


Abstract
Clustered DNA damages—two or more lesions on opposing strands and within one or two helical turns—are formed in cells by ionizing radiation or radiomimetic antitumor drugs. They are hypothesized to be difficult to repair, and thus are critical biological damages. Since individual abasic sites can be cytotoxic or mutagenic, abasic DNA clusters are likely to have significant cellular impact. Using a novel approach for distinguishing abasic clusters that are very closely spaced (putrescine cleavage) or less closely spaced (Nfo protein cleavage), we measured induction and processing of abasic clusters in 28SC human monocytes that were exposed to ionizing radiation. γ-rays induced ∼1 double-strand break: 1.3 putrescine-detected abasic clusters: 0.8 Nfo-detected abasic clusters. After irradiation, the 28SC cells rejoined double-strand breaks efficiently within 24 h. In contrast, in these cells, the levels of abasic clusters decreased very slowly over 14 days to background levels. In vitro repair experiments that used 28SC cell extracts further support the idea of slow processing of specific, closely spaced abasic clusters. Although some clusters were removed by active cellular repair, a substantial number was apparently decreased by ‘splitting’ during DNA replication and subsequent cell division. The existence of abasic clusters in 28SC monocytes, several days after irradiation suggests that they constitute persistent damages that could lead to mutation or cell killing.
INTRODUCTION
Clustered damages—two or more closely spaced abasic sites, oxidized bases or strand breaks on opposing DNA strands—are hypothesized to be highly significant biological lesions because they may be difficult for cells to repair (1,2). An attempted simultaneous repair of the constituent opposing lesions of the cluster could produce a double-strand break (DSB). In fact, Escherichia coli and rodent cells do generate de novo DSBs that could result from processing of bistranded clustered DNA lesions (3–6). Alternatively, clusters could be resistant to processing by glycosylases or endonucleases, as shown for synthetic oligonucleotides that contain clusters of specific composition and configuration (7–10). Such repair-resistant clusters could persist for a substantial time after irradiation.
Abasic sites (apurinic and apyrimidinic sites, AP) are frequent cellular DNA lesions that are formed by spontaneous base loss, by exposure to radiation or antitumor drugs (11–13), or as intermediates during base excision repair (BER) of oxidized, deaminated or alkylated bases (14). Human cells repair isolated abasic DNA sites by either ‘short-patch’ or ‘long-patch’ BER (15). Since unrepaired AP sites can be both toxic and highly mutagenic (16,17), persistent clusters that contain AP sites could have serious consequences for the cell. Previous studies showed that X- or γ-rays induce different types of clustered DNA damages such as oxypyrimidine, oxypurine and abasic clusters in human cells (18,19). Approximately twice as many non-DSB clustered DNA damages (oxidized base clusters and abasic clusters) as DSB are induced in cells by sparsely ionizing radiation (X- or γ-rays).
To determine the fate of abasic clusters in human cells, we determined first their induction by γ-rays. We then followed cellular processing of these clusters by human monocytes as well as their in vitro repair by cell extracts. We used a novel approach that distinguishes between clusters that contain very closely spaced lesions and lesions that were less densely spaced. Specifically, putrescine (PUTR) can cleave high-density abasic clusters (AP sites on opposite strands 0–3 bp apart). In contrast, Nfo protein cleaves such high-density clusters poorly, although it does cleave as well as PUTR at less dense clusters (opposing AP sites ≥5 bp apart) (10). Some of the clusters appear to be removed by active cellular repair. However, a large fraction was not repaired but was apparently decreased by ‘splitting’ upon DNA replication. A fraction of both abasic cluster classes could be detected even after several days. Thus abasic clusters comprise persistent clusters that could either be lethal or lead to genomic instability.
MATERIALS AND METHODS
Cell culture
Human monocytes (28SC, American Type Culture Collection CRL-9855) were grown on Iscove's modified Dulbecco's medium (IMDM, Gibco/BRL, Grand Island, NY), supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) without antibiotics as described previously (20). They were ascertained to be free of mycoplasma by periodic testing (Bionique, Saranac Lake, NY).
Irradiation and preparation of human DNA
Cells (∼1 × 106 cells/ml) in complete medium were exposed to 137Cs γ-rays at the Brookhaven Controlled Environment Radiation Facility (dose rates, 1–2 Gy/min) either (i) for measurement of initial damage levels—they were chilled on ice for 15 min to minimize repair, irradiated on ice and harvested immediately after irradiation by immersion in liquid nitrogen or (ii) for measurements of repair—they were irradiated at room temperature, placed at 37°C immediately after irradiation for further incubation, then harvested as described. For measurements of extended repair, media were changed every 4 days. A certified operator of the facility performed dosimetry by using thermoluminescent lithium fluoride chips.
Frozen cells were thawed rapidly and EDTA was added to 50 mM; the cells were then mixed with an equal volume of 1.8% InCert agarose [(FMC, Rockland, ME) prepared in lysis buffer L (100 mM EDTA, 20 mM NaCl, 10 mM Tris–HCl, pH 8.3) and kept at 65°C], and then formed into plugs. To the solidified plugs (∼1.5 μg DNA each), lysis solution [1 mg/ml proteinase K (Roche Molecular Biochemicals, Indianapolis, IN) and 1.5% n-lauroylsarcosine in L-buffer] were added. The plugs were incubated for 2 h at 4°C, then for 72 h at 37°C with daily changes of lysis solution, and 0.5% reductions of n-lauroylsarcosine concentration. All solutions were sterile, freshly prepared and bubbled with argon for 20 min, where practical, before use (18). The plugs were washed twice (30 min each) with 3 ml of NTE buffer (150 mM NaCl, 10 mM Tris–HCl and 0.1 mM EDTA, pH 8) that contained 40 μg/ml of phenylmethylsulfonyl fluoride. They were washed five times (1 h each) with 3 ml of ice-cold NTE buffer, then six times with 3 ml of AscI reaction buffer (20 mM Tris–acetate, 10 mM magnesium acetate and 50 mM potassium acetate, pH 7.9) for 24 h at 4°C. They were finally placed in 300 μl of reaction buffer that contained 1 mM DTT. AscI (6 U/plug, New England Biolabs, Beverly, MA) was added; plugs were then incubated for 2 h on ice, then for 16 h at 37°C. Two units of additional AscI were added, and the reactions were incubated for 2 h at 37°C. The AscI reaction buffer was removed, 1 ml of ice-cold mix of 70 mM HEPES–KOH, 100 mM KCl and 100 mM EDTA, pH 7.6 were added, and the plugs were washed twice (1 h each) with this buffer on ice.
