

Abstract
Cisplatin nephropathy can be regarded as a mitochondrial disease. Intervention to halt such deleterious injury is under investigation. Recently, the flavanol (–)-epicatechin emerges as a novel compound to protect the cardiovascular system, owing in part to mitochondrial protection. Here, we have hypothesized that epicatechin prevents the progression of cisplatin-induced kidney injury by protecting mitochondria. Epicatechin was administered 8 h after cisplatin injury was induced in the mouse kidney. Cisplatin significantly induced renal dysfunction and tubular injury along with an increase in oxidative stress. Mitochondrial damages were also evident as a decrease in loss of mitochondrial mass with a reduction in the oxidative phosphorylation complexes and low levels of MnSOD. The renal damages and mitochondrial injuries were significantly prevented by epicatechin treatment. Consistent with these observations, an in vitro study using cultured mouse proximal tubular cells demonstrated that cisplatin-induced mitochondrial injury, as revealed by a decrease in mitochondrial succinate dehydrogenase activity, an induction of cytochrome c release, mitochondrial fragmentation, and a reduction in complex IV protein, was prevented by epicatechin. Such a protective effect of epicatechin might be attributed to decreased oxidative stress and reduced ERK activity. Finally, we confirmed that epicatechin did not perturb the anticancer effect of cisplatin in HeLa cells. In conclusion, epicatechin exhibits protective effects due in part to its ability to prevent the progression of mitochondrial injury in mouse cisplatin nephropathy. Epicatechin may be a novel option to treat renal disorders associated with mitochondrial dysfunction.
Keywords: AKI, cocoa, complex IV, dark chocolate, MnSOD
cisplatin is one of the most widely used chemotherapeutic agents in patients with cancer of the testes, head, neck, ovary, cervix, lung, and other organs. Cisplatin is particularly beneficial in testicular cancer, where the cure rate exceeds 90% and is nearly 100% for early stages (7, 34, 38). However, side effects of cisplatin, especially in the kidney, limit its use. Specifically, tubular epithelial cells in the kidney are one of the major targets for cisplatin (2, 30). Many research groups have been intensively investigating how cisplatin injures tubular cells to find a way to protect the kidney.
One mechanism could be by impairing mitochondrial function. Previous studies demonstrated that cisplatin induces activation of the Bcl2-family protein Bax, which forms pores on the mitochondrial outer membrane (24, 40). The p53/PUMA-α pathway is also activated by cisplatin and leads to mitochondrial membrane damage (19, 20). Recent reports have suggested that cisplatin also directly binds mitochondrial DNA and the voltage-dependent anion channel in cancer cells (43). The injury to the mitochondrial membrane causes a loss of membrane potential and an impaired function in mitochondrial oxidative phosphorylation (22, 23). Similarly, cisplatin-induced apoptosis could be caused by a release of cytochrome c from mitochondria as a consequence of an increase in mitochondrial membrane permeability (5). This anticancer agent also impairs mitochondrial structure, leading to mitochondrial fragmentation (5).
(–)-Epicatechin is a flavonoid (a subclass of polyphenols) and is predominantly contained in cacao. Therefore, cocoa powder or dark chocolate is typically rich in epicatechin (8, 13). Our attention to the favorable effect of cocoa-derived flavonoid was generated from an epidemiological observation in the Kuna Indians, a native population living on islands off the coast of Panama (16, 17). The island-living Kuna belong to one of the few cultures that are protected against age-related cardiovascular disease, including an increase in blood pressure and a decline in kidney function (16, 17). Interestingly, this native population regularly consumes significant amounts of cocoa drink (16). For those Kuna who moved to mainland Panama, where the homemade cocoa was not available, the protection from cardiovascular disease was lost (16).
Recently, Villarreal's research group (28, 31, 32, 36, 41, 42) has examined the effects of epicatechin in the setting of cardiovascular disease. They showed that the beneficial effect of epicatechin could be due in part to protection of mitochondria (36). Huttemann et al. (18) also recently reported that epicatechin could maintain exercise-induced mitochondrial capacity in the hindlimb muscles of mice even after discontinuation of exercise.
Given evidence that cisplatin induces mitochondrial injury in tubular cells, we tested the hypothesis that epicatechin might alleviate cisplatin-induced renal damage by protecting mitochondria.
METHODS
Experimental protocols.
All animal experiments were performed in accordance with the Animal Care and Use Committee of the University of Colorado. Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) at 8 wk of age were used in the present study. The mice were fed a standard pellet laboratory chow ad libitum. Mice were divided into four subgroups: 1) a control (saline) group, 2) an epicatechin-treated control group, 3) a cisplatin group, and 4) an epicatechin-treated cisplatin group (n = 8–12 for each group). Cisplatin [cis-diamminedichloro-platinum (II); Aldrich Chemical, Milwaukee, WI] was dissolved in normal (0.9%) saline at a concentration of 1 mg/ml immediately before administration. Mice were given 30 mg/kg of cisplatin or normal saline (as a vehicle control) intraperitoneally to induce acute kidney injury (AKI). As previously reported (12), this model shows overt renal dysfunction and severe tubular injury on day 3 after cisplatin injection. First of all, (–)-epicatechin (Sigma-Aldrich, St. Louis, MO) was prepared in normal saline every time before injection, and the dosage of 1 mg/kg was given intraperitoneally twice a day until the day before death. Epicatechin treatment started 8 h after cisplatin injection (Fig. 1A). All the mice were euthanized 72 h after the injection of cisplatin, and blood samples and kidney tissues were obtained.
