

Summary
Transected axons fail to regrow in the mature central nervous system (CNS). Astrocyte scars are widely regarded as causal in this failure. Here, using three genetically targeted loss-of-function manipulations in adult mice, we show that preventing astrocyte scar formation, attenuating scar-forming astrocytes, or deleting chronic astrocyte scars all failed to result in spontaneous regrowth of transected corticospinal, sensory or serotonergic axons through severe spinal cord injury (SCI) lesions. In striking contrast, sustained local delivery via hydrogel depots of required axon-specific growth factors not present in SCI lesions, plus growth-activating priming injuries, stimulated robust, laminin-dependent sensory axon regrowth past scar-forming astrocytes and inhibitory molecules in SCI lesions. Preventing astrocyte scar formation significantly reduced this stimulated axon regrowth. RNA sequencing revealed that astrocytes and non-astrocyte cells in SCI lesions express multiple axon-growth supporting molecules. Our findings show that contrary to prevailing dogma, astrocyte scar formation aids rather than prevents CNS axon regeneration.
Introduction
Transected axons fail to regrow spontaneously across severe tissue lesions in the mature mammalian central nervous system (CNS). Potential mechanisms include (i) reduced intrinsic growth capacity of mature CNS neurons1–3, (ii) absence of external growth stimulating and supporting factors1,4,5, and (iii) presence of external inhibitory factors associated with myelin6,7, fibrotic tissue8 or astrocyte scars9. Alleviating cellular and molecular mechanisms underlying axon regeneration failure is fundamental to improving CNS repair after traumatic injury, stroke or degenerative disease.
Astrocyte scars have been regarded as barriers to CNS axon regrowth since the mid 20th century based on their appearance and early reports that attenuating astrocyte scar formation enabled spontaneous axon regrowth10,11. Although axon growth promoting effects of early scar attenuators proved illusory, reports correlating failed axon regrowth with presence of mature astrocytes12 or astrocyte scars9, plus evidence that astrocytes produce chondroitin sulfate proteoglycans (CSPGs) that inhibit axon growth in vitro9, led to widespread views that astrocyte scars are critical inhibitors of CNS axons and that nullifying this inhibition will lead to spontaneous axon regeneration.
Here, we tested the hypothesis that astrocyte scar formation plays a causal role in the failure of transected mature CNS axons to regenerate across severe tissue lesions. We used multiple transgenic loss-of-function strategies to either ablate scar-forming astrocytes, genetically attenuate scar-forming astrocytes or delete chronic astrocyte scars after severe spinal cord injury (SCI) in adult mice. We quantified the effects of these manipulations on (i) spontaneous regeneration of three major types of CNS axons, (ii) total CSPG levels, (iii) genome-wide expression by astrocytes and non-astrocytes in SCI lesions of molecules associated with axon growth, and (iv) axon regeneration stimulated by conditioning lesions plus delivery via synthetic hydrogel depots of known axon-required growth factors missing from SCI lesions.
Results
No axon regrowth after preventing scars
We first determined effects of preventing astrocyte scar formation on the potential for spontaneous unstimulated axon regeneration through severe CNS lesions. After focal traumatic tissue damage, CNS lesions comprise central areas of non-neural lesion core tissue surrounded by narrow astrocyte scar borders13,14. Astrocyte scar formation is complete by two weeks after adult murine SCI and is critically dependent on astrocyte proliferation and STAT3 signaling15–18. We prevented astrocyte scar formation with two loss-of-function transgenic mouse models that either (i) selectively kills proliferating scar-forming astrocytes15,16 or (ii) deletes STAT3 signaling selectively from astrocytes17,18, referred to respectively as TK+GCV or STAT3-CKO mice (Supplementary Information).
After severe crush SCI, wild-type (WT) mice formed dense astrocyte scars by two weeks that persisted for eight weeks, whereas TK+GCV, and STAT3-CKO mice failed to form scars and instead exhibited larger areas of non-neural tissue around lesion centers that were essentially devoid of astrocytes from two to eight weeks after SCI (Fig. 1b–d; Extended Data Fig. 1; Supplementary Information).
Figure 1. Preventing astrocyte scar formation does not lead to spontaneous regrowth of CST, AST, or 5HT axons after SCI.
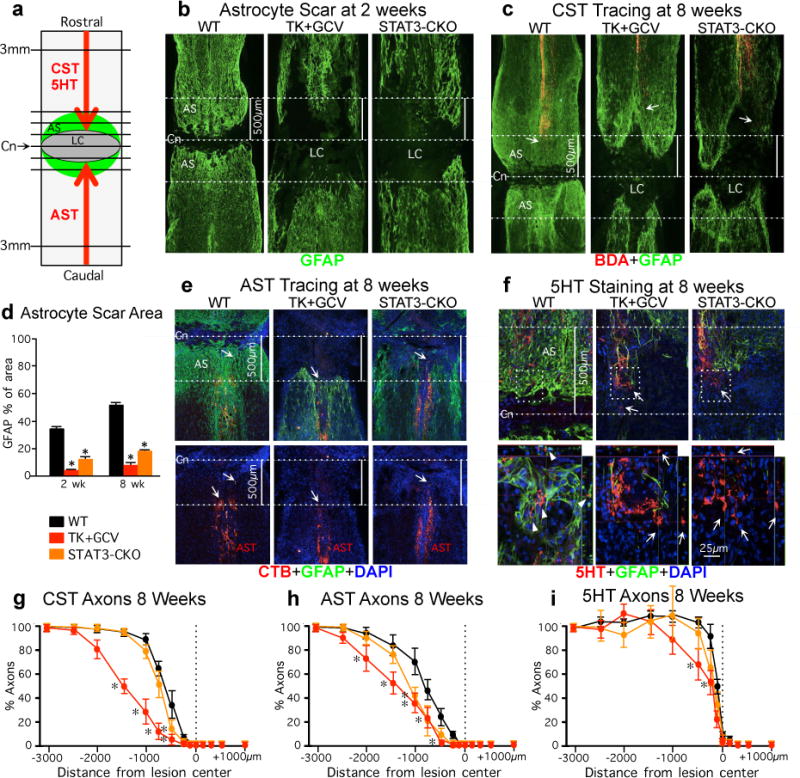
(a) Experiment summary schematic. Horizontal view of lesion core (LC) and astrocyte scar (AS) after SCI. Intercepts of CST, AST or 5HT axons with lines drawn at various distances from lesion center (Cn) were counted and expressed as a percent of axons 3mm proximal. (b,c,e,f) Dotted lines demarcate Cn and 500μm on either side. (b,d) Area occupied by GFAP-positive scar-forming astrocytes within 500μm on either side of Cn at 2 or 8 weeks after SCI. n = 6; (c, e) Arrows depict most caudal CST axons (BDA-tracing) or most rostral AST axons (CTB-tracing). (f) Arrows in top images depict most caudally penetrating 5HT axons, boxed areas are shown below. In WT mice, 5HT axons are surrounded by AS (arrowheads). In TK+GCV and STAT3-CKO mice, many 5HT axons are not in contact with AS (arrows), but have not regrown. (g–i) Numbers of CST (g), AST (h) and 5HT (i) axons at various distances from the SCI lesion center (Cn) as a percent of the number of axons present 3mm proximal. n = 6 CST; n = 5 AST and 5HT; * p<0.05 versus WT (ANOVA with Newman-Keuls).
Effects of preventing astrocyte scar formation on axon regeneration were quantified in three axonal systems, (i) descending corticospinal tract (CST), (ii) ascending sensory tract (AST) and (iii) descending serotonergic (5HT) tract, visualized either by axonal tract tracing or immunohistochemistry (Fig. 1c,e–i; Supplementary Information). As expected after severe SCI in adult WT mice, transected CST and AST axons both exhibited moderate dieback away from lesion centers (Fig. 1c,e,g,h). Preventing astrocyte scar formation in both TK+GCV or STAT3-CKO mice failed to result in spontaneous regrowth of transected CST or AST axons through SCI lesions, and instead significantly increased axonal dieback (Fig. 1c,e,g,h). As expected19,20, transected 5HT axons exhibited little dieback from lesion centers (Fig. 1f,i). Preventing scar formation in TK+GCV mice or STAT3-CKO mice did not exacerbate dieback of 5HT axons. Nevertheless, although many 5HT axons remained in lesion centers devoid of astrocytes, they also failed to regrow (Fig. 1f,i).
Thus, in spite of the essential absence of scar-forming astrocytes from SCI lesions for eight weeks after SCI in TK+GCV mice or STAT3-CKO mice, there was no spontaneous regeneration of transected CST, AST or 5HT axons through the lesions. This regrowth failure was particularly apparent for AST and 5HT axons whose axonal tips were often present along or within large areas devoid of astrocytes but did not regrow spontaneously through such areas (Fig. 1e,f).
No axon regrowth after deleting chronic scars
Acute astrocyte scar formation restricts inflammation and preserves neural tissue14–16,18. It has been proposed that after inflammation has resolved, chronic astrocyte scars are expendable and detrimental because they continually prevent axon regeneration. To test this hypothesis, we deleted chronic astrocyte scars five weeks after SCI with genetically targeted diphtheria toxin receptor and ultralow doses of diphtheria toxin21 (Fig.2; Supplementary Information). Distribution and specificity of targeting to mature astrocyte scars was verified with the genetic reporter, tdTomato (Fig. 2b; Extended Data Fig. 1c; Supplementary Information). GFAP immunohistochemistry verified efficient deletion of chronic astrocyte scars (Fig. 2c). Axon quantitation ten weeks after SCI showed that transected CST, AST or 5HT axons all failed to regrow spontaneously through areas depleted of chronic astrocyte scars (Fig. 2d–i). Again, this failure was particularly striking for AST and 5HT axons in or along areas devoid of scar-forming astrocytes that did not regrow through these areas (Fig. 2e,f). We also deleted chronic astrocyte scars and adjacent astrocytes over larger areas to reach ‘died-back’ CST and AST axons, but this approach caused pronounced tissue degeneration and large lesions (Extended Data Fig. 1e) that contained essentially no detectable CST, AST or 5HT axons. These findings show that deleting chronic astrocyte scars fails to result in spontaneous regrowth of CST, AST or 5HT axons through SCI lesions, and that chronic astrocyte scars remain critical for sustaining tissue integrity.
Figure 2. No spontaneous regrowth of CST, AST or 5HT axons after deleting chronic astrocyte scars.
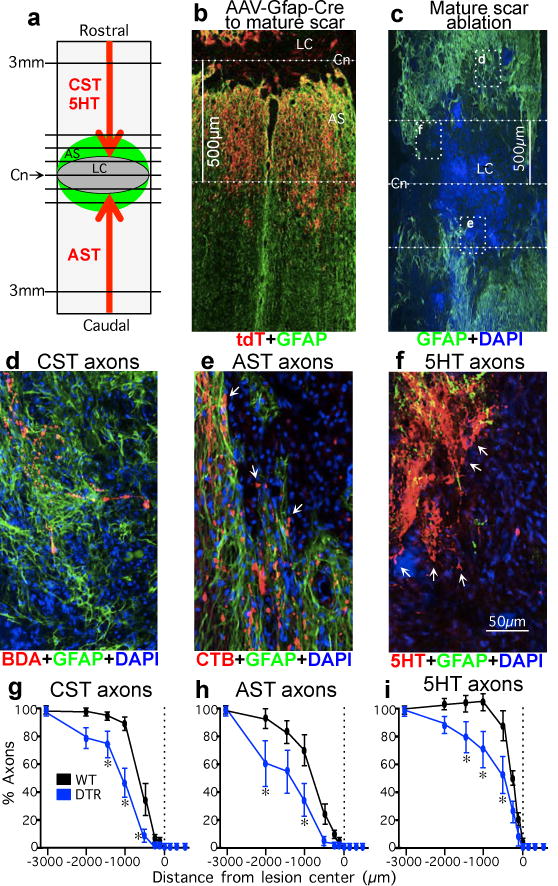
(a) Experiment summary schematic. (b) Selective targeting of tdT reporter to GFAP-positive scar-forming astrocytes in 500μm zone occupied by astrocyte scar (see Extended Data Figure 1c). (c) DTR-DTX mediated ablation of chronic astrocyte scar after severe SCI. Dotted lines indicate Cn and 500μm on either side normally occupied by scar-forming astrocytes in WT mice. Boxes show locations of d–f in adjacent sections. (d) CST axons are found only among GFAP-positive astrocytes proximal to ablated scar. (e) AST axons at margins of large area depleted of scar but have not regrown (arrows). (f) 5HT axons are within area depleted of scar but have not regrown (arrows). (g–i) Numbers of CST (F), AST (G), or 5HT (H) axons at various distances from SCI lesion centers as a percent of the number of axons present at 3mm proximal. n = 6 CST; n = 5 AST and 5HT; * p<0.05 versus WT (ANOVA with Newman-Keuls).
