

Abstract
Understanding how complex tissues are formed, maintained, and regenerated through local growth, differentiation, and remodeling requires knowledge on how single‐cell behaviors are coordinated on the population level. The self‐renewing hair follicle, maintained by a distinct stem cell population, represents an excellent paradigm to address this question. A major obstacle in mechanistic understanding of hair follicle stem cell (HFSC) regulation has been the lack of a culture system that recapitulates HFSC behavior while allowing their precise monitoring and manipulation. Here, we establish an in vitro culture system based on a 3D extracellular matrix environment and defined soluble factors, which for the first time allows expansion and long‐term maintenance of murine multipotent HFSCs in the absence of heterologous cell types. Strikingly, this scheme promotes de novo generation of HFSCs from non‐HFSCs and vice versa in a dynamic self‐organizing process. This bidirectional interconversion of HFSCs and their progeny drives the system into a population equilibrium state. Our study uncovers regulatory dynamics by which phenotypic plasticity of cells drives population‐level homeostasis within a niche, and provides a discovery tool for studies on adult stem cell fate.
Keywords: differentiation, hair follicle stem cells, niche, reprogramming, stem cell cultures
Subject Categories: Development & Differentiation, Stem Cells, Systems & Computational Biology
Introduction
Adult somatic stem cells (SCs) fuel tissue renewal, repair, and remodeling. The ability of SCs to tune their proliferation and differentiation rates to the changing needs of their resident tissues is central for the maintenance of organ homeostasis. Given their potency, even incremental alterations in SC behavior should lead to substantial changes in tissue size and architecture. Yet, such changes are strikingly rare, strongly implying that SCs are under tight homeostatic regulation allowing rapid adaptation of the system to disturbances to efficiently restore tissue functions. However, the mechanisms of such population‐level regulation are poorly understood.
Hair follicle stem cells (HFSCs) fuel cyclical bouts of adult hair follicle regeneration. Due to their well‐defined quiescence‐activation cycle, HFSCs represent an excellent paradigm for studying somatic adult SC lineage commitment (Blanpain & Fuchs, 2014). HFSCs are activated in a two‐step process: First, quiescent HFSCs are activated to generate primed HFSCs that in a second step establish a pool of transit‐amplifying cells (TACs; Greco et al, 2009; Hsu et al, 2014b). TACs are a transition state between SCs and their differentiated progeny, and their generation is a rate‐limiting step in SC differentiation (Hsu et al, 2014b).
SCs reside in spatially distinct microenvironments termed niches that consist of neighboring cells, extracellular matrix and signals derived from these compartments. Niches integrate signals to adjust SC behavior to the needs of organisms, to prevent SC depletion and at the same time restrict excessive SC expansion into the surrounding tissue (Morrison & Spradling, 2008; Blanpain & Fuchs, 2014; Scadden, 2014). Even though the critical importance of niches in SC regulation has been established, their complexity in mammals has prevented identification of the precise nature and combination of niche‐derived signals, and hindered mechanistic studies of adult SC regulation. Interestingly, lineage tracing and ablation studies have demonstrated that HFSCs are dispensable for regeneration and that activated progeny re‐populate the ablated SC niche to sustain hair regeneration (Hsu et al, 2011; Rompolas et al, 2013). This suggests that the niche instructs reprogramming of committed progenitors to a SC state, providing a mechanism to ensure robustness of tissue homeostasis. However, the mechanisms of this niche‐directed reprogramming are completely unknown.
Thus, there is a fundamental need to unravel the complex signaling circuitry governing HFSC identity and behavior and to define how the niche instructs HFSC homeostasis. One major obstacle to uncovering these fundamentals has been the lack of a system for ex vivo maintenance of HFSCs in the absence of other heterologous cell types and that also allows precise manipulation and monitoring of HFSC fate decisions. While various 2D cell culture systems for epidermal keratinocytes exist (Barrandon & Green, 1987; Trempus et al, 2003; Blanpain et al, 2004; Jensen et al, 2010; Bilousova & Roop, 2013), methods to maintain and expand bona fide multipotent HFSCs in culture in the absence of feeder cells are lacking, as are in vitro methods to capture the dynamic behavior of HFSCs and their progeny. In the current study, we identify a specific combination of niche factors that for the first time allow expansion and long‐term maintenance of HFSCs. Utilizing this system, we uncover self‐organizing phenotypic plasticity and dynamic bidirectional interconversion between HFSCs and their progeny, providing a cellular mechanism for homeostatic regulation of a SC niche.
Results
Establishment of a HFSC culture system
We aimed at reconstituting the essential components of the HFSC niche in vitro by applying knowledge gained from in vivo studies on signaling within the HFSC niche. Freshly isolated epidermal cells from telogen‐stage mice (P21) contained 5.6 ± 1.2% (± SD) CD34+α6+ HFSCs (Fig 1A). These isolated epidermal cell suspensions were subsequently cultured in standard 2D culture conditions either in a keratinocyte growth medium (KGM) or in FAD medium on a fibroblast feeder layer, which are widely used culture conditions for murine keratinocytes (Watt & Green, 1982; Morgner et al, 2015). Flow cytometry analyses of cells grown under these conditions demonstrated that the CD34+α6+ HFSC population was depleted within 14 days (Fig 1A and B, and Appendix Fig S1A).
Figure 1. Establishment of a HFSC culture system.
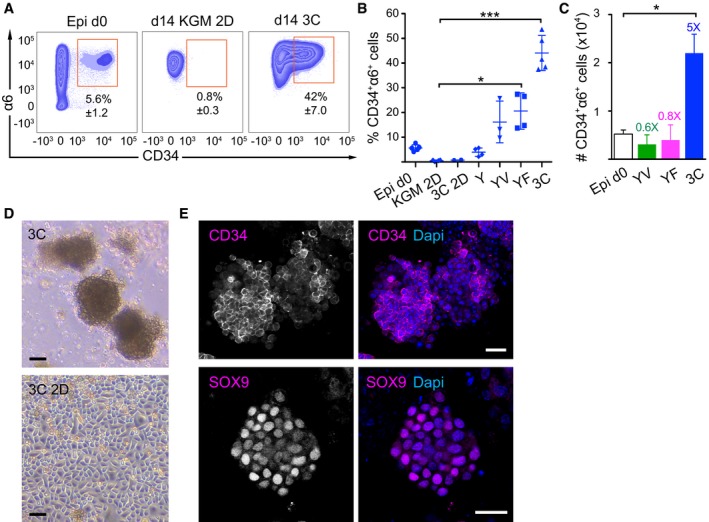
- Representative FACS plots of freshly isolated epidermal cells (Epi d0) and day 14 (d14) cultures of epidermal cells in different growth conditions and media. KGM 2D: 2D in basal KGM; 3C: 3D‐Matrigel in KGM + Y27632 + VEGF‐A + FGF‐2.
- CD34+α6+ cells were quantified by flow cytometry from d14 as in panel (A) (mean ± SD; n = 3–5; *P ≤ 0.05; ***P ≤ 0.001, Kruskal–Wallis/Dunn's post‐test). 3C 2D: 2D in KGM + Y27632 + VEGF‐A + FGF‐2; Y: 3D‐Matrigel in KGM + Y27632; YV: Y + VEGF‐A; YF: Y + FGF‐2; 3C: Y + VEGF‐A + FGF‐2.
- Absolute numbers of CD34+α6+ cells from Epi d0 and d14 cultures as in (A and B). Fold enrichment over Epi d0 is shown (mean ± SEM, n = 3–5; *P ≤ 0.05, Mann–Whitney U‐test).
- Epidermal cells grown for 14 days in the indicated conditions. 3C: 3D‐Matrigel in KGM + Y27632 + VEGF‐A + FGF‐2. 3C 2D: 2D in 3C medium. Scale bars 30 μm.
- Immunofluorescence analysis of 3C cultures showing the expression of the HFSC markers CD34 and SOX9 in a subset of cells. Scale bars 25 μm. See also Appendix Fig S1.
As laminins are important for HFSC maintenance (DeRouen et al, 2010; Morgner et al, 2015), and laminin‐rich basement membrane extracts such as Matrigel have been successfully used to support growth of other epithelial SCs (Sato & Clevers, 2013), we tested whether providing a basement membrane‐like extracellular matrix 3D environment would support HFSC growth in culture. We embedded freshly isolated epidermal cells in Matrigel and cultured them in KGM. As cells failed to survive under these conditions, we included a ROCK inhibitor (Y27632) to prevent anoikis that occurs due to deprivation of cell–cell contacts (Hofmann et al, 2007). These culture conditions provided a small but consistent increase in the population of CD34+α6+ HFSCs compared to cells cultured in 2D (Fig 1B).