Fig. 1.
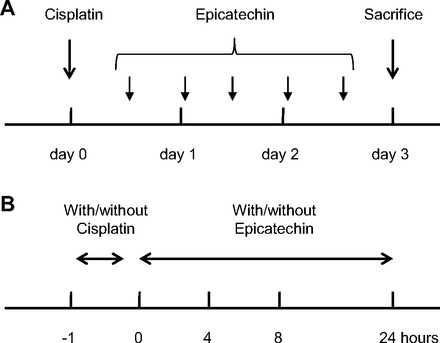
Experimental designs for in vivo and in vitro study. A: experimental design for mouse study. Eight hours after cisplatin injection, epicatechin treatment was started every 12 h by day 3. B: experimental design for cell culture study. After incubation of mouse proximal tubular cells with 100 μM cisplatin for 1 h, cisplatin-containing culture medium was removed and replaced with new medium with/without epicatechin and incubated for 4–24 h.
Renal function.
Blood urea nitrogen (BUN) was measured using an Alfa Wassermann VetACE autoanalyzer (Schiaparelli Biosystems). Serum creatinine concentration was analyzed with HPLC-MS/MS (Applied Biosystems 3200 Qtrap). Creatinine and [2H3]creatinine were detected in multiple reaction monitoring mode, monitoring the transitions of the m/z from 114 to 44.2 and from 117 to 47.2, respectively.
Histological analysis.
Formalin-fixed, paraffin-embedded sections (3 μm) were stained with the periodic acid-Schiff reagent (PAS) for light microscopy. On coronal sections of the kidney, acute tubular injuries were evaluated and quantified as the acute tubular necrosis (ATN) score, which was determined by the percentage of tubules displaying detachment of epithelium, loss of brush border, or cast formation, as follows: 0 = none, 1 = 10%, 2 = 10–25%, 3 = 26–50%, 4 = 51–75%, and 5 = >75%. Ten fields on each section at ×200 magnification were examined. Kidney sections were observed by two investigators in a blinded manner.
Apoptosis.
For the detection of apoptotic cells in the kidney, terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed on frozen section (10 μm) using a TACS 2 TdT-Blue Label In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD) according to the manufacturer's protocol. Digital images were obtained with a Scan Scope CS Digital Slide Scanner (Aperio, Vista, CA) at ×200 magnification. Ten fields were examined to determine the average number of TUNEL-positive nuclei in each section.
Immunohistochemistry.
Formalin-fixed, paraffin-embedded sections (3 μm) were used for immunohistochemistry as previously described (35). The following antibodies were used as primary antibodies: 1) rabbit anti-4-hydroxynonenal (4-HNE) antibody (Abcam, Cambridge, MA); and 2) rabbit anti-nitrotyrosine antibody (Chemicon International, Temecula, CA). Briefly, after deparaffinization, the sections were treated with 3% H2O2 for 10 min to inactivate endogenous peroxidase activity. The sections were treated with 10 mM citrate buffer (pH 6.0) for 30 min in a steamer for antigen retrieval. After incubation with a background “sniper” (Biocare Medical, Concord, CA) for 15 min, sections were incubated with primary antibodies overnight at 4°C. Then, the sections were incubated with Mach2 rabbit horseradish peroxidase (HRP) polymer (Biocare Medical). Diaminobenzidine (DAB) was used as a chromogen. Slides were counterstained with hematoxylin. Digital images were obtained with a Scan Scope CS Digital Slide Scanner (Aperio) at ×200 magnification.
Mitochondrial DNA content.
Total DNA was extracted from kidney tissue and purified using DNeasy Blood and Tissue Kit (Qiagen, Chatsworth, CA). For the detection of cytochrome c oxidase subunit (COX)-II, a mitochondrial gene and UCP-2 (nuclear gene encoding mitochondrial protein), the following oligonucleotide primers were used: COX-II, 5′- TTTTCAGGCTTCACCCTAGATGA-3′ (forward) and 5′-GAAGAATGTTATGTTTACTCCTACGAATATG-3′ (reverse); and UCP-2, 5′-GCGTTCTGGGTACCATCCTAAC-3′ (forward) and 5′-GCGACCAGCCCATTGTAGA-3′ (reverse). Diluted DNA was added to SYBR Green JumpStart (Sigma-Aldrich) with the specific primer to make reaction mixes. The PCR program was optimized and performed as denaturation at 95°C for 3 min followed by 50 cycles of amplification (COX-II, 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min; and UCP-2, 95°C for 30 s, 57°C for 30 s, and 72°C for 1 min, respectively) using the MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad). The amount of COX-II PCR products was normalized with UCP-2 to determine the ratio for mitochondrial DNA/genomic DNA.
