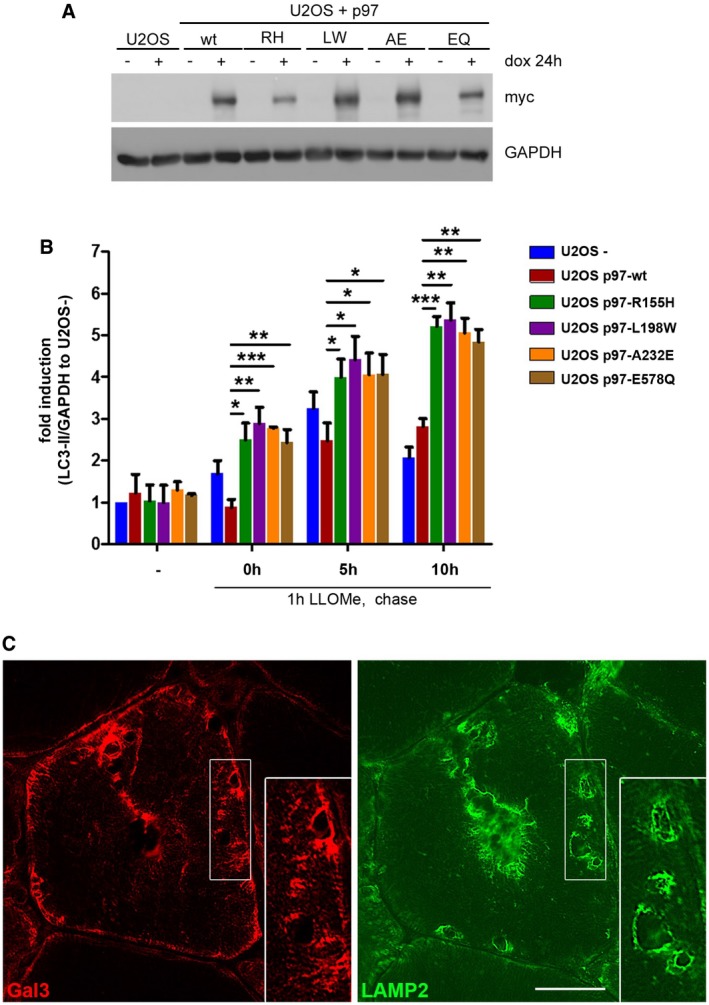
- Induction and expression levels of myc‐tagged p97 and variants in cells used in Fig 2B were determined via immunoblotting.
- Quantification of LC3‐II level normalized to GAPDH from three independent experiments (mean ± SD, Student's t‐test). LC3‐II level in U2OS cells treated with doxycycline (U2OS‐) were set to fold change = 1. *P < 0.05; **P < 0.01; ***P < 0.001.
- Skeletal muscle biopsy from a p97‐R155H patient. Serial sections were immunostained for Gal3 (red) and LAMP2 (green) and demonstrate colocalization in areas of lysosomal membrane accumulation within vacuolated fibers. Scale bar, 25 μm.
Source data are available online for this figure.
A subset of p97 cofactors cooperates in the response to lysosomal damage
To explore the molecular basis for p97 activity in lysophagy, we aimed to identify the relevant cofactors that cooperate with p97 in the process. We speculated that the lysosomal damage response involves the p97 cofactors UBXD1 and PLAA, because they had previously been linked to the endolysosomal system or autophagy in different systems and are affected by p97 disease mutations (Ren et al, 2008; Ossareh‐Nazari et al, 2010; Ritz et al, 2011; Wu et al, 2016). Moreover, we considered the DUB, YOD1, whose homologue OTU1 physically and genetically interacts with p97 and PLAA/Ufd3 in yeast (Rumpf & Jentsch, 2006). Of note, siRNA‐mediated depletion of UBXD1, PLAA, and YOD1 led to significant persistence of Gal3 vesicles compared to negative control depletions 12 h after LLOMe treatment (Fig 3A and B). Depletion efficiency was verified (Fig EV3A). As expected, control depletion of p97 or the general autophagy factors ATG5, ATG7, and p62 also affected Gal3 clearance. In contrast, depletion of the Ufd1 cofactor, which cooperates with p97 in facilitating proteasome‐mediated degradation of ubiquitinated substrates at the ER, on the outer mitochondrial membrane and on chromatin (Stolz et al, 2011), with an established siRNA, had no effect on Gal3 clearance.
Figure 3. A subset of p97 cofactors is directly involved in the lysosomal damage response and forms a substrate‐stabilized complex.
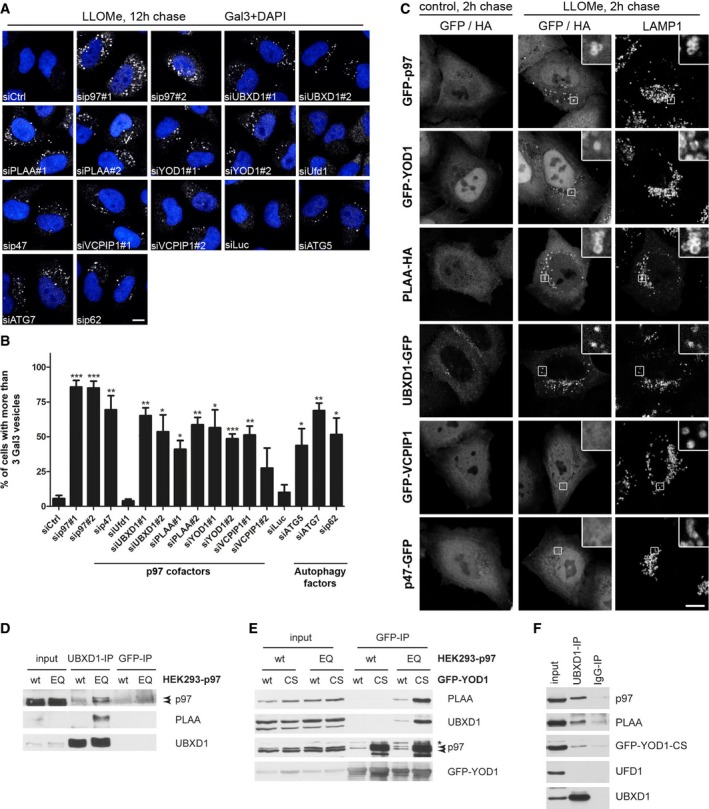
- Candidate screen for p97 cofactors involved in autophagy of damaged lysosomes. HeLa cells were transfected with indicated siRNA oligos and LLOMe‐treated for 1 h. Cells were chased for 12 h, fixed, and stained for Gal3. See Fig EV3 for depletion efficiency.
- Quantification of the percentage of cells with more than three Gal3 vesicles per cell for each condition. This threshold was chosen so that < 1% of untreated cells were considered positive. Data represent mean ± SEM of three independent experiments with ≥ 30 cells quantified per condition. *P < 0.05; **P < 0.01; ***P < 0.001 (one‐sided Welch's t‐tests).
- YOD1, UBXD1, and PLAA are translocated along with p97 to ruptured lysosomes. Cells expressing indicated tagged proteins were LLOMe‐treated for 1 h or with vehicle alone (control), fixed 2 h after washout, and stained for endogenous LAMP1 and, in the case of PLAA, with Alexa 488‐conjugated anti‐HA antibodies. See Fig EV3B for quantification.
- An ATP‐stabilized complex of p97 containing both UBXD1 and PLAA. Stable HEK293 cell lines were doxycycline‐induced to express p97 wild‐type (wt) or the ATPase mutant E578Q (EQ) at near endogenous level. Endogenous UBXD1 was immunoprecipitated and associated proteins detected by immunoblotting with indicated antibodies. Arrowheads indicate endogenous (lower band) and induced p97 (upper band).
- Stable HEK293 cells treated as in (D) and transiently expressing GFP‐tagged YOD1‐wt or the YOD1‐C160S catalytic mutant (CS) were processed for co‐immunoprecipitation with GFP nanobodies. Asterisk, unspecific band. Arrowheads indicate endogenous (lower band) and induced p97 (upper band). Note that the YOD1‐CS mutant stabilized p97 binding and, in the p97‐EQ background, also associated with UBXD1 and PLAA.
- Control (IgG) and UBXD1 co‐immunoprecipitation with indicated associated proteins from HeLa cells expressing GFP‐YOD1‐CS after formaldehyde cross‐linking. Ufd1 served as a negative control.