Several growth factors, including FGFs, EGF and VEGF‐A, are expressed in cells of the HF and have been shown to regulate their growth (Kozlowska et al, 1998; Ozeki & Tabata, 2002; Doma et al, 2013; Plikus & Chuong, 2014). Combining Y27632 with either FGF‐2 or VEGF‐A in KGM‐3D Matrigel resulted in a further relative increase in CD34+α6+ cells (Fig 1B), but these media did not increase the absolute numbers of CD34+α6+ cells (Fig 1C), suggesting that both conditions promoted HFSC survival and/or growth to a limited extent. In contrast, culturing epidermal cells in KGM‐3D Matrigel containing Y27632, FGF‐2, and VEGF‐A (from now on referred to as 3C) significantly expanded CD34+α6+ cells within the cultured keratinocyte population (Fig 1A–C), inducing a ~sevenfold increase in the relative amount and a fivefold increase in absolute numbers of CD34+α6+ cells after 14 days of culture (Fig 1A–C). FGFs 7, 10, and 18 promoted enrichment of CD34+α6+ cells to the same extent as FGF‐2 (Appendix Fig S1B). The 3D configuration was essential, as there was no enrichment of CD34+α6+ cells when cells were cultured in the 3C medium in standard 2D conditions (3C 2D; Fig 1B). Interestingly, the commonly used keratinocyte medium FAD supplemented with 3C failed to support cell growth even in the presence of 3D Matrigel (Appendix Fig S1C).
Next, we analyzed the morphology of cells grown in 3C conditions. Cultures containing CD34+α6+ cells grew into spheroid cell clusters that did not generate a lumen (Fig 1D), in contrast to other epithelial‐cell 3D culture systems (Lee et al, 1984; Sato et al, 2009). Interestingly, the complex laminin‐rich mixture of matrix proteins present in Matrigel was dispensable as CD34+α6+ cells could also be grown in type I collagen gels using 3C medium (Appendix Fig S1D), most likely due to the expression and deposition of a laminin‐332 meshwork that was observed in both Matrigel and collagen I cultures (Appendix Fig S1E).
Immunofluorescence analysis of 3C cultures revealed expression of the HFSC markers CD34, keratin‐15, and SOX9 (Fig 1E and Appendix Fig S1F and G), the latter a pioneer transcription factor required to establish HFSC identity (Adam et al, 2015). Expression of these markers was not detected in 2D cultures even in the presence of 3C medium (Appendix Fig S1G). Importantly, the cultures comprised of 97.7 ± 0.63% epithelial cells as indicated by staining for the epithelial cell adhesion molecule (EpCAM; Appendix Fig S1H). Altogether, these data show that a 3D environment together with FGF‐2, VEGF‐A, and Y27632 provide an essential set of HFSC‐niche factors that are both necessary and sufficient to enable growth and expansion of cells with HFSC characteristics.
Cells in 3C cultures retain self‐renewing capacity and multipotency
To assess whether cells cultured in 3C conditions maintain their proliferative potential characteristic for HFSCs, colony‐forming assays were performed (Jensen et al, 2010). Cells from 3C cultures subsequently plated on feeders at clonal density gave rise to more colonies that were also larger in size compared to freshly isolated epidermal cells containing 5.6 ± 1.2% HFSCs (Fig 2A). In addition, these colonies contained small, tightly packed, cobblestone‐like colonies (Appendix Fig S2A), characteristic of holoclones observed in feeder‐dependent 2D cultures of purified HFSCs (Blanpain et al, 2004; Greco et al, 2009). These findings indicate that the 3C conditions enrich for epidermal cells with high proliferative potential, a hallmark of HFSCs and their immediate progeny (Blanpain et al, 2004; Greco et al, 2009).
Figure 2. Cells in 3C cultures retain self‐renewing capacity and multipotency.
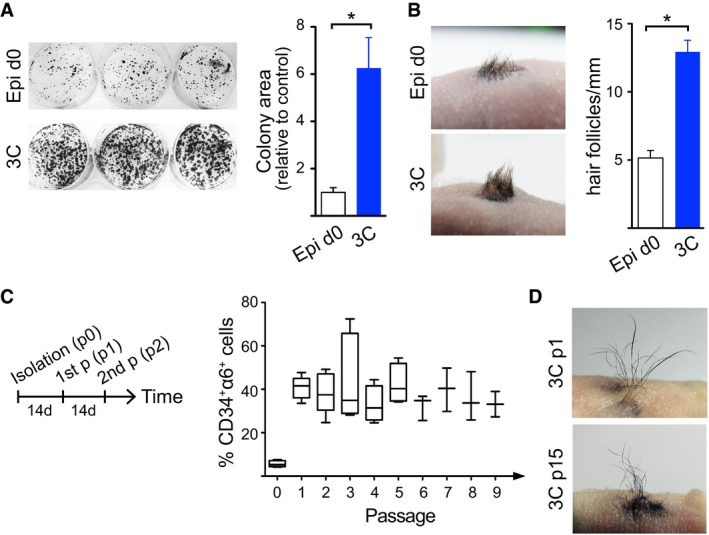
- Colony‐forming assays show increased proliferative potential of cells cultured in 3C compared to freshly isolated epidermal cells (Epi d0; control; mean ± SEM; n = 5; *P ≤ 0.05, Mann–Whitney U‐test). 3C: 3D‐Matrigel in KGM + Y27632 + VEGF‐A + FGF‐2.
- Full‐thickness skin reconstitution assay with freshly isolated epidermal cells (Epi d0) or 3C cultures shows that cells cultured in 3C retain their self‐renewal capacity and multipotency. A representative recipient of four mice/condition is shown. Right panel shows quantification of hair follicle density. Note that 3C cultures give rise to more hair compared to controls (mean ± SEM; n = 4; *P ≤ 0.05, Mann–Whitney U‐test).
- Long‐term culture of cells in 3C was performed as shown in the left panel. 3C cultures maintain a stable population of CD34+α6+ long term (data are shown using a box‐and‐whisker plot: box indicates 25th and 75th percentiles; error bars represent 10th and 90th percentiles; middle line is the median; n = 3–5 independent experiments). p: passage.
- Full‐thickness skin reconstitution assay using 3C cells from passage 1 (p1) and passage 15 (p15) shows that cells cultured in 3C maintain their multipotency and self‐renewal capacity in long‐term cultures. A representative recipient of four mice/condition is shown. See also Appendix Fig S2.
In order to evaluate whether the ex vivo expanded CD34+α6+ cells represented functional HFSCs, we examined their self‐renewal and multipotency in vivo. To this end, we performed full‐thickness skin reconstitution assays (Lichti et al, 1995; Blanpain et al, 2004; Jensen et al, 2010). Cells cultured in 3C fully reconstituted the epidermis and produced hair, and as expected, were slightly more efficient in their regenerative capacity compared to freshly isolated epidermal cells reflecting the difference in HFSC quantity (5.6 ± 1.2% CD34+α6+ in epidermis compared to 42 ± 7% CD34+α6+ in 3C cultures; Fig 2B). This demonstrates that cells cultured in 3C preserve their multipotency and capacity to self‐renew.
Strikingly, 3C cultures could be passaged every 2 weeks into fresh 3D Matrigel for a period of at least 20 weeks without changes in the levels of CD34+α6+ cells (Fig 2C) or their proliferative potential (Appendix Fig S2B). The proportion of CD34+α6+ cells within the 3C cultures remained largely stable from the first passage onward and the cells propagated long‐term were still fully competent to generate hair in full‐thickness skin reconstitution assays (Fig 2D). Moreover, freeze–thaw experiments demonstrated that 3C cultures could be stored frozen and cultured again without loss of the CD34+α6+ population or its ability to generate hair (Appendix Fig S2C and D). Collectively, these data demonstrate that the 3C conditions recapitulate the conditions of a SC niche, enabling maintenance and enrichment of functionally competent cells that display HFSC characteristics. In addition, long‐term 3C cultures sustain a stable equilibrium between CD34+α6+ and CD34−α6+ cells that can be stored frozen without loss of multipotency.
Transcriptomes of cells cultured in 3C resemble in vivo HFSCs
In order to provide an optimal model to uncover novel HFSC biology, cells cultured in 3C should share high resemblance to in vivo HFSCs also on the molecular level. To examine the molecular identity of cells cultured in 3C, we performed RNA sequencing (RNAseq) and compared the transcriptomes of epidermal cells cultured in 3C to freshly isolated epidermal cells that were used to establish the 3C cultures as well as to FACS‐purified in vivo CD34+α6+ HFSCs (Fig 3A and B; Dataset EV1). 3C cultures more closely resembled in vivo HFSCs than the epidermal cell mixtures they were derived from, as shown by Euclidian distance (Fig 3B), Pearson's correlation, and principal component analysis (Appendix Fig S3A and B).
Figure 3. Transcriptomes of cells cultured in 3C resemble in vivo HFSCs.