Cell culture.
Conditionally immortalized mouse proximal tubular cells (TKPTS) (10) were cultured with DMEM/F-12 50/50 medium (Mediatech, Manassas, VA) supplemented with 5% FBS, penicillin (100 U/l), streptomycin (100 μg/l), and SITE-liquid media supplement (Sigma-Aldrich) at 37°C. HeLa cells were cultured with MEM (Mediatech) supplemented with 10% FBS, penicillin (100 U/l), and streptomycin (100 μg/l). At 90% confluence, the cells were stimulated by 100 μM cisplatin in complete medium for 1 hour at 37°C before the cisplatin-containing medium was removed. Then, the cells were cultured in complete medium with or without 1 × 10−7, 5 × 10−7, or 1 × 10−6 M epicatechin, or 1 mM tempol (Enzo Life Science, Plymouth Meeting, MA) for appropriate time in each experiment (Fig. 1B).
Cell survival assay.
To determine the number of live cells, a CyQUANT NF Cell Proliferation Assay Kit (Invitrogen), which can measure nuclear DNA content via fluorescent dye binding, was used. Mouse proximal tubular cells or HeLa cells were seeded in 96-well plates at 7,500 or 1,500 cells/well and allowed to adherent at 37°C overnight. One hour after stimulation by 100 μM cisplatin, this medium was replaced with new medium containing various concentrations of epicatechin. Cells were incubated for 4 or 24 h before the culture medium was replaced with fluorescent dye-containing HBSS, and incubated for 1 h at 37°C. Fluorescent intensity was measured using a Synergy 2 Multi-Mode Microplate Reader (Bio-Tek, Winooski, VT) with excitation at 485 nm and emission detection at 528 nm. A strongly positive correlation was shown between this fluorescence intensity and the real number of cells on 96-well plates (R2 = 0.98, P < 0.05). Cell number was expressed as an estimate (cells/well) using this trendline equation.
Succinate dehydrogenase activity.
A conventional MTT assay was used to assess the activity of succinate dehydrogenase, complex II as a mitochondrial oxidative phosphorylation protein (6). Mouse proximal tubular cells were stimulated in the same way as in the above-noted cell survival assay. The cells were treated with epicatechin in 100 μl of complete medium for 4 h after the cisplatin-containing medium was removed. Then, 20 μl of thiazolyl blue tetrazolium bromide (Sigma-Aldrich) dissolved in PBS at 5 mg/ml was added to each well, and the plate was incubated for 3 h at 37°C. Then, culture medium was replaced with MTT solvent (4 mM HCl and 0.1% NP-40 in isopropanol), and cells were agitated on an orbital shaker for 15 min in the dark. Absorbance was read at 590 nm with a reference filter of 620 nm. Data were expressed as values relative to control.
Western blotting.
Kidney cortex tissues were homogenized in RIPA buffer at 4°C. Mitochondria were isolated from kidney tissues using a Mitochondria Isolation Kit for Tissue (Thermo Scientific, Waltham, MA) and lysed in RIPA buffer at 4°C. Cultured cell lysate was prepared in cell lysis buffer (Cell Signaling, Danvers, MA). Samples were processed for SDS-PAGE and electrotransferred onto a nitrocellulose membrane. After being blocked with 5% nonfat dry milk in 1× TBS containing 0.1% Tween 20 for 1 h, membranes were incubated overnight with the following antibodies at 4°C: 1) mouse anti-nitrotyrosine antibody (Chemicon International); 2) mouse anti- bovine Complex I antibody (Abcam); 3) rabbit anti-human CYC1 (component of Complex III antibody, Abcam); 4) rabbit anti-human COX-IV antibody (Abcam); 5) mouse anti-rat MnSOD antibody (Enzo Life Science); 6) mouse monoclonal β-actin antibody (Sigma-Aldrich); 7) rabbit anti-human phospho-p53 (Ser15) antibody (Cell Signaling); 8) mouse monoclonal anti-p53 antibody (Cell Signaling); 9) rabbit anti-human phospho-cyclin-dependent kinase 2 (CDK2; Thr160 antibody, Cell Signaling); 10) rabbit monoclonal anti-CDK2 antibody (Cell Signaling); 11) mouse monoclonal anti-p-ERK antibody (Santa Cruz Biotechnology, Santa Cruz, CA); and 12) rabbit anti-human ERK1 antibody (Santa Cruz Biotechnology). After incubation with an HRP-labeled anti-rabbit or anti-mouse IgG antibody (Cell Signaling) for 1 h, signals were detected using Immune Star HRP (Bio-Rad, Hercules, CA). The membranes were reprobed with a rabbit anti-β-actin antibody (Sigma-Aldrich) or COX-IV antibody to serve as controls for equal loading. The density of each band was determined using National Institutes of Health Image software and expressed relative to the density of the corresponding band of β-actin or COX-IV, with an arbitrary unit value of 1 assigned to the normal control.