Enzyme or polyamine treatment of human DNA
For abasic cluster measurement (10), plugs were incubated in 500 μl of the appropriate reaction buffer (see below) for 24 h with six changes of buffer at 4°C. E.coli Nfo protein (Endonuclease IV) was purified from a strain that overexpressed the Nfo gene on the plasmid pET24-Eco-Nfo (the kind gift of Dr Richard Cunningham) by streptomycin and ammonium sulfate precipitations, heating (21), centrifugation, and chromatography of the supernatant on Q Sepharose and Heparin HiTrap (both from Amersham, Piscataway, NJ) columns. Its specific activity (1.2 × 1014 AP sites/μg/min) was determined by cleavage of supercoiled pUC18 DNA (2.68 kb, Bayou Biolabs, Harahan, LA) that contained an average of 0.7 abasic sites/molecule. Its non-specific activity that was assessed on undamaged pUC18 DNA, was found to be negligible. Recombinant human Ape1 protein was purified and used essentially as described (22).
Duplicate plug slices (∼500 ng DNA) were incubated in 200 μl of 70 mM HEPES–KOH, 100 mM KCl, 0.1 mM EDTA and 1 mM DTT, pH 7.6, then with 100 ng of Nfo protein (or without enzyme) for 1 h at 4°C, then for 1 h at 37°C. The enzyme solution was then removed, 1 mg/ml of proteinase K, 1% n-lauroylsarcosine in L-Buffer was added, and the mixture was incubated for 1 h at 4°C, then for 16 h at 37°C. Plugs were rinsed with TE buffer (six times, 1 h each), and then equilibrated into 0.5× TBE (45 mM Tris base, 45 mM boric acid and 1 mM Na2–EDTA, pH 7.8).
Putrescine-HCl (Sigma, St Louis, MO) was dissolved in double distilled, sterile water at 0.5 M. Duplicate plugs slices (∼500 ng DNA) were incubated in 200 μl of PUTR reaction buffer P (5 mM HEPES–KOH, 0.1 mM EDTA and 1 mM KCl, pH 7.25) with 100 mM PUTR, or without PUTR, for 1 h at 4°C and then for 1 h at 37°C. Ice-cold 5 M NaCl was added to a final concentration of 0.5 M (48), and incubation continued for 1 h at 28°C. Plugs were rinsed with TE (0.5 ml, six times, 1 h each) equilibrated for 24 h in 0.5× TBE.
Bistranded abasic cluster measurement
Samples and molecular length standards were electrophoresed in a 0.85% neutral agarose gel [Sea Kem Gold (BioWhittaker Molecular Applications, Rockland, ME) in 0.5× TBE] in a Bio-Rad CHEF (contour-clamped homogeneous electric field) DR-II apparatus. The length standards included yeast chromosomal DNAs (Schizosaccharomyces pombe, Hansenula wingei and Saccharomyces cerevisiae, Bio-Rad, Hercules, CA) and T4, T7 and T7 BglI digest fragments (170, 39.9 and 4–22 kb respectively). The samples were electrophoresed by using a dual pulsing regime (65 V, 9°C; 12 h, by using a linearly ramping pulsing regime from 10 to 4000 s), optimized for separation of DNA fragments ranging from 5.7 Mb to 13 kb (23). The upper portion of the gel that contained the wells was removed and the main portion of the gel was further electrophoresed for 72 h at 9°C in a linearly ramping pulsing regime from 3400 to 10 s (24 h at 60 V; 24 h, 70 V; 1 h, 80 V; 4 h, 85V). Gels were stained with ethidium bromide (1 μg/ml in double distilled water) for 1 h, destained overnight, and an electronic image was obtained (24). A DNA dispersion curve that related DNA length to electrophoretic mobility was determined, and from the profiles of irradiated and unirradiated samples (or enzyme, polyamine-treated and untreated DNA populations), the number average length, ![]() , of each DNA distribution and then the frequencies of DSBs (φDSB) and of abasic clusters (φCL-N for Nfo-clusters and φCL-P for PUTR-clusters) were calculated (10). Since electrophoresis is carried out under non-denaturing conditions, this method is specific for measurement of bistranded abasic clusters.
, of each DNA distribution and then the frequencies of DSBs (φDSB) and of abasic clusters (φCL-N for Nfo-clusters and φCL-P for PUTR-clusters) were calculated (10). Since electrophoresis is carried out under non-denaturing conditions, this method is specific for measurement of bistranded abasic clusters.
Clonogenic assay
Cells (104 in 3 ml of complete medium) were mixed with 6 ml of agarose mix [0.55% Sigma Type II agarose in IMDM at 45°C] and 1 ml of FBS at 37°C (HyClone, Logan UT); 3 ml of this mixture was plated on a 12 ml-base [0.5% Type II agarose in IMDM, 10% bovine calf serum (HyClone)] (25). The plates were incubated for 21 days (37°C, 5% CO2); 1 ml of complete medium was added periodically to each plate. Colonies of >50 cells were counted.
Detection of cell death
Hoechst 33258 (HO, Sigma, St Louis MO), propidium iodide (PI, Sigma, St. Louis, MO) and erythrosin B (EB, Sigma) were used for detection of apoptotic, late apoptotic/dead cells and dead cells respectively (26,27). Cells (∼2 × 106) were washed with Dulbecco's phosphate buffered saline (PBS, Gibco, Grand Island, NY) and resuspended in 0.5 ml of PBS. The cell suspensions (100 μl) were mixed with dye stock solutions of 10 μg/ml of HO, 30 μg/ml of PI or 0.04% EB in PBS. For HO and PI, the cells were incubated with dye for 30 min at room temperature in the dark. For visualization and analysis, a standard light microscope (Nikon Diaphot) was used for EB. For PI and HO, a fluorescence microscope (Zeiss GFL) was used (HO, 365 nm excitation and 450 nm emission; PI, 546 nm excitation and 580 nm emission). Electronic images were recorded by a charge-coupled device video camera (Cohu 4910), and the frequencies of cells stained with each dye was determined. In each case the percentage of dead and/or dying cells is expressed as the ratio of number of cells stained by the dye (intense red for PI positive and intense blue for HO positive cells) to the total number of cells (∼500 cells were scored per measurement).