Figure EV3. Characterization of ELDR component depletion and overexpression (related to Fig 3).
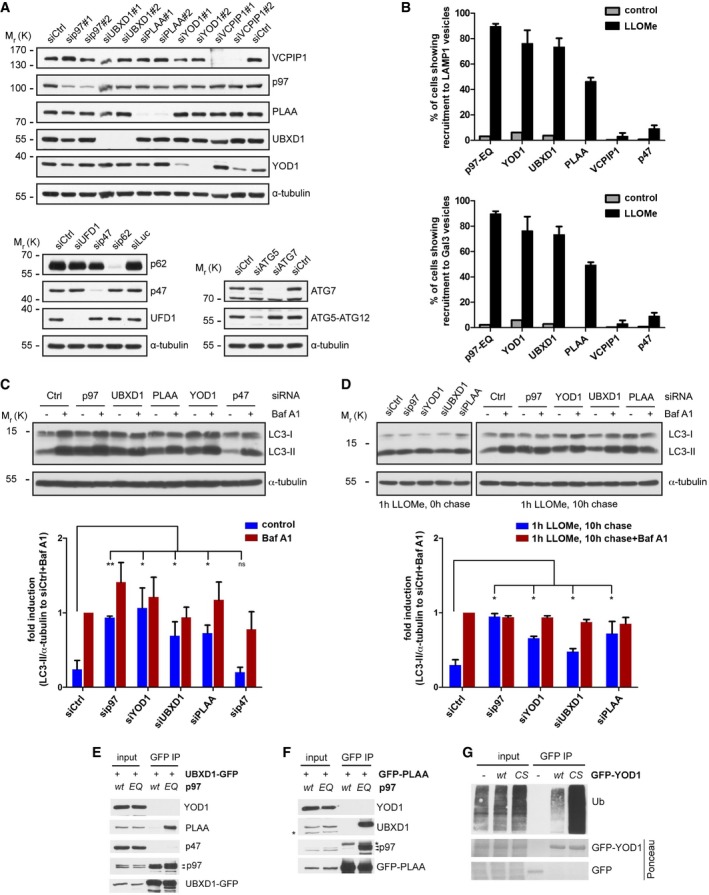
- Depletion efficiency of siRNA oligos used in Fig 3 was confirmed in HeLa cells by immunoblotting of total lysates with specific antibodies as indicated.
- YOD1, UBXD1, and PLAA localize to lysosomes upon damage. HeLa cells co‐transfected with mCherry‐Gal3 and GFP‐p97‐EQ, YOD1‐HA, UBXD1‐HA, PLAA‐HA, VCPIP1‐HA, or p47‐HA were treated with vehicle alone (control) or LLOMe for 1 h and chased for 2 h. After staining with anti‐HA and anti‐LAMP1 antibodies, the percentage of cells showing colocalization of ELDR components with LAMP1 or Gal3 was calculated from three independent experiments (mean ± SD).
- HeLa cells were transfected with indicated siRNA oligos and treated with 200 nM bafilomycin A1 (Baf A1) for 4 h or left untreated (control). Lysates were analyzed for LC3 and α‐tubulin. Quantification of LC3‐II levels normalized to α‐tubulin from three independent experiments. LC3‐II level in control‐depleted cells (siCtrl) treated with Baf A1 was set to fold change = 1 (mean ± SD, Student's t‐test for control treated samples is shown, and one‐sample t‐test for Baf A1‐treated samples revealed no significant changes). ns, not significant. *P < 0.05; **P < 0.01.
- HeLa cells transfected with indicated siRNA oligos were treated with LLOMe for 1 h and lysed directly or 10 h after LLOMe washout in the presence of Baf A1 or without. LC3‐II levels were analyzed as in (C).
- The ATP‐bound state of p97 stabilizes interaction with UBXD1 and PLAA. Stable HEK293 cell lines were doxycycline‐induced to express p97 wild‐type (wt) or the ATPase mutant E578Q (EQ), and transiently transfected with UBXD1‐GFP. UBXD1‐GFP was isolated and co‐precipitating proteins were detected by immunoblotting with indicated antibodies. Arrowheads indicate endogenous and induced p97‐myc‐strep.
- Cells were processed for co‐immunoprecipitation as in (E), but with GFP‐PLAA instead of UBXD1‐GFP. The asterisks indicate non‐specific bands. Arrowheads indicate endogenous and induced p97‐myc‐strep.
- Trapping of ubiquitin conjugates using the catalytically inactive YOD1 mutant. Cells were transfected with empty vector (−), GFP‐YOD1‐wt, or the YOD1‐C160S catalytic inactive mutant (CS). GFP or GFP‐YOD1 was isolated (stained by Ponceau S) and co‐precipitating ubiquitin conjugates (Ub) were detected by immunoblotting.
Source data are available online for this figure.
To evaluate whether the involvement of cofactors in lysophagy is direct, we monitored recruitment of the candidates to damaged lysosomes. Indeed, UBXD1 and YOD1—like p97—readily translocated to lysosomes upon LLOMe treatment in the majority of cells, and also PLAA was recruited in more than 40% of cells, whereas the proteins distributed throughout the cytoplasm and were not localized to lysosomes in untreated cells (Figs 3C and EV3B for quantification). To understand at what level autophagic clearance of Gal3‐positive lysosomes was inhibited, we examined levels of lipidated LC3 (LC3‐II). Depletion of p97, UBXD1, YOD1, or PLAA resulted in accumulation of LC3‐II (Fig EV3C), which was not increased by bafilomycin A1 treatment compared to control‐depleted cells, suggesting impairment of autophagosome clearance. Consistently, LC3‐II levels persisted in UBXD1‐, PLAA‐, YOD1‐, and p97‐depleted cells 10 h after LLOMe washout, whereas they were cleared in control‐depleted cells (Fig EV3D). Again, bafilomycin A1 treatment did not further increase LC3‐II levels compared to control cells after LLOMe washout (Fig EV3D).
These data indicate a direct involvement of the UBXD1, PLAA, and YOD1 cofactors in the clearance of damaged lysosomes. Intriguingly, the alternative cofactor p47, or the p97‐p47‐associated DUB VCPIP1, also delayed Gal3 clearance (Fig 3A and B). However, in contrast to YOD1, UBXD1, or PLAA, neither p47 nor VCPIP1 localized to damaged lysosomes independently on whether tagged on the N‐ or C‐terminus (Figs 3C and EV3B, and data not shown). Moreover, p47 depletion even led to a decrease rather than increase of LC3‐II accumulation (Fig EV3C) indicating an independent role upstream during LC3 activation consistent with previous results (Zhang et al, 2015).
YOD1, UBXD1, and PLAA can all bind p97 individually through dedicated interaction domains (Stolz et al, 2011; see Appendix Fig S1A for domain structures). We next asked whether they can bind p97 at the same time and therefore first analyzed proteins co‐immunoprecipitating with UBXD1. Because p97‐cofactor complexes can dissociate quickly after cell lysis (Xue et al, 2016), we used stable cells expressing the ATPase‐deficient trapping mutant p97‐E578Q (p97‐EQ) at near endogenous levels in addition to p97‐wt cells. Indeed, UBXD1 co‐precipitated p97 and also co‐isolated PLAA in the p97‐EQ background (Fig 3D). This was confirmed for overexpressed UBXD1‐GFP and the reciprocal GFP‐PLAA pulldowns (Fig EV3E and F), suggesting that a UBXD1‐p97‐PLAA complex can form at least transiently in an ATPase‐dependent manner. YOD1 also only weakly interacted with p97 in co‐immunoprecipitations. However, binding was largely increased in the case of the catalytic mutant C160S (YOD1‐CS) (Fig 3E), which stabilizes associated ubiquitin conjugates (Fig EV3G). When expressed in the p97‐EQ background, YOD1‐CS robustly co‐isolated p97, UBXD1, and PLAA. The YOD1‐CS‐stabilized complex could also be isolated with endogenous UBXD1 in the absence of p97‐EQ expression, but this required chemical cross‐linking (Fig 3F). The UBXD1 pulldowns also confirmed that the complex did not contain the alternative cofactors Ufd1 or p47 (Figs 3F and EV3E). Thus, UBXD1, PLAA, and YOD1 not only colocalize and cooperate with p97 on damaged lysosomes but also interact at least transiently in a substrate‐dependent manner. For simplicity and to distinguish their activity from other p97‐mediated processes, we would therefore like to term them the Endo‐Lysosomal Damage Response (ELDR) components.