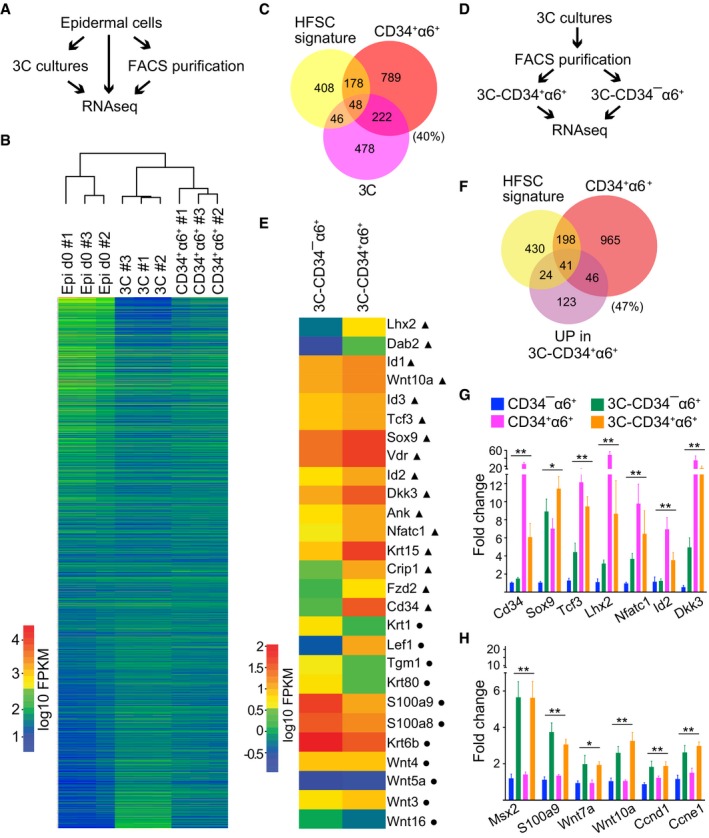
- Schematic workflow of the RNAseq experiment from FACS‐purified CD34+α6+ HFSCs (CD34+α6+), cells cultured in 3C and freshly isolated epidermal cells (Epi d0).
- Heatmap and Euclidian distance dendrogram of quantified transcripts from RNAseq data generated as shown in panel (A). 3C cultures cluster with purified CD34+α6+ HFSCs (n = 3 biological replicates).
- Venn diagram of genes most significantly upregulated in 3C and in purified CD34+α6+ (log2FC > 2, padj < 0.05) together with published HFSC signature shows significant overlap between the three signatures (P = 0.00252451 and P = 3.058928e‐51, hypergeometric distribution).
- Schematic workflow of the RNAseq experiment from FACS‐purified CD34+α6+ and CD34−α6+ cells from 3C cultures.
- Heatmap of selected genes differentially expressed in 3C‐CD34+α6+ and 3C‐CD34−α6+ cells. Note upregulation of multiple key HFSC lineage identity transcription factors and bulge markers (triangles) in 3C‐CD34+α6+ cells, and upregulation of genes implicated in lineage progression in 3C‐CD34−α6+ cells (circles).
- Venn diagram of genes upregulated in 3C‐CD34+α6+ and in purified CD34+α6+ cells (log2FC > 2, padj < 0.05) together with a published HFSC signature shows significant overlap between the three groups (P = 1.381896e‐09 and P = 0.01636903, hypergeometric distribution).
- RT–qPCR analyses of FACS‐purified CD34+α6+ and CD34−α6+ cells from either 3C cultures or from freshly isolated epidermis show upregulation of HFSC identity genes in both freshly isolated and 3C‐CD34+α6+ cells compared to freshly isolated CD34−α6+ progenitors. 3C‐CD34−α6+ cells show intermediated expression (mean ± SEM; n = 4; *P ≤ 0.05, **P ≤ 0.01, Kruskal–Wallis).
- Both 3C‐CD34+α6+ and 3C‐CD34−α6+ cells show upregulation of a panel of cell cycle genes and genes shown to be enriched in HFSC progeny (mean ± SEM; n = 4; *P ≤ 0.05, **P ≤ 0.01, Kruskal–Wallis). See also Appendix Fig S3 and Datasets EV1, EV2, and EV3.
We next examined in which respects the gene expression profiles of 3C cultures resembled and differed from in vivo HFSCs. For this, we computed genewise Euclidian distance calculations of the three conditions (Epi d0, 3C, CD34+α6+ HFSCs) to identify clusters of genes that explained most of the variance in gene expression among the three groups. Gene ontology (GO) term analysis of the top 5 clusters revealed three clusters of genes (clusters 1, 2, and 5; Appendix Fig S3C) that showed comparable, lower expression levels in 3C and CD34+α6+ HFSCs and higher expression levels in Epi d0 (Appendix Fig S3C). These clusters contained GO terms for protein translation, protein transport, and metabolism. This was intriguing, as low protein translation rates have been recently linked to HFSC identity and function (Blanco et al, 2016). Cluster 3 contained genes that displayed an intermediate expression level in 3C (Appendix Fig S3C). These genes were enriched in GO terms for metabolism, translation, and oxidative phosphorylation. Only one cluster (cluster 4; Appendix Fig S3C) was identified for genes that were different in 3C when compared to CD34+α6+ HFSCs, which enriched for genes involved in transcription, tRNA processing, and DNA replication, possibly reflecting the slower replication rates of the quiescent CD34+α6+ HFSCs isolated from telogen‐stage mice compared to cells in culture.
We further analyzed the genes that were similarly expressed in 3C cultures and purified CD34+α6+ HFSCs in comparison with total epidermal cells in more detail. The most significantly upregulated genes (log2FC > 2, padj < 0.05; Dataset EV2) were compiled to generate gene expression enrichment profiles for 3C cultures and purified CD34+α6+ HFSCs, respectively, which were then compared to each other and to a compiled HFSC signature from published datasets (Morris et al, 2004; Tumbar et al, 2004; Lien et al, 2011; Dataset EV2). Despite being a ~50:50 mixture of CD34+α6+ and CD34−α6+ cells, genes enriched in 3C cultures showed significant overlap with genes enriched in purified CD34+α6+ HFSCs (P = 3.058928e‐51) and, importantly, with the published HFSC signature (P = 0.00252451) including key HFSC lineage identity genes such as Cd34, Sox9, Tcf3 (Fig 3C). This provided further support that cells propagated in 3C cultures resemble HFSCs.
We proceeded to explore the differences in gene expression between 3C cultures and purified CD34+α6+ HFSCs. GO term and Gene set enrichment (GSEA; (Subramanian et al, 2005) analyses revealed that genes involved in Wnt signaling, regulation of cell proliferation, extracellular matrix organization, and keratinocyte differentiation were among the most significantly overrepresented among the differentially expressed genes (Appendix Fig S3D–F). Closer examination of the specific genes revealed that negative regulators of Wnt such as Dkk3 and Sfrp1, as well as Wnt ligands shown to be regulated by the BMP signaling pathway (Wnt 5b, 16), were up in CD34+α6+ HFSCs whereas Wnts shown to be enriched in activated HFSCs or their progeny (Wnt 4, 9a, 10a, 10b) were upregulated in 3C (Appendix Fig S3F). Furthermore, positive regulators of the cell cycle such as cyclin D1 and G1 (Ccnd1 and Ccng1), as well as genes associated with keratinocyte differentiation were upregulated in 3C (Appendix Fig S3F). A number of ECM genes were upregulated in CD34+α6+ HFSCs possibly reflecting the excess of ECM proteins present in the Matrigel of the 3C cultures (Appendix Fig S3E and F).
As the transcriptome‐wide expression analyses indicated that cells cultured in 3C conditions closely resembled in vivo HFSCs, but that differences in Wnt and BMP signaling pathway regulators and targets as well as cell cycle regulators were observed, we hypothesized that the 3C cultures consist of a ~50:50 mixture of HFSCs (CD34+α6+ cells) and their progeny (CD34−α6+ cells). To test this, we FACS‐purified CD34+α6+ and CD34−α6+ cells from 3C cultures and performed RNAseq (Fig 3D and E; Dataset EV3). Remarkably, 47% of the genes upregulated in CD34+α6+ cells from 3C cultures (3C‐CD34+α6+) were components of HFCS signatures (Fig 3E and F), including several key lineage identity genes such as Lhx2, Id2, Sox9, Nfatc1, Tcf3, as well as bulge markers Krt15 and Cd34 (Fig 3E). In contrast, genes downregulated in the 3C‐CD34+α6+ population contained markers of early lineage commitment (Fig 3E and Appendix Fig S3I), previously shown to be upregulated in HFSC progeny (TACs, hair germ or hair matrix cells; Greco et al, 2009; Lien et al, 2011).