Cleavage of abasic clusters by cell extracts, human AP endonuclease and putrescine
Oligonucleotide substrates
The 23mer double-stranded oligonucleotides (Operon, Alameda, CA) that contained abasic clusters with inter-lesion spacings from 0 (opposing) to 5 bp apart (Table 1), were prepared as described previously (10). Briefly, the double-stranded oligonucleotides that contained uracil in one or both strands were incubated at 37°C with 1 U of uracil–DNA glycosylase (UDG; New England Biolabs, Beverly, MA) in 10 μl of STE buffer (10 mM Tris–HCl, 1 mM EDTA and 50 mM NaCl, pH 7.8) for 30 min. The efficiency of AP site formation was >98%, as determined from the strand breaks introduced by heating the duplexes to 95°C for 10 min in 0.5 M NaOH, and electrophoresis in a non-denaturing 15.2% polyacrylamide gel for 2.5 h at 90 V. The UDG-treated DNA duplexes were digested with 1 mg/ml of proteinase K, 1% n-lauroylsarcosine in L Buffer (30 min at 4°C, then 6 h at 37°C) and precipitated by the addition of an equal volume of isopropanol, incubation at room temperature for 20 min and centrifugation at 14 000 g (30 min, 4°C). The supernatant was removed, the precipitated DNA was resuspended in (for treatment with cell extract or enzyme) 10 mM MgCl2, 50 mM HEPES–KOH, 50 mM KCl and 5% glycerol or Buffer P for PUTR treatment and equilibrated for 15 min at 4°C. The DNA was then precipitated (as above), the supernatant was removed and the precipitated DNA chilled on ice, air-dried for 5 h and resuspended in the appropriate buffer as above.
Table 1. Sequences of 23mer oligonucleotide duplexes containing AP sites (X) at specific positions.
| Duplex | Sequence | Positions |
|---|---|---|
| E | 5′-AGAGGAXATGTATGTATGGAGAG-3′ | −5 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| D | 5′-AGAGGATAXGTATGTATGGAGAG-3′ | −3 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| C | 5′-AGAGGATATGXATGTATGGAGAG-3′ | −1 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| B | 5′-AGAGGATATGTXTGTATGGAGAG-3′ | 0 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| F | 5′-AGAGGATATGTAXGTATGGAGAG-3′ | +1 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| G | 5′-AGAGGATATGTATGXATGGAGAG-3′ | +3 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| H | 5′-AGAGGATATGTATGTAXGGAGAG-3′ | +5 |
| A | 3′-TCTCCTATACAXACATACCTCTC-5′ | |
| B | 5′-AGAGGATATGTXTGTATGGAGAG-3′ | Control (C) |
| AC | 3′-TCTCCTATACATACATACCTCTC-5′ |
Cell extracts
The cells (∼107) were washed twice in PBS, mixed on ice with 100 μl of 5× cell extract sonication buffer (5× CESB: 500 mM Tris–HCl, 500 mM KCl, 1 mM EDTA and 5% glycerol, pH 7.5). They were sonicated on ice (4 × 20 s) by using a sonifier cell disruptor model W185 (Heat Systems Ultrasonics Inc., Plainview, NY). About 2 ml of 1× CESB were added, the extract was vortexed, incubated for 5 min on ice and then centrifuged for 15 min (12 000 g, 4°C) (28). The supernatant was removed and its protein concentration was determined by using a Bradford assay (Bio-Rad, Hercules CA). Reactions [10 μl; 10 mM MgCl2, 50 mM HEPES–KOH, 50 mM KCl, 5% glycerol, 1 mM DDT and 0.01% Triton X-100, pH 7.6, that contained 100 ng of oligonucleotide (29) and 3 μg of cell extract] were incubated at 4°C for 30 min, then at 37°C for 12 min. The reactions were treated with 1 mg/ml of proteinase K, 1% n-lauroylsarcosine in L-Buffer for 30 min at 4°C, then for 6 h at 37°C. The oligonucleotides were precipitated by adding an equal volume of 4 M ammonium acetate, mixed, incubated for 10 min at room temperature and ice-cold ethanol was added to 67% (30). The solution was vortexed, incubated on ice for 5 min, centrifuged (5 min, 13 000 g, room temperature), the supernatant removed and the pellets dissolved in 100 μl TE. After the addition of 1 ml of 70% ethanol at room temperature and gentle shaking, the solution was centrifuged as above and the supernatant was removed. The pellets were dried on ice, then, resuspended in TE. The oligonucleotides were electrophoresed in a 15.2% polyacrylamide gel and cleavage was quantified (10).
Human AP endonuclease
Recombinant human Ape1 protein was purified and used essentially as described (22). The amount (2.5 ng) of hAPE1 [specific activity ∼1 pmol AP sites/ng/min] was optimized from titration studies on oligonucleotides that contained abasic clusters (−5, +5, see Table 1) (data not shown). Reactions were as for cell extracts but were incubated for 30 min at 37°C. The reactions were terminated and the oligonucleotides were recovered as above.
Putrescine
Oligonucleotides in buffer P were incubated with 1 mM PUTR for 30 min at 37°C. The reactions were terminated by the addition of ice-cold 5 M NaCl to 0.5 M and incubated for 30 min at 28°C (10). The oligonucleotides were recovered as described above.
RESULTS
Abasic cluster quantitation using Nfo protein or PUTR
We first determined the optimum conditions for Nfo protein or PUTR cleavage of abasic clusters recognized by these agents in DNA isolated from human cells that were exposed to 0 or 20 Gy of γ-rays. Figure 1A and B show the results of such titrations for Nfo protein and PUTR, respectively. Approximately 100 ng of Nfo protein per 500 ng of human DNA gave maximal cleavage of Nfo-recognized abasic clusters, with substantially less cleavage of unirradiated DNA; this ratio was chosen for all further experiments. Titration of PUTR cleavage (Figure 1B) showed that maximum cleavage of irradiated DNA required ∼100 mM PUTR/500 ng of DNA. PUTR also showed very little cleavage of unirradiated DNA.
Figure 1.

Abasic clusters in human monocytes exposed to 0–20 Gy of γ-rays. (A and B) Titration of human DNA cleavage by Nfo protein or PUTR. DNA in agarose plugs from cells irradiated with 0 or 20 Gy of γ-rays was treated with increasing amounts of Nfo protein (A) or PUTR (B) and the cluster frequencies determined. About 100 ng Nfo protein and 100 mM PUTR per 500 ng of DNA were chosen for all further experiments. The curves were fit to the points by eye. (C) DSB, (D) Nfo-abasic cluster and (E) PUTR-abasic cluster levels as a function of dose. Solid symbols correspond to individual data points and open symbols to averages. Error bars, SEM, in some cases are smaller than the corresponding symbol. Lines represent the least squares fit of the average points.