Cytochrome c release.
The cytoplasmic fraction was separated from the mitochondrial fraction in mouse proximal tubular cells using a Mitochondrial Isolation Kit for Cultured Cells (Thermo Scientific). Released cytochrome c in cytoplasm from mitochondria was detected by Western blotting with a mouse monoclonal anti-cytochrome c antibody (BD Biosciences, San Jose, CA) as a primary antibody. The membranes were reprobed with a mouse monoclonal anti-α-tubulin antibody (Sigma-Aldrich) to serve as a control for equal loading.
Mitochondrial membrane potential.
Mitochondrial membrane potential was evaluated using a JC-1 Mitochondrial Potential Sensor (Invitrogen). In this assay, potential dependent accumulation of JC-1 causes the fluorescence to shift from green to red. On 96-well plates, adherent mouse proximal tubular cells were treated with 100 μM cisplatin for 1 h and 10−6 M epicatechin for 8 h. The cells were then incubated with medium containing JC-1 at 1:2,000 for 30 min at 37°C. Fluorescent intensity was measured using a Synergy 2 Multi-Mode Microplate Reader with excitation at 485 nm and emission at 528 nm for green and excitation at 530 nm and emission at 590 nm for red. Data were expressed as ratio of red to green fluorescence.
Oxidative stress detection assay.
An Image-iT LIVE Green Reactive Oxygen Species Detection Kit (Invitrogen) was used for the detection of increased oxidative stress in vitro. Adherent mouse proximal tubular cells seeded on 96-well plates were treated with cisplatin for 1 h. Then, the cells were incubated with medium containing 10−6 M epicatechin or 1 mM tempol for 4 h. Culture medium was replaced with 25 μM carboxy-H2DCFDA-containing HBSS and incubated for 1 h at 37°C. Cells were washed with HBSS two times, and then fluorescent intensity was measured using a Synergy 2 Multi-Mode Microplate Reader with excitation at 485 nm and emission at 528 nm. Data were expressed as values relative to control.
Mitochondrial imaging in vitro.
To obtain mitochondrial images in vitro, mitochondria on unfixed murine proximal tubular cells were stained with Mitotracker Mitochondrion-Selective Probes (Invitrogen). After incubation in the complete medium with or without 10−6 M epicatechin or 1 mM tempol, the cells were incubated with 500 nM Mitotracker in PBS at 37°C for 45 min. After washes with PBS three times, the cells were observed by a laser-scanning confocal microscope (LSM 510 META, Carl Zeiss Microimaging, Thornwood, NY) to obtain images. Fragmented mitochondria looked punctate and rounded, whereas normal mitochondria showed a threadlike structure. Cells with mitochondrial fragmentation were defined as those containing a majority (>70%) of fragmented mitochondria as in a previous report (4), and they were counted to determine the percentage in 30–35 cells/sample.
Statistical analysis.
All values are expressed as means ± SD. Statistical analysis was performed with one-way ANOVA using Tukey's method to compare the groups. A level of P < 0.05 was considered a statistically significant difference.
RESULTS
Renal function.
Cisplatin significantly increased serum creatinine and BUN on day 3 compared with vehicle (saline) treatment while these elevations were significantly blocked by epicatechin treatment (Fig. 2, A and B).
Fig. 2.
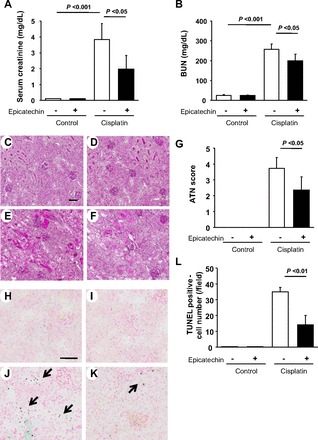
Effect of epicatechin on renal function and histology in mouse cisplatin nephropathy at death. The level of serum creatinine (A) and blood urea nitrogen (BUN; B) are shown. Periodic acid-Schiff (PAS) staining demonstrates the representative histological features in the sham group (C), sham group with epicatechin treatment (D), cisplatin group (E), and cisplatin with epicatechin group (F). Quantification of acute tubular necrosis (ATN) score from these groups is shown (G) as well as terminal uridine deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining in the sham group (H), sham with epicatechin group (I), cisplatin group (J), and cisplatin with epicatechin group (K). Blue dot indicates positive cells, while red color indicates nuclear staining. A quantification of TUNEL-positive cells from these groups is also shown (G). Bar = 50 μm.
Tubular injury.