The ELDR components p97, UBXD1, PLAA, and YOD1 specifically target K48‐linked ubiquitin conjugates on K63‐decorated damaged lysosomes
The involvement of YOD1 in lysophagy is intriguing, because it is a DUB with specificity for K48‐linked as well as K11‐linked and atypical ubiquitin chains, but has very low activity toward K63‐linked chains (Ernst et al, 2009; Mevissen et al, 2013). We therefore assessed the ubiquitination pattern on damaged lysosomes by staining with available linkage‐specific antibodies. As expected, we detected a strong signal for K63‐linked ubiquitin chains on the majority of LAMP1‐positive compartments after LLOMe treatment that were also positive for Gal3 and p62 (Fig 4A), consistent with a role of K63 chains in p62 recruitment to damaged lysosomes for autophagy. K11 staining did not result in any specific signal on lysosomes (data not shown). In contrast, we detected a robust signal for K48 chains (Fig 4A). Of note, K48‐linked ubiquitin conjugates were present on a smaller fraction of damaged lysosomes (Fig 4A). Because co‐staining of K48 and K63 chains with antibodies was technically not possible, we expressed the GFP‐TAB2‐NZF sensor to detect K63 chains (van Wijk et al, 2012). K48‐specific antibodies confirmed that individual lysosomes were decorated with K63 and K48 chains at the same time (Fig 4B), suggesting that K48 conjugation occurred on a subpopulation of K63‐labeled lysosomes.
Figure 4. The ELDR components p97, UBXD1, PLAA, and YOD1 selectively target K48‐linked ubiquitin conjugates on K63‐decorated damaged lysosomes.
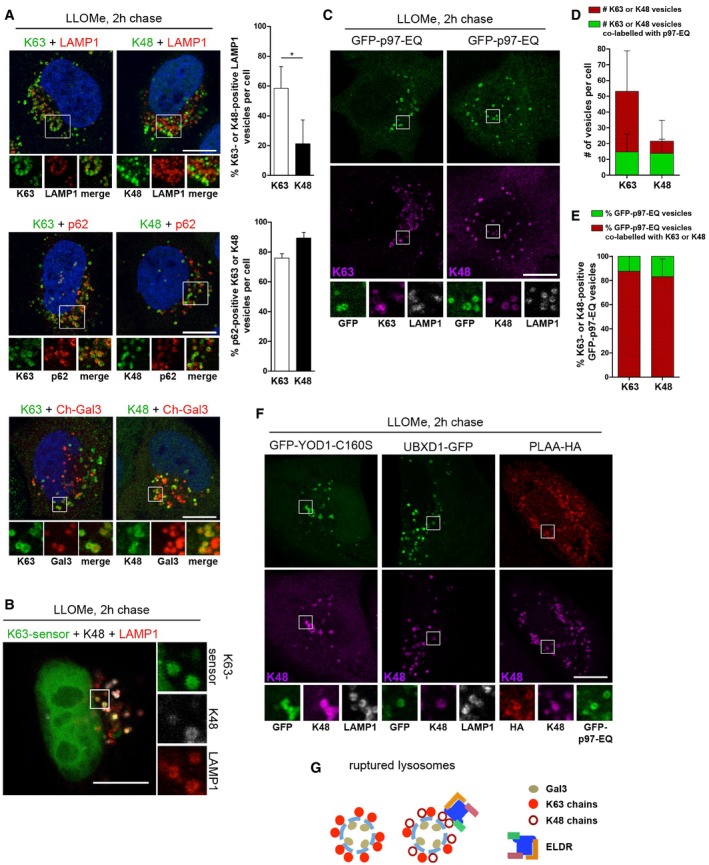
- K63 and K48 ubiquitination of damaged lysosomes. Untransfected HeLa cells, or cells expressing mCherry‐Gal3 (Ch‐Gal3) were LLOMe‐treated for 1 h. Two hours after washout, cells were stained with antibodies specific for K63 or K48 chains, and LAMP1 or p62 as indicated. Percentage of K63‐ or K48‐ubiquitinated LAMP1 vesicles and percentage of p62‐positive K63 or K48 vesicles were quantified from three independent experiments (mean ± SD, Student's unpaired t‐test for LAMP1). *P < 0.05.
- Colocalization of K63‐ and K48‐linked ubiquitin conjugates on individual lysosomes. Cells expressing GFP‐TAB2‐NZF (K63‐sensor) were treated as in (A) and stained for K48 chains and LAMP1. Note that the apparent nuclear localization of the sensor is a consequence of the extraction procedure (van Wijk et al, 2012).
- Cells expressing GFP‐p97‐EQ were treated as in (A) and stained with antibodies as indicated.
- Quantification of the total number of K63‐ or K48‐ubiquitinated vesicles per cell, and number of p97‐EQ positive K63‐ or K48‐ubiquitinated vesicles (mean ± SD).
- Percentage of GFP‐p97‐EQ vesicles positive for K63 or K48 chains was determined (mean ± SD).
- ELDR components target K48 conjugates on lysosomes. Detection of GFP‐YOD1‐CS or UBXD1‐GFP alone, or co‐transfected with PLAA‐HA and GFP‐p97‐EQ, 2 h after LLOMe washout and co‐stained with indicated antibodies.
- Scheme of ubiquitination of lysosomes after damage. K48‐linked ubiquitination occurs on a subpopulation of lysosomes decorated with K63‐linked chains. The ELDR components specifically target K48‐linked ubiquitin conjugates.
To investigate whether the ELDR components target these K48‐linked conjugates, we first tested GFP‐p97‐EQ, because it traps substrate proteins. Indeed, GFP‐p97‐EQ localized to the majority of K48‐positive lysosomes (Fig 4C and D). The fact that it localized only to a fraction of all K63‐positive lysosomes, but the vast majority of p97‐positive lysosomes were labeled with both K48 and K63 chains (Fig 4C and E), concurs with the notion that p97 selectively localized with K48 conjugates on K63‐positive lysosomes. Furthermore, p97‐positive lysosomes were negative for LysoTracker and cathepsin activity, and colocalized with Gal3 and LAMP1 (Fig EV4A–C), showing that these lysosomes were damaged and lost acidification. Colocalization with K48 chains was confirmed for endogenous p97 (Fig EV4D), as well as for UBXD1, PLAA, and the YOD1‐CS variant (Figs 4F and EV4F), suggesting that all ELDR components targeted K48‐linked conjugates. Consistently, the lysosomes that recruited ELDR components were also positive for K63 chains (Figs 4C and EV4E and F, and EV5C). Thus, a subset of ruptured lysosomes is decorated with K48‐linked ubiquitin chains in addition to K63‐linked ubiquitin and the ELDR components are recruited to these particular lysosomes, as summarized in Fig 4G.
Figure EV4. The ELDR components localize to damaged lysosomes (related to Fig 4).
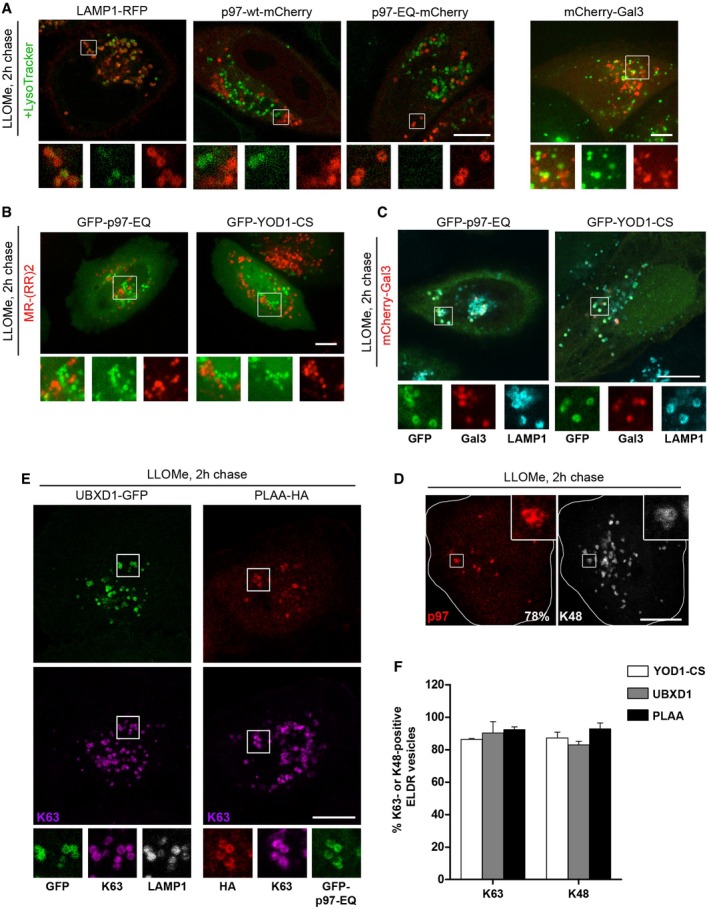
- p97 localizes to non‐acidified, damaged lysosomes. HeLa cells transfected with LAMP1‐RFP, p97‐wt‐mCherry or EQ, or mCherry‐Gal3 marker proteins were treated with LLOMe for 1 h and chased for 2 h. Cells were incubated with the pH‐sensitive LysoTracker probe (100 nM, Invitrogen) for 1 h and imaged live.