Multicellular CSPG production
We next looked for molecular mechanisms that might explain why ablating or attenuating astrocyte scars failed to enable spontaneous axon regrowth through severe lesions. CSPGs produced by astrocyte scars are regarded as principal inhibitors of axon regeneration9. Total CSPG levels determined by dot blot with CS56 antibody9,22 were, as expected9, significantly higher in our WT SCI lesions, but were not significantly reduced by transgenic ablation or disruption of astrocyte scar formation (Fig. 3a). Because diverse cells in SCI lesions including pericytes, fibroblast lineage cells and inflammatory cells13 can produce CSPGs23, we examined cellular production of CSPG and GFAP and quantified immunohistochemically stained tissue areas. In SCI lesions of TK+GCV and STAT3-CKO mice, GFAP area was significantly reduced in both grey and white matter compared with WT, whereas CSPG area was not significantly reduced in lesion core tissue or in regions of ablated astrocyte scar, which were filled with CSPG-positive, GFAP-negative, cells (Figs. 3b,c; Extended Data Fig. 2; Supplementary Information). These findings show that non-astrocyte cells in SCI lesions produce substantive CSPGs and that preventing astrocyte scar formation fails to reduce total CSPG production in SCI lesions.
Figure 3. CSPG production by non-astrocyte cells in SCI lesions after ablation or attenuation of astrocyte scars.
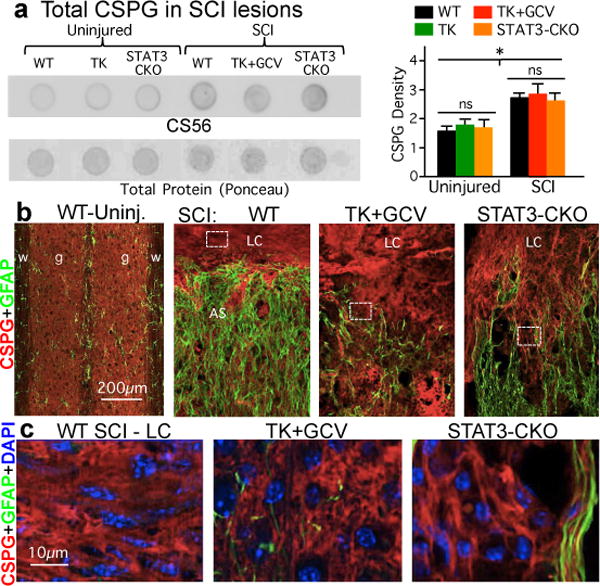
(a) Dot blots and quantitation of total CSPG detected by CS56 antibody relative to total protein (Ponceau). n = 4; * p<0.05 versus uninjured (ANOVA with Newman-Keuls). (b) CS56 and GFAP immunohistochemistry (see Extended Data Fig. 2). (c) Details of boxed areas in b showing CSPG production by GFAP-negative cells.
Genomic Dissection of SCI lesions
Numerous molecules attract, repel, support or inhibit axon growth during development, and many of these are present in CNS lesions7,24,25. To look broadly at molecules produced by astrocytes or non-astrocyte cells in SCI lesions that might impact on axon regrowth, we conducted genome-wide RNA sequencing of (i) astrocyte-specific ribosome-associated RNA (ramRNA) precipitated via a hemagglutinin tag26 transgenically targeted to either WT or STAT3-CKO astrocytes, and (ii) non-precipitated (flow-through) RNA deriving from non-astrocyte cells in the same tissue samples (Fig. 4; Extended Data Fig. 4; Supplementary Information).
Figure 4. Genomic dissection of astrocytes and non-astrocyte cells in SCI lesions.
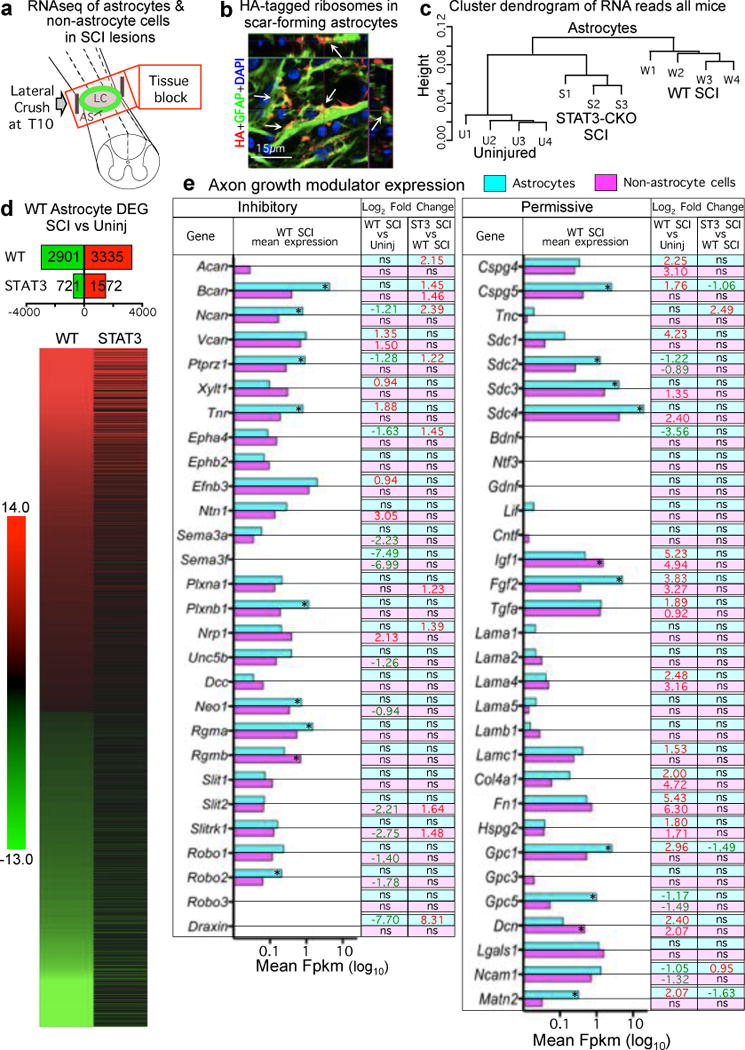
(a) Schematic of tissue harvested for RNA-sequencing. (b) Transgenically-targeted HA-tagged ribosome clusters (arrows) among GFAP filaments in scar-forming astrocytes (see Extended Data Fig. 3a). (c) Unsupervised hierarchical clustering dendrogram based on Pearson correlations shows that transcriptome profiles of STAT3-CKO SCI astrocytes cluster nearer to (and are more similar to) those of uninjured as compared to WT SCI astrocytes. (d) Numbers and heat map of significantly differentially expressed genes (DEGs) in WT astrocytes after SCI and the comparative differential expression profile of that specific cohort of genes in STAT3-CKO SCI astrocytes. Differential expression relative to uninjured, FDR<0.1. (e) Histogram of astrocyte and non-astrocyte expression of axon inhibitory or permissive molecules after SCI shown as mean FPKM. * significant difference astrocytes versus non-astrocytes, FDR<0.1. Numbers show Log2 fold significant differences. Red upregulated, green downregulated, ns non-significant.
At two weeks after SCI, astrocytes and non-astrocyte cells in SCI lesions exhibited significantly altered expression of many genes in both WT and STAT3-CKO mice, and WT astrocytes exhibited expected known changes28 (Extended Data Fig. 4a–d; Supplementary Information). Notably, 63% of genes significantly regulated by WT astrocytes were not significantly altered by STAT3-CKO astrocytes after SCI, and STAT3-CKO SCI astrocyte transcriptomes clustered more similarly towards uninjured astrocytes than towards WT SCI astrocytes (Fig. 4c,d).
We analyzed 59 molecules reported to negatively or positively modulate axon growth in SCI lesions (Extended Data Table 1). In SCI lesions from WT mice, both astrocytes and non-astrocyte cells expressed a majority not only of 28 known axon inhibitors, including specific CSPGs, ephrins, netrins, neuropillins, plexins, slits and others, but also a majority of 31 known axon permissive molecules, including specific CSPGs, laminins, syndecans, glypicans, decorin and others (Fig. 4e). Remarkably, both astrocytes and non-astrocytes down-regulated more axon inhibitory molecules than were upregulated, and upregulated more than three times the number of axon permissive molecules than were downregulated. Preventing astrocyte scar formation in STAT3-CKO mice (i) did not significantly decrease the expression by astrocytes or non-astrocytes of a single reported inhibitor, whereas eleven were upregulated, and (ii) significantly increased the expression of two permissive molecules, and decreased three, compared to WT SCI (Fig. 4e).
In agreement with our immunoblot and immunohistochemical CSPG findings (above), various individual CSPG ramRNAs were expressed by both astrocytes and non-astrocyte cells in SCI lesions (Fig. 4e). Interestingly, aggrecan, the prototypical CSPG used in axon growth inhibition studies in vitro9,29, was not detectably expressed by scar-forming astrocytes at either the ramRNA or immunohistochemistry of protein levels (Fig. 4e; Extended Data Fig. 5a). Other axon inhibitory CSPGs, brevican, neurocan, versican and phosphacan, were all expressed by both scar-forming astrocytes and non-astrocyte cells in SCI lesions as revealed by RNA analysis (Fig. 4e) and confirmed by immunohistochemistry of protein for brevican and neurocan (Extended Data Figs. 5b,6a).
CSPGs are diverse with respect to inhibiting or supporting axon growth at both protein and sugar-epitope levels30,31. Of the five growth-inhibitory CPSGs, WT scar-forming astrocytes increased only versican ramRNA, and significantly decreased neurocan and phosphacan (Fig. 4e). In contrast, ramRNAs of two axon growth-supportive CSPGs, Cspg4 (NG2) and Cspg5 (neuroglycan C) (Extended Data Table 1), were significantly upregulated by scar-forming astrocytes (Fig. 4e) and both NG2 and CSPG5 clearly decorated scar-forming astrocytes as revealed by immunohistochemistry (Extended Data Fig. 6b,c).
Our genomic findings show that (i) STAT3-CKO prevents or attenuates a majority of genome-wide changes in astrocytes associated with astrogliosis and scar formation in WT mice; (ii) astrocytes and non-astrocyte cells in SCI lesions express a large, diverse mix of axon inhibitory and permissive molecules; (iii) non-astrocyte cells in SCI lesions substantively express CSPGs; (iv) preventing astrocyte scar formation with STAT3-CKO does not reduce expression of CSPGs or other inhibitory molecules in SCI lesions; (v) scar-forming astrocytes upregulate and substantively express axon growth supporting CSPGs, indicating that CS56 immune detection of total CSPG levels22 need not indicate a purely axon-inhibitory environment; and (vi) scar-forming astrocytes and non-astrocyte cells in SCI lesions upregulate multiple axon growth permissive matrix molecules, including laminins.
Axon regrowth in spite of scar formation
We next stimulated axon growth after SCI in the presence or absence of astrocyte scar formation. Developing axons do not grow by default but require stimulatory cues32. This requirement may apply also to regrowth of transected mature axons. Some transected mature AST axons can be stimulated to regrow in severe SCI lesions by activating neuron intrinsic growth programs with peripheral conditioning lesions33–35, and this regrowth can be significantly augmented by cell grafts that provide supportive matrix plus the neurotrophic factors NT3 and BDNF that attract AST axon growth during development36. We noted the essential absence of Nt3 and Bdnf expression in our WT SCI lesions, combined with expression of permissive matrix molecules including laminins known to support developing AST axons37 (Fig. 4e; Extended Data Table 1). We therefore tested effects of conditioning lesions plus local delivery of NT3 and BDNF on AST axon regeneration stimulated in the presence or absence of astrocyte scar formation (Fig. 5; Extended Data Figs. 7–9). Because cell grafts modify astrocyte scars36 and provide permissive substrates for regrowing axons, we delivered NT3 and BDNF via synthetic hydrogel depots that do not modify astrocyte scar formation and provide prolonged neurotrophin delivery38–40 (Fig. 5d; Supplementary Information).
Figure. 5. Robust regrowth of AST axons can be stimulated after WT SCI and is significantly attenuated by preventing astrocyte scar formation.