Tubular epithelial cells are the primary site for cisplatin injury. Hence, we examined tubular injury including detachment of epithelium, loss of brush border, or cast formation. Compared with control, epicatechin treatment had no effects on tubular epithelial cells (Fig. 2, C and D). In contrast, cisplatin markedly caused tubular injury while such injuries were partially ameliorated by epicatechin treatment. Similarly, the cisplatin group had a significantly higher ATN score (Fig. 2, E–G), suggesting this anticancer drug caused widespread severe injury. However, such tubular damage was alleviated by epicatechin. In addition, TUNEL staining was performed to detect apoptotic cells. The cisplatin group significantly exhibited a higher number of apoptotic cells compared with the control group and epicatechin group (Fig. 2, H–L). However, epicatechin partially but significantly blocked the formation of apoptosis (Fig. 2, K and L).
Oxidative stress.
Oxidative stress is a major factor causing renal injury in response to cisplatin. We examined the level of oxidative stress using two markers, 4-hydroxynonenal (4-HNE) and nitrotyrosine (NT) (Fig. 3, A–F). By immunohistochemistry, both markers were highly expressed in the kidney treated by cisplatin compared with the control. In particular, damaged tubules showed strong signals for both markers (Fig. 3, B and E). However, treatment with epicatechin reduced these signals (Fig. 3, C and F). These data suggest that cisplatin induces oxidative stress in damaged tubules while such stress was significantly blocked by epicatechin treatment. This finding was next confirmed by Western blotting for NT using the mouse kidney. NT expression was significantly induced by cisplatin whereas such induction was significantly blocked by epicatechin (Fig. 3G). Since mitochondria are a source of oxidative stress, we also isolated mitochondria to examine the NT level. As shown in Fig. 3H, cisplatin treatment resulted in higher levels of NT in isolated mitochondria while it was significantly reduced by epicatechin in this model.
Fig. 3.
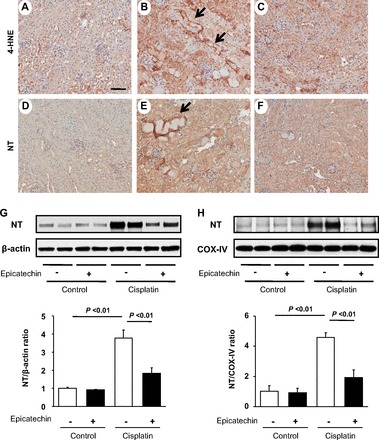
Effect of epicatechin on oxidative stress in renal cortex and mitochondria. Immunohistochemistry for 4-hydroxynonenal (4-HNE; A–C) and nitrotyrosine (NT; D–F) in the saline group (A and D), cisplatin group (B and E), and cisplatin with epicatechin group (C and F). Immunoblot with renal cortex for NT and its quantification (G). as well as the immunoblot with mitochondria isolated from renal cortex for NT and its quantification are also shown (H). COX, cytochrome c oxidase. Bar = 50 μm.
Mitochondrial effect of epicatechin in the mouse kidney.
Since we found that epicatechin reduced oxidative stress in mitochondria in this model, we further examined the mitochondrial effect of epicatechin. First, we examined mitochondrial DNA content, which is supposed to correspond to mitochondrial number. Consistent with mitochondrial oxidative stress, cisplatin significantly reduced mitochondrial DNA whereas epicatechin significantly blocked such a reduction (Fig. 4A). Similarly, the protein expression of mitochondrial oxidative phosphorylation complexes and MnSOD was significantly reduced by cisplatin while such a reduction was ameliorated by epicatechin (Fig. 4, B–D).
Fig. 4.
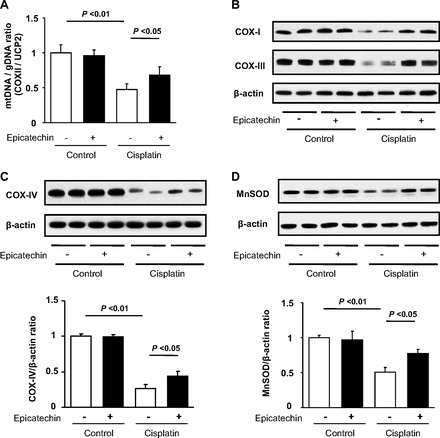
Effect of epicatechin on mitochondria in mouse cisplatin nephropathy at death. Mitochondrial DNA content corrected by genomic DNA in the renal cortex is shown (A). Immunoblotting for complex I and III in the renal cortex (B) and immunoblotting for complex IV (C) and MnSOD (D) and their quantifications are also shown.
Mitochondrial effect of epicatechin in cultured mouse proximal tubular cells:.
To confirm the benefit of epicatechin on mitochondria, we next performed an in vitro study using mouse proximal tubular cells (TKPTS) cells. In this experiment, we tested the effect of epicatechin after cells had been treated with 100 μM cisplatin for 1 h. First, we examined the activity of succinate dehydrogenase in complex II of the mitochondrial phosphorylation complex. A MTT reduction, as an index of this enzyme activity, was significantly low in the cells without epicatechin compared with control cells at 8 h. In contrast, epicatechin-treated cells exhibited significantly higher level of MTT reduction compared with cells without epicatechin, suggesting that succinate dehydrogenase was functionally activated by epicatechin (Fig. 5A). Because MTT reduction is also associated with cell number, we determined whether the higher level of reduced MTT could be due to an increase in cell number. To address this issue, we examined DNA content in the culture cell dish as a surrogate of cell number. As shown in Fig. 5B, we did not find any difference in DNA content, suggesting that cisplatin as well as subsequent epicatechin treatment did not alter cell number at this time point.