Induction of abasic clusters in human monocytes by γ-rays
We then used PUTR and Nfo protein to determine the relative levels of γ-ray induced Nfo- and PUTR-abasic clusters. A reasonably linear dose response was found for Nfo- and PUTR-clusters as well as for radiation-induced DSBs (Figure 1C–E). From these dose–response lines, the DSB yield was calculated to be 11.9 DSB/Gb/Gy, in good agreement with the data of Gulston et al. (19). The yield of Nfo-abasic clusters (9.5 Nfo-clusters/Gb/Gy) was lower than that of PUTR-abasic clusters (15.4 PUTR-clusters/Gb/Gy) by a factor of ∼1.6, similar to the factor of ∼1.5 found previously for T7 DNA irradiated under radioquenching conditions (10). The ratio of frank DSBs to abasic clusters found here (1 DSB: ∼0.8 Nfo-clusters) is also similar to that found previously for X-irradiated human cells of 1 DSB: ∼0.75 Nfo-clusters (18). For PUTR-clusters the ratio is 1 DSB: ∼1.3 PUTR-clusters.
Double-strand breaks in human monocytes
To follow the processing of radiation-induced complex damages by human cells, we measured their levels 14 days after radiation exposure. Figure 2A shows segments of a CHEF gel that contained DNA isolated from unirradiated cells (lanes 1–3), as well as from cells exposed to 5 Gy, then incubated at 37°C for 0, 0.1, 2 or 14 days (lanes 4–15). We first measured DSB rejoining by electrophoresing DNA without enzyme or putrescine treatment (–, lanes 1, 4, 7, 10 and 13). The cells incubated at 37°C resealed DSBs, observed here as an increase in DNA size from 0.1 to 14 days (cf. lanes 4, 7, and 13). On day 2, increased DNA fragmentation was detected with distinctive DNA bands between 3 Mb and 200 kb (lanes 10–12). These high molecular length DNA fragments (which were also seen to a lesser extent at 1 and 1.5 days, data not shown) could be attributed to the initiation of apoptosis (31).
Figure 2.

Processing of DNA lesions by human 28SC monocytes exposed to 5 Gy of γ-rays as a function of post-exposure time. (A) Electronic image of a neutral electrophoretic gel containing DNA from human 28SC monocytes exposed to 5 Gy of γ-rays, and harvested as a function of time post-exposure. 0 Gy, lanes 1–3; 5 Gy at increasing times (0–14 days), lanes 4–15; molecular size markers, lanes 16–19: T4, T7 and BglI digest of T7, showing here only the 22 kb fragment, lane 16; S.cerevisae, lane 17; H.wingei, lane 18; S.pombe, lane 19. The molecular sizes of these DNAs are shown in kb at the right of the gel image. For each time point three samples are shown: untreated, −; Nfo-treated, N; PUTR-treated, P. (B) DSB levels: 0 Gy, circles; 5 Gy, squares. (C) Nfo-detected abasic cluster levels: 0 Gy, circles; 5 Gy, diamonds; and (D) PUTR-detected cluster levels: 0 Gy, circles; 5 Gy, inverted triangles. Solid symbols correspond to individual data points and open symbols to average values. Error bars, SEM, where not shown are smaller than the corresponding symbol.
DNA fragments that are apparently the result of radiation-induced apoptosis are shown in Figure 2A (lanes 10–12). However, since 28SC cells contain multiple mutations in p53 (32), it was not clear if they could undergo apoptosis. We therefore used three cytostaining dyes taken up by apoptotic cells (HO), late apoptotic and dying cells (PI), and by cells unable to exclude vital dyes (EB). The quantitative results for each dye, for irradiated (5 Gy) and unirradiated (0 Gy) cells, as well as representative images of the cell samples that are used for analysis are shown in Figure 3A–C. In the early post-exposure period (day ‘0’: 10–15 min after irradiation), the percentage of HO-apoptotic cells was almost double (∼14%) that of PI or EB-positive cells (5–8%). This difference results from the fact that HO can enter cells even at early stages of apoptosis, with significantly less uptake by viable cells (33). With increasing time (1–8 days), the percentage of late apoptotic or dead cells with ruptured membranes (PI- and EB-positive cells) also increased. For days 8–14, the percentage of HO positive cells decreased significantly (equal to those found shortly after irradiation), corresponding to substantial increases in viable cell numbers, whereas PI- and EB-stained cell levels remained substantially higher (ranging from 20–45%), which suggests the existence of some late apoptotic/dead cells and the emergence of survivors. Therefore, 28SC cells undergo apoptosis as a result of exposure to 5 Gy of γ-rays.
Figure 3.

Staining of unirradiated or irradiated 28SC human monocytes by Hoechst 33258, propidium iodide, or erythrosin B. Percent of cells stained with (A) Hoechst 33258 (apoptotic cells, intense blue), (B) Propidium iodide (late apoptotic/dead cells, intense red), and (C) Erythrosin B (dead cells, intense black), after exposure of cells to 0 Gy (circles) or 5 Gy, (triangles) of γ-rays and incubation for the times indicated. Solid symbols correspond to individual data points and open symbols to average values. Error bars, SEM, where not shown are smaller than the corresponding symbol. The curves were fit to the points by eye. Images correspond to irradiated samples (5 Gy) at 0, 7, or 14 days after irradiation.
Processing of abasic clusters versus DSBs in human monocytes
To evaluate the paths in human cells for dealing with complex damages, we quantified the levels of and Nfo- and PUTR-detected abasic clusters. The results are shown in Figure 2B–D. In unirradiated cells, abasic clusters levels were low which indicates that handling of cells and incubation did not produce significant levels of clusters (lanes 1–3 in Figure 2A and circles in Figure 2B–D). In irradiated cells, DSB levels decreased significantly after 1 h and DSBs were completely rejoined within 1 day. The increase in DSB levels between the second and eighth day probably results from apoptotic fragmentation. After an apparent initial drop, both Nfo- and PUTR-detected clusters, increased in the first three hours after irradiation (0–0.1 day) and remained higher than the controls for about 10 days. During the initial period after irradiation, PUTR treatment led to higher cleavage than Nfo protein. After day 1, the levels of these cluster types were similar (Figure 2A, 14 d: lanes 14 and 15).
Cellular processes that decrease abasic cluster levels
The gradual disappearance of abasic clusters after 6 h (Figure 2C and D) could be the result of active repair of these complex damages. However, even without repair, DNA replication on strands containing a lesion that is a component of a cluster, would produce new DNA molecules without measurable clusters. DNA replication would split the bistranded cluster into unistranded lesions, and in addition, increase the total DNA mass, thus diluting the damages. The effect of such splitting (assuming no cluster removal occurred by actual repair) and dilution is given by Equation 1:
![]()
where Ct is the cluster frequency at time t, Ci is the initial cluster frequency and α is the fraction of cells at time t that have divided (determined by the cell counts as shown in Figure 4A). To evaluate the role of such cluster splitting and dilution on their apparent disappearance, we measured both (i) the increase in cell numbers during the critical post-exposure period of 0–3 days where irradiated cells (5 Gy) have approximately doubled (Figure 4A) and (ii) the clonogenic survival (Table 2). The survival data show that ∼40% of the cells exposed to 5 Gy maintained the ability to divide and form colonies, whereas this percentage decreased to ∼5% for cells that were exposed to 10 Gy. Similar values were found if 104 cells were plated (data not shown).