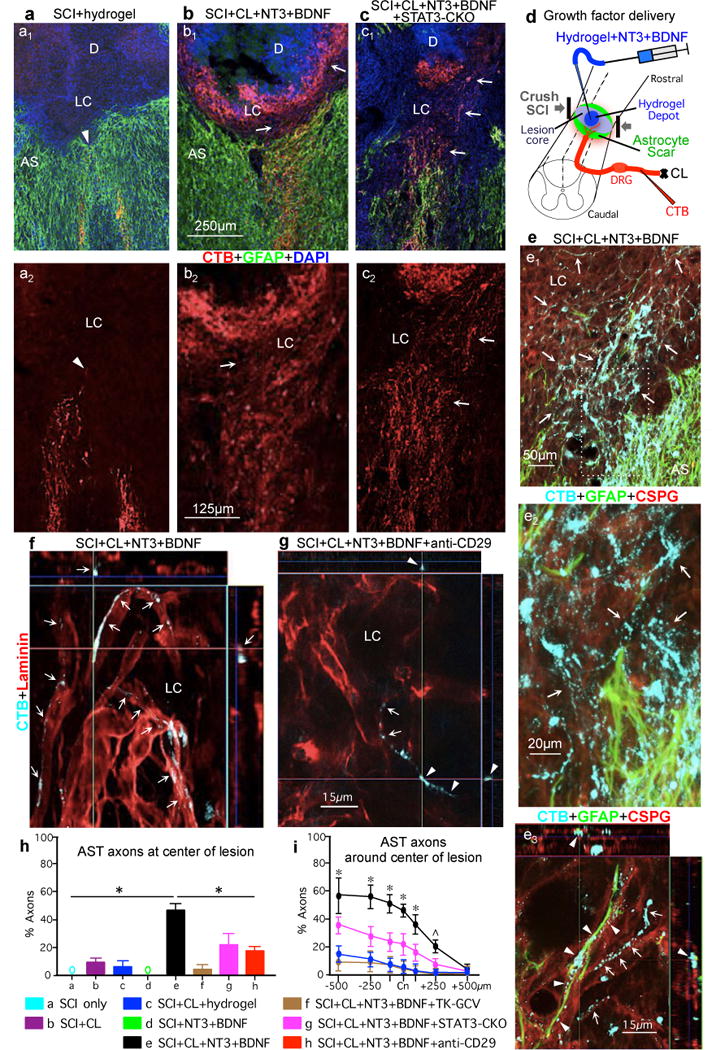
(a1–c1) AST axons (CTB-tracing) plus GFAP immunohistochemistry. (a2–c2) AST axons alone. (a) WT mouse, SCI and hydrogel only (no growth factors). Arrowhead denotes most rostrally penetrating axons that do not pass beyond AS. (b) WT mouse, SCI plus conditioning lesions (CL) and hydrogel depot (D) with NT3+BDNF. Arrows denote robust regrowth of AST axons past AS into LC and along, but not into, the depot that releases NT3+BDNF but provides no adhesive matrix. (c) STAT3-CKO mouse, SCI plus CL and NT3+BDNF depot. Arrows denote regrowth of AST axons into LC. (d) Experiment summary schematic. (e–f) WT mice. (e1–e3) AST plus GFAP and CSPG (CS56) immunohistochemistry. Box in e1 is shown in e2. (e1 and e2) Arrows denote robust regrowth of stimulated AST axons past AS into LC through CSPG. (e3) Regrowing AST axons track along CSPG-positive GFAP-negative structures (arrows) or along CSPG-positive GFAP-positive astrocyte processes (arrowheads) (See Extended Data Figures 7,8). (f,g) AST axons plus laminin immunohistochemistry. (f) Arrows denote regrowing stimulated AST axons tracking along laminin. (g) Arrowheads denote stimulated AST axons exposed to anti-CD29 antibody and failing to maintain contact with laminin. (h,i) Numbers of AST axons at SCI Cn (h) or on either side (i) expressed as percent of axons 3mm proximal. n = 5; * p<0.05 versus all other groups, ˆ p<0.05 versus all groups except STAT3-CKO (ANOVA with Newman-Keuls).
No AST fibers regrew past astrocyte scars into lesion cores after SCI alone or with hydrogel without growth factors (Fig. 5a,h; Extended data Fig. 7c). Small numbers of AST axons regrew into lesion cores in mice with SCI plus conditioning lesions alone, or SCI plus NT3 and BDNF without conditioning lesions (Fig. 5h,i; Extended data Fig. 7c). In striking contrast, mice receiving both conditioning lesions plus NT3 and BDNF exhibited robust axon regrowth through and beyond astrocyte scars, and the number of axon intercepts counted at lesion centers averaged over 45% of that in intact sensory tracts three mm proximal to lesions (Fig. 5b,h,i; Extended data Fig. 7c). Remarkably, these stimulated AST axons regrew profusely through, and past, dense astrocyte scars in spite of substantial CSPG (Figs. 5b,e, Extended Data Figs. 7b,c,8). AST axons stimulated to regrow in SCI lesions were thin and uniformly tracked along laminin surfaces, turning and even reversing direction along these surfaces as expected of regenerating axons41, whereas AST axons in intact gracile-cuneate tracts were coarsely beaded and were not in direct contact with laminin (Figs. 5f; Extended Data Fig. 9a–f). Hydrogel delivery of anti-CD29 laminin-integrin function-blocking antibodies37 together with NT3 and BDNF significantly reduced AST axon regrowth in lesion cores by 73%, demonstrating that laminin interactions were critical for stimulated AST axon regrowth (Figs. 5g–h; Extended Data Fig. 9g–I; Supplementary Information). Lastly, preventing astrocyte scar formation did not augment AST axon regrowth stimulated by conditioning lesions plus NT3 and BDNF, but instead significantly attenuated axon regrowth in both TK+GCV mice and STAT3-CKO mice (Fig. 5c,h,i), demonstrating that astrocyte scar formation aids, rather than inhibits appropriately stimulated AST axon regeneration after SCI.
Discussion
Our findings show that contrary to prevailing dogma, astrocyte scar formation is not a principal cause for the failure of injured mature CNS axons to regrow across severe CNS lesions and that scar-forming astrocytes permit and support robust amounts of appropriately stimulated CNS axon regeneration. Although our observations with stimulated AST axon regeneration needs extension to other axonal systems, our findings are consistent with evidence that (i) astrocytes can support growth of different CNS axons in vivo during development42,43 or after mature CNS injury19,44, (ii) genetic activation of axonal growth programs by mature neurons leads to axon regeneration across CNS lesions only when scar-forming astrocyte bridges are present2,45, and (iii) grafts of progenitor-derived astrocytes support axon regeneration through non-neural SCI lesion cores46,47.
The predominant mechanistic proposal for astrocyte scar inhibition of axon regeneration is CSPG production9. However, specific CSPGs can support or repel axon growth (see Extended Data Table 1). We show that scar-forming astrocytes upregulate growth supportive CSPG4 and CSPG5, and that both astrocytes and non-astrocytes in SCI lesions express multiple axon-growth supportive molecules, including laminins. Axon growth and guidance depend both on intrinsic growth potential2,3,32,33,48 and on a balance of extrinsic regulatory cues that modulate one another’s effects on growth cones25. Our findings show that (i) sustained delivery of required axon-specific growth factors not adequately expressed in SCI lesions, combined with activation of neuron-intrinsic growth programs, can stimulate robust regrowth of transected axons along specifically required supportive matrix cues in spite of the presence of inhibitory cues, and (ii) this stimulated axon regrowth can occur either in direct contact with scar-forming astrocyte processes or independently of astrocyte processes. These observations provide direct evidence that the requirements for achieving axon regeneration across severe CNS lesions, where transected axons lack intrinsic and extrinsic conditions for long distance regrowth, are fundamentally different from requirements to achieve local neurite outgrowth in perilesion intact but reactive grey matter, where conditions compatible with axon terminal growth and remodeling are present and where blocking inhibitory regulators such as CSPGs and others produced by astrocytes and other cells may be sufficient to promote axon sprouting that might also improve function9,15,24,29,41,49. Our findings have important implications for CNS repair strategies by demonstrating that rather than being hostile to axon growth, newly generated immature scar-forming astrocytes derived after SCI from endogenous progenitors18,42,45, and potentially from grafted progenitors46,47, aid axon regeneration and may represent exploitable bridges for regrowing axons across severe CNS lesions.
METHODS
Mice
All non-transgenic, transgenic and control mice used in this study were derived from in house breeding colonies backcrossed >12 generations onto C57/BL6 backgrounds. All mice used were young adult females between two and four months old at the time of spinal cord injury. All transgenic mice used have been previously well characterized or are the progeny of crossing well-characterized lines: (1) mGFAP-TK transgenic mice line 7.115,16,50. (2) mGFAP-Cre-STAT3-loxP mice generated by crossing STAT3-loxP mice with loxP sites flanking exon 22 of the STAT3 gene51 with mGFAP-Cre mice line 73.1217,18. (3) loxP-STOP-loxP-DTR (diphtheria toxin receptor) mice21. (4) mGFAP-Cre-RiboTag mice generated by crossing mice with loxP-STOP-loxP-Rpl22-HA (RiboTag)26 with mGFAP-Cre mice line 73.1217,18. (5) mGFAP-tdT reporter mice generated by crossing loxP-STOP-loxP-tdT (td-tomato) reporter mice52 with mGFAP-Cre mice line 73.1217,18. All mice were housed in a 12-hour light/dark cycle in a specific pathogen-free facility with controlled temperature and humidity and were allowed free access to food and water. All experiments were conducted according to protocols approved by the Animal Research Committee of the Office for Protection of Research Subjects at University of California Los Angeles.
Surgical procedures
All surgeries were performed under general anesthesia with isoflurane in oxygen-enriched air using an operating microscope (Zeiss, Oberkochen, Germany), and rodent stereotaxic apparatus (David Kopf, Tujunga, CA). Laminectomy of a single vertebra was performed and severe crush SCI were made at the level of T10 using to expose the spinal cord. For severe spinal cord injury (SCI), No. 5 Dumont forceps (Fine Science Tools, Foster City, CA) without spacers and with a tip width of 0.5mm were used to completely compress the entire spinal cord laterally from both sides for 5 seconds16–18. For pre-conditioning lesions, sciatic nerves were transected and ligated one week prior to SCI. Hydrogels were injected stereotaxically into the center of SCI lesions 0.6 mm below surface at 0.2μl per minute using glass micropipettes (ground to 50 to 100 μm tips) connected via high-pressure tubing (Kopf) to 10μl syringes under control of microinfusion pumps, two days after SCI53. Tract-tracing was performed by injection of (i) biotinylated dextran amine 10,000 (BDA, Invitrogen) 10% wt/vol in sterile saline injected 4 × 0.4μl into the left motor cerebral cortex 14 days prior to perfusion to visualize corticospinal tract (CST) axons, or (ii) choleratoxin B (CTB) (List Biological Laboratory, Campbell, CA) 1μl of 1% wt/vol in sterile water injected into both sciatic nerves three days prior to perfusion to visualize ascending sensory tract (AST) axons34. AAV2/5-GfaABC1D-Cre (see below) was injected either 3 or 6 × 0.4μl (1.29 × 1013 gc/ml in sterile saline) into and on either side of mature SCI lesions two weeks after SCI, or into uninjured spinal cord after T10 laminectomy. All animals received analgesic prior to wound closure and every 12 hours for at least 48 hours post-injury. Animals were randomly assigned numbers and evaluated thereafter blind to genotype and experimental condition.
AAV2/5-GfaABC1D-Cre
Adeno-associated virus 2/5 (AAV) vector with a minimal Gfap promoter (AAV2/5 GfaABC1D) was used to target Cre-recombinase expression selectively to astrocytes54–56.
Hydrogel with growth factors and antibodies
Diblock co-polypeptide hydrogel (DCH) K180L20 was fabricated, tagged with blue fluorescent dye (AMCA-X) and loaded with growth factor and antibody cargoes as described39,40,53. Cargo molecules comprised: Human recombinant NT3 and BDNF were gifts (Amgen, Thousand Oaks, CA, (NT3 Lot#2200F4; BDNF Lot#2142F5A) or were purchased from PeproTech (Rocky Hill, NJ; NT3 405-03, Lot#060762; BDNF 405-02 Lot#071161). Function blocking anti-CD29 mouse monoclonal antibody was purchased from BD Bioscience (San Diego, CA) as a custom order at 10.25mg/ml (product #BP555003; lot#S03146). Freeze dried K180L20 powder was reconstituted on to 3.0% or 3.5% wt/vol basis in sterile PBS without cargo or with combinations of NT3 (1.0μg/μl), BDNF (0.85μg/μl) and anti-CD29 (5μg/μl). DCH mixtures were prepared to have G′ (storage modulus at 1Hz) between 75 and 100 Pascal (Pa), somewhat below that of mouse brain at 200 Pa39,40.
Ganciclovir (GCV), BrdU or DTX injections
GCV (Cytovene-IV ® Hoffman LaRoche, Nutley, NJ), 25mg/kg/day dissolved in sterile physiological saline was administered as single daily subcutaneous injections starting immediately after surgery and continued for the first 7 days after SCI. Bromodeoxyuridine (BrdU, Sigma), 100 mg/kg/day dissolved in saline plus 0.007N NaOH, was administered as single daily intraperitoneal injections on days 2 through 7 after SCI. Diphtheria toxin A (DTX, Sigma #DO564) 100ng in 100μl in sterile saline was administered twice daily as intraperitoneal injections for 10 days starting three weeks after injection of AAV2/5-GfaABC1D-Cre to loxP-DTR mice (which was 5 weeks after SCI) (see timeline in Extended Data Fig. 1d).
Hindlimb locomotor evaluation, animal inclusion criteria, randomization and blinding
Two days after SCI, all mice were evaluated in open field and mice exhibiting any hindlimb movements were not studied further. Mice that passed this pre-determined inclusion criterion were randomized into experimental groups for further treatments and were thereafter evaluated blind to their experimental condition. At 3,7,14 days and then weekly after SCI, hindlimb movements were scored using a simple six-point scale in which 0 is no movement and 5 is normal walking17.