Fig. 5.
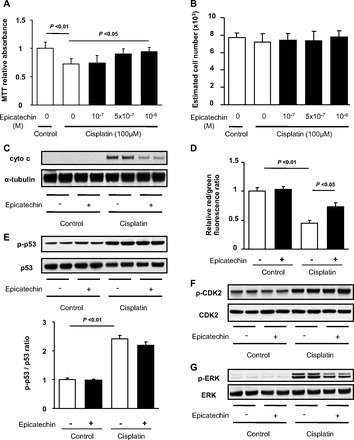
Effect of epicatechin on mitochondria in mouse cultured proximal tubular cells. The activity of succinate dehydrogenase is measured by a reduction of MTT to formazan at 8 h (A). In the same condition, DNA content was measured (B). Released cytochrome c into cytoplasmic fraction separated from mitochondrial fraction was detected by Western blotting (C). Loss of mitochondrial membrane potential was evaluated by JC-1 Mitochondrial Potential Sensor assay (D). Data are expressed as the ratio of red to green fluorescence. Western blots for p53 and phosphorylated p53 and its quantification are shown (E). Western blots for cyclin-dependent kinase 2 (CDK2) and phosphorylated (activated) CDK2 (F) and ERK and phosphorylated-ERK (G) are also shown.
We next examined whether epicatechin protects the mitochondrial membrane from cisplatin injury. Cisplatin caused a marked release of cytochrome c from mitochondria to cytoplasm while epicatechin treatment prevented such injury at 24 h (Fig. 5C). Similarly, cisplatin induced a significant loss of mitochondrial membrane potential, as shown in the JC-1 mitochondrial potential sensor assay, and epicatechin also suppressed this alteration (Fig. 5D).
To determine mechanisms by which epicatechin protects mitochondria from cisplatin injury, three representative signalings were examined. Phosphorylation of p53, which has been recently implicated in cisplatin-induced mitochondrial membrane damage, was markedly accelerated by cisplatin treatment whereas epicatechin did not affect it (Fig. 5E). Similarly, activation of CDK2 caused by cisplatin was not inhibited by epicatechin (Fig. 5F). In contrast, phosphorylation of ERK MAPK, which is a key pathway involved in cisplatin nephropathy, was prevented by epicatechin (Fig. 5G).
Antioxidative effect of epicatechin might be a key mechanism for mitochondrial protection.
As shown in the in vivo experiments, oxidative stress was found to be induced by cisplatin treatment in proximal tubular cells, suggesting that oxidative stress might be a key mediator of cisplatin injury in mitochondria. To confirm this hypothesis, we examined whether blocking oxidative stress with the potent antioxidant tempol could prevent mitochondrial injury in response to cisplatin. As expected, an increased in oxidative stress was significantly prevented by tempol as potently as epicatechin (Fig. 6A). Furthermore, a cisplatin-mediated reduction in complex IV protein expression was also ameliorated by epicatechin as well as tempol at 24 h (Fig. 6B).
Fig. 6.
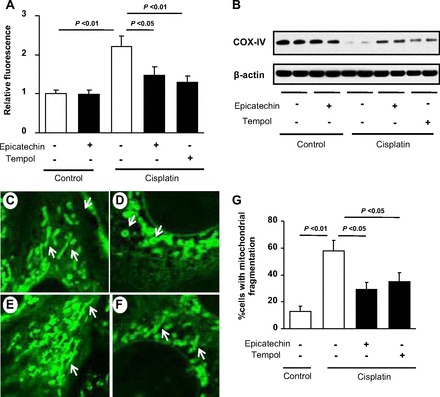
Effects of epicatechin on oxidative stress and mitochondrial fragmentation. The increased production of reactive oxygen species (ROS) in cisplatin-treated mouse proximal tubular cells was detected by the reaction with carboxy-H2DCFDA (A). Immunoblotting for complex IV protein is shown (B). Increased ROS and the reduced complex IV level caused by cisplatin (100 μM) were prevented by both epicatechin (10−6 M) and the antioxidant tempol (1 mM). Images by using a laser confocal microscope show mitochondrial structure detected by Mitotracker (green) staining on control (C), cisplatin (100 μM) alone (D)-, cisplatin+10−6 M epicatechin (E)-, and cisplatin+1 mM tempol (F)-treated mouse proximal tubular cells. Mitochondria in control conditionally immortalized mouse proximal tubular cells (TKPTS) exhibit a reticulotubular appearance (white arrow in C). Cisplatin fragmented them into multiple small rounded organelles (mitochondrial fragmentation; white arrow in D), whereby subsequent 10−6 M epicatechin improved such structural perturbation at 8 h (E) as well as did 1 mM tempol (F). The percentage of cells with mitochondrial fragmentation is shown (G).
