

Abstract
Removal of ruptured lysosomes by autophagy is one of the mechanisms by which cells alleviate detrimental consequences of lysosome leakage and may prevent the initiation of signaling cascades that lead to cell death. In this issue of The EMBO Journal, Papadopoulos et al (2017) report an essential role of p97 and its cofactors in autophagic clearance of damaged lysosomes and provide evidences for the relevance of p97 in neurodegenerative conditions.
Subject Categories: Autophagy & Cell Death; Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Lysosomes constitute the main compartment for terminal degradation of macromolecules from secretory, endocytic, autophagic, and phagocytic pathways. Their degradative properties are executed by various soluble acid hydrolases, whose combined actions maintain a constant removal of unwanted cellular material (Settembre et al, 2013). However, potent hydrolytic capacity of lysosomal enzymes can also cause a threat to the cellular integrity when released to the cytosol upon rupture of the lysosomal membrane, leading to cathepsin‐induced cell death. Lysosome membrane permeabilization (LMP) can occur due to endogenous stressors such as ROS, osmotic lysis, as a result of endosome to cytosol host cell invasion by bacteria and viruses, or as a consequence of direct membranolytic activity of incorporated chemical compounds and mineral crystals (Repnik et al, 2014). Autophagy is one of the potential mechanisms implicated in lysosome quality control and may prevent excessive cell death during LMP by sequestering membrane remnants (Hung et al, 2013; Maejima et al, 2013; Khaminets et al, 2016).
Upon endosomal or lysosomal damage, β‐galactoside conjugates, which normally are located within lumen‐oriented membrane leaflets, get exposed to the cytosol and are recognized by galectins. Galectin‐3 (Gal3) marks membranes damaged by lysosomotropic agent L‐leucyl‐L‐leucine methyl ester (LLOMe) and, together with ubiquitin, recruit the autophagy receptor p62 and assembles core autophagy factors Atg16L, ULK1, and Atg9L1 to promote engulfment of cargo by LC3‐decorated autophagosomes (Maejima et al, 2013). Recently, also tripartite motif (TRIM) proteins have been implicated in selective autophagy. In particular, TRIM16 was reported to be involved in the response to endolysosomal damage by activating the autophagy machinery and being an instrumental component for optimal ubiquitination of the lysosomal membrane (Chauhan et al, 2016). This suggests that different factors cooperate in recognition of membrane remnants upon endolysosomal damage and regulation of their autophagic removal. In this issue, Papadopoulos et al (2017) provide an additional layer of complexity to the cellular response to endolysosomal damage, termed by the authors Endo‐Lysosomal Damage Response, ELDR. They describe an essential role of p97 and its cofactors UBX domain containing protein 1 (UBXD1), phospholipase A‐2‐activating protein (PLAA), and the deubiquitinating enzyme (DUB) YOD1 in autophagic clearance of ruptured lysosomes (Fig 1).
Figure 1. p97 with its cofactors drive clearance of damaged lysosomes by autophagy.
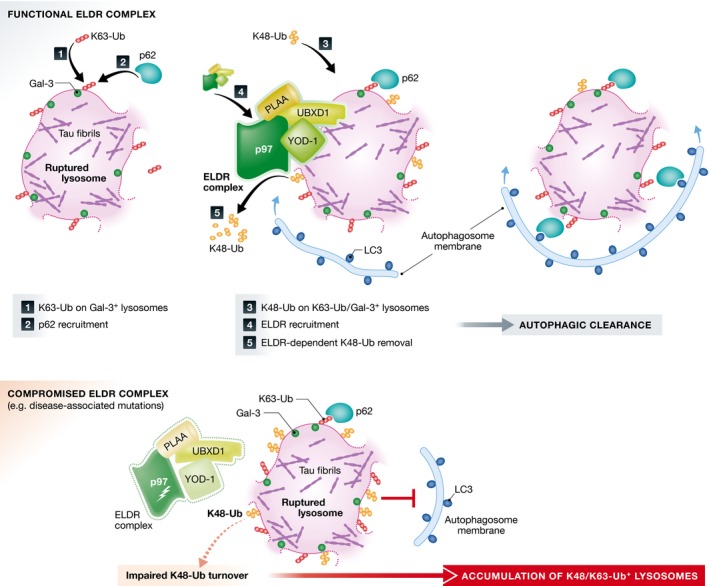
Upon membrane rupture, for example, caused by endocytosed tau fibrils, galectin‐3 marks membrane remnants and K63‐Ub of membrane proteins mediate p62 recruitment to initiate the autophagic response. A subset of lysosomes is also marked by K48‐Ub that needs to be removed by ELDR components to promote autophagosome formation and lysosome clearance. Compromised ELDR function, for example, due to p97 mutations that cause inclusion body myopathy and neurodegeneration, results in impaired turnover of K48‐Ub and failure of lysosome clearance.