Histology and immunohistochemistry
After terminal anesthesia by barbiturate overdose mice were perfused transcardially with 10% formalin (Sigma). Spinal cords were removed, post-fixed overnight, and cryoprotected in buffered 30% sucrose for 48 hours. Frozen sections (30μm horizontal) were prepared using a cryostat microtome (Leica) and processed for immunofluorescence as described16–18. Primary antibodies were: rabbit anti-GFAP (1:1000; Dako, Carpinteria, CA); rat anti-GFAP (1:1000, Zymed Laboratories); goat anti-CTB (1:1000, List Biology Lab); rabbit anti-5HT (1:2000, Immunostar); goat anti-5HT (1:1000, Immunostar); mouse anti-CSPG22 (1:100, Sigma); rabbit-anti hemagglutinin (HA) (1:500 Sigma); mouse-anti HA (1:3000 Covance); sheep anti-BrdU (1:6000, Maine Biotechnology Services, Portland, ME); rabbit anti-laminin (1:80, Sigma, Saint Louis, MO); guinea pig anti-NG2 (CSPG4) (Drs. E.G. Hughes and D.W. Bergles57, Baltimore, MA); goat anti-aggrecan (1:200, NOVUS); rabbit anti-brevican (1:300, NOVUS); mouse anti-neurocan (1:300, Milipore); mouse anti-phosphacan (1:500, Sigma); goat anti-versican (1:200, NOVUS); rabbit anti-neurglycan C (CSPG5) (1:200, NOVUS). Fluorescence secondary antibodies were conjugated to: Alexa 488 (green) or Alexa 350 (blue) (Molecular Probes), or to Cy3 (550, red) or Cy5 (649, far red) all from (Jackson Immunoresearch Laboratories). Mouse primary antibodies were visualized using the Mouse-on-Mouse detection kit (M.O.M. ®, Vector). BDA tract-tracing was visualized with streptavidin-HRP plus TSB Fluorescein geen or Tyr-Cy3 (Jackson Immunoresearch Laboratories). Nuclear stain: 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 2ng/ml; Molecular Probes). Sections were coverslipped using ProLong Gold anti-fade reagent (InVitrogen, Grand Island, NY). Sections were examined and photographed using deconvolution fluorescence microscopy and scanning confocal laser microscopy (Zeiss, Oberkochen, Germany).
Axon quantification
Axons labeled by tract tracing or immunohistochemistry were quantified using image analysis software (NeuroLucida®, MicroBrightField, Williston, VT) operating a computer-driven microscope regulated in the x, y and z axes (Zeiss) by observers blind to experimental conditions. Using NeuroLucida ®, lines were drawn across horizontal spinal cord sections at SCI lesion centers and at regular distances on either side (Fig. 1a) and the number of axons intercepting lines was counted at 63× magnification under oil immersion by observers blind to experimental conditions. Similar lines were drawn and axons counted in intact axon tracts three mm proximal to SCI lesions and the numbers of axon intercepts in or near lesions were expressed as percentages of axons in the intact tracts in order to control for potential variations in tract-tracing efficacy or intensity of immunohistochemistry among animals. Two sections at the level of the CST or AST, and three sections through the middle of the cord for 5HT, were counted per mouse and expressed as total intercepts per location per mouse. To determine efficacy of axon transection after SCI, we examined labeling three mm distal to SCI lesion centers, with the intention of eliminating mice that had labeled axons at this location on grounds that these mice may have had incomplete lesions. However, all mice that had met the strict behavioral inclusion criterion of no hindlimb movements two days after severe crush SCI, exhibited no detectable axons three mm distal to SCI lesions regardless of treatment group.
Quantification of immunohistochemically stained areas
Sections stained for GFAP, CSPG or laminin were photographed using constant exposure settings. Single channel immunofluorescence images were converted to black and white and thresholded (Fig. 1d; Extended Data Figure 2b) and the amount of stained area measured in different tissue compartments using NIH Image J software.
Statistics, power calculations and group sizes
Statistical evaluations of repeated measures were conducted by ANOVA with post hoc, independent pair wise analysis as per Newman-Keuls (Prism®, GraphPad, San Diego, CA). Power calculations were performed using G*Power Software V 3.1.9.258. For quantification of histologically-derived neuroanatomical outcomes such as numbers of axons or percent of area stained for GFAP or CSPG, group sizes were used that were calculated to provide at least 80% power when using the following parameters: probability of type I error (alpha) = .05, a conservative effect size of 0.25, 2–8 treatment groups with multiple measurements obtained per replicate. Using main Figure 5h as an example, evaluation of n = 5 biological replicates (with multiple measurements per replicate) in each of 8 treatment groups provided greater than 88% power.
Dot blot
For dot blot immunoassay of chondroitin sulfate proteoglycans (CSPG), spinal cord tissue blocks were lysed and homogenized in standard RIPA (radio-immunoprecipitation-assay) buffer. LDS (lithium dodecyl sulfate) buffer (Life Technologies) was added to the post-mitochondrial supernatant and 2μL containing 2μg/uL protein was spotted onto a nitrocellulose membrane (Life Technologies), set to dry and incubated overnight with mouse anti-chondroitin sulfate antibody (CS56, 1:1000, Sigma Aldrich), an IgM-monoclonal antibody that detects glyco-moieties of all CSPGs22. CS56 immunoreactivity was detected on X-ray film with alkaline phosphatase-conjugated secondary antibody and chemiluminescent substrate (Life Technologies). Densitometry measurements of CS56 immunoreactivity were obtained using ImageJ software (NIH) and normalized to total protein (Poncau S) density59.
Isolation, sequencing and analysis of RNA from astrocytes and non-astrocyte cells
Two weeks after SCI, spinal cords of wild type control (GFAP-RiboTag) and STAT3-CKO (GFAP-STAT3CKO-RiboTag) mice were rapidly dissected out of the spinal canal. The central 3mm of the lower thoracic lesion including the lesion core and 1mm rostral and caudal were then rapidly removed and snap frozen in liquid nitrogen. Hemagglutinin (HA) immune-precipitation (HA-IP) of astrocyte ribosomes and ribosome-associated mRNA (ramRNA) was carried out as described26. The non-precipitated flow through (FT) from each IP sample was collected for analysis of non-astrocyte total RNA. HA and FT samples underwent on-column DNA digestion using the RNase-Free Dnase Set (Qiagen) and RNA purified with the RNeasy Micro kit (Qiagen). Integrity of the eluted RNA was analyzed by a 2100 Bioanalyzer (Agilent) using the RNA Pico chip, mean sample RIN = 8.0±0.95. RNA concentration determined by RiboGreen RNA Assay kit (Life Technologies). cDNA was generated from 5ng of IP or FT RNA using the Nugen Ovation® 2 RNA-Seq Sytstem V2 kit (Nugen). 1 ug of cDNA was fragmented using the Covaris M220. Paired-end libraries for multiplex sequencing were generated from 300 ng of fragmented cDNA using the Apollo 324 automated library preparation system (Wafergen Biosystems) and purified with Agencourt AMPure XP beads (Beckman Coulter). All samples were analyzed by an Illumina NextSeq 500 Sequencer (Illumina) using 75-bp paired-end sequencing. Reads were quality controlled using in-house scripts including picard-tools, mapped to the reference mm10 genome using STAR60, and counted using HT-seq61 with mm10 refSeq as reference, and genes were called differentially expressed using edgeR62. Individual gene expression levels in the Figure 4e histogram are shown as mean FPKM (Mean fragments per kilobase of transcript sequence per million mapped fragments). Additional details of differential expression analysis are described in figure legends of Figure 4 and Extended Data Figures 3 and 4. Raw and normalized data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE76097 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76097).
Extended Data
Extended data figure 1. SCI model schematic, locomotor behavioral effects, and AAV vector targeting specificity and effects.
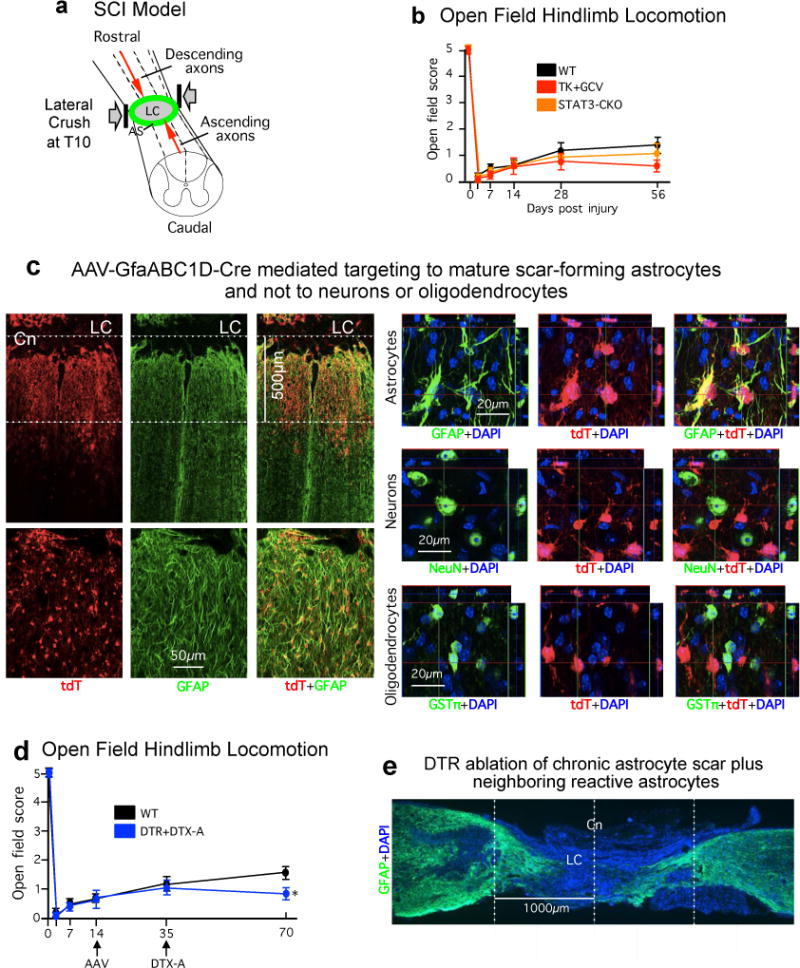
(a) Schematic of severe lateral crush SCI at thoracic level T10 that generates a large lesion core (LC) of non–neural tissue surrounded by an astrocyte scar (AS) and completely transects descending and ascending axons. (b) Open field hindlimb locomotor score at various times after SCI assessed using a 5 point scale where 5 is normal and 0 is no movement of any kind17. No significant differences were observed among any of the experimental groups at any time point. n = 6 ns at all time points p>0.5 (ANOVA with Newman-Keuls post hoc analysis). (c) Horizontal sections through a severe SCI lesion of a representative tdT reporter mouse52 injected with an AAV vector with a minimal Gfap promoter regulating Cre (AAV2/5-GfaABC1D-Cre) into the lesion at two weeks after SCI and perfused at three weeks. tdT labeling demonstrates that this AAV2/5-GfaABC1D-Cre efficiently and specifically targets GFAP-positive astrocytes. In this mouse, the amount AAV2/5-GfaABC1D-Cre injected was intentionally titrated on the basis of previous trial and error to target primarily the astrocyte scar border in an approximately 500μm zone immediately abutting the SCI lesion core (LC). High magnification analysis of individual fluorescence channels stained for tdT plus various cell markers shows the specificity of Cre-activity targeting to cells expressing the astrocyte marker, GFAP, but not to cells expressing either the neuronal marker, NeuN, or the mature oligodendrocyte marker, GSTπ. AAV2/5-GfaABC1D-Cre was prepared using a previously described and well-characterized cloning strategy54. (d) Open field hindlimb locomotor scores at various times after SCI. There was no difference in scores of control mice and loxP-DTR mice that received AAV2/5-GfaABC1D-Cre prior to injections of DTX. Five weeks after DTX injections, loxP-DTR mice that received AAV2/5-GfaABC1D-Cre exhibited a slightly, but significantly, lower locomotor score. Hindlimb locomotion was assessed using a 5 point scale where 5 is normal and 0 is no movement of any kind17. n = 6 per group, * p<0.05 versus WT (ANOVA with Newman-Keuls). (e) GFAP immunohistochemistry of a sagittal section after ablation of a chronic astrocyte scar plus adjacent astrocytes. DTX was administered to a transgenic mouse expressing DTR targeted selectively to astrocytes around a severe SCI. In this case, the amount of AAV2/5-GfaABC1D-Cre injected was titrated to target not only primarily the astrocyte scar border but also adjacent astrocytes spread over approximately two mm on either side of the center (Cn) of the SCI lesion core (LC). Note the profound degeneration of neural tissue resulting from the selective ablation of the chronic astrocyte scar plus adjacent astrocytes after SCI.
Extended data figure 2. Single channel CSPG and GFAP immunofluorescence and stained area quantification.