Figure 4.

Growth of irradiated (5 Gy) and unirradiated 28SC cells, and potential effects on abasic cluster processing. (A) Cell concentration for 0–3 days after irradiation, during which the irradiated cells approximately doubled: 0 Gy (circles) 5 Gy, (triangles). Inset: cell concentration measured for 0–14 days. (B and C) Abasic cluster levels; experimental averages (open symbols), calculated for ‘splitting effect’ (solid symbols); (B) Nfo protein, diamonds and (C) putrescine, squares. Curves, power best fittings (y = axb) for the experimental points (solid lines), Nfo protein, a = 47.21 b = 0.60; PUTR, a = 67.04, b = 0.56; fit to the points by eye (dotted lines) for the calculated points. Solid symbols correspond to individual data points and open symbols to average values. Error bars, SEM, where not shown are smaller than the corresponding symbol.
Table 2. Survival of colony-forming ability of 28SC cells exposed to 0, 5 or 10 Gy of 137Cs γ-raysa.
| Dose (Gy) | Colonies per ∼3 × 103 cells plated | Survival (%) |
|---|---|---|
| 0 | 844 ± 15 | 100 |
| 5 | 352 ± 21 | 42 |
| 10 | 42 ± 6 | 5 |
aErrors correspond to SEM.
Assuming an average G2 period of ∼4 h (34), we plotted the resulting expected cluster frequencies (solid symbols, dotted lines) along with the experimental averages (open symbols, taken from Figure 2), as shown in Figure 4B and C. The actual measured levels of PUTR-recognized abasic clusters (plotted as the averages and continuous curves, Figure 4C) are lower than the values expected if clusters were removed only by cluster splitting through DNA replication and dilution through increase in DNA mass. The experimental data for Nfo-cleaved clusters are closer to the calculated values (Figure 4B). These data suggest that substantial levels of abasic clusters formed by these radiation doses are not repaired by these human cells, but ‘disappear’ by splitting during DNA replication. However, they also suggest that at least some PUTR-clusters appear to be repaired actively, and some Nfo-clusters may be subject to repair as well.
In vitro repair of abasic clusters by cell extracts
To gain insight into the putative mechanisms of repair of abasic clusters by human monocytes, we studied the cleavage of defined abasic clusters by 28SC cell extracts. Figure 5A shows the cleavage of specific abasic cluster configurations by increasing quantities of cell extract. The cell extract cleaved the −5 and +5 clusters well, but acted comparatively poorly on the closely spaced −1 and +1 clusters. The cleavage efficiency of the extract on different clusters is shown in Figure 5A. Based on these results we chose 3 μg of cell extract for all further experiments.
Figure 5.
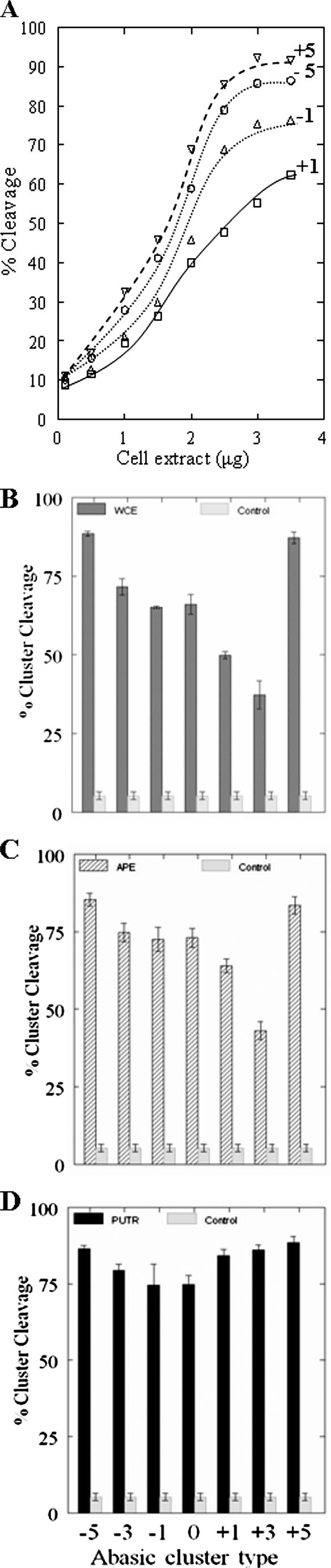
Cleavage of abasic clusters by extracts of 28SC cells, PUTR or hAPE1. (A) Oligonucleotides (23 bp) containing bistranded AP sites at different interlesion spacings (Table 1) were incubated (37°C, 12 min) with increasing quantities of extract. Cleavage efficiency is given as the percentage of conversion of the intact oligonucleotides to specific product fragments of lower size. (B) Cleavage efficiency by cell extract (WCE, 3 μg), (C) hAPE1 (2.5 ng) or (D) PUTR (1 mM) of specific abasic clusters (Table 1). The controls reflect the average ‘background’ cleavage by the extract of the control oligonucleotide containing only one abasic site (see Table 1). In neutral electrophoretic gels, no double-strand cleavage is expected for an oligonucleotide containing a single AP site. Similar cleavage was found for the substrate containing a single abasic site by PUTR and hAPE1 (data not shown).
In addition, we tested hAPE1 and PUTR cleavage of these clusters (Figure 5B). As previously found for PUTR (10), cleavage is practically independent of the interlesion distance with a decreased activity on 0, −1 and +1 clusters. The human AP endonuclease cleaves poorly at several closely spaced clusters (−3, −1, 0, +1, +3), and especially at the +1 and +3 clusters. The straight line in Figure 5B corresponds to the average value (∼4%) found for background cleavage for the control oligonucleotide that contained a single abasic site (see Table 1) substrate by cell extracts. (This negative control was chosen to test whether neutral electrophoresis under our conditions of an oligonucleotide with a single-strand break could produce smaller fragments that could be mistaken for products of double-strand cleavage at a cluster.) The corresponding values for the other two agents (hAPE1 and putrescine) were also similar (data not shown). All the above results suggest that (i) cleavage of abasic clusters by the 28SC extract is qualitatively similar to that by purified recombinant hAPE1, (ii) these activities are strikingly different from that of PUTR and (iii) 28SC extracts as well as hAPE1 cleave closely spaced abasic clusters (−3, −1, 0, +1, +3) relatively poorly, but cleave spaced clusters with larger inter-lesion separations (−5, +5) efficiently. Both hAPE1 and extracts cleaved +1 and +3 clusters most poorly. On the other hand PUTR cleaves all these clusters well.