- Cells transfected with GFP‐p97‐EQ or GFP‐YOD1‐CS were treated as in (A), incubated with the cathepsin B substrate MR‐(RR)2 (Magic Red Cathepsin B detection kit, ImmunoChemistry Technologies, LLC), and imaged live by confocal spinning disk microscopy.
- Cells co‐transfected with mCherry‐Gal3 and GFP‐p97‐EQ or GFP‐YOD1‐CS were treated as in (A) and stained for LAMP1.
- Endogenous p97 localizes to K48‐decorated lysosomes after damage. LLOMe‐treated HeLa cells were chased for 2 h, fixed, and stained with antibodies specific for K48 chains and p97. Quantification of K48‐positive p97 vesicles from three independent experiments (77.8% ± 9.6, mean ± SD).
- Lysosomes that recruit ELDR components are also positive for K63 chains. Detection of UBXD1‐GFP alone, or PLAA‐HA co‐transfected with GFP‐p97‐EQ, 2 h after LLOMe washout and co‐stained with indicated antibodies.
- Quantification of results in Fig 4F and panel (E). Percentage of YOD1, UBXD1, or PLAA vesicles positive for K63 or K48 chains was determined (mean ± SD).
Figure EV5. The ELDR components remove K48‐linked ubiquitin conjugates from damaged lysosomes (related to Fig 5).
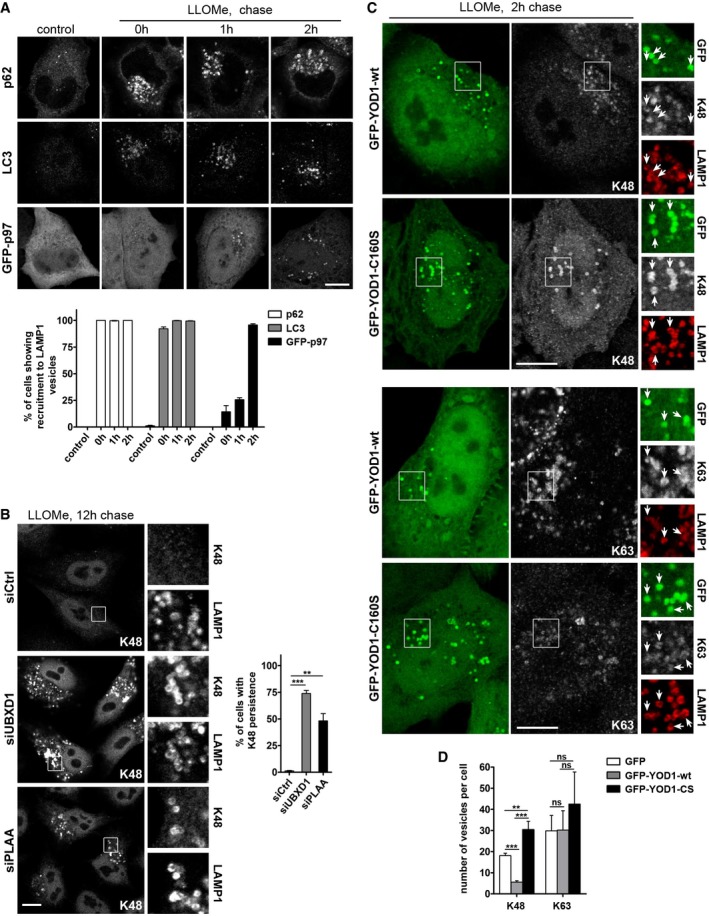
- Timing of p62, LC3, and p97 recruitment correlates with K63 and K48 ubiquitination, respectively. HeLa cells were transfected with GFP‐p97 (lower panel) or left untransfected (upper and middle panel), treated with LLOMe, and chased for the indicated time points. Cells were fixed and stained for endogenous p62 or LC3. Percentage of cells showing recruitment of p62, LC3, or GFP‐p97 to LAMP1 vesicles was determined (mean ± SD from three independent experiments).
- K48 conjugates persist upon depletion of ELDR components. HeLa cells were transfected with control (Ctrl), UBXD1, or PLAA siRNA oligos and treated with LLOMe, chased for 12 h, and then fixed and stained with antibodies specific for K48 chains. Percentage of cells with persistence of K48 vesicles was determined (mean ± SD from three independent experiments, Student's t‐test).
- The catalytically inactive YOD1 mutant is trapped on K48‐positive lysosomes. HeLa cells expressing GFP‐YOD1‐wt or C160S were LLOMe‐treated, chased for 2 h, and stained with antibodies specific for K48 or K63 chains. Arrows indicate LAMP1‐positive vesicles colocalizing with YOD1‐wt or C160S. Note increased colocalization of the K48 signal, but not K63, with the catalytically inactive YOD1‐C160S compared to wt.
- Number of K63‐positive lysosomes 2 h after washout in cells treated as in (C) were quantified from three independent experiments. Values on K48‐positive lysosomes 2 h after washout (taken from Fig 5F) were plotted for direct comparison.
The ELDR components turn over K48‐linked ubiquitin conjugates on damaged lysosomes
We followed the dynamics of ubiquitin chains by determining the fraction of K63‐ and K48‐decorated lysosomes after lysosomal damage over time (Fig 5A). In control cells, K63 chains emerged early on lysosomes and peaked already after 1 h LLOMe treatment (corresponding to 0 h after washout) with more than 60% of lysosomes labeled at the given LLOMe concentration. In contrast, K48‐linked conjugates appeared later peaking at around 2–4 h after LLOMe washout with about 25% lysosomes affected indicating that K63 and K48 ubiquitination represent temporally separated events. Both chain types disappeared within 12 h. Consistent with their colocalization, p97 recruitment coincided with the K48 subpopulation peak (2 h after washout), whereas translocation of p62 and initial recruitment of LC3 was already observed earlier (Fig EV5A). Importantly, depletion of either YOD1 or p97 had a marked effect specifically on K48 chains, leading to an increased number of K48‐positive lysosomes and persistence over time (Fig 5B). In contrast, the level of K63 chains and their initial rate of turnover during the first hours were not affected, although a fraction of K63‐labeled lysosomes persisted along with K48 conjugates.
Figure 5. The ELDR components specifically remove K48‐linked ubiquitin conjugates from damaged lysosomes.
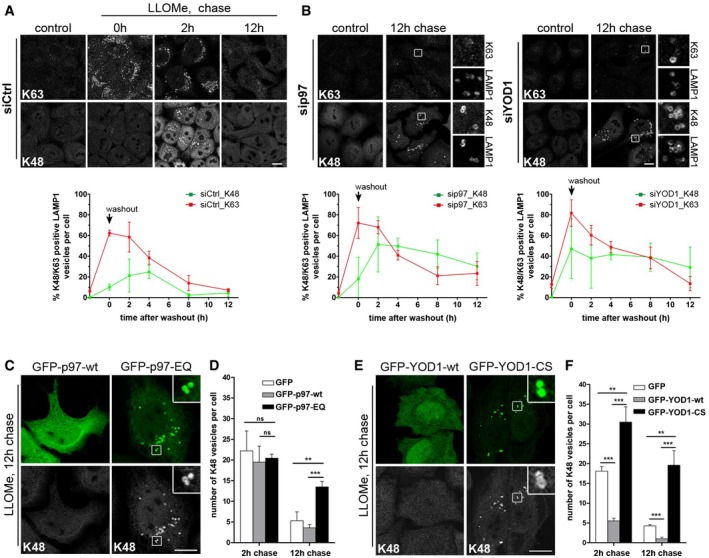
- Time course of K63 and K48 ubiquitination. Control‐depleted cells were fixed at indicated time points after LLOMe washout and stained with chain‐specific antibodies and LAMP1. Automated quantification of LAMP1 vesicles positive for K63 or K48 chains. Data represent mean ± SD of three independent experiments.