In particular, the authors utilize two models of endolysosomal membrane damage: LLOMe treatment of cultured cells and neurotoxic aggregates formed from fragments of tau fibrils that are capable of inducing aggregation of endogenous tau when added to the culture medium. Following LLOMe treatment, p97, UBXD1, YOD1, and to some extent PLAA were recruited to lysosomes. Depletion of p97 resulted in a slower rate of autophagic clearance of damaged lysosomes as assessed by an increased number of Gal3‐labeled vesicles. The persistent accumulation of Gal3‐positive vesicles was also observed in the neurotoxic aggregates‐mediated model of lysosome rupture and in LLOMe‐treated cells when other ELDR components were depleted. This effect seems to be a consequence of impairment of later steps of autophagy rather than autophagy initiation or LC3 activation, which is consistent with previous reports of defective maturation of autophagosomes upon p97 knockdown (Ju et al, 2009; Tresse et al, 2010).
To better understand the ELDR process, the authors analyzed the formation of the p97/UBXD1/PLAA/YOD1 complex and showed that it strongly depends on YOD1 binding to ubiquitin.
K63‐linked ubiquitin chains (K63‐Ub) have been reported to be a prevalent signal for the recruitment of the selective autophagy machinery to cargoes including damaged organelles (Khaminets et al, 2016). Thus, the observation of dysfunctional autophagic clearance of lysosomes upon YOD1 depletion, a DUB with activity toward K48‐linked chains (K48‐Ub), but not K63‐Ub, has driven the authors to monitor ubiquitination patterns on ruptured lysosomes over time after LLOMe treatment. They found that a subset of damaged lysosomes is decorated with both K48‐ and K63‐Ub. Interestingly, these events are temporally separated as K63‐Ub chains are detected early after damage and precede the appearance of K48‐Ub. While K63‐Ub species coincide with the recruitment of the canonical autophagy machinery members, p62 and LC3, ELDR components are recruited selectively to lysosomes decorated with both K48‐ and K63‐Ub suggesting that ELDR is recognizing K48‐Ub chains.
In the final set of experiments, the authors demonstrate that the recruitment of the ELDR components leads to the removal of K48‐Ub, which depends on both the ATPase activity of p97 and the deubiquitinating activity of YOD1. The removal of K48‐Ub is a prerequisite for the engulfment of damaged lysosomes by LC3‐decorated autophagosomes and for their efficient clearance.
Interestingly, a requirement of p97‐dependent extraction of ubiquitinated membrane proteins for efficient autophagic removal seems to be a common feature for both lysosomes and mitochondria. Upon mitochondrial depolarization, the E3 ligase Parkin prevents or delays re‐fusion of mitochondria by ubiquitinating Mitofusins, large GTPases that mediate mitochondrial fusion (Tanaka et al, 2010). p97 mediates extraction of these ubiquitinated outer mitochondrial membrane (OMM) proteins promoting their subsequent degradation, even though different p97 cofactors are involved. The delayed re‐fusion of mitochondria allows for efficient autophagic removal of the organelles. An interesting question raised by the work of Papadopoulos et al (2017) is whether the K48‐Ub on the lysosomal membrane modulates lysosome dynamics similarly as it does for mitochondria. Moreover, the identity of potential targets and of the E3 Ub ligase would also allow a better understanding of this process.
Findings of Papadopoulos et al (2017) not only reveal an essential function of p97 and its cofactors in the autophagic clearance of ruptured lysosomes, providing new insight into the molecular pathway, but also point out toward physiological consequences of its impairment. Mutations in p97 cause inherited pleiotropic degenerative disorders affecting muscles, bones, and the nervous system and associated with inclusion body myopathy, Paget disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis (Meyer & Weihl, 2014). The authors report defects in lysosome clearance in mouse embryonic fibroblasts expressing a disease‐associated p97 allele as well as in stable inducible cell lines in which other mutant p97 variants were expressed. Also, muscle tissue from patients carrying p97 mutations, in contrast to control muscle, accumulates ruptured lysosomes. Thus, failure of lysosome clearance seems to contribute to pathogenesis of multisystem proteinopathies. Since defective lysosome integrity has also been proposed to be a prominent feature in other classes of neurodegenerative conditions like tauopathies and Alzheimer disease (Piras et al, 2016), the new pathway described by Papadopoulos et al (2017) may be of much more general importance.
See also: C Papadopoulos et al (January 2017)
References
- Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, Choi SW, Peters R, Mandell M, Bruun J‐A, Johansen T, Deretic V (2016) TRIMs and galectins globally cooperate and TRIM16 and galectin‐3 co‐direct autophagy in endomembrane damage homeostasis. Dev Cell 39: 13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung Y‐H, Chen LM‐W, Yang J‐Y, Yuan Yang W (2013) Spatiotemporally controlled induction of autophagy‐mediated lysosome turnover. Nat Commun 4: 2111 [DOI] [PubMed] [Google Scholar]
- Ju J‐S, Fuentealba RA, Miller SE, Jackson E, Piwnica‐Worms D, Baloh RH, Weihl CC (2009) Valosin‐containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol 187: 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32: 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127: 3877–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, Poehler R, Dressler A, Fengler S, Arhzaouy K, Lux V, Ehrmann M, Weihl CC, Meyer H (2017) VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36: 135–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piras A, Collin L, Grüninger F, Graff C, Rönnbäck A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of post‐mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnik U, Hafner Česen M, Turk B (2014) Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion 19(Pt A): 49–57 [DOI] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D‐F, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao T‐P, Dantuma NP, Taylor JP (2010) VCP/p97 is essential for maturation of ubiquitin‐containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]