To validate the sequencing data and to dissect the gene expression differences in 3C cells (3C‐CD34+α6+ and 3C‐CD34−α6+) and freshly isolated in vivo CD34−α6+ (basal progenitors) and CD34+α6+ HFSCs in more detail, we performed RT–qPCR analyses. Importantly, these analyses corroborated the RNAseq results and demonstrated that key HFSC lineage identity genes (Sox9, Tcf3, Lhx2, Nfatc1, Id2, and Dkk3) were highly enriched in 3C‐CD34+α6+ compared to progenitors in vivo (CD34−α6+) or in 3C (3C‐CD34−α6+), and in most cases expressed at comparable levels as in in vivo CD34+α6+ HFSCs, confirming that the 3C‐CD34+α6+ cells are HFSCs (Fig 3G). In line with the RNAseq data, a panel of cell cycle regulators and hair germ/matrix markers showed upregulation in both 3C populations compared to freshly isolated cells (Fig 3H), most likely reflecting their active expansion compared to the telogen‐stage in vivo CD34+α6+ cells (Fig 1C). Interestingly, 3C‐CD34−α6+ cells showed mild upregulation of early lineage commitment genes (Fig 3E and Appendix Fig S3G), but displayed higher expression of the key HFSCs lineage identity genes compared to in vivo CD34−α6+ cells (Fig 3G), suggesting that these cells could represent immediate progeny of 3C‐CD34+α6+ cells.
3C cultures evolve into a stable population equilibrium of HFSCs and non‐HFSCs
Our data so far showed that long‐term 3C cultures established and maintained a stable mixture of CD34+α6+ HFSCs and CD34−α6+ progenitors (Fig 2C). To understand how this was generated, we first analyzed the dynamic evolution of the population equilibrium in 3C cultures generated from freshly isolated epidermal cell mixtures. Time course analyses revealed that after an initial drop in both CD34+α6+ and CD34−α6+ cell numbers, both populations steadily expanded starting from day 2 with CD34−α6+ showing a more rapid increase (Fig 4A). However, after both populations reached peak values by day 8, the numbers of CD34−α6+ declined to establish an approximate 50:50 equilibrium of HFSCs and non‐HFSCs (Fig 4A). This was accompanied by a plateau in the size of the 3C cell clusters (Appendix Fig S4A). The population equilibrium remained constant during passaging (Appendix Fig S4B), similar to what was observed in long‐term cultures (Fig 2C). Interestingly, determination of proliferation rates by EdU incorporation after the cultures had reached equilibrium (d10‐d14) showed that cultured CD34+α6+ cells cycled slightly faster than the CD34−α6+ population (Fig 4B), whereas the apoptosis rates were comparable between the two populations (Appendix Fig S4C).
Figure 4. 3C cultures evolve into a stable population equilibrium of HFSCs and non‐HFSCs.
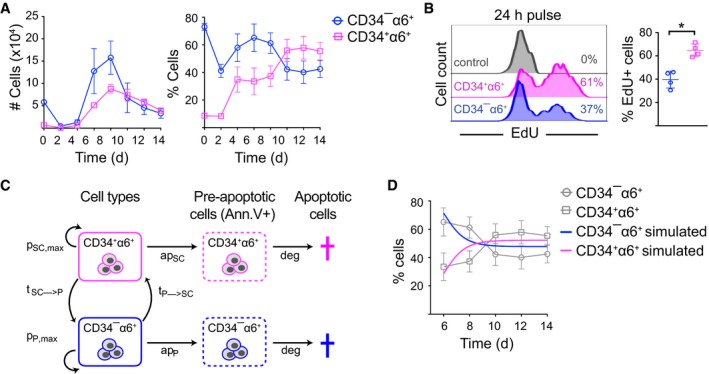
- FACS analysis of CD34+α6+ cells over time shows that 3C cultures reach a ˜50:50 population equilibrium after 10 days of culture (mean ± SEM; n = 3).
- Cells were EdU‐labeled for 24 h (day 10–11) prior to analysis by flow cytometry to quantify proliferating cells. Percentage of EdU+ cells is indicated (n = 4; mean ± SD; *P ≤ 0.05, Mann–Whitney U‐test).
- Schematic illustration of the parameters used in the mathematical model. HFSCs (CD34+α6+) and non‐HFSCs (CD34−α6+) have a maximum proliferation rate of pSC,max and pP,max, respectively. They enter apoptosis with rates apSC and apP and are both degraded with a rate deg. Non‐HFSCs can convert to HFSCs with a rate tP→SC and vice versa with rate tSC→P.
- Simulated population proportions of CD34+α6+ HFSCs and CD34−α6+ non‐HFSCs derived from the mathematical model describing steady‐state cultures. Experimental data from (A) are shown as reference (in gray) (mean ± SEM; n = 3). See also Appendix Fig S4 and Dataset EV4.
To understand how the system would self‐evolve into equilibrium, we modeled the steady‐state dynamics of 3C cultures. To this end, we used a mathematical model based on ordinary differential equations (ODE) to describe the behavior of CD34+α6+ HFSCs and CD34−α6+ non‐HFSCs. In both states, cells are able to proliferate (up to a limiting capacity) and to undergo apoptosis with rates determined by our experiments. Furthermore, cells are capable of switching from one state to the other with conversion rates tSC→P (conversion of HFSCs to non‐HFSCs) and tP→SC (conversion of non‐HFSCs to HFSCs), respectively (Fig 4C). Conversion rates were estimated together with proliferation rates by fitting the model to the time course data (Fig 4A and Appendix Fig S4B) using a maximum‐likelihood (ML) approach (Fig 4D and Appendix Fig S4D and E). The model reflected the observed growth dynamics and, surprisingly, indicated conversion of non‐HFSCs to HFSCs in addition to the expected HFSC to progenitor transition (Appendix Fig S4E; Dataset EV4).
Bidirectional interconversion drives a dynamic self‐organizing equilibrium between HFSCs and non‐HFSCs
To experimentally test the predicted bidirectional interconversion, we FACS‐purified CD34+α6+ and CD34−α6+ (> 98–99% purity) from total epidermal cells and cultured the two populations independently (Fig 5A). Strikingly, both freshly purified CD34+α6+ and CD34−α6+ populations were able to generate a mixture of CD34+α6+ and CD34−α6+ cells (Fig 5A). To analyze the dynamics of the interconversion in more detail, we performed time course experiments where we quantified CD34+α6+ and CD34−α6+ cells and their proliferation rates in 3C cultures established from FACS‐purified CD34+α6+ or CD34−α6+ cells. To prevent the extensive cell loss due to stress caused by the combination of tissue disruption and FACS purification, we used cells precultured in 3C. Interestingly, both purified cell populations self‐organized into ~50:50 equilibrium cultures of CD34+α6+ and CD34−α6+ cells, further substantiating bidirectional interconversion (Fig 5B and C). Pure CD34+α6+ cells established the equilibrium earlier than CD34−α6+ cells (Fig 5B and C), indicating that the CD34+α6+ to CD34−α6+ conversion occurs more frequently than vice versa. Quantification of proliferation and apoptosis rates over time in 3C cultures derived from pure populations of CD34+α6+ or CD34−α6+ cells revealed that, like in the 3C cultures derived from mixed, freshly isolated cells, the apoptosis rates of both populations were similar, whereas after an initial phase of exponential proliferation of both populations, the CD34+α6+ cells proliferated slightly faster, irrespective of their cell type of origin (Appendix Fig S5A and B). Importantly, the generation of a population equilibrium from pure CD34+α6+ cells or pure CD34−α6+ cells could be recapitulated by the mathematical model only under the assumption of bidirectional interconversion (Fig 5D and E, and Appendix Fig S5C–E), although the interconversion appeared slower than in the primary, steady‐state cultures. This indicates that dynamic bidirectional lineage transitions can explain a stable, self‐organizing population equilibrium and that this equilibrium can also be established from pure populations of HFSCs or non‐HFSCs.
Figure 5. Bidirectional interconversion drives a dynamic self‐organizing equilibrium between HFSCs and non‐HFSCs.