DISCUSSION
Abasic clusters are induced by γ rays in human monocytes
We have measured the induction by γ-rays and repair of abasic DNA clusters in human monocytes, by employing two agents that cleave at AP sites, an E.coli AP endonuclease (Nfo protein) and a polyamine (PUTR). These agents cleave abasic clusters differently; PUTR can cleave at high-density clusters that are resistant to cleavage by Nfo protein (10). Nfo protein is a type II AP endonuclease that cleaves DNA at the 5′ side of the AP site, generating 3′-hydroxyl and 5′ deoxyribose phosphate termini. This enzyme recognizes regular as well as oxidized AP sites (35). Polyamines also cleave regular and oxidized AP sites, but act via a different mechanism, nicking the phosphodiester bond 3′ to the AP site by a β-elimination reaction (12,36). In addition, polyamines including PUTR cleave AP endonuclease-resistant abasic sites, e.g., opposite a single-strand break (SSB) or another closely spaced AP site (0–5 bp apart) (10,37,38). Our results show that PUTR detects ∼1.6 times more clusters than Nfo protein (Figure 1). Nfo-resistant clusters constitute approximately a third of the total abasic clusters, and can either be closely spaced (high-density clusters) or specific conformations not easily recognized by Nfo protein. The percentage of PUTR-clusters is ∼130% that of DSBs, i.e., ∼1.3 PUTR-clusters per DSB. This ratio, plus that of 2 radiation-induced oxybase clusters/DSB (18,19), indicate that more than 3 non-DSB (oxybase plus abasic) clusters are formed in cells by radiation exposure.
Processing of DSBs and abasic clusters by human cells
An attempted repair of a bistranded cluster could produce de novo DSBs. Previous studies showed an increase in DSBs during repair of radiation-induced DNA damage in E.coli (5,6) and mammalian (3,4,39) cells respectively. To determine whether de novo DSBs could be produced during the processing of oxidative DNA damage in 28SC cells (radiation-resistant, repair proficient human monocytes), we followed the post-exposure levels of DSBs and abasic clusters in cells that were exposed to a radiotherapeutic dose of γ-rays. The cells rapidly rejoined the radiation-induced DSBs within a few hours. Although some additional DSBs were detected after 2 days, the characteristic DNA laddering that was detected on gels and the appearance of dye-positive cells indicates that they are probably apoptotic DNA fragments. This does not preclude the possibility that DSBs could be generated during cluster processing. However, if they are produced, they are short-lived transients that do not accumulate to detectable levels in these cells.
Replication of bistranded DNA lesions forming a cluster could result in their conversion to a single lesion in one of the daughter strands (‘cluster splitting’) and thus their disappearance. This mechanism is apparently a significant contributor to the elimination of damage clusters in irradiated 28SC cells. Comparison of the experimental and calculated levels (at each time point) of abasic clusters (Figure 4B–C) indicates that splitting accounts for the disappearance of 50–80% of the initial clusters. Thus, human monocytes repair these complex damages poorly. By replication using the damaged strands as templates, they convert a cluster to lesions that affect only one strand, which can then be handled more efficiently by cellular repair. Replication of DNA that contained unrepaired lesions (abasic sites or base lesions like 8-oxoguanine or thymine glycol) is mainly performed by translesion synthesis, i.e., direct synthesis of DNA past altered bases, probably carried out by the Y-family of polymerases (40). The identity of the polymerase that was recruited to handle clustered damages is unknown. Our survival data also support the likelihood that some clustered damages are diluted through replication since a relatively large percentage (∼42%) of the cells exposed to 5 Gy survive and divide (Table 2).
Cellular processing of abasic clusters was quite different from that of DSBs. First, the cluster level increased by ∼50% within the first 3 h, whereas DSB levels decreased to ∼60% of the initial value. Second, abasic clusters persisted for a substantial time after irradiation. The increase of abasic clusters probably represents the creation of intermediate AP sites as the result of the release of clustered base damages by DNA glycosylases (14). Post-exposure increases of isolated (non-clustered) AP sites have also been reported in human cells exposed to H2O2 (13,41).
Previous data show that about two oxidized base clusters are induced per DSB in human cells that were exposed to sparsely ionizing radiation (18). These clusters could be converted by glycosylase action to abasic clusters (including the creation of a SSB at one oxidized base through sequential action of glycosylase/lyase or endonuclease). This idea also supports the fact that the major human glycosylases possess strong glycosylase activity relative to their lyase activity, especially in the presence of an AP endonuclease (42–44). The data in Figure 2 show an increase in abasic clusters of ∼40–60 clusters/Gb. If these de novo clusters resulted from repair of OxyBase clusters, only a minority of the initially induced OxyBase clusters were detectable as accumulated abasic clusters. The remainder may undergo rapid repair of lesions on both strands, conversion to rapidly resealed DSBs or they could remain as persistent lesions. Clusters could also disappear through cell death (Figure 3) and consequent loss of DNA upon cellular disintegration.
Persistence of abasic clusters
Since PUTR cleaves high-density abasic clusters and specific configurations that were not cleaved by Nfo protein, use of the two agents should reveal differences in the repair of these cluster classes. Indeed, more PUTR-clusters were detected for all the time periods up to 1 day. From day 1 to 14, the levels of Nfo- and PUTR-detected clusters were not significantly different (at the 5% level). The apparently higher means of PUTR-clusters at early times were consistent either with an existence of a persistent population of abasic clusters and/or compensating generation of de novo clusters by processing of radiation-induced clustered lesions. Since PUTR detected most abasic clusters that were refractory to Nfo cleavage, i.e., high-density clusters, this population may be primarily high-density abasic clusters.
Our in vitro repair experiments suggest that cells deal poorly with closely spaced abasic clusters (Figure 5). The +1 and +3 clusters may be repaired especially slowly in cells. The major AP endonuclease, hAPE1, that processes abasic clusters in human cells (45), was also strongly inhibited in processing the +1 and +3 clusters (Figure 5B), in accord with previous results (7,9). These data suggest that many abasic clusters in γ-irradiated cells that were detected long after irradiation may constitute +1 and +3 clusters. Previous data showed that Nfo protein and PUTR detect +1 and +3 abasic cluster configurations almost equally well (10). This may explain why the levels of abasic clusters detected using Nfo or PUTR after 1 day do not differ significantly (Figure 2C and D, respectively). Our results suggest that BER may be significantly slower when processing a lesion within a damage cluster (46,47).