Finally, we examined the effect of epicatechin in mitochondrial structure using a laser-scanning confocal microscope. The typical reticulotubular appearance of mitochondria in healthy TKPTS cells (Fig. 6C) had disintegrated into multiple small rounded organelles in response to cisplatin at 8 h (Fig. 6D). However, epicatechin treatment prevented these structural changes in mitochondria (Fig. 6, E and G). Interestingly, tempol showed similar protection on mitochondrial structural changes (Fig. 6, F and G), suggesting these morphological alterations of mitochondria were at least partially mediated by oxidative stress.
Anticancer effect of cisplatin is not disturbed by epicatechin in HeLa cells.
It is clinically important that supplemental treatment should not block the anticancer effect of cisplatin, but should selectively protect the kidney. Hence, we next examined whether epicatechin could disturb cell toxicity in HeLa cells, which are derived from a patient with cervical adenocarcinoma. In the TKPTS cells, cisplatin significantly reduced DNA content whereas epicatechin (10−6 M) prevented such toxicity of this anticancer agent at 24 h (Fig. 7A). In contrast, epicatechin failed to protect HeLa cells from cisplatin toxicity in cell number (Fig. 7B).
Fig. 7.
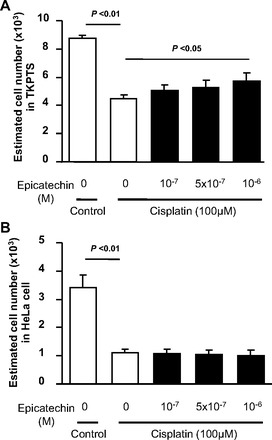
Anticancer effect of cisplatin is not disturbed by epicatechin in the HeLa cells. The anticancer activity of cisplatin is examined by cell number, which corresponds to survived cell number at 24 h. Cisplatin significantly reduced cell number in both TKPTS cells (A) and HeLa cells (B). However, epicatechin treatment prevented a loss of cell number in TKPTS at 24 h, but not in HeLa cells.
DISCUSSION
Consistent with previous studies, we found that cisplatin-caused renal dysfunction and morphological changes were associated with mitochondrial injury in the mouse kidney. These findings was also confirmed by using immortalized mouse proximal tubular epithelial cells in which we documented that cisplatin caused mitochondrial dysfunction as revealed by a reduction in protein expression of the mitochondrial oxidative phosphorylation complex and MnSOD. Here, we tested whether epicatechin prevents such renal injury because of mitochondrial protection. In particular, we examined whether epicatechin administration after pretreatment with cisplatin could prevent cisplatin-induced renal injury. Our primary finding is that epicatechin exhibited protective effects, and such protection was likely owing to an ability of epicatechin to protect mitochondria. In fact, both in vivo and in vitro studies demonstrated that cisplatin-mediated injuries, including a reduction in mitochondrial protein, its functional perturbation, and morphological alterations, were significantly prevented by epicatechin. Importantly, epicatechin selectively protect tubular epithelial cells, but not cancer (HeLa) cells.
Recently, the cardiovascular benefits of epicatechin have been shown in several studies. Villarreal's research group (41) has performed a series of studies and found that epicatechin potently ameliorated myocardial injury in the setting of myocardial ischemia-reperfusion injury in rats. Subsequent studies using rats also documented that myocardial infarction caused by permanent coronary occlusion was also significantly alleviated by this compound (42). In turn, clinical studies have indicated that the underlying mechanisms for epicatechin benefits could be endothelial protection since epicatechin is capable of inducing vasorelaxation as well as activation of endothelial nitric oxide synthase (14, 33). Recently, epicatechin was also shown to improve exercise performance due to an increase in capacity for muscle aerobic metabolism (18, 28). While several mechanisms may be involved, a study in skeletal muscle documented that a potential mechanism is likely a stimulation of mitochondrial biogenesis (28, 36).
To confirm mitochondrial protection by epicatechin, cultured mouse proximal tubular cells were stimulated by cisplatin for 1 h, cisplatin was removed, and the medium was replaced with new conditioned medium with/without epicatechin. This protocol was designed to test whether epicatechin could be protective after injection of cisplatin injury. In this experiment, mitochondrial function, as revealed by oxidative phosphorylation protein expression as well as its activity, remained low at 8 h but was rescued by epicatechin treatment. Similarly, other markers of mitochondrial injury, including a release of cytochrome c from mitochondria and loss of mitochondrial membrane potential, were also prevented by epicatechin treatment. These data suggest that epicatechin protects mitochondria from cisplatin injury.