- Depletion of p97 or YOD1 results in increase and persistence of K48 chains on damaged lysosomes. Experiment and quantification as in (A) in cells treated with p97 or YOD1 siRNA oligos.
- The p97‐EQ mutant stabilizes K48 conjugates. Lysosome damage was induced as in (A) in cells transfected with GFP‐tagged p97‐wt or p97‐EQ. Cells were chased after washout for 12 h and stained for K48 chains.
- Quantification of K48‐positive lysosomes 2 h and 12 h after damage in cells treated as in (A). Data represent mean ± SD of three independent experiments.
- The catalytically inactive mutant of YOD1 stabilizes K48 conjugates. Cells transfected with GFP‐tagged YOD1‐wt or the YOD1‐CS were treated and stained as in (C).
- Quantification as in (D) of cells treated as in (E). Data represent mean ± SD of three independent experiments.
Also depletion of PLAA or UBXD1 led to persistence of K48‐positive lysosomes (Fig EV5B), suggesting that all four ELDR components cooperate in turning over K48 conjugates. Moreover, overexpression of the p97‐EQ mutant impaired clearance of K48‐labeled lysosomes, and p97‐EQ strongly colocalized with these lysosomes even 12 h after washout (Fig 5C and D). Overexpression of the YOD1‐CS mutant had an even stronger effect and resulted in accumulation of K48‐linked ubiquitin conjugates already at 2 h after washout (Figs 5E and F, and EV5C), demonstrating that both the ATPase activity of p97 and the deubiquitinating activity of YOD1 are essential for removing K48‐linked ubiquitin conjugates from damaged lysosomes. In agreement, YOD1‐wt localization to lysosomes correlated with decreased K48 signal on these membranes at 2 and 12 h after washout (Figs 5F and EV5C), suggesting that YOD1 is the major DUB in this process. As a consequence of stabilizing the K48 chains, localization of YOD1‐CS to lysosomes was strongly increased compared to wild‐type. Importantly, overexpression of YOD1‐wt or YOD1‐CS did not affect the levels of K63‐linked ubiquitin conjugates (Fig EV5C and D).
YOD1 functions as a p97‐activated ubiquitin sensor
YOD1 contains a ubiquitin‐binding site, termed S2, outside of the catalytic center (Mevissen et al, 2013). However, in ubiquitin‐GST pulldown experiments, YOD1 alone did not detectably interact with ubiquitin (Fig 6A, see Appendix Fig S1 for protein purity). Also PLAA (full length), which has ubiquitin‐binding sites in its WD40 repeat and PFU domains (Mullally et al, 2006; Pashkova et al, 2010), did not bind at the given conditions, and p97 interacted only weakly (Fig 6A). When combined with p97, however, YOD1, but not PLAA, associated with ubiquitin‐GST, indicating that YOD1 and p97 cooperatively interact with ubiquitin. This was dependent on the S2 site, because when using a previously characterized mutant of this site, termed MutS2 (Mevissen et al, 2013), the interaction of YOD1 and p97 with ubiquitin was largely reduced (Fig 6B). Since PLAA was not detected in the pulldowns, we repeated the experiments with p97‐EQ and also included UBXD1. Consistent with co‐immunoprecipitations from cells, PLAA and UBXD1 integrated into the ubiquitin‐bound complex of YOD1 and p97‐EQ (Fig 6C), showing that YOD1 can act as a ubiquitin sensor for the ELDR components. We next assessed the relevance of the YOD1 S2 ubiquitin‐binding site in cells. Whereas YOD1‐CS was efficiently recruited to damaged lysosomes as described above, the localization of YOD1‐CS‐MutS2 was greatly reduced (Fig 6D). Moreover, overexpression of YOD1‐wt again largely reduced the K48 signal on membranes after LLOMe treatment (Fig 6E). In contrast, YOD1‐MutS2 failed to do so, demonstrating that YOD1 needs to bind ubiquitin via the S2 site to efficiently target and turn over K48‐linked ubiquitin conjugates.
Figure 6. YOD1 functions as a p97‐activated ubiquitin sensor through its ubiquitin‐binding site S2.
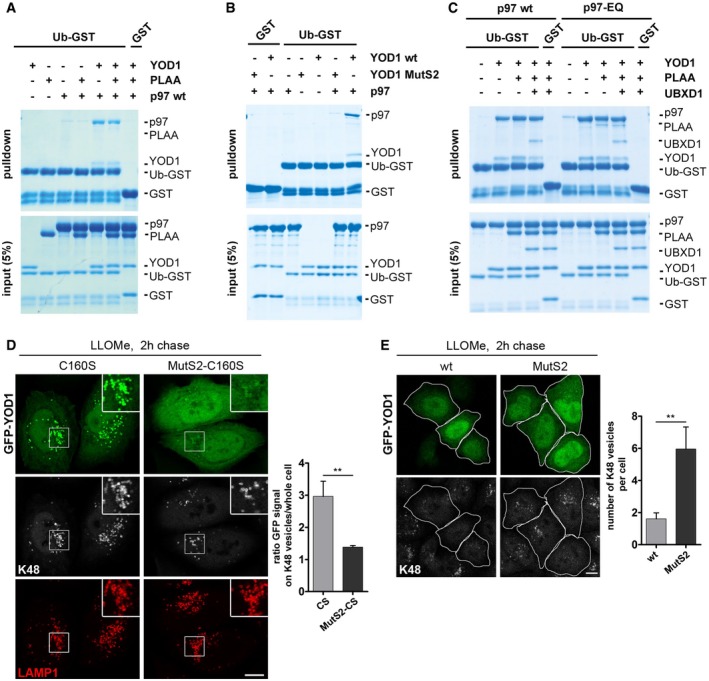
- YOD1 can link p97 to ubiquitin. Ubiquitin‐GST (Ub‐GST) or GST alone was incubated with the indicated purified proteins and isolated. Co‐isolated proteins (pulldown) were detected in SDS gels. Note binding of p97 and YOD1 to Ub‐GST only in combination.
- The S2 site of YOD1 is critical for ubiquitin binding. Experiments as in (A) with indicated combinations of p97 and YOD1‐wt or the ubiquitin‐binding mutant, MutS2.
- Reconstitution of a complex of ELDR components on ubiquitin. Experiments as in (A) with combinations of p97‐wt or p97‐EQ, YOD1, PLAA, and UBXD1 as indicated. Note that PLAA associates with p97 and YOD1 on ubiquitin only in the case of p97‐EQ.
- Translocation of YOD1 to lysosomes depends on ubiquitin binding by the S2 site. LLOMe‐treated cells expressing GFP‐YOD1‐CS or the double mutant GFP‐YOD1‐CS‐MutS2 were chased for 2 h and stained for K48 chains and LAMP1. Ratio of GFP signal on K48 vesicles to GFP signal in the whole cell was determined.
- The S2 site is essential for K48 conjugate removal. GFP‐YOD1‐wt‐ or GFP‐YOD1‐MutS2‐overexpressing cells treated as in (D) were stained for K48 chains. Quantification of the number of K48‐positive vesicles per cell.
ELDR‐mediated removal of K48‐linked ubiquitin conjugates is required for autophagosome formation
We next assessed the functional relevance of ELDR‐mediated turnover of K48 conjugates for lysophagy. Because p97 and the ELDR components did not regulate p62 recruitment, we asked whether K48 removal by p97 and YOD1 was linked to LC3 recruitment and autophagosome formation. Of note, GFP‐LC3 efficiently colocalized with K63‐labeled lysosomes in control cells 2 h after damage with high correlation according to the Pearson's coefficient, as expected (Fig 7A). In contrast, LC3 colocalized only to a smaller extent and lower intensity with lysosomes that were decorated with K48 chains (Fig 7A), suggesting that the presence of K48 conjugates on a subpopulation of damaged lysosomes may be incompatible with LC3 recruitment.
Figure 7. ELDR components remove K48 conjugates from damaged lysosomes to promote autophagosome formation.