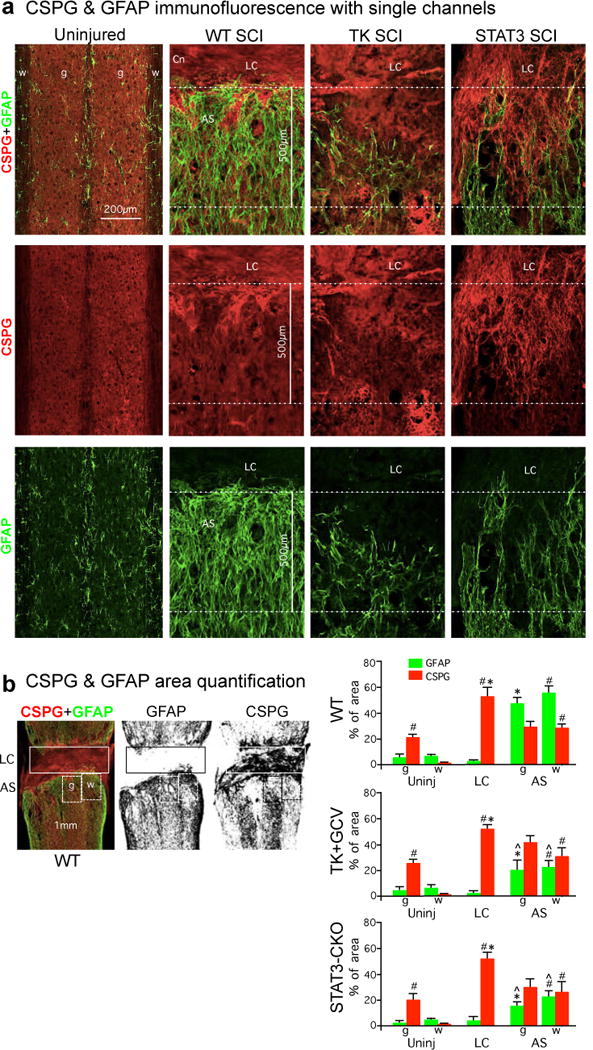
(a) Individual fluorescence channels of CS56 and GFAP immunohistochemistry from horizontal sections of uninjured mice and at two weeks after severe SCI shown in main text figure 3b. Sections are taken from WT mice and mice with transgenic ablation (TK+GCV) or attenuation (STAT3-CKO) of astrocyte scar formation. (b) Example of black and white thresholding of single channels of immunofluorescence staining for image analysis to quantify (using NIH Image J software) the amount of CSPG or GFAP stained area in different tissue compartments in SCI lesions. Boxes denote areas quantified to obtain values for lesion core (LC) and grey (g) or white (w) matter in astrocyte scar (AS) or equivalent regions in uninjured tissue. Graphs show percent of areas stained for CSPG or GFAP determined using Image J. n = 4 WT, n = 6 TK+GCV and STAT3-CKO; # p<0.05 versus uninjured white matter, * p<0.05 versus uninjured grey matter in same experimental group (ANOVA with Newman-Keuls); ˆ p<0.05 versus equivalent anatomical region in WT (ANOVA with Newman-Keuls).
Extended data figure 3. Specificity of HA-targeting to astrocytes and enrichment of HA-IP for astrocyte specific RNA transcripts.
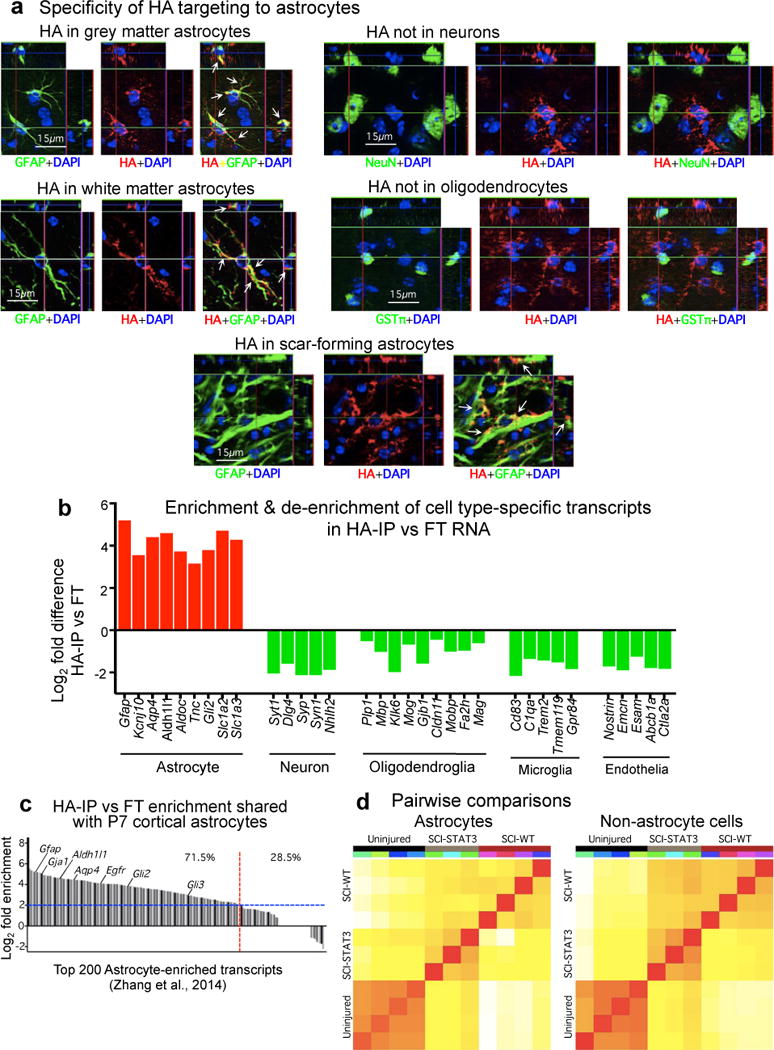
(a) Individual fluorescence channels of immunohistochemistry for transgenically-targeted hemagglutinin (HA) plus various cell markers showing the specificity of HA targeting to cells expressing the astrocyte marker, GFAP, and not to cells expressing either the neuronal marker, NeuN, or the mature oligodendrocyte marker, GSTπ, in uninjured grey and white matter and in astrocyte scar at 2 weeks after SCI. (b) CNS cell type-specific gene transcript enrichment of ribosome-associated mRNA (ramRNA) isolated from WT uninjured spinal cord by HA immune-precipitation (HA-IP). Differential expression analysis by RNA-sequencing (RNA-Seq) indicates significant enrichment (red) for astrocyte-specific gene transcripts, and de-enrichment (green) for gene transcripts enriched in other CNS cell types, FDR<0.1. A log2 scale is used so that positive and negative differences are directly comparable. The mean numerical enrichment of three quintessential astrocyte genes, Gfap, Aldh1l1 and Aqp4, is 25 fold greater in HA samples than in flow through samples. (c) Gene transcript enrichment of HA-IP ramRNA relative to P7 mouse primary cortical astrocytes27. Of the 200 most highly expressed genes previously described27 for post-natal mouse cortical astrocytes, 71.5% (red line) are at least 4-fold enriched (blue line) in HA-IP ramRNA isolated from uninjured spinal cord relative to flow through RNA from non-astrocyte cells. (d) Pearson correlation plots of total normalized RNA-Seq reads from individual biological replicates for each treatment condition. Correlation coloring indicates little (white) to high (red) similarity. n = 4 for uninjured controls and wild type SCI (SCI-WT); n = 3 for STAT3-CKO SCI (SCI-STAT3). FDR<0.1 for differential expression and enrichment analysis. Raw and normalized data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE76097 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76097).
Extended data figure 4. Comparison of genomic data from astrocytes and non-astrocyte cells from WT and STAT3-CKO mice after SCI.
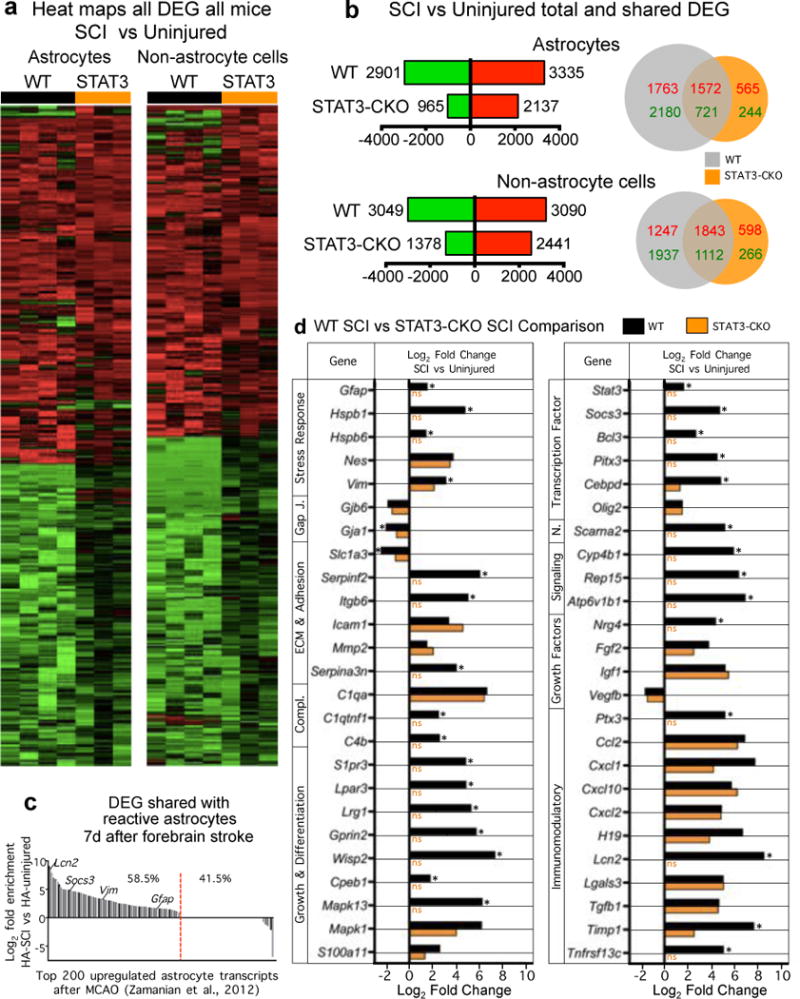
(a) Heat maps depicting all significantly differentially expressed genes (DEG), as determined by RNA-Seq, for WT and STAT3-CKO astrocytes and non-astrocytes from independent biological replicates two weeks after SCI relative to uninjured WT control. Red upregulated, green downregulated. (b) Total numbers and Venn diagrams of significant DEGs in WT and STAT3-CKO astrocytes and non-astrocytes two weeks after SCI relative to uninjured control. Red and green numerical values indicate significantly upregulated and downregulated genes, respectively. (c) Comparison of altered gene expression in our SCI-reactive astrocytes and previously reported forebrain stroke-reactive astrocytes28. Of the 200 most highly elevated genes in forebrain astrocytes 1 week following stroke28, 58.5% (red line) are also significantly elevated in astrocytes after SCI, relative to uninjured. (d) Comparison of expression by WT SCI and STAT3-CKO SCI reactive astrocytes of a selected cross-section of genes that are highly regulated after SCI by WT reactive astrocytes. Many of the regulated genes exhibit changes that are expected and implicated in WT reactive astrogliosis mechanisms and roles, and some of the changes appear to be newly identified in this context. Note that many of the genes are not regulated or exhibit attenuated changes in STAT3-CKO SCI astrocytes. n = 4 for uninjured and WT SCI; n = 3 for STAT3-CKO SCI (SCI-STAT3). FDR<0.1 for differential expression and enrichment analysis.
Extended data figure 5. Immunohistochemistry of specific CSPGs.