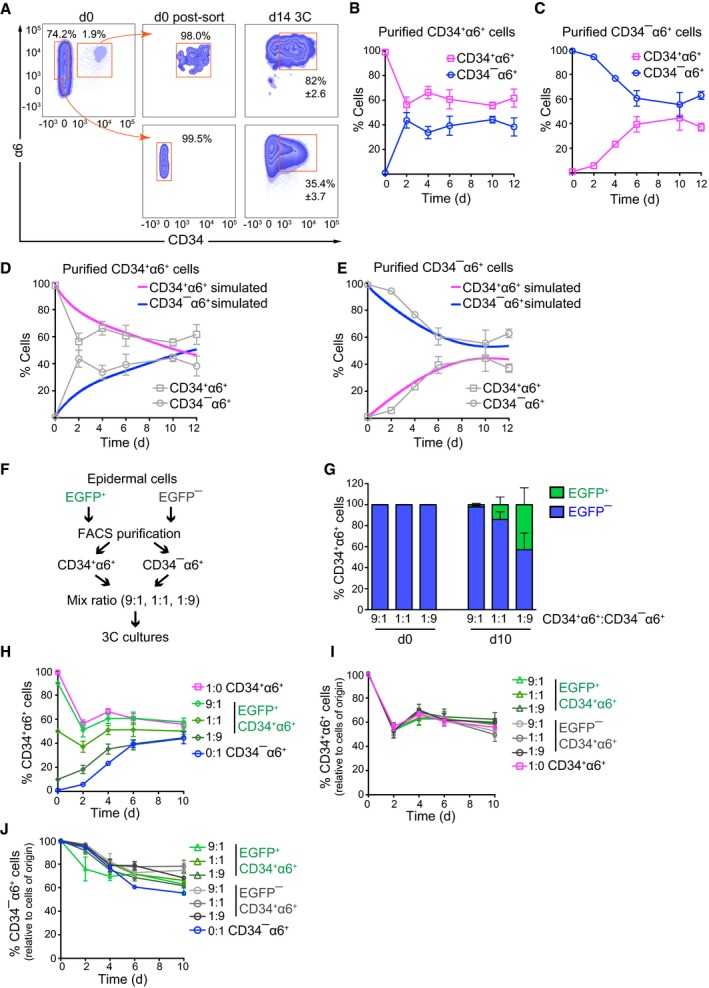
-
ACD34+α6+ HFSCs and CD34−α6+ non‐HFSCs were FACS‐purified from total epidermis (d0) to ≥ 98% purity post‐sorting (d0 post‐sort), and grown 14 days in 3C cultures (d14 3C). A significant population of CD34+α6+ HFSCs and CD34−α6+ non‐HFSCs is generated from both purified populations after 14 days. A representative of three independent experiments is shown (mean ± SD).
-
B, CTime course analyses of 3C cultures established from FACS‐purified populations of CD34+α6+ HFSCs (B) or CD34−α6+ non‐HFSCs (C) from 3C cultures. Both cultures reach population equilibrium between day 6 and 10 (n = 4; mean ± SD).
-
D, ESimulated population proportions of CD34+α6+ HFSCs (D) and CD34−α6+ non‐HFSCs (E) from the mathematical model describing behavior of pure cell populations. The model parameters were estimated from the experimental data in (B, C; shown in gray as reference).
-
FSchematic illustration of lineage‐tracing experiments.
-
GLineage tracing of FACS‐purified CD34+α6+ HFSCs and CD34−α6+ non‐HFSCs from EGFP− and EGFP+ 3C cultures, respectively. CD34+α6+ and CD34−α6+ were mixed at the indicated ratios at d0, and EGFP expression was used to trace the cells of origin over time by flow cytometry. Presence of CD34+α6+ EGFP+ cells in cultures at d10 indicates non‐HFSC to HFSC interconversion.
-
HLineage tracing as in (F) shows that 3C cultures reach equilibrium between day 6 and 10 independent of the initial proportions of HFSCs and non‐HFSCs.
-
IQuantification of the proportion of CD34+α6+ cells that maintain their fate over time (experiments with both EGFP+ and EGFP− labeled cells are indicated).
-
JQuantification of the proportion of CD34−α6+ cells that maintain their fate over time (experiments with both EGFP+ and EGFP− labeled cells are indicated).
Finally, we assessed whether the interconversion rates would depend on the relative amounts of CD34+α6+ and CD34−α6+ cells within the cultures. To this end, we performed lineage‐tracing experiments using the LifeAct‐EGFP transgenic mice (Riedl et al, 2010) as a stable genetic marker for the cell type of origin, and FACS‐purified CD34+α6+ and CD34−α6+ cells from both EGFP‐positive and EGFP‐negative mice. The purified populations were subsequently mixed in various ratios (9:1, 1:1, 1:9) and placed in 3C cultures (Fig 5F). The numbers of EGFP‐expressing cells within the CD34+α6+ and CD34−α6+ populations were subsequently quantified as a function of time. These experiments further substantiated bidirectional interconversion as indicated by the presence of EGFP‐positive CD34+α6+ cells in cultures generated from mixtures of EGFP‐negative CD34+α6+ cells and EGFP‐positive CD34−α6+ cells as well as vice versa (Fig 5G and Appendix Fig S5F). As predicted by the model, these experiments further revealed that the rate by which the ~50:50 equilibrium was reached was independent of the ratios of HFSCs and non‐HFSCs in the initial mixture (Fig 5H and Appendix Fig S5G). Furthermore, the two populations behaved independently of each other, as regardless of the fraction of CD34−α6+ cells present in the initial cell mixtures, ~50% of the CD34+α6+ population converted to CD34−α6+ cells over time (Fig 5I). Likewise, irrespective of the initial fraction of CD34+α6+ cells, ~30% of CD34−α6+ cells converted to CD34+α6+ cells (Fig 5J). This independence of single‐cell conversion rates from the composition of the cell mixture was consistent with the observations that the individual cell clusters were generated by a single cell as revealed by live cell imaging (Appendix Fig S5H and Movie EV1) and that each cluster contained both CD34+α6+ and CD34−α6+ cells (Fig 1E), suggesting that each cell cluster within the 3C cultures behaves in an independent manner.
Intriguingly, de novo generation of CD34+α6+ cells was also observed in 3C cultures when epidermal cells were first completely depleted of CD34+α6+ cells by culturing them for 2 weeks in 2D conditions (Appendix Figs S1A and S5I), followed by passaging and growth in 3C cultures (Appendix Fig S5I). Together, our results indicate that the 3C conditions can drive reprogramming of non‐HFSCs to generate a stable HFSC population, as has previously been achieved by in vivo transplantation (Adam et al, 2015). Our results further indicate that each cell cluster within the 3C cultures represents a single‐cell‐derived system that is capable of generating both HFSCs and non‐HFSCs independently of the relative abundance of these cells in the initial mixture.
Shh and BMP signaling mediate transitions between HFSCs and progeny to control population dynamics
We next sought to dissect the underlying signaling pathways that mediate the bidirectional interpopulation conversion. As TACs have been shown to signal through Shh to maintain HFSC activation during hair growth (Hsu et al, 2014b), we predicted that blocking this pathway should favor the HFSCs state, and therefore lead to decreased numbers of CD34−α6+ cells. Addition of the Shh inhibitor cyclopamine (Taipale et al, 2000) led to a drastic increase in the proportion of CD34+α6+ cells (Fig 6A–C). On the other hand, as BMP signaling is required to maintain quiescent HFSCs (Genander et al, 2014) we hypothesized that inhibiting BMP signaling would activate CD34+α6+ cells to generate differentiated progeny. Inhibition of BMP signaling with dorsomorphin (Yu et al, 2008) led to substantial depletion of the CD34+α6+ population in 3C cultures (Fig 6A–C).
Figure 6. Shh and BMP signaling mediate transitions between HFSCs and progeny to control population dynamics.
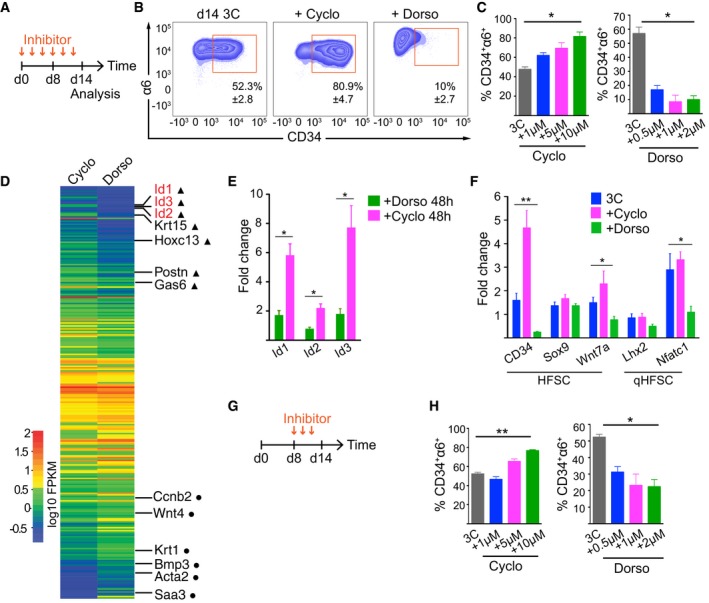
-
A, BInhibitor treatment scheme (A) and FACS plots (B) of 3C cultures treated with the Shh inhibitor cyclopamine or the BMP inhibitor dorsomorphin (mean ± SD; n = 3).
-
CQuantification of experiments as in (B) shows enrichment of HFSCs upon cyclopamine (Cyclo) treatment, whereas dorsomorphin (Dorso) treatment leads to depletion of HFSCs (mean ± SD; n = 3; *P ≤ 0.05, Kruskal–Wallis).