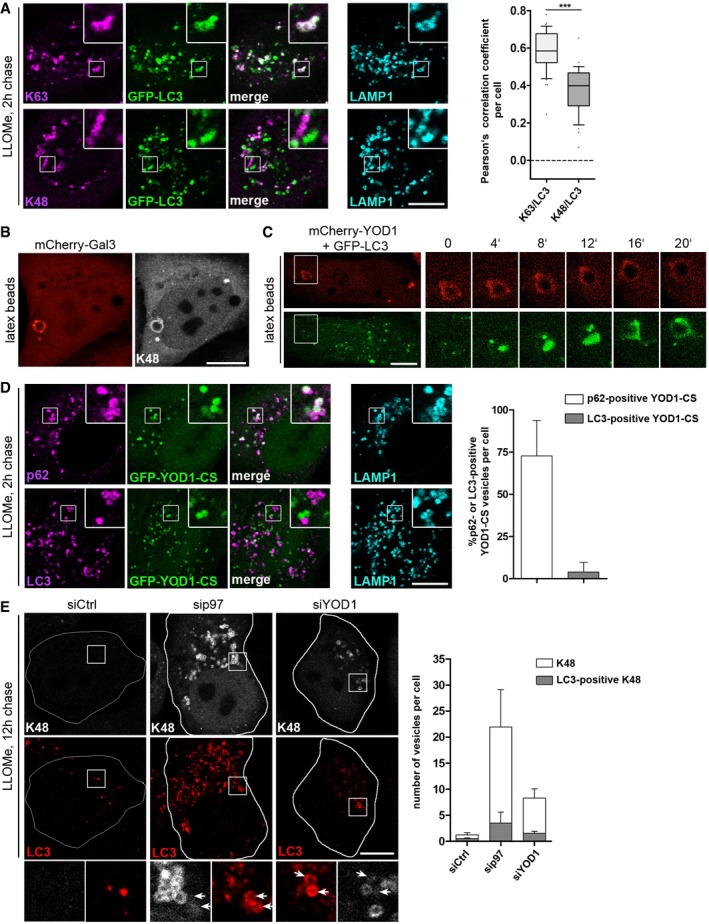
- Largely mutually exclusive localization of LC3 and K48 chains. GFP‐LC3‐expressing HeLa cells were fixed 2 h after LLOMe washout and stained for endogenous LAMP1 and K48 or K63 chains. Correlation coefficients were calculated for colocalization of LC3 with K63 or K48. The whiskers represent 10th and 90th percentile. Points below and above the whiskers are drawn as individual dots. The single line indicates the median. ***P < 0.001 (Mann–Whitney U‐test).
- Lipofectamine‐coated latex beads damage lysosomes and induce their K48 ubiquitination. Cells expressing mCherry‐Gal3 were incubated with Lipofectamine‐coated latex beads for 3 h and stained with K48 chain‐specific antibodies.
- YOD1 association and dissociation precede LC3 recruitment. Live cell video microscopy stills of cells co‐expressing mCherry‐YOD1‐wt and GFP‐LC3 after treatment with Lipofectamine‐coated latex beads for 30 min at 2 min image intervals.
- Stabilization of K48 conjugates on lysosomes impairs recruitment of LC3. Cells expressing GFP‐YOD1‐CS were treated as in (A) and stained for LAMP1 and p62 or LC3. Percentage of GFP‐YOD1 vesicles positive for p62 or LC3 was determined (mean ± SD).
- Depletion of ELDR components impairs LC3 recruitment for autophagosome formation. Cells were transfected with control (Ctrl), p97, or YOD1 siRNA oligos, treated with LLOMe, fixed after 12‐h chase, and stained for endogenous LC3 and K48 chains. Quantification of the number of K48‐positive vesicles per cell and number of LC3‐positive K48 vesicles per cell was determined. Data represent mean ± SD of three independent experiments. Arrows indicate position of LC3‐positive membranes.
We therefore next analyzed the temporal relationship between YOD1 targeting of K48 chains and LC3 recruitment. We made use of latex beads that are incorporated by cells and, when coated with transfection reagent, can rupture lysosomes and induce an autophagic response (Kobayashi et al, 2010). We first confirmed in fixed cells that the beads were taken up in LAMP1 compartments and induced Gal3 influx into bead‐containing endolysosomal compartments, which recruited p62 and were K48‐ubiquitinated (Figs 7B and Appendix Fig S2A and B). In time‐lapse microscopy of live cells expressing mCherry‐YOD1‐wt and GFP‐LC3, we observed targeting of YOD1 to beads followed by slow dissociation (Fig 7C and Movie EV1), indicating that YOD1 bound and then mediated the turnover of K48 conjugates on the bead‐containing lysosomes. Notably, as the YOD1 signal disappeared, it was gradually replaced by LC3 that first associated on one side of the bead and then fully covered it, consistent with removal of K48‐modified substrate proteins by YOD1 being required for LC3 recruitment.
To test this notion, we first blocked removal of K48‐linked ubiquitin conjugates by overexpression of YOD1‐CS. Overexpressed YOD1‐CS, which we showed above significantly accumulates and colocalizes with K48‐positive vesicles (Figs 4F and EV5C), overlapped with p62‐labeled lysosomes (Fig 7D). However, despite the presence of the LC3‐adapter p62, these lysosomes failed to recruit LC3 (Fig 7D). In a second approach, we inhibited removal of K48 conjugates by depletion of p97 or YOD1 and analyzed the persisting K48‐labeled lysosomes at 12 h after LLOMe‐induced damage. While LC3‐decorated membranes accumulated in the vicinity of K48‐labeled lysosomes, they failed to coalesce even at this late time point (Fig 7E) showing that removal of K48 conjugates by p97 and the ELDR components is required to allow LC3 recruitment and autophagosome formation.
Discussion
The data presented here reveal an essential function of p97 in the autophagic clearance of damaged lysosomes. p97 cooperates with a distinct set of cofactors, including UBXD1, PLAA, and the DUB, YOD1, which we term ELDR components (for Endo‐Lysosomal Damage Response) to distinguish this activity from other cellular roles of p97 such as ER‐ or chromatin‐associated degradation. Moreover, we demonstrate that the pathway specifically targets and turns over K48‐linked ubiquitin conjugates on damaged lysosomes to promote autophagosome formation. Thus, our findings shed light on the role of p97 in the cellular stress response, autophagy, and disease. In addition, they reveal an important functional distinction of different ubiquitin chain types during autophagic clearance of lysosomes.
Our data are consistent with K63 chains initiating the autophagic response and mediating p62 recruitment (Tan et al, 2008; Matsumoto et al, 2011; van Wijk et al, 2012), because K63 chains and p62 coincide temporally and spatially early during lysophagy, and are not regulated by the ELDR components. In contrast, the K48 conjugates occur later during the process and only on a subpopulation of K63‐decorated damaged lysosomes. Importantly, our data suggest that, in contrast to K63 chains, K48 conjugates need to be turned over by p97 with help of the ELDR factors to allow formation of autophagosomes. We find that YOD1 specifically cleaves K48 chains, which is in line with previous biochemical studies (Ernst et al, 2009; Mevissen et al, 2013), and that overexpression of YOD1‐CS inhibits removal of K48 conjugates on damaged lysosomes and suppresses association of LC3‐positive membranes. Consistently, depletion of any of the ELDR components leads to persistence of K48‐decorated lysosomes that then fail to associate with LC3. This is supported by our finding that YOD1 recruitment and dissociation precedes LC3 association in live cell microscopy.
Because p97 is generally thought to extract ubiquitinated proteins from cellular structures, an obvious explanation for our observations is that K48‐linked ubiquitination and the ELDR‐mediated removal of one or more factors that interfere with autophagosome formation. A similar model has also been proposed for the role of p97 in mitophagy, where p97‐mediated extraction of factors on the outer membrane, such as mitofusins, is thought to facilitate the clearance of damaged mitochondria or mitophagy (Tanaka et al, 2010; Chan et al, 2011). Of note, extraction of mitofusins involves the Ufd1‐Npl4 cofactor (Kim et al, 2013; Wu et al, 2016), whereas we identified a distinct set of cofactors in lysophagy including the DUB, YOD1. DUBs such as YOD1 were also shown to contribute to extraction of proteins from the ER membrane during ER‐associated degradation (Ernst et al, 2009; Liu & Ye, 2012; Stein et al, 2014). It is therefore likely that YOD1 assists protein extraction from lysosomes. On lysosomes, a whole range of membrane proteins is ubiquitinated upon damage (Fujita et al, 2013). Removal of the K48 conjugates may therefore facilitate access of LC3‐decorated autophagic membranes to K63‐p62 assemblies on damaged lysosomes. Extraction may be followed by degradation, but this would not be essential for the process, because proteasome inhibitors do not affect lysophagy. Identification of the relevant ubiquitin ligase(s) in lysophagy will help clarify these questions.