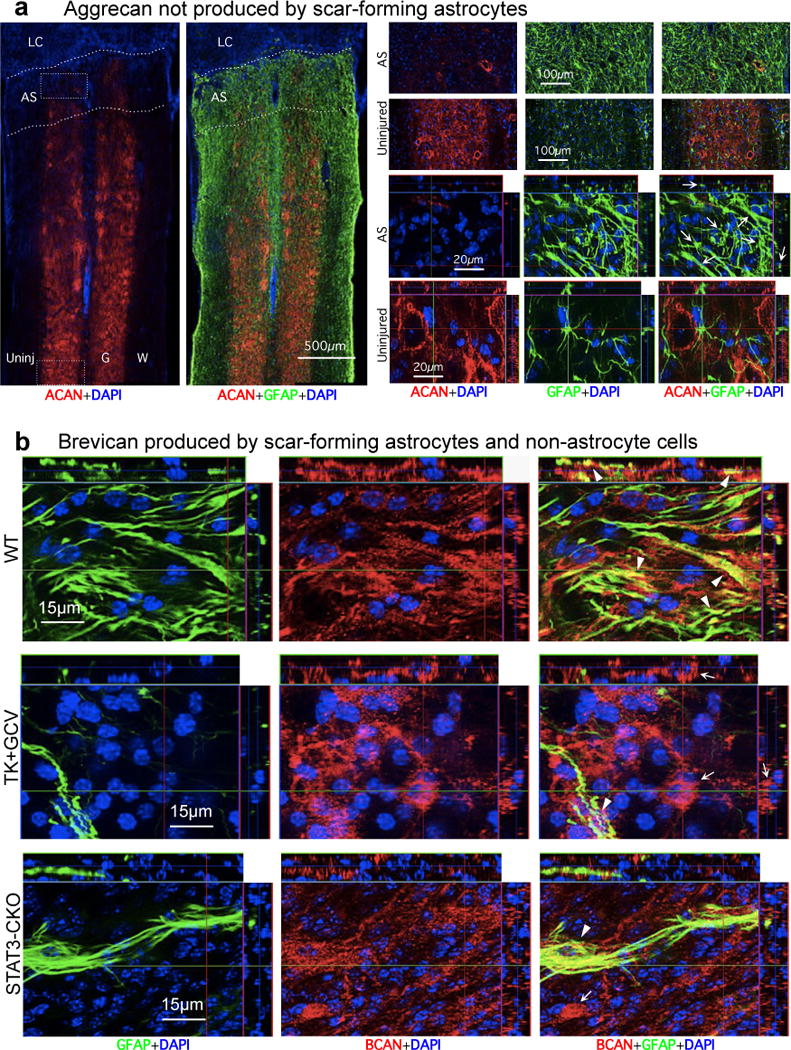
(a) Absence of aggrecan (ACAN) production by scar-forming astrocytes. Images show individual fluorescence channels of ACAN and GFAP immunohistochemistry from horizontal sections two weeks after severe SCI in a representative WT mouse. Boxes denote areas of astrocyte scar (AS) or uninjured tissue (Uninj) shown at higher magnification. Note that ACAN (i) is heavily present in the perineuronal nets that surround neurons in uninjured tissue, (ii) is almost absent from AS and lesion core (LC), and (iii) is not detectably produced by newly generated scar-forming astrocytes (arrows). (b) Brevican (BCAN) production by scar-forming astrocytes and non-astrocyte cells. Images show individual fluorescence channels of BCAN and GFAP immunohistochemistry from horizontal sections two weeks after severe SCI, in WT mice and mice with transgenic ablation (TK+GCV) or attenuation (STAT3-CKO) of astrocyte scar formation. Note that BCAN is produced both by GFAP-positive scar-forming astrocytes (arrowheads) and by non-astrocyte cells (arrows).
Extended data figure 6. Immunohistochemistry of specific CSPGs.
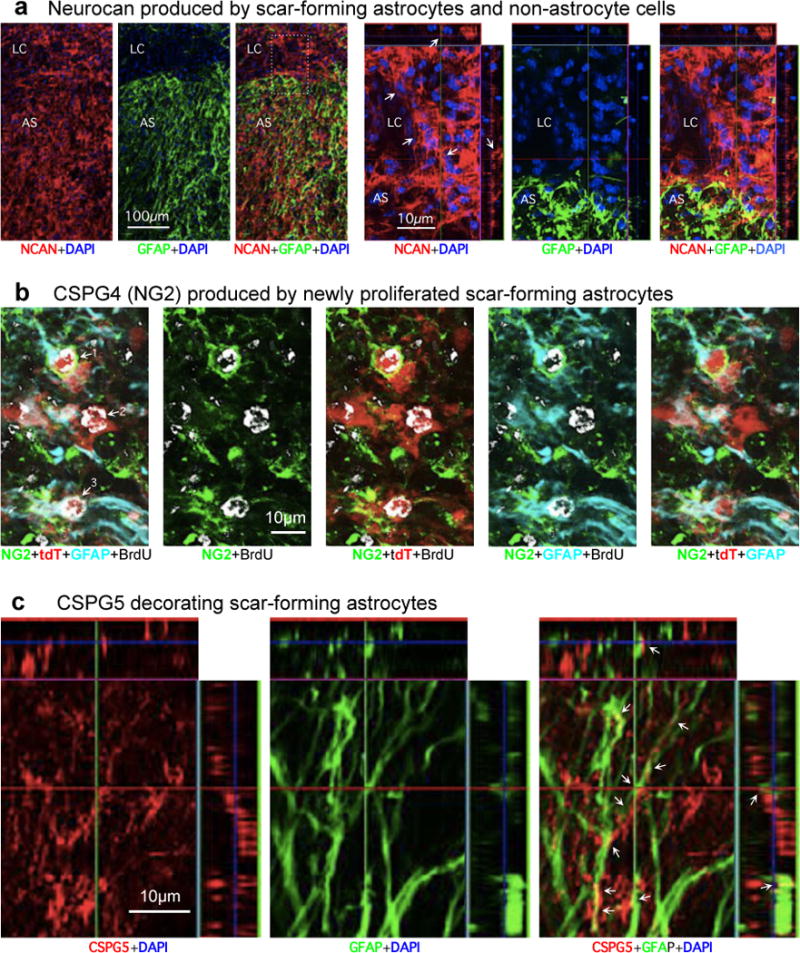
(a) Neurocan (NCAN) production by scar-forming astrocytes and non-astrocyte cells. Images show individual fluorescence channels of NCAN and GFAP immunohistochemistry from horizontal sections two weeks after severe SCI, in a representative WT mouse. Box denotes area of lesion core (LC) and astrocyte scar (AS) shown at higher magnification. Note that NCAN is produced both by GFAP-positive scar-forming astrocytes and by non-astrocyte cells (arrows) in the lesion core (LC). (b) NG2 (CSPG4) production by newly proliferated scar-forming astrocytes. Images show individual channels and various combinations of immunofluorescence staining for NG2, GFAP, tdT, BrdU (proliferation marker) and DAPI showing astrocytes in a mature SCI scar. The images are representative of findings from tdT-reporter mice52 injected with AAV2/5-GfaABC1D-Cre vector54 into multiple sites of the uninjured spinal cord to label mature astrocytes. Three weeks after AAV2/5-GfaABC1D-Cre injection, the mice received a severe SCI and were administered BrdU from days 2–7 after SCI. The mice were perfused after two weeks after SCI. Images comparing individual fluorescence channels show that astrocytes labeled 1 and 3: (i) incorporated BrdU and thus are newly proliferated after SCI, (ii) express tdT reporter, (iii) express GFAP, the prototypical marker of reactive and scar-forming astrocytes, and (iv) express NG2 both intracellularly and along their cell surfaces. In contrast, astrocyte number 2 is also BrdU-labeled and expresses both tdT and GFAP, but does not appear to express detectable levels of NG2. (c) CSPG5 (Neuroglycan C) production by scar-forming astrocytes. Images show individual channels and various combinations of immunofluorescence staining for CSPG5 or GFAP. Note that CSPG5 is present within and along the processes of GFAP-positive scar-forming astrocytes (arrows).
Extended data figure 7. Specificity and effects of treatments to stimulate AST axon regrowth after SCI.
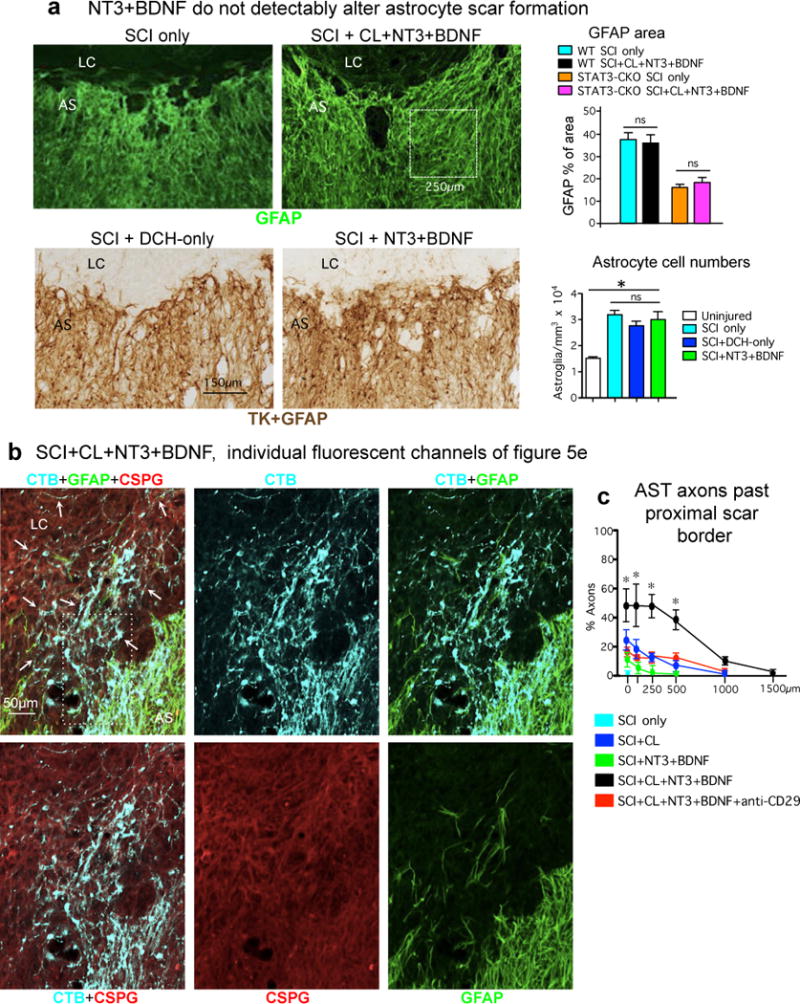
(a) BDNF and NT3 treatment does not alter the appearance or density of astrocyte scars in WT or STAT3-CKO mice. Images show horizontal sections of mice at two weeks after SCI or after SCI followed by delayed injection of hydrogel only (as a control) or hydrogel releasing NT3 and BDNF. Top images show GFAP immunofluorescence; boxed area denotes size of areas taken from multiple locations in the astrocyte scar (AS) for GFAP area quantification shown in graph. n = 5 mice per group, ns p>0.05 (ANOVA with Newman-Keuls). Bottom images show brightfield immunohistochemistry simultaneously of GFAP+TK to stain both astrocyte cell processes (GFAP) and cell bodies (TK) in mGFAP-TK transgenic mice for quantification of astrocyte cell numbers shown in graph. For these experiments the transgene derived TK is used as a reporter protein that efficiently labels astrocyte cell bodies and thereby improves cell quantification18 and the mice were not given GCV. n = 4 mice per group, * p<0.05 versus uninjured (ANOVA with Newman-Keuls); ns p>0.05 (ANOVA with Newman-Keuls). (b) AST axon regrowth through scar-forming astrocytes and CSPGs in SCI lesions. Images show individual channels and various combinations of immunofluorescence staining for CTB, GFAP and CS56 to detect total CSPGs from a WT mouse after SCI followed by delayed injection of a hydrogel depot releasing NT3 and BDNF, shown as multichannel image in main text figure 5e1. Arrows denote robust regrowth of many AST axons along, through and past scar-forming astrocytes into and through the lesion core. Note that the stimulated axons are regrowing through CSPG containing areas in the astrocyte scar and lesion core. Boxed area is shown at higher magnification in Extended data figure 8. (c) Graph shows numbers of AST axons at various distances past the proximal border of the astrocyte scar under different conditions. n = 5 per group. * p<0.001 significant difference SCI+CL+BDNF+NT3 versus all other groups (ANOVA with post-hoc Newman-Keuls).
Extended data figure 8. AST axon regrowth through scar-forming astrocytes and CSPGs in SCI lesions.
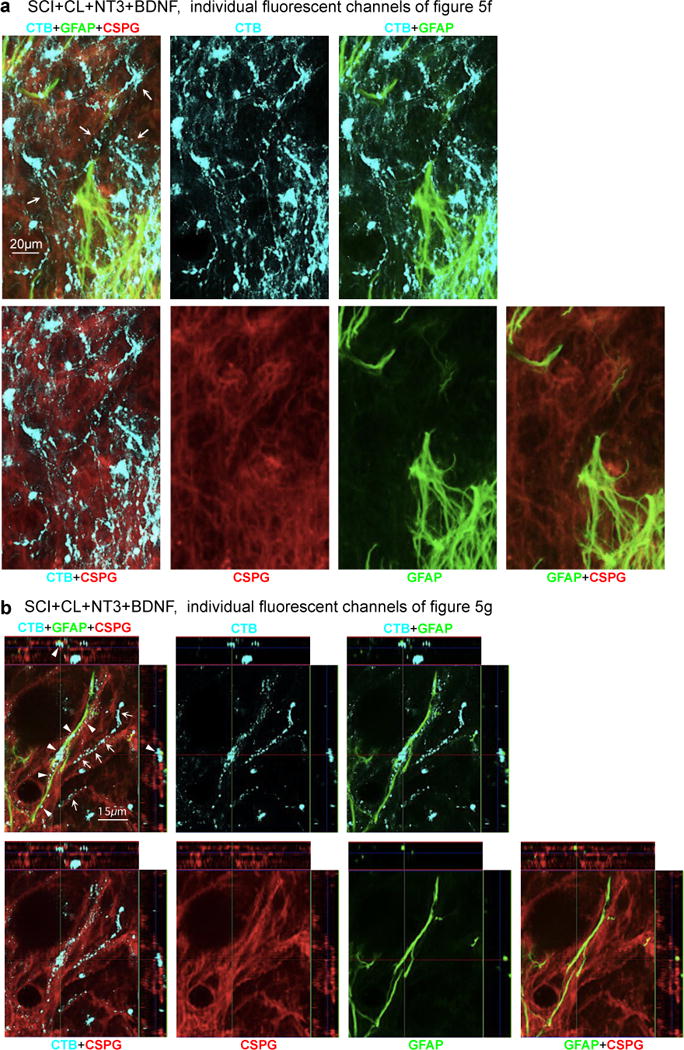
(a,b) Images show individual channels and various combinations of immunofluorescence staining for CTB, GFAP and CS56 to detect total CSPGs from a WT mouse after SCI followed by delayed injection of a hydrogel depot releasing NT3 and BDNF, shown as multichannel images in main text Figures 5e2 and 5e3. Arrows in (a) denote robust regrowth of many AST axons along, through and past scar-forming astrocytes into and through the lesion core; note that the stimulated axons are regrowing through CSPG containing areas in the astrocyte scar and lesion core. (b) High magnification orthogonal images of axons in three visual planes. Arrows in (b) denote AST regrowing axons tracking along CSPG-positive and GFAP-negative structures. Arrowheads in (b) denote AST axons tracking along GFAP-positive and CSPG-positive astrocyte processes, passing from one astrocyte process to another.