-
DHeatmap of significantly (padj value < 0.05) differentially expressed genes from RNAseq analysis of 3C cultures treated with Cyclo or Dorso for 48 h. HFSC identity genes (triangles) and genes implicated in HFSC lineage progression (circles) are indicated.
-
ERT–qPCR analysis of 3C cultures treated as in (D) confirms that Id 1,2, and 3 are significantly upregulated in Cyclo‐treated cells (mean ± SEM; n = 4; *P ≤ 0.05, Mann–Whitney U‐test).
-
FRT–qPCR analysis of 3C cultures treated as in (A) shows upregulation of HFSC identity genes upon cyclopamine treatment whereas regulators of HFSC quiescence are downregulated upon dorsomorphin treatment (mean ± SEM; n = 4; *P ≤ 0.05, **P ≤ 0.01, Kruskal–Wallis).
-
GInhibitor treatment scheme of 3C cultures treated from day 8 onward.
-
HQuantification of experiments as in (G) (mean ± SD; n = 3–4; *P ≤ 0.05, **P ≤ 0.01, Kruskal–Wallis). See also Appendix Fig S6 and Dataset EV5.
To substantiate that inhibition of Shh and BMP induced fate changes of HFSCs and to identify underlying factors of these changes, we performed RNAseq experiments of 3C cultures treated for 48 h with cyclopamine or dorsomorphin (Fig 6D and Dataset EV5). GO term analysis of differentially expressed genes revealed that genes involved in keratinocyte differentiation, angiogenesis, and wound healing were most significantly altered (Appendix Fig S6A). Interestingly, among a panel of upregulated HFSC identity genes (Gas6, Krt15, Postn, Hoxc13), the Id genes Id1, Id2, Id3 were found to be one of the most significantly upregulated genes in cyclopamine‐treated 3C cultures, also confirmed by RT–qPCR analyses (Fig 6D and E). This was intriguing as these genes represent direct BMP and Smad 1/5 targets, are highly transcribed in HFSCs, downregulated during telogen to anagen transition, as well as functionally implicated in HFSC activation and TAC specification (Genander et al, 2014), confirming that the 3C system recapitulates complex in vivo signaling networks of HFSCs. These early transcriptional changes were accompanied by later changes in the expression of lineage identity genes and markers of HFSC quiescence, as observed by RT–qPCR analyses of 3C cultures treated with the inhibitors for 14 d (Fig 6F). Consistent with the depletion of HFSCs, dorsomorphin‐treated 3C cultures showed downregulation of the lineage identity genes Cd34 and Wnt7a as well as Lhx2 and Nfatc1 (Fig 6F), two master regulators of HFSC quiescence.
We further validated the observed fate changes using functional assays. Importantly, cyclopamine‐treated 3C cultures showed increased colony‐forming capacity compared to untreated cultures (Appendix Fig S6B). These colonies showed densely packed morphology typical of HFSCs (Appendix Fig S6C), confirming that Shh inhibition enriched for HFSCs. Substantiating that BMP inhibition promoted HFSC differentiation, colony‐forming assays from dorsomorphin‐treated 3C cultures showed reduced colony area (Appendix Fig S6B) with loosely packed, larger cells, typical for committed HFSC progeny (Appendix Fig S6C). The specificity of the BMP inhibition was confirmed using another compound, K02288 (Sanvitale et al, 2013), which showed a similar effect (Appendix Fig S6D).
We next asked whether the system could be dynamically tuned to switch between the HFSC and non‐HFSC states by interfering with Shh and BMP signaling once the equilibrium was established. To this end, we cultured epidermal cells for 8 d before adding the inhibitors and analyzed the proportion of CD34+α6+ cells. Once the equilibrium had been established, inhibition of Shh or BMP still shifted the balance toward the CD34+α6+ population in case of Shh inhibition, and toward the CD34−α6+ population in the case of BMP inhibition (Fig 6G and H), indicating that the steady‐state equilibrium critically depends on the activity of these two pathways.
To further test whether Shh and BMP pathways could dynamically reprogram the cell populations, we first treated 3C cultures with dorsomorphin or cyclopamine to enrich for CD34−α6+ or CD34+α6+ cells, respectively, and then passaged the cells into fresh 3C conditions without inhibitors. In both cases, the cultures re‐established the steady‐state equilibrium after removal of the inhibitors (Appendix Fig S6E and F). Remarkably, we could also obtain cultures enriched for CD34+α6+ cells with cyclopamine in dorsomorphin‐treated cells and vice versa (Appendix Fig S6E and F), demonstrating that changes triggered by Shh and BMP inhibition are fully reversible and that the cells grown in 3C maintain their phenotypic plasticity even after 4 weeks in culture.
Altogether our results demonstrated that HFSCs and their immediate progeny can be de novo generated and expanded in 3C cultures, which evolve into a dynamic equilibrium between HFSCs and their progeny through Shh‐ and BMP‐mediated signaling mechanisms.
Discussion
Lineage tracing and ablation studies have demonstrated that HFSCs are dispensable for regeneration and that activated progeny re‐populate the ablated SC compartment to sustain hair regeneration (Hsu et al, 2011; Rompolas et al, 2013). This indicates that the niche can instruct committed progenitors to be reprogrammed to a SC state through currently unknown mechanisms. We have identified a set of factors sufficient for this reprogramming and established an ex vivo niche that allows, for the first time, long‐term maintenance of HFSCs without loss of their multipotency. HFSCs grow as 3D cell clusters without forming any obvious anatomical structure. The absence of lineage commitment and HF‐like morphogenesis points to an indispensible role of dermal resident cells, in particular dermal papilla fibroblasts, in HF morphogenesis (Hsu et al, 2014a; Morgan, 2014), or a requirement of a topological cue from the local tissue environment. But on the other hand it indicates that the minimal essential components of the HFSC niche are ECM proteins, epidermal non‐HFSCs, and a distinct set of growth factors.
Beyond important implications for regenerative medicine, our ex vivo niche enables systematic molecular dissection of SC fate decisions due to the stable proportions of HFSCs and non‐HFSCs. We show that inhibition of BMP signaling leads to a rapid depletion of the HFSC population, whereas inhibition of Shh signaling enriches for these cells, as would be predicted from genetic ablation studies in the mouse (Hsu et al, 2014a). The fact that our 3C culture system recapitulates complex in vivo signaling networks highlights the potential of the system to uncover physiologically relevant signaling networks that govern SC fate.
One of the remarkable features of tissue resident adult somatic SC populations is their ability to remain constant over the majority of adult life. This is particularly striking in high turnover rate tissues, such as the intestine or skin, where SCs are constantly activated to produce differentiated progeny to fuel tissue self‐renewal. We observe that HFSCs and non‐HFSCs establish a self‐evolving stable equilibrium, providing a mechanism to enable tissue robustness.
Strikingly, we observe that a stable HFSC–non‐HFSC equilibrium can evolve from a pure population of non‐HFSCs. This not only corroborates previous studies showing that activated progeny re‐populate an ablated SC niche and subsequently adopt SC fate (Hsu et al, 2011; Rompolas et al, 2013), but for the first time defines a set of factors that can drive this reprogramming. The tunable nature of the 3C culture system now allows precise dissection of the genetic and epigenetic requirements of the reprogramming process. The fact that the cultures of de novo reprogrammed HFSCs evolve into a similar phenotypic equilibrium state as mixed cultures implies that stemness could represent a population trait rather than a trait of individual cells that could be defined by the precise molecular composition of the niche. Experimental and theoretical exploration of this concept will be an important future research avenue.
Materials and Methods
Mice
Epidermal cells were isolated from 21‐day‐old or 48‐day‐old to 60‐day‐old C57BL/6J mice or LifeAct‐EGFP mice (Riedl et al, 2010). 7‐ to 9‐week‐old BALB/c nude mice (CAnN.Cg‐Foxn1nu/Crl; Charles River, Germany) were used as recipients in transplantation experiments. Primary dermal fibroblasts were isolated from 2‐day‐old C57BL/6J mice. Animals were housed and maintained according to FELASA guidelines in the animal facility of the Max Planck Institute for Biology of Ageing, Cologne, Germany. All experiments were approved by local authorities.