Paths for eliminating clustered DNA damage in human cells
Repair of clustered damages has been expected to generate double-strand breaks from simultaneous attempted repair of opposing lesions, and indeed heavily irradiated rodent cells and TK6 human cells do produce de novo DSBs after radiation exposure (3,4,39). In vitro studies of oligonucleotides that contained specific clusters suggested production of DSBs at some clusters, but also suggested that sequential repair of lesions constituting the cluster could be an alternate repair path that could avoid the generation of DSBs (7,9).
Radiation-resistant, repair-proficient 28SC human monocytes, however, appear to process complex damage clusters by quite different and unanticipated paths. First, the level of abasic clusters increases early after radiation exposure, presumably from partial processing of oxidized base clusters. Second, the only increase in DSB observed—at ∼2 days after radiation exposure—appears to be associated with apoptotic laddering rather than cluster processing. Third, in these cells, the disappearance of the majority of abasic clusters is apparently temporally coincident with DNA replication. Thus DNA replication ‘splits’ the cluster: the parental DNA that contained a bistranded clustered damage is converted by replication and translesion synthesis into two daughter DNAs, each containing one parental strand with unistranded lesion(s) and one newly replicated strand without any lesions. Thus, in these repair-proficient cells, abasic clusters appear to be very poorly repaired, if at all. Clusters thus comprise persistent, potentially-mutagenic and genome-destabilizing damages.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr C. de los Santos for helpful discussions and critical reading of the manuscript. We also thank Dr K. Thompson for valuable suggestions on the statistical evaluation of the data, Dr J. Sutherland for use of ImageSystem, Mr J. Trunk and Ms D. Monteleone of his group for help with microscopy and with image processing and Mr R. Sautkulis for his help in dosimetry and irradiations. We thank Mr Dan McNeill (NIA) for his effort in purifying the Ape1 protein. This work was supported by grants from the Low Dose Program of the Office of Biological and Environmental Research of the US Department of Energy co-sponsored by the Office of Biological and Physical Research of the National Aeronautic and Space Administration to B.M.S. (BO-086), from the National Institutes of Health to B.M.S. (R01 CA 86897) and National Space Biomedical Research Institute to A. Gewirtz (University of Pennsylvania) and B.M.S. (IIH00207), and by a grant from the Office of Biological and Physical Research of the US National Aeronautics and Space Administration to B.M.S.
REFERENCES
- 1.Ward J.F. (1981) Some biochemical consequences of the spatial distribution of ionizing radiation produced free radicals. Radiat. Res., 86, 185–195. [PubMed] [Google Scholar]
- 2.Goodhead D.T. (1994) Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Rad. Biol., 65, 7–17. [DOI] [PubMed] [Google Scholar]
- 3.Dugle D., Gillespie,C. and Chapman,J.D. (1976) DNA strand breaks, repair and survival in X-irradiated mammalian cells. Proc. Natl Acad. Sci. USA., 73, 809–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahnstrom G. and Bryant,P.E. (1982) DNA double-strand breaks generated by the repair of X-ray damage in Chinese hamster cells. Int. J. Radiat. Biol., 41, 671–676. [DOI] [PubMed] [Google Scholar]
- 5.Bonura T. and Smith,K.C. (1975) Enzymatic production of deoxyribonucleic acid double-strand breaks after ultraviolet irradiation of Escherichia coli K12. J. Bacteriol., 121, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaisdell J.O. and Wallace,S. (2001) Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli. Proc. Natl Acad. Sci. USA, 98, 7426–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhry M.A. and Weinfeld,M. (1997) Reactivity of human apurinic/apyrimidinic endonuclease and Escherichia coli exonuclease III with bistranded abasic sites in DNA. J. Biol. Chem., 272, 15650–15655. [DOI] [PubMed] [Google Scholar]
- 8.Harrison L., Hatahet,Z. and Wallace,S. (1999) In Vitro Repair of Synthetic Ionizing Radiation-induced Multiply Damaged DNA Sites. J. Mol. Biol., 290, 667–684. [DOI] [PubMed] [Google Scholar]
- 9.David-Cordonnier M.H., Cunniffe,S.M.T., Hickson,I.D. and O'Neill,P. (2002) Efficiency of incision of an AP site within clustered DNA damage by the major human AP endonuclease. Biochemistry, 41, 634–642. [DOI] [PubMed] [Google Scholar]
- 10.Georgakilas A.G., Bennett,P.V. and Sutherland,B.M. (2002) High efficiency detection of bistranded abasic clusters in γ-irradiated DNA by putrescine. Nucleic Acids Res., 30, 2800–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Sonntag C. (1987) The Chemical Basis of Radiation Biology. Taylor & Francis, London. [Google Scholar]
- 12.Nakamura J. and Swenberg,A.J. (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res., 59, 2522–2526. [PubMed] [Google Scholar]
- 13.Atamna H., Cheung,I. and Ames,B.N. (2000) A method of detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proc. Natl Acad. Sci. USA, 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCullough A.K., Dodson,M.L. and Lloyd,R.S. (1999) Initiation of base excision repair: Glycosylase mechanisms and structures. Annu. Rev. Biochem., 68, 255–285. [DOI] [PubMed] [Google Scholar]
- 15.Klungland A. and Lindahl,T. (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J., 16, 3341–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou W. and Doetsch,P.W. (1993) Effects of abasic sites and DNA single-strand breaks on procaryotic RNA polymerases. Proc. Natl Acad. Sci. USA, 90, 6601–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu S.L., Lee,S.K., Johnson,R.E., Prakash,L. and Prakash,S. (2003) The stalling of transcription at abasic sites is highly mutagenic. Mol. Cell. Biol., 23, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutherland B.M., Bennett,P.V., Sutherland,J.C. and Laval,J. (2002) Clustered DNA damages induced by X-rays in human cells. Radiat. Res., 157, 611–616. [DOI] [PubMed] [Google Scholar]