Intriguingly, we find that only a subpopulation of damaged lysosomes is conjugated with K48 chains and that some autophagosomes form early without involvement of K48 chains and independently of the EDLR components. One possible explanation is that K48‐labeled lysosomes represent a particular type of late endosomes or lysosomes and therefore require an additional layer of regulation. Alternatively, the populations may be distinct with respect to the type or degree of damage. Consistent with this notion, diverse bacteria that damage endolysosomal membranes in different ways also induce distinct autophagic responses (Gomes & Dikic, 2014).
The p97‐governed response involves a distinct set of cofactors that assist p97 during lysophagy. Even though these cofactors bind at the same time to p97 only transiently, it is likely that they tightly cooperate during lysophagy because they all colocalize on damaged lysosomes and are essential for their clearance. Among the three p97 cofactors, UBXD1 appears to be the defining component because it binds p97 in a mutually exclusive manner with other major cofactors such as p47 or Ufd1‐Npl4, and has been associated with endolysosomes and trafficking before (Ritz et al, 2011; Stolz et al, 2011; Haines et al, 2012). YOD1 harbors the essential catalytic DUB activity, but can also function as a ubiquitin sensor by means of its S2 ubiquitin‐binding site. We find that ubiquitin binding by YOD1 appears to stimulate interaction of YOD1 with p97 along with UBXD1 and PLAA and, in turn, p97 activates ubiquitin binding of YOD1. However, targeting of p97 is likely more complex because YOD1 depletion does not impair p97 recruitment to damaged lysosomes (data not shown), whereas it clearly affects p97‐mediated lysophagy. So far, the role of PLAA remains unclear. It appears to be a versatile factor because it also teams up with diverse p97 complexes in other processes at least in yeast (Richly et al, 2005; Balakirev et al, 2015; Wu et al, 2016). It has ubiquitin‐binding domains (Mullally et al, 2006; Pashkova et al, 2010) but binds only transiently to p97 depending on the nucleotide state, suggesting that it may coordinate ATP hydrolysis within the process of lysophagy.
Disease mutations in p97 cause a late onset myodegenerative and neurodegenerative disease termed MSP. MEFs expressing a disease‐associated p97 allele fail to clear damaged lysosomes and MSP patient tissue accumulates damaged lysosomes. This raises the possibility that compromised clearance of damaged lysosomes contributes to the pathogenesis of p97‐associated disease. One intriguing aspect of p97‐associated MSP is the variable penetrance of muscle or CNS pathology even within the same family. This phenotypic heterogeneity has led to speculation that either secondary genetic or environmental factors dictate disease penetrance. The emerging role for p97 in mediating “damage responses”—whether it is DNA damage repair, mitochondrial clearance, or lysosomal homeostasis—suggests that environmental stressors that occur with age play a key role in p97‐associated diseases (Meyer & Weihl, 2014; Johnson et al, 2015).
Our findings also have implications for the relevance of p97 in other diseases where lysosomal membrane permeabilization is an element, such as lysosomal storage diseases and neurodegenerative disorders (Boya & Kroemer, 2008; Nixon, 2013; Repnik et al, 2013). Intriguingly, tau fibrils, which occur and can be released from cells in a number of pathologies, can damage lysosomes when endocytosed. Tau‐induced damage could further decrease lysosomal fitness and thereby affect the capacity of cells to maintain protein homeostasis. The fact that p97 clears damaged lysosomes and also responds to the tau‐induced lysosomal damage adds an unanticipated aspect to the emerging picture of p97 as a critical modulator of neurodegeneration (Rubinsztein, 2006; Ling et al, 2013).
Materials and Methods
Cell culture
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) in the presence of penicillin/streptomycin. Immortalized primary mouse embryonic fibroblasts (MEFs) were generated from p97R155H/wt mice as described in Appendix Supplementary Methods. Stable inducible p97‐myc‐strep HEK293 cells were cultured and induced as described previously (Ritz et al, 2011). Cells were transfected with plasmids using Lipofectamine 2000 (Invitrogen), JetPEI (Polyplus), or JetPRIME (Polyplus), or with siRNA (12.5 nM) using RNAiMax (Invitrogen) according to the manufacturer's instructions. Transfected cells were changed to regular growth medium 4 h after transfection and analyzed after 24 h (plasmids) or 48 h (siRNA). Plasmids, siRNA, and reagents used in this study are described in Appendix Supplementary Methods. To induce lysosomal damage, HeLa cells were treated with 250 μM Leu‐Leu methyl ester hydrobromide (LLOMe, Sigma). Cell viability was determined with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega). Latex beads (NH2‐, 17145‐5; PolySciences, Inc.) were coated with Lipofectamine 2000 for 45 min and then processed as described previously (Fujita et al, 2013).
Patient skeletal muscle biopsies
Patient muscle was obtained under an approved IRB protocol at Washington University School of Medicine. Frozen tissue was sectioned (10 μm), acetone‐fixed, and processed for immunodetection. For some antibodies, a biotinylated secondary antibody was used and then detected using a Vectastain ABC kit (Vector Labs). Specimens were examined using a fluorescence microscope (Nikon 80i upright) and a Roper Scientific EZ monochrome CCD (charge‐coupled device) camera with deconvolution software analysis (NIS Elements, Nikon) at room temperature.
Fluorescence microscopy
Confocal laser scanning and confocal spinning disk microscopy were performed as described in Appendix Supplementary Methods. Live cell imaging was performed at 37°C in imaging medium (P04‐05545, PAN‐Biotech) with 10% FCS. Antibodies used in this study are described in Appendix Supplementary Methods. Images were processed using ImageJ software (https://imagej.nih.gov/ij/) and Photoshop (Adobe Photosystems). Automated quantifications were performed using Cell Profiler (Carpenter et al, 2006). Generation of graphs and statistical analysis were performed using Excel (Microsoft Corporation), R (www.r-project.org), or GraphPad Prism (GraphPad Software).
Immunoprecipitation and immunoblotting
Cells were harvested 24 h after transient transfection and/or 20 h after induction of p97‐wt/EQ in IP buffer (150 mM KCl, 5 mM MgCl2, 50 mM Tris–HCl pH 7.4, 1% Triton X‐100, 5% glycerol, 2 mM β‐mercaptoethanol, Roche Complete™ EDTA‐free protease inhibitors) and incubated on ice for 20 min. Lysates were cleared by centrifugation (15 min, 16,000 × g, 4°C). GFP‐tagged proteins were affinity‐isolated using anti‐GFP nanobodies. As described previously (Eiteneuer et al 2014), His‐tag nanobodies, purified from BL21(DE3), were coupled to NHS‐activated Sepharose 4 Fast Flow (GE) at 2 mg per ml bead slurry according to the manufacturer's instructions. Immunoprecipitations were performed for 2 h at 4°C rotating. Beads were washed 4 times in IP buffer and eluted with 2× SDS sample buffer (95°C for 5 min) for Western blot analysis. For immunoprecipitations upon chemical cross‐linking, the cells were incubated for 10 min at RT in 0.4% PFA and subsequently quenched by the addition of glycine to a final concentration of 125 mM. Samples were harvested in IP buffer as above with additional 0.5% Na‐deoxycholate. During the 20‐min lysis incubation on ice, the samples were sonicated twice (15 s). Cleared lysates were incubated with the indicated antibodies for 2 h rotating at 4°C and additionally with Protein G Agarose for 1 h. Beads were washed as above and eluted in 2× SDS sample buffer at 95°C for 10 min to also reverse the chemical cross‐link. Samples were resolved by SDS–PAGE and transferred to nitrocellulose membranes (Amersham, GE Healthcare). Immunoblot analysis was performed with the indicated antibodies and visualized with SuperSignal West Pico Chemiluminescent substrate (Pierce). Antibodies used in this study are described in Appendix Supplementary Methods.