Cell culture media and reagents
The following components were used to prepare a keratinocyte growth medium (KGM): MEM (Spinners modification, Sigma), 5 μg/ml insulin (Sigma), 10 ng/ml EGF (Sigma), 10 μg/ml transferrin (Sigma), 10 μM phosphoethanolamine (Sigma), 10 μM ethanolamine (Sigma), 0.36 μg/ml hydrocortisone (Calbiochem), 2 mM glutamine (Gibco), 100 U/mL penicillin and 100 μg/ml streptomycin (Gibco), 8% chelated fetal calf serum (Gibco). The following components were used to prepare the FAD medium: 2 parts of DMEM (Gibco) and 1 part of Ham's F12 (Gibco), 2 mM glutamine (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), 10% chelated fetal calf serum (Gibco), 50 μg/ml vitamin C (sodium L‐ascorbate; Sigma), 10 ng/ml EGF (Sigma), 5 μg/ml insulin (Sigma), 0.5 μg/ml hydrocortisone (Calbiochem), 0.1 nM cholera toxin (Sigma), 180 μM adenine (Sigma). The following media compositions were used: KGM basal medium, medium Y (KGM, 5 μM Y27632; Sigma‐Aldrich), medium YV (KGM, 5 μM Y27632, 20 ng/ml mouse recombinant VEGF‐A; Miltenyi Biotec), medium YF (KGM, 5 μM Y27632, 20 ng/ml human recombinant FGF‐2; Miltenyi Biotec), 3C (KGM, 5 μM Y27632, 20 ng/ml mouse recombinant VEGF‐A, 20 ng/ml human recombinant FGF‐2), 3C‐FAD (FAD, 5 μM Y27632, 20 ng/ml mouse recombinant VEGF‐A, 20 ng/ml human recombinant FGF‐2). Cyclopamine hydrate (Sigma), dorsomorphin (Miltenyi Biotec), and K02288 (Tocris) were added where indicated.
Cell isolation and culture
Keratinocytes were isolated from back skin of mice by incubating skin pieces in 0.8% trypsin (Gibco) for 50 min at 37°C. After separating the epidermis from the underlying dermis, cells were passed through 70‐μm and 45‐μm cell strainers (BD Biosciences) and pelleted at 900 rpm for 3 min. For 2D culture of epidermal cells, tissue culture Petri dishes were coated with a mixture of collagen I (30 μg/ml) and fibronectin (10 μg/ml; both from Millipore) in MEM for 1 h at 37°C after which cells resuspended in KGM or FAD medium were seeded on the coated dishes. For 3D culture, 8 × 104 cells were suspended in 40 μl ice‐cold 1:1 mixture of KGM and growth factor‐reduced Matrigel (Corning) that was dispensed as a droplet in 24‐well cell culture dishes. The suspension was allowed to solidify for 15 min after which it was overlaid with 500 μl of medium. All cultures were incubated at 37°C, 5% CO2. Medium was exchanged the next day after initial seeding and thereafter every second day. For cell passaging, cells were extracted from Matrigel by mechanical homogenization and incubation in 0.5% Trypsin (Gibco), 0.5 mM EDTA, or Accutase (Gibco) for 10 min at 37°C. Cells were passaged every 10–14 days in a 1:3–1:5 ratio in fresh 1:1 KGM:Matrigel.
Proliferative potential assay
Proliferative potential of stem and progenitor cells was assayed as previously described (Jensen et al, 2010). Briefly, 2,000–4,000 cells were plated on 6‐well plates containing Mitomycin C‐treated J2 feeder cells. Cultures were incubated at 37°C, 5% CO2 for 12–14 days during which the medium was replaced every 2 days. Experiments were terminated when colonies reached a sufficient size to be visually identified and quantified. Colonies were fixed with 4% paraformaldehyde for 10 min and stained with 1% crystal violet. Colonies were quantified using the Fiji software. All conditions were performed in triplicates.
Flow cytometry
Single‐cell suspensions were prepared from murine back skin or from cultured cells as described above. Cells were rinsed once with KGM and stained with fluorescently labeled antibodies for 30 min on ice. After two washes with FACS Buffer (2% FCS, 2 mM EDTA, PBS), cells were analyzed in a BD FACSCanto II or sorted in either a BD FACSAria II or a BD FACSAria Fusion. Data were analyzed using FlowJo software version 10. Expression of cell surface markers was analyzed on live cells after exclusion of cell doublets and dead cells using 7AAD (eBioscience) or Fixable Viability Dye eFluor506 (eBioscience). Apoptotic cells were labeled with BV421‐Annexin V (BD Biosciences). The following antibodies were used: eFluor660‐ or FITC‐CD34 (clone RAM34, eBioscience), BV421‐CD34 (clone RAM34, BD), eFluor450‐, PE‐Cy7‐ or FITC‐α6 Integrin (clone GoH3, eBioscience), PE‐Cy7‐EpCAM (clone G8.8, eBioscience).
EdU incorporation assay
Cells were grown in the presence or absence of 9.4 μm EdU (LifeTechnologies) for 24 h before analysis. After preparing single‐cell suspensions, cells were stained with a fixable viability dye eFluor506 followed by antibody staining before fixation in 2% PFA for 10 min at room temperature. Cells were subsequently permeabilized in 0.025% Triton X‐100, PBS for 10 min, and incubated 30 min in EdU reaction cocktail (100 mM Tris pH 8.5, 1 mM CuSO4, 0.5 μM AlexaFluor 488 or 647‐Azide (A10266 or A10277, Life Technologies), 100 mM ascorbic acid). After two washes with PBS, cells were analyzed by flow cytometry.
Immunofluorescence and immunohistochemistry
Cultured HFSCs were rinsed once in PBS, followed by fixation in 2% PFA, PBS for 30 min at RT. Fixed cells were rinsed three times with 100 mM glycine, PBS, then permeabilized and blocked for 2 h at 37°C in 0.3% Triton X‐100, 5% BSA, PBS. Cells were stained with unlabeled or fluorescent primary antibodies in 0.3% Triton X‐100, 1% BSA, PBS overnight at RT. Secondary, fluorescent antibodies were used to detect primary antibody binding, and nuclei were visualized with DAPI. Slides were mounted with Elvanol mounting medium. The following antibodies were used: SOX9 (H‐90, Santa Cruz Biotechnology), keratin‐15 (LHK15, NeoMarkers), eFluor660‐CD34, laminin 332 (gift from R.E. Burgeson). Secondary antibodies anti‐mouse AlexaFluor 488 and anti‐rabbit AlexaFluor 568 were from LifeTechnologies.
Microscopy and image analysis
Fluorescent images were collected by laser scanning confocal microscopy (TCS SP5X; Leica) with 63×, 40× or 20× immersion objectives using LAS X software. All images were recorded sequentially and averaged at least twice. Live imaging was performed using an inverted Axio Observer microscope (Zeiss) equipped with an environment chamber set to 5% CO2, 37°C. Images were acquired using a 10× objective with DIC optics with a rate of 1 frame/10 min for 4 days. Image acquisition was controlled by VisiView Software (Visitron). Image processing (linear brightness and contrast enhancement) was performed with Fiji Software version 2.0.0 or Adobe Photoshop CS5.
Full‐thickness skin reconstitution assay
Transplantation of epidermal cells to assess their multipotency and self‐renewal capacity was performed as previously described (Blanpain et al, 2004; Jensen et al, 2010). Briefly, a mixture of 1–5 × 105 freshly isolated epidermal cells or 3C cells together with 5 × 106 freshly isolated neonatal fibroblasts was injected into a silicon chamber inserted into a full‐thickness wound on the back skin of nude mice. The chamber was removed a week after transplantation, and hair growth was monitored for the following 2–4 weeks.
RNA sequencing
Total RNA was isolated using the RNeasy Plus Micro Kit after which the RNA quality was determined using an Agilent 2200 TapeStation. Libraries were prepared with NEBNext Ultra Directional RNA Library Prep Kit (New England Biolabs) followed by sequencing with HiSeq 2500 (Illumina) from three biological replicates/condition.
After quality control and preprocessing, reads were mapped to the Mus musculus reference genome (build GRCm38_79), followed by differential gene expression analysis using Cufflinks (version 2.2.1). Transcripts regulated ≥ 2 log2 fold change and with q‐value ≤ 0.05 were defined as “gene expression enrichment profiles” for a given condition. Genewise Euclidean distances were calculated in R v3.2.4. A cutoff of 5 was applied to select the five top clusters. Gene ontology term enrichment analyses were performed using DAVID (Huang da et al, 2009). Gene set enrichment analysis was performed on a pre‐ranked gene list (ranked according to log2 fold change) and compared to the Broad Institute Molecular Signatures Database collection of chemical and genetic perturbations (C2) using the Web‐based tool available from the Broad Institute (Subramanian et al, 2005). Enrichments with an FDR value < 0.25 were considered significant. The data have been submitted to NCBI‐GEO (GSE76779).
qPCR
RNA was isolated using the RNeasy Plus Mini Kit (Qiagen), after which cDNA was synthesized using the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems) or SuperScript VILO (LifeTechnologies). qPCR was performed on the StepOne Plus Real Time PCR System (Applied Biosystems) or CFX384 Touch Real Time PCR Detection System (Bio‐Rad) using the DyNAmo ColorFlash SYBR Green Mix (Thermo Fisher). Gene expression changes were calculated following normalization to Actb or to ERCC spike‐in reference RNA (Ambion‐LifeTechnologies) using the comparative Ct (cycle threshold) method. Primer sequences used are available upon request.