- 19.Gulston M., Fulford,J., Jenner,T., de Lara,C. and O'Neill,P. (2002) Clustered DNA damage induced by γ radiation in human fibroblasts (HF 19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res., 30, 3464–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutherland B., Bennett,P.V., Sidorkina,O. and Laval,J. (2000) DNA damage clusters induced by ionizing radiation in isolated DNA and in human cells. Proc. Natl Acad. Sci. USA, 97, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ljungquist S. (1977) A new endonuclease from Escherichia coli acting at apurinic sites in DNA. J. Biol. Chem., 252, 2808–2814. [PubMed] [Google Scholar]
- 22.Erzberger J.P., Barsky,D., Scharer,O.D., Colvin,M.E. and Wilson,D.M.R. (1998) Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res., 26, 2771–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland B.M., Georgakilas,A.G., Bennett,P.V., Laval,J. and Sutherland,J.C. (2003) Quantifying clustered DNA damage induction and repair by gel electrophoresis, electronic imaging and number average length analysis. Mutat. Res., 531, 93–107. [DOI] [PubMed] [Google Scholar]
- 24.Sutherland J.C., Monteleone,D.C., Trunk,J.G., Bennett,P.V. and Sutherland,B.M. (2001) Quantifying DNA damage by gel electrophoresis, electronic imaging and number average length analysis. Electrophoresis, 22, 843–854. [DOI] [PubMed] [Google Scholar]
- 25.Sutherland B.M. and Bennett,P.V. (1984) Human cell transfection with skin cancer DNA. Photodermatology, 2, 186–191. [PubMed] [Google Scholar]
- 26.Cohen J.J., Duke,R.C., Fadok,V.A. and Sellins,K. (1992) Apoptosis and programmed cell death in immunity. Annu. Rev. Immunol., 10, 267–293. [DOI] [PubMed] [Google Scholar]
- 27.Clifford J.L., Menter,D.G., Wang,M., Lotan,R. and Lippman,S.M. (1999) Retinoid receptor-dependent and independent effects of N-(4-hydroxyphenyl)-retinamide in F9 embryonal carcinoma cells. Cancer Res., 59, 14–18. [PubMed] [Google Scholar]
- 28.Kreklau E.L., Limp-Foster,M., Liu,N., Xu,Y., Kelley,M.R. and Erickson,L.C. (2001) A novel fluorometric oligonucleotide assay to measure O6-methylguanine DNA methyltransferase, methylpurine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res., 29, 2558–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson D.M. III, Takeshita,M., Grollman,A.P. and Demple,B. (1995) Incision activity of human apurinic endonuclease (Ape) at abasic sites analogs in DNA. J. Biol. Chem., 270, 16002–16007. [DOI] [PubMed] [Google Scholar]
- 30.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1999) Short Protocols in Molecular Biology, 4th edn. John Wiley and Sons, Inc., NY. [Google Scholar]
- 31.Rogakou E.V., Nieves-Neira,W., Boon,C., Pommier,Y. and Bonner,W.M. (2000) Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX Histone at Serine 139. J. Biol. Chem., 275, 9390–9395. [DOI] [PubMed] [Google Scholar]
- 32.Bennett P.V., Cintron,N.S., Gros,L., Laval,J. and Sutherland,B.M. (2004) Are endogenous clustered DNA damages induced in human cells? Free Radic. Biol. Med., 37, 488–499. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz D., Almog,N., Peled,A., Goldfinger,N. and Rotter,V. (1997) Role of wild-type p53 and in the G2 phase: regulation of the gamma-irradiation-induced delay and DNA repair. Oncogene, 15, 2597–2607. [DOI] [PubMed] [Google Scholar]
- 34.Alberts B., Johnson,A., Lewis,J., Raff,M., Roberts,K. and Walter,P. (2002) Molecular Biology of The Cell, 4th edn. Garland Science, NY. [Google Scholar]
- 35.Haring M., Rudiger,H., Demple,B., Boiteux,S. and Epe,B. (1994) Recognition of oxidized abasic sites by repair endonucleases. Nucleic Acids Res., 22, 2010–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailly V. and Verly,W. (1988) Possible role of beta-elimination and delta-elimination reactions in the repair of DNA containing AP (apurinic/apyrimidinic) sites in mammalian cells. Biochem. J., 253, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindahl T. and Andersson,A. (1972) Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry, 11, 3618–3623. [DOI] [PubMed] [Google Scholar]
- 38.Povirk L.F. and Houlgrave,C.W. (1988) Effect of apurinic/apyrimidinic endonucleases and polyamines on DNA treated with bleomycin and neocarzinostatin: specific formation and cleavage at closely opposed lesions in complementary strands. Biochemistry, 27, 3850–3857. [DOI] [PubMed] [Google Scholar]
- 39.Yang N., Galick,H. and Wallace,S.S. (2004) Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst), 3, 1323–1334. [DOI] [PubMed] [Google Scholar]
- 40.Lehmann A.R. (2002) Replication of damaged DNA in mammalian cells: new solutions to an old problem. Mutat. Res., 509, 23–34. [DOI] [PubMed] [Google Scholar]
- 41.Nakamura J., La,D.K. and Swenberg,J.A. (2000) 5′-nicked apurinic/apyrimidinic sites are resistant to β-elimination by β-polymerase and are persistent in human cultured cells after oxidative stress. J. Biol. Chem., 275, 5323–5328. [DOI] [PubMed] [Google Scholar]
- 42.Klungland A., Hoss,M., Gunz,D., Constantinou,A., Clarkson,S.G., Doetsch,P.W., Bolton,P.H., Wood,R.D. and Lindahl,T. (1999) Base excision repair of oxidative DNA damage activated by XPG protein. Mol. Cell, 3, 33–42. [DOI] [PubMed] [Google Scholar]
- 43.Demple B. and DeMott,M.S. (2002) Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene, 21, 8926–8934. [DOI] [PubMed] [Google Scholar]
- 44.Marenstein D.R., Chan,M.K., Altaminaro,A., Basu,A.K., Boorstein,R.J., Cunningham,R.P. and Teebor,G.W. (2003) Substrate specificity of human endonuclease III (hNTH1). Effect of human APE1 on hNTH1 activity. J. Biol. Chem., 278, 9005–9012. [DOI] [PubMed] [Google Scholar]
- 45.Wilson D.M.R. and Barsky,D. (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res., 485, 283–307. [DOI] [PubMed] [Google Scholar]
- 46.McKenzie A.A. and Strauss,P.R. (2001) Oligonucleotides with bistranded abasic sites interfere with substrate binding and catalysis by human apurinic/apyrimidinic endonuclease. Biochemistry, 40, 13254–13261. [DOI] [PubMed] [Google Scholar]
- 47.Budworth H. and Dianov,G.L. (2003) Mode of inhibition of short-patch base excision repair by thymine glycol within clustered DNA lesions. J. Biol. Chem., 278, 9378–9381. [DOI] [PubMed] [Google Scholar]
- 48.Male R., Fosse,V.M. and Kleppe,K. (1982) Polyamine-induced hydrolysis of apurinic sites in DNA and nucleosomes. Nucleic Acids Res., 10, 6305–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]