Extended data figure 9. AST axon regrowth in SCI lesions is dependent on laminin.
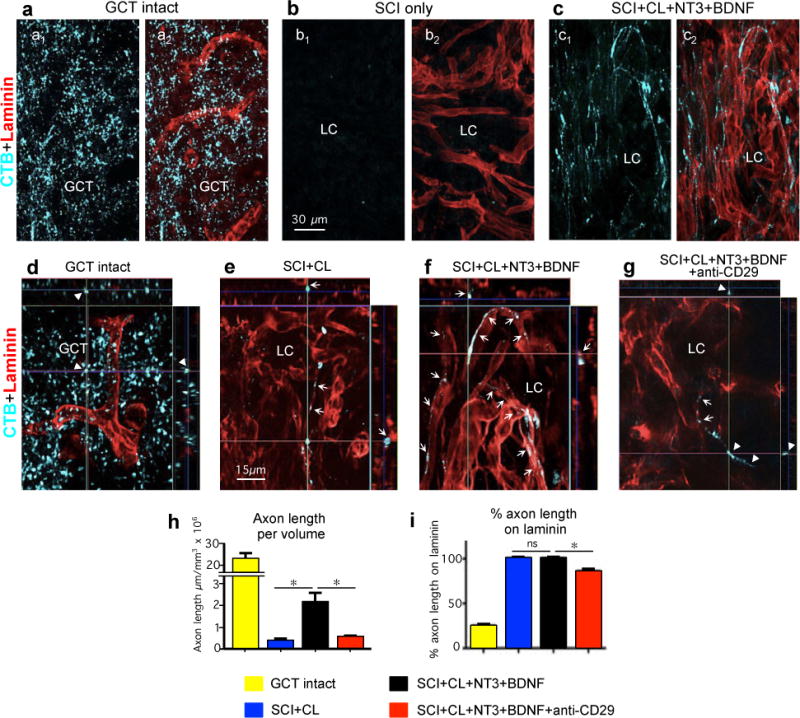
(a–g) Tract-tracing for of AST axons using CTB and laminin immunohistochemistry. (a–c) Same fields imaged for CTB alone (a1–c1), or CTB plus laminin (a2–c2). (a,d) Intact gracile cuneate tract (GCT). (b) SCI only. (c,f) SCI plus conditioning lesion (CL) plus hydrogel with growth factors. (e) SCI plus conditioning lesion. (g) SCI plus conditioning lesion plus hydrogel with growth factors and anti-CD29. (d–g) High magnification orthogonal images of axons in three visual planes. Arrows indicate regrowing axons in direct contact with laminin. Arrowheads indicate axons not in direct contact with laminin in the intact GCT (d) or with anti-CD29 treatment (g). Note the difference in appearance of axons in the intact gracile cuneate tract (GCT), which are independent of laminin, compared with regrowing axons in lesion core (LC), which track along laminin. (h) Axon length per tissue volume in intact GCT or in SCI lesions under different conditions. (Intact GCT values were not included in ANOVA comparison of other 3 groups.) (i) Percent of AST axon length in direct contact with laminin under different conditions. n = 5 per group. * p<0.001 (ANOVA with post-hoc Newman-Keuls); ns non-significant (ANOVA with post-hoc Newman-Keuls).
Extended data Table 1. Axon growth inhibitory molecules and axon growth permissive molecules.
This table lists the gene abbreviations and full names and of the 59 axon growth modulating molecules whose gene expression levels are presented in figure 4. The table also summarizes literature providing evidence for the axon growth inhibitory or permissive effects of each molecule.
| Gene abbreviation | Molecule name | References for function | |
|---|---|---|---|
| Axon growth inhibitory molecule | Acan | Aggrecan | 9,29 |
| Bcan | Brevican | 9,29 | |
| Ncan | Neurocan | 9,29,62,63 | |
| Vcan | Versican | 9,29 | |
| Ptprz1 | Phosphacan | 9,63 | |
| Xylt1 | Xylosyltransferase 1 | 64 | |
| Tnr | Tenascin R | 65 | |
| Epha4 | Ephrin A4 | 7,66 | |
| Ephb2 | Ephrin B2 | 7,66 | |
| Efnb3 | Ephrin B3 | 7,66 | |
| Ntn1 | Netrin 1 | 7,66,67 | |
| Sema3a | Semaphorin 3a | 7,66 | |
| Sema3f | Semaphorin 3f | 7,66 | |
| Plxna1 | Plexin A1 | 7,68 | |
| Plxnb1 | Plexin B1 | 7,69 | |
| Nrp1 | Neuropilin 1 | 7,70 | |
| Unc5b | Netrin receptor Unc5b | 7,66,71 | |
| Dcc | Deleted in Colorectal Cancer | 7,72,73 | |
| Neo1 | Neogenin 1 | 7,74 | |
| Rgma | Repulsive guidance molecule A | 7,75 | |
| Rgmb | Repulsive guidance molecule B | 7,75 | |
| Slit1 | Slit 1 | 7,66 | |
| Slit2 | Slit 2 | 7,66 | |
| Slitrk1 | SLIT and NTRK-like family, member 1 | 76 | |
| Robo1 | Robo 1 | 7,66 | |
| Robo2 | Robo 2 | 7,66 | |
| Robo3 | Robo 3 | 7,66 | |
| Draxin | Draxin | 73,77 | |
| Axon growth permissive molecule | Cspg4 | NG2 | 78–81 |
| Cspg5 | Neuroglycan C | 82 | |
| Tnc | Tenascin C | 83,84 | |
| Sdc1 | Syndecan 1 | 85,86 | |
| Sdc2 | Syndecan 2 | 85,86 | |
| Sdc3 | Syndecan 3 | 85,86 | |
| Sdc4 | Syndecan 4 | 85,86 | |
| Bdnf | Brain derived neurotrophic factor | 35,87 | |
| Ntf3 | Neurotrophin 3 | 35,88 | |
| Gdnf | Glial derived neurotrophic factor | 89 | |
| Lif | Leukemia inhibitory factor | 90,91 | |
| Cntf | Ciliary neurotrophic factor | 92 | |
| Igf1 | Insulin-like growth factor-1 | 93 | |
| Fgf2 | Fibroblast growth factor 2 | 94 | |
| Tgfa | Transforming growth factor alpha | 95 | |
| Lama1 | Laminin A1 | 36 | |
| Lama2 | Laminin A2 | 36 | |
| Lama4 | Laminin A4 | 36 | |
| Lama5 | Laminin A5 | 36 | |
| Lamb1 | Laminin B1 | 36 | |
| Lamc1 | Laminin C1 | 36 | |
| Col4a1 | Collagen 4a1 | 8 | |
| Fn1 | Fibronectin 1 | 96 | |
| Hspg2 | Perlecan | 86,97 | |
| Gpc1 | Glypican 1 | 86,98 | |
| Gpc3 | Glypican 3 | 86,98 | |
| Gpc5 | Glypican 5 | 86,98 | |
| Dcn | Decorin | 99 | |
| Lgals1 | Galectin 1 | 100 | |
| Ncam1 | Neural cell adhesion molecule 1 | 101 | |
| Matn2 | Matrilin | 102 |
Supplementary Material
Acknowledgments
We thank Dr. D.W. Bergles for NG2 antibody, and the Microscopy Core Resource of the UCLA Broad Stem Cell Research Center-CIRM Laboratory. This work was supported by the US National Institutes of Health (NS057624 and NS084030 to M.V.S.; and NS060677, MH099559A, MH104069 to B.S.K.), and the Dr. Miriam and Sheldon G. Adelson Medical Foundation (M.V.S. and T.J.D.), and Wings for Life (M.V.S.).
Footnotes
Author contributions M.A.A., J.E.B., B.S.K., T.J.D., M.V.S. designed experiments; M.A.A., J.E.B., Y.R., Y.A. conducted experiments; M.A.A., J.E.B., Y.A., T.M.O’S, R.K., G.C, M.V.S. analyzed data. M.A.A., J.E.B., T.M.O’S, B.S.K, T.J.D., M.V.S. prepared the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author information Raw and normalized genomic data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE76097 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE76097).
The authors declare no competing financial interests.