Mathematical modeling and simulations
A coupled ordinary differential equation (ODE) model was used to describe the cell numbers within four compartments, namely HFSCs and non‐HFSCs, as well as their pre‐apoptotic counterparts (corresponding to the Annexin V+ subpopulations). HFSCs and non‐HFSCs follow a logistic growth dynamic with maximum proliferation rates pSC,max and pP.max, respectively, and a carrying capacity K (see Appendix Supplementary Methods for details). Cells of both compartments enter into apoptosis with rates apSC and apP, respectively, before they are finally removed from the system after an average residence time in the pre‐apoptotic state of 12 h (corresponding to a rate of deg = 1/12 cells/h). Conversion rates tSC→P and tP→SC between the HFSCs and non‐HFSCs states as well as the carrying capacity K and the proliferation rates were estimated from the time course data in Fig 4A and Appendix Fig S4A by minimizing the residual sum of squares of simulated and measured data. Simulations were implemented and calculated using the software R (version 3.2.2, 2015‐08‐14) and the R package “bbmle” (R Development Core Team, 2008; Ben Bolker and R Development Core Team, 2016). For a detailed description and model parameters see Appendix Supplementary Methods and Dataset EV4.
Statistical analysis
Statistical analyses were performed using R or GraphPad Prism software (GraphPad, version 5.0). Statistical significance was determined by the Mann–Whitney U‐test, Kruskal–Wallis with Dunn's post‐test or hypergeometric distribution probability as indicated in the corresponding figure legends.
Author contributions
CAC‐M designed and performed most of the experiments and analyzed data, MK and IG designed and performed the mathematical modeling, CN advised with transplantation experiments, SAW conceived and supervised the study, designed experiments, and analyzed data. CAC‐M and SAW wrote the paper. All authors commented and edited the manuscript.
Conflict of interest
A patent application describing the HFSC culture system has been filed to the European Patent Office.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Movie EV1
Review Process File
Acknowledgements
We thank Carien Niessen and Saman Honarnejad for critical reading of the manuscript, Nadine Bremicker, Sarah Löck, and Ramona Hoppe for technical assistance, Jorge Boucas for help with bioinformatics analyses, and the FACS & Imaging, Comparative Biology, and the Bioinformatics Core Facilities of MPI for Biology of Ageing and the Max Planck Genome Centre Cologne for support. We also thank members of the Wickström laboratory for their input. This work was supported by the Max Planck Society, the Max Planck Förderstiftung (to SAW), the Deutsche Forschungsgemeinschaft through WI 4177 (to SAW) and through SFB 829 (to CN and SAW), and the German Federal Ministry of Research and Education (grant number 031A315 “MessAge”, to MK and IG).
The EMBO Journal (2017) 36: 151–164
See also: A Sánchez‐Danés & C Blanpain (January 2017)
References
- Adam RC, Yang H, Rockowitz S, Larsen SB, Nikolova M, Oristian DS, Polak L, Kadaja M, Asare A, Zheng D, Fuchs E (2015) Pioneer factors govern super‐enhancer dynamics in stem cell plasticity and lineage choice. Nature 521: 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrandon Y, Green H (1987) Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA 84: 2302–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Bolker and R Development Core Team (2016) bbmle: Tools for General Maximum Likelihood Estimation. R package version 1.0.18. URL http://CRAN.R-project.org/package=bbmle
- Bilousova G, Roop DR (2013) Generation of functional multipotent keratinocytes from mouse induced pluripotent stem cells. Methods Mol Biol 961: 337–350 [DOI] [PubMed] [Google Scholar]
- Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortes‐Garrido R, Gkatza N, Dietmann S, Frye M (2016) Stem cell function and stress response are controlled by protein synthesis. Nature 534: 335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E (2004) Self‐renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118: 635–648 [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E (2014) Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 344: 1242281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRouen MC, Zhen H, Tan SH, Williams S, Marinkovich MP, Oro AE (2010) Laminin‐511 and integrin beta‐1 in hair follicle development and basal cell carcinoma formation. BMC Dev Biol 10: 112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma E, Rupp C, Baccarini M (2013) EGFR‐ras‐raf signaling in epidermal stem cells: roles in hair follicle development, regeneration, tissue remodeling and epidermal cancers. Int J Mol Sci 14: 19361–19384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genander M, Cook PJ, Ramskold D, Keyes BE, Mertz AF, Sandberg R, Fuchs E (2014) BMP signaling and its pSMAD1/5 target genes differentially regulate hair follicle stem cell lineages. Cell Stem Cell 15: 619–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco V, Chen T, Rendl M, Schober M, Pasolli HA, Stokes N, Dela Cruz‐Racelis J, Fuchs E (2009) A two‐step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 4: 155–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann C, Obermeier F, Artinger M, Hausmann M, Falk W, Schoelmerich J, Rogler G, Grossmann J (2007) Cell‐cell contacts prevent anoikis in primary human colonic epithelial cells. Gastroenterology 132: 587–600 [DOI] [PubMed] [Google Scholar]
- Hsu YC, Pasolli HA, Fuchs E (2011) Dynamics between stem cells, niche, and progeny in the hair follicle. Cell 144: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Li L, Fuchs E (2014a) Emerging interactions between skin stem cells and their niches. Nat Med 20: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Li L, Fuchs E (2014b) Transit‐amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 157: 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Jensen KB, Driskell RR, Watt FM (2010) Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc 5: 898–911 [DOI] [PubMed] [Google Scholar]
- Kozlowska U, Blume‐Peytavi U, Kodelja V, Sommer C, Goerdt S, Majewski S, Jablonska S, Orfanos CE (1998) Expression of vascular endothelial growth factor (VEGF) in various compartments of the human hair follicle. Arch Dermatol Res 290: 661–668 [DOI] [PubMed] [Google Scholar]
- Lee EY, Parry G, Bissell MJ (1984) Modulation of secreted proteins of mouse mammary epithelial cells by the collagenous substrata. J Cell Biol 98: 146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichti U, Scandurro AB, Kartasova T, Rubin JS, LaRochelle W, Yuspa SH (1995) Hair follicle development and hair growth from defined cell populations grafted onto nude mice. J Invest Dermatol 104: 43S–44S [DOI] [PubMed] [Google Scholar]
- Lien WH, Guo X, Polak L, Lawton LN, Young RA, Zheng D, Fuchs E (2011) Genome‐wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 9: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BA (2014) The dermal papilla: an instructive niche for epithelial stem and progenitor cells in development and regeneration of the hair follicle. Cold Spring Harb Perspect Med 4: a015180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgner J, Ghatak S, Jakobi T, Dieterich C, Aumailley M, Wickström SA (2015) Integrin‐linked kinase regulates the niche of quiescent epidermal stem cells. Nat Commun 6: 8198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RJ, Liu Y, Marles L, Yang Z, Trempus C, Li S, Lin JS, Sawicki JA, Cotsarelis G (2004) Capturing and profiling adult hair follicle stem cells. Nat Biotechnol 22: 411–417 [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Spradling AC (2008) Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 132: 598–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeki M, Tabata Y (2002) Promoted growth of murine hair follicles through controlled release of vascular endothelial growth factor. Biomaterials 23: 2367–2373 [DOI] [PubMed] [Google Scholar]
- Plikus MV, Chuong CM (2014) Macroenvironmental regulation of hair cycling and collective regenerative behavior. Cold Spring Harb Perspect Med 4: a015198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2008) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; ISBN 3‐900051‐07‐0, URL http://www.R-project.org [Google Scholar]
- Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, Bradke F, Massberg S, Aszodi A, Sixt M, Wedlich‐Söldner R (2010) Lifeact mice for studying F‐actin dynamics. Nat Methods 7: 168–169 [DOI] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Greco V (2013) Spatial organization within a niche as a determinant of stem‐cell fate. Nature 502: 513–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, Wang Y, Triffitt JT, Cuny GD, Yu PB, Hill CS, Bullock AN (2013) A new class of small molecule inhibitor of BMP signaling. PLoS ONE 8: e62721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Sato T, Clevers H (2013) Growing self‐organizing mini‐guts from a single intestinal stem cell: mechanism and applications. Science 340: 1190–1194 [DOI] [PubMed] [Google Scholar]
- Scadden DT (2014) Nice neighborhood: emerging concepts of the stem cell niche. Cell 157: 41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406: 1005–1009 [DOI] [PubMed] [Google Scholar]
- Trempus CS, Morris RJ, Bortner CD, Cotsarelis G, Faircloth RS, Reece JM, Tennant RW (2003) Enrichment for living murine keratinocytes from the hair follicle bulge with the cell surface marker CD34. J Invest Dermatol 120: 501–511 [DOI] [PubMed] [Google Scholar]
- Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, Fuchs E (2004) Defining the epithelial stem cell niche in skin. Science 303: 359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FM, Green H (1982) Stratification and terminal differentiation of cultured epidermal cells. Nature 295: 434–436 [DOI] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 4: 33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Movie EV1
Review Process File