Protein interaction assays
Proteins were expressed and purified as described in Appendix Supplementary Methods. For GST pulldown assays, proteins were mixed in 50 μl buffer (20 mM Hepes pH 7.2, 150 mM KCl, 5% glycerol, 5 mM MgCl2, 2 mM ATP, 5 mM DTT, 0.01% Triton X‐100). Samples were cleared (17,000 × g, 2 min), supplemented with 10 μl bead slurry (Glutathione Sepharose 4B, GE Healthcare), and incubated rotating for up to 90 min at room temperature. Beads were washed and eluted with buffer containing 20 mM glutathione for 10 min at room temperature. Eluates were analyzed by SDS–PAGE and stained with colloidal Coomassie.
Tau fibril treatment
Expression and heparin‐induced fibrillization of recombinant tau was done as described before (Poepsel et al, 2015). Fibrils were labeled with DyLight™ 488 NHS Ester (Thermo Fisher Scientific) according to the manufacturer's protocol. HeLa cells were fed for 24 h with 300 nM sonicated fibrils in regular growth medium.
Author contributions
CP, PK, and HM were involved in conceptualization; NS was involved in formal analysis; CP, PK, MB, DG, LK, RP, AD, SF, KA, and VL were involved in investigation; HM wrote the original draft of the manuscript; CP, PK, MB, ME, CCW, and HM wrote, reviewed, and edited the manuscript; CP was involved in visualization; and ME and HM were involved in funding acquisition.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank Graham Warren and James Shorter for critical reading. The work was funded by DFG grant Me 1626/4‐1 to HM and SFB 1093 subprojects to HM and ME.
The EMBO Journal (2017) 36: 135–150
See also: M Seczynska & I Dikic (January 2017)
References
- Balakirev MY, Mullally JE, Favier A, Assard N, Sulpice E, Lindsey DF, Rulina AV, Gidrol X, Wilkinson KD (2015) Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. eLife 4: e06763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Kroemer G (2008) Lysosomal membrane permeabilization in cell death. Oncogene 27: 6434–6451 [DOI] [PubMed] [Google Scholar]
- Buchan JR, Kolaitis RM, Taylor JP, Parker R (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153: 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM (2006) Cell Profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7: R100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Lacas‐Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F (2009) Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6: 137–149 [DOI] [PubMed] [Google Scholar]
- Ernst R, Mueller B, Ploegh HL, Schlieker C (2009) The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol Cell 36: 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiteneuer A, Seiler J, Weith M, Beullens M, Lesage B, Krenn V, Musacchio A, Bollen M, Meyer H (2014) Inhibitor‐3 ensures bipolar mitotic spindle attachment by limiting association of SDS22 with kinetochore‐bound protein phosphatase‐1. EMBO J 33: 2704–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz A, Ackermann L, Hoppe T (2014) Create and preserve: proteostasis in development and aging is governed by Cdc48/p97/VCP. Biochim Biophys Acta 1843: 205–215 [DOI] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O'Connor C, Wiethoff CM, Campbell EM (2013) Alpha‐synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 8: e62143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, Haraguchi T, Guan JL, Iwai K, Tokunaga F, Saito K, Ishibashi K, Akira S, Fukuda M, Noda T, Yoshimori T (2013) Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Dikic I (2014) Autophagy in antimicrobial immunity. Mol Cell 54: 224–233 [DOI] [PubMed] [Google Scholar]
- Haines DS, Lee JE, Beauparlant SL, Kyle DB, den Besten W, Sweredoski MJ, Graham RL, Hess S, Deshaies RJ (2012) Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC‐53 trafficking. Mol Cell Proteomics 11 M111.016444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YH, Chen LM, Yang JY, Yang WY (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Johnson AE, Shu H, Hauswirth AG, Tong A, Davis GW (2015) VCP‐dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo . eLife 4: e07366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju JS, Fuentealba RA, Miller SE, Jackson E, Piwnica‐Worms D, Baloh RH, Weihl CC (2009) Valosin‐containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78: 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Schulman BA (2014) Dynamic regulation of macroautophagy by distinctive ubiquitin‐like proteins. Nat Struct Mol Biol 21: 336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Kojidani T, Osakada H, Yamamoto A, Yoshimori T, Hiraoka Y, Haraguchi T (2010) Artificial induction of autophagy around polystyrene beads in nonphagocytic cells. Autophagy 6: 36–45 [DOI] [PubMed] [Google Scholar]
- Krick R, Bremer S, Welter E, Schlotterhose P, Muehe Y, Eskelinen EL, Thumm M (2010) Cdc48/p97 and Shp1/p47 regulate autophagosome biogenesis in concert with ubiquitin‐like Atg8. J Cell Biol 190: 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ye Y (2012) Roles of p97‐associated deubiquitinases in protein quality control at the endoplasmic reticulum. Curr Protein Pept Sci 13: 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A et al (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9: 548–556 [DOI] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 44: 279–289 [DOI] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, Freund SM, Ovaa H, Komander D (2013) OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154: 169–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol 14: 117–123 [DOI] [PubMed] [Google Scholar]
- Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127: 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B (2010) Autophagy in mammalian development and differentiation. Nat Cell Biol 12: 823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally JE, Chernova T, Wilkinson KD (2006) Doa1 is a Cdc48 adapter that possesses a novel ubiquitin binding domain. Mol Cell Biol 26: 822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997 [DOI] [PubMed] [Google Scholar]
- Ossareh‐Nazari B, Bonizec M, Cohen M, Dokudovskaya S, Delalande F, Schaeffer C, Van Dorsselaer A, Dargemont C (2010) Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11: 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S, Piper RC (2010) WD40 repeat propellers define a ubiquitin‐binding domain that regulates turnover of F box proteins. Mol Cell 40: 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poepsel S, Sprengel A, Sacca B, Kaschani F, Kaiser M, Gatsogiannis C, Raunser S, Clausen T, Ehrmann M (2015) Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat Chem Biol 11: 862–869 [DOI] [PubMed] [Google Scholar]
- Ramanathan HN, Ye Y (2011) The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res 22: 346–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Pashkova N, Winistorfer S, Piper RC (2008) DOA1/UFD3 plays a role in sorting ubiquitinated membrane proteins into multivesicular bodies. J Biol Chem 283: 21599–21611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U, Cesen MH, Turk B (2013) The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol 5: a008755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005) A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120: 73–84 [DOI] [PubMed] [Google Scholar]
- Ritz D, Vuk M, Kirchner P, Bug M, Schütz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, Meyer H (2011) Endolysosomal sorting of ubiquitinated caveolin‐1 is regulated by VCP/p97 and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 13: 1116–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178 [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC (2006) The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature 443: 780–786 [DOI] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S (2006) Functional division of substrate processing cofactors of the ubiquitin‐selective Cdc48 chaperone. Mol Cell 21: 261–269 [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A (2012) Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging 4: 166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaid S, Brandts CH, Serve H, Dikic I (2013) Ubiquitination and selective autophagy. Cell Death Differ 20: 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, Rapoport TA (2014) Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158: 1375–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Hilt W, Buchberger A, Wolf DH (2011) Cdc48: a power machine in protein degradation. Trends Biochem Sci 36: 515–523 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ohsumi Y (2010) Current knowledge of the pre‐autophagosomal structure (PAS). FEBS Lett 584: 1280–1286 [DOI] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL (2008) Lysine 63‐linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17: 431–439 [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin‐containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I (2012) Fluorescence‐based sensors to monitor localization and functions of linear and K63‐linked ubiquitin chains in cells. Mol Cell 47: 797–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Herman M, Liu L, Simoes S, Acker CM, Figueroa H, Steinberg JI, Margittai M, Kayed R, Zurzolo C, Di Paolo G, Duff KE (2013) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288: 1856–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li L, Jiang H (2016) Doa1 targets ubiquitinated substrates for mitochondria‐associated degradation. J Cell Biol 213: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Blythe EE, Freiberger EC, Mamrosh J, Hebert AS, Reitsma JM, Hess S, Coon JJ, Deshaies RJ (2016) VCP‐adaptor interactions are exceptionally dynamic and subject to differential modulation by a VCP inhibitor. Mol Cell Proteomics 15: 2970–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gui L, Zhang X, Bulfer SL, Sanghez V, Wong DE, Lee Y, Lehmann L, Lee JS, Shih PY, Lin HJ, Iacovino M, Weihl CC, Arkin MR, Wang Y, Chou TF (2015) Altered cofactor regulation with disease‐associated p97/VCP mutations. Proc Natl Acad Sci USA 112: E1705–E1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3