References
- 1.Ramon Y, Cajal S. Degeneration and regeneration of the nervous system. Oxford University Press; 1928. [Google Scholar]
- 2.Sun F, et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu K, Tedeschi A, Park KK, He Z. Neuronal intrinsic mechanisms of axon regeneration. Annu Rev Neurosci. 2011;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PM, McGuinness UM, Aguayo AJ. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284:264–265. doi: 10.1038/284264a0. [DOI] [PubMed] [Google Scholar]
- 5.David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- 6.Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci. 2010;11:799–811. doi: 10.1038/nrn2936. [DOI] [PubMed] [Google Scholar]
- 7.Harel NY, Strittmatter SM. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat Rev Neurosci. 2006;7:603–616. doi: 10.1038/nrn1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klapka N, Muller HW. Collagen matrix in spinal cord injury. J Neurotrauma. 2006;23:422–435. doi: 10.1089/neu.2006.23.422. [DOI] [PubMed] [Google Scholar]
- 9.Silver J, Miller JH. Regeneration beyond the glial scar. Nature Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 10.Windle WF, Clemente CD, Chambers WW. Inhibition of formation of a glial barrier as a means of permitting a peripheral nerve to grow into the brain. J Comp Neurol. 1952;96:359–369. doi: 10.1002/cne.900960207. [DOI] [PubMed] [Google Scholar]
- 11.Windle WF. Regeneration of axons in the vertebrate central nervous system. Physiol Rev. 1956;36:427–440. doi: 10.1152/physrev.1956.36.4.427. [DOI] [PubMed] [Google Scholar]
- 12.Liuzzi FJ, Lasek RJ. Astrocytes block axonal regeneration in mammals by activating the physiological stop pathway. Science. 1987;237:642–645. doi: 10.1126/science.3603044. [DOI] [PubMed] [Google Scholar]
- 13.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bush TG, et al. Leukocyte infiltration, neuronal degeneration and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 16.Faulkner JR, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrmann JE, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wanner IB, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JK, et al. Combined genetic attenuation of myelin and semaphorin-mediated growth inhibition is insufficient to promote serotonergic axon regeneration. J Neurosci. 2010;30:10899–10904. doi: 10.1523/JNEUROSCI.2269-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawthorne AL, et al. The unusual response of serotonergic neurons after CNS Injury: lack of axonal dieback and enhanced sprouting within the inhibitory environment of the glial scar. J Neurosci. 2011;31:5605–5616. doi: 10.1523/JNEUROSCI.6663-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buch T, et al. A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nature methods. 2005;2:419–426. doi: 10.1038/nmeth762. [DOI] [PubMed] [Google Scholar]
- 22.Avnur Z, Geiger B. Immunocytochemical localization of native chondroitin-sulfate in tissues and cultured cells using specific monoclonal antibody. Cell. 1984;38:811–822. doi: 10.1016/0092-8674(84)90276-9. [DOI] [PubMed] [Google Scholar]
- 23.Mikami T, Kitagawa H. Biosynthesis and function of chondroitin sulfate. Biochimica et biophysica acta. 2013;1830:4719–4733. doi: 10.1016/j.bbagen.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Mironova YA, Giger RJ. Where no synapses go: gatekeepers of circuit remodeling and synaptic strength. Trends Neurosci. 2013;36:363–373. doi: 10.1016/j.tins.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin AC, Holt CE. Local translation and directional steering in axons. The EMBO journal. 2007;26:3729–3736. doi: 10.1038/sj.emboj.7601808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanz E, et al. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamanian JL, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang BT, et al. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature. 2015;518:404–408. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamaguchi Y. Lecticans: organizers of the brain extracellular matrix. Cellular and molecular life sciences: CMLS. 2000;57:276–289. doi: 10.1007/PL00000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller GM, Hsieh-Wilson LC. Sugar-Dependent Modulation of Neuronal Development, Regeneration, and Plasticity by Chondroitin Sulfate Proteoglycans. Exp Neurol. 2015 doi: 10.1016/j.expneurol.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg JL, et al. Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron. 2002;33:689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- 33.Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- 34.Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- 35.Omura T, et al. Robust Axonal Regeneration Occurs in the Injured CAST/Ei Mouse CNS. Neuron. 2015;86:1215–1227. doi: 10.1016/j.neuron.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alto LT, et al. Chemotropic guidance facilitates axonal regeneration and synapse formation after spinal cord injury. Nat Neurosci. 2009;12:1106–1113. doi: 10.1038/nn.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plantman S, et al. Integrin-laminin interactions controlling neurite outgrowth from adult DRG neurons in vitro. Mol Cell Neurosci. 2008;39:50–62. doi: 10.1016/j.mcn.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 38.Nowak AP, et al. Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 39.Yang CY, et al. Biocompatibility of amphiphilic diblock copolypeptide hydrogels in the central nervous system. Biomaterials. 2009;30:2881–2898. doi: 10.1016/j.biomaterials.2009.01.056. [DOI] [PubMed] [Google Scholar]
- 40.Song B, et al. Sustained local delivery of bioactive nerve growth factor in the central nervous system via tunable diblock copolypeptide hydrogel depots. Biomaterials. 2012;33:9105–9116. doi: 10.1016/j.biomaterials.2012.08.060. [DOI] [PubMed] [Google Scholar]
- 41.Tuszynski MH, Steward O. Concepts and methods for the study of axonal regeneration in the CNS. Neuron. 2012;74:777–791. doi: 10.1016/j.neuron.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brosius Lutz A, Barres BA. Contrasting the glial response to axon injury in the central and peripheral nervous systems. Developmental cell. 2014;28:7–17. doi: 10.1016/j.devcel.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Mason CA, Edmondson JC, Hatten ME. The extending astroglial process: development of glial cell shape, the growing tip, and interactions with neurons. J Neurosci. 1988;8:3124–3134. doi: 10.1523/JNEUROSCI.08-09-03124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawaja MD, Gage FH. Reactive astrocytes are substrates for the growth of adult CNS axons in the presence of elevated levels of nerve growth factor. Neuron. 1991;7:1019–1030. doi: 10.1016/0896-6273(91)90346-2. [DOI] [PubMed] [Google Scholar]
- 45.Zukor K, et al. Short hairpin RNA against PTEN enhances regenerative growth of corticospinal tract axons after spinal cord injury. J Neurosci. 2013;33:15350–15361. doi: 10.1523/JNEUROSCI.2510-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih CH, Lacagnina M, Leuer-Bisciotti K, Proschel C. Astroglial-derived periostin promotes axonal regeneration after spinal cord injury. J Neurosci. 2014;34:2438–2443. doi: 10.1523/JNEUROSCI.2947-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S, et al. Thermoresponsive copolypeptide hydrogel vehicles for CNS cell delivery. ACS Biomat Sci Eng. 2015;1:705–717. doi: 10.1021/acsbiomaterials.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruschel J, et al. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science. 2015 doi: 10.1126/science.aaa2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush TG, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 51.Takeda K, et al. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- 52.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang S, et al. Tunable diblock copolypeptide hydrogel depots for local delivery of hydrophobic molecules in healthy and injured central nervous system. Biomaterials. 2014;35:1989–2000. doi: 10.1016/j.biomaterials.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shigetomi E, et al. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. The Journal of general physiology. 2013;141:633–647. doi: 10.1085/jgp.201210949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang R, Haustein MD, Sofroniew MV, Khakh BS. Imaging Intracellular Ca2+ Signals in Striatal Astrocytes from Adult Mice Using Genetically-encoded Calcium Indicators. J Vis Exp. 2014 doi: 10.3791/51972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tong X, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behavior research methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 59.Romero-Calvo I, et al. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Analytical biochemistry. 2010;401:318–320. doi: 10.1016/j.ab.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 60.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Friedlander DR, et al. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sango K, et al. Phosphacan and neurocan are repulsive substrata for adhesion and neurite extension of adult rat dorsal root ganglion neurons in vitro. Exp Neurol. 2003;182:1–11. doi: 10.1016/s0014-4886(03)00090-6. [DOI] [PubMed] [Google Scholar]
- 65.Hurtado A, Podinin H, Oudega M, Grimpe B. Deoxyribozyme-mediated knockdown of xylosyltransferase-1 mRNA promotes axon growth in the adult rat spinal cord. Brain. 2008;131:2596–2605. doi: 10.1093/brain/awn206. [DOI] [PubMed] [Google Scholar]
- 66.Becker CG, Schweitzer J, Feldner J, Becker T, Schachner M. Tenascin-R as a repellent guidance molecule for developing optic axons in zebrafish. J Neurosci. 2003;23:6232–6237. doi: 10.1523/JNEUROSCI.23-15-06232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- 68.Masuda T, et al. Netrin-1 acts as a repulsive guidance cue for sensory axonal projections toward the spinal cord. J Neurosci. 2008;28:10380–10385. doi: 10.1523/JNEUROSCI.1926-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winberg ML, et al. Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell. 1998;95:903–916. doi: 10.1016/s0092-8674(00)81715-8. [DOI] [PubMed] [Google Scholar]
- 70.Hu H, Marton TF, Goodman CS. Plexin B mediates axon guidance in Drosophila by simultaneously inhibiting active Rac and enhancing RhoA signaling. Neuron. 2001;32:39–51. doi: 10.1016/s0896-6273(01)00453-6. [DOI] [PubMed] [Google Scholar]
- 71.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997;90:739–751. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 72.Lu X, et al. The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature. 2004;432:179–186. doi: 10.1038/nature03080. [DOI] [PubMed] [Google Scholar]
- 73.Keino-Masu K, et al. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87:175–185. doi: 10.1016/s0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- 74.Ahmed G, et al. Draxin inhibits axonal outgrowth through the netrin receptor DCC. J Neurosci. 2011;31:14018–14023. doi: 10.1523/JNEUROSCI.0943-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rajagopalan S, et al. Neogenin mediates the action of repulsive guidance molecule. Nature cell biology. 2004;6:756–762. doi: 10.1038/ncb1156. [DOI] [PubMed] [Google Scholar]
- 76.Monnier PP, et al. RGM is a repulsive guidance molecule for retinal axons. Nature. 2002;419:392–395. doi: 10.1038/nature01041. [DOI] [PubMed] [Google Scholar]
- 77.Kajiwara Y, Buxbaum JD, Grice DE. SLITRK1 binds 14-3-3 and regulates neurite outgrowth in a phosphorylation-dependent manner. Biol Psychiatry. 2009;66:918–925. doi: 10.1016/j.biopsych.2009.05.033. [DOI] [PubMed] [Google Scholar]
- 78.Islam SM, et al. Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science. 2009;323:388–393. doi: 10.1126/science.1165187. [DOI] [PubMed] [Google Scholar]
- 79.Yang Z, et al. NG2 glial cells provide a favorable substrate for growing axons. J Neurosci. 2006;26:3829–3839. doi: 10.1523/JNEUROSCI.4247-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hossain-Ibrahim MK, Rezajooi K, Stallcup WB, Lieberman AR, Anderson PN. Analysis of axonal regeneration in the central and peripheral nervous systems of the NG2-deficient mouse. BMC neuroscience. 2007;8:80. doi: 10.1186/1471-2202-8-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lu P, Jones LL, Tuszynski MH. Axon regeneration through scars and into sites of chronic spinal cord injury. Exp Neurol. 2007;203:8–21. doi: 10.1016/j.expneurol.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 82.Busch SA, et al. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J Neurosci. 2010;30:255–265. doi: 10.1523/JNEUROSCI.3705-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakanishi K, et al. Identification of neurite outgrowth-promoting domains of neuroglycan C, a brain-specific chondroitin sulfate proteoglycan, and involvement of phosphatidylinositol 3-kinase and protein kinase C signaling pathways in neuritogenesis. J Biol Chem. 2006;281:24970–24978. doi: 10.1074/jbc.M601498200. [DOI] [PubMed] [Google Scholar]
- 84.Gotz B, et al. Tenascin-C contains distinct adhesive, anti-adhesive, and neurite outgrowth promoting sites for neurons. J Cell Biol. 1996;132:681–699. doi: 10.1083/jcb.132.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andrews MR, et al. Alpha9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. J Neurosci. 2009;29:5546–5557. doi: 10.1523/JNEUROSCI.0759-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edwards TJ, Hammarlund M. Syndecan promotes axon regeneration by stabilizing growth cone migration. Cell reports. 2014;8:272–283. doi: 10.1016/j.celrep.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farhy Tselnicker I, Boisvert MM, Allen NJ. The role of neuronal versus astrocyte-derived heparan sulfate proteoglycans in brain development and injury. Biochemical Society transactions. 2014;42:1263–1269. doi: 10.1042/BST20140166. [DOI] [PubMed] [Google Scholar]
- 88.Lu P, Jones LL, Tuszynski MH. BDNF-expressing marrow stromal cells support extensive axonal growth at sites of spinal cord injury. Exp Neurol. 2005;191:344–360. doi: 10.1016/j.expneurol.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 89.Grill R, Murai K, Blesch A, Tuszynski MH. Cellular delivery of neurotrophin-3 promotes coricospinal axonal growth and partial functional recovery after spinal cord injury. J Neurosci. 1997;17:5560–5572. doi: 10.1523/JNEUROSCI.17-14-05560.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blesch A, Tuszynski MH. Cellular GDNF delivery promotes growth of motor and dorsal column sensory axons after partial and complete spinal cord transections and induces remyelination. J Comp Neurol. 2003;467:403–417. doi: 10.1002/cne.10934. [DOI] [PubMed] [Google Scholar]
- 91.Blesch A, et al. Leukemia inhibitory factor augments neurotrophin expression and corticospinal axon growth after adult CNS injury. J Neurosci. 1999;19:3556–3566. doi: 10.1523/JNEUROSCI.19-09-03556.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cafferty WB, et al. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci. 2001;21:7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Muller A, Hauk TG, Fischer D. Astrocyte-derived CNTF switches mature RGCs to a regenerative state following inflammatory stimulation. Brain. 2007;130:3308–3320. doi: 10.1093/brain/awm257. [DOI] [PubMed] [Google Scholar]
- 94.Ozdinler PH, Macklis JD. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci. 2006;9:1371–1381. doi: 10.1038/nn1789. [DOI] [PubMed] [Google Scholar]
- 95.Szebenyi G, et al. Fibroblast growth factor-2 promotes axon branching of cortical neurons by influencing morphology and behavior of the primary growth cone. J Neurosci. 2001;21:3932–3941. doi: 10.1523/JNEUROSCI.21-11-03932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.White RE, Yin FQ, Jakeman LB. TGF-alpha increases astrocyte invasion and promotes axonal growth into the lesion following spinal cord injury in mice. Exp Neurol. 2008;214:10–24. doi: 10.1016/j.expneurol.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tom VJ, Doller CM, Malouf AT, Silver J. Astrocyte-associated fibronectin is critical for axonal regeneration in adult white matter. J Neurosci. 2004;24:9282–9290. doi: 10.1523/JNEUROSCI.2120-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qin J, Liang J, Ding M. Perlecan antagonizes collagen IV and ADAMTS9/GON-1 in restricting the growth of presynaptic boutons. J Neurosci. 2014;34:10311–10324. doi: 10.1523/JNEUROSCI.5128-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hill JJ, Jin K, Mao XO, Xie L, Greenberg DA. Intracerebral chondroitinase ABC and heparan sulfate proteoglycan glypican improve outcome from chronic stroke in rats. Proc Natl Acad Sci U S A. 2012;109:9155–9160. doi: 10.1073/pnas.1205697109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Minor K, et al. Decorin promotes robust axon growth on inhibitory CSPGs and myelin via a direct effect on neurons. Neurobiol Dis. 2008;32:88–95. doi: 10.1016/j.nbd.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 101.Horie H, et al. Galectin-1 regulates initial axonal growth in peripheral nerves after axotomy. J Neurosci. 1999;19:9964–9974. doi: 10.1523/JNEUROSCI.19-22-09964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walsh FS, Doherty P. Neural cell adhesion molecules of the immunoglobulin superfamily: role in axon growth and guidance. Annual review of cell and developmental biology. 1997;13:425–456. doi: 10.1146/annurev.cellbio.13.1.425. [DOI] [PubMed] [Google Scholar]
- 103.Malin D, et al. The extracellular-matrix protein matrilin 2 participates in peripheral nerve regeneration. Journal of cell science. 2009;122:995–1004. doi: 10.1242/jcs.040378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.