Cisplatin injury was found to be regulated by several pathways. The activation of p53, a proapoptotic protein, was initiated as a consequence of DNA damage in response to cisplatin, and it occurs before a release of cytochrome c, indicating that activation of p53 could be upstream of mitochondrial injury (9, 19). Similarly, CDK2, a cell cycle protein, seems to be activated by cisplatin and mediates cell injury independently of the p53 pathway (26). However, the current study documented that epicatechin had no effects on these pathways. Another pathway involved in this injury could be the MAPK pathway, which regulates a variety of cell responses, including proliferation, differentiation, and apoptosis (29). Interestingly, this pathway seems to be involved both upstream and downstream of mitochondrial injury. In fact, pharmaceutical inhibition of ERK was shown to be capable of preventing cytochrome c release and Bax activation (21) whereas mitochondria-induced oxidative stress could cause ERK activation (39). In the current study, we documented that epicatechin potently blocked the activation of ERK, thereby providing a potential mechanism for the beneficial effect of epicatechin.
We also found that cisplatin causes mitochondrial fragmentation as well as a reduction in mitochondrial number, and such findings are consistent with previous papers (5). Given the fact that mitochondria are a major source of oxidative stress, it is intuitively assumed that mitochondrial dysfunction could result in a reduction of reactive oxygen species (ROS) production. However, as opposed to such an assumption, fragmented mitochondria are found to be not bioenergically functional (3). Moreover, blocking oxidative phosphorylation with either pharmaceutical maneuvers or molecular manipulation can accelerate ROS production (11, 25, 37). Hence, an increase in mitochondrial oxidative stress may be due in part to cisplatin-induced mitochondrial injury. In contrast, we also found that blocking oxidative stress with tempol potently inhibited mitochondrial injury, suggesting that oxidative stress may also drive mitochondrial injury under cisplatin stimulation. Consistent with our results, a recent study has demonstrated that a reduction in mitochondrial-specific oxidative stress ameliorated kidney injury in a mouse cisplatin nephropathy model (27). Since mitochondrial dysfunction is usually tightly connected with oxidative stress, it seems likely that ROS can be a cause and a consequence of mitochondrial injury.
While we found that epicatechin exhibits antioxidative effects as potently as tempol, it remains unclear as to how this compound blocks ROS production. One potential mechanism may be by enhancing mitochondrial antioxidative function by activating MnSOD and glutathione (1). Epicatechin can also block NADPH oxidase activity in the model of cyclosporine nephropathy (15). Taking this together, we now speculate that epicatechin may have several pathways for blocking oxidative stress, and these pathways should be explored in a future study.
In conclusion, we demonstrated that epicatechin is protective in mouse cisplatin nephropathy even if treatment starts after cisplatin injury had occurred in tubular cells. Mechanisms are likely that epicatechin exhibits mitochondrial protection. Given the fact that epicatechin is a derivative of cocoa, it might be safe for us to use this compound in clinical nephrology studies.
GRANTS
This study is also supported by a Japan Heart Foundation/Bayer Yakuhin Research Grant (K. Tanabe).
DISCLOSURES
G. Schreiner is a member of Cardero, Inc., which is developing epicatechin as a treatment for various disorders. R. J. Johnson and T. Nakagawa have stock in Cardero, Inc. All other investigators declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Author contributions: K.T. and T.N. provided conception and design of research; K.T., Y.T., M.A.L., M.M., N.S., W.S., and T.N. performed experiments; K.T., M.A.L., M.M., and T.N. analyzed data; K.T., Y.M., G.F.S., R.J.J., and T.N. interpreted results of experiments; K.T. prepared figures; K.T. and T.N. drafted manuscript; K.T., F.J.V., and R.J.J. edited and revised manuscript; K.T., Y.T., M.A.L., M.M., N.S., W.S., Y.M., G.F.S., F.J.V., R.J.J., and T.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Francisco Villarreal (University of California, San Diego) for a great discussion.
REFERENCES
- 1.Al-Malki AL, Moselhy SS. The protective effect of epicatchin against oxidative stress and nephrotoxicity in rats induced by cyclosporine. Hum Exp Toxicol 30: 145–151, 2011. [DOI] [PubMed] [Google Scholar]
- 2.Arany I, Safirstein RL. Cisplatin nephrotoxicity Semin Nephrol 23: 460–464, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25: 3900–3911, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol 300: C447–C455, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119: 1275–1285, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowanadisai W, Bauerly KA, Tchaparian E, Wong A, Cortopassi GA, Rucker RB. Pyrroloquinoline quinone stimulates mitochondrial biogenesis through cAMP response element-binding protein phosphorylation and increased PGC-1alpha expression. J Biol Chem 285: 142–152, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol 67: 93–130, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Corti R, Flammer AJ, Hollenberg NK, Luscher TF. Cocoa and cardiovascular health. Circulation 119: 1433–1441, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in cisplatin-induced renal cell apoptosis. J Pharmacol Exp Ther 308: 921–928, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Ernest S, Bello-Reuss E. Expression and function of P-glycoprotein in a mouse kidney cell line. Am J Physiol Cell Physiol 269: C323–C333, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci USA 96: 4820–4825, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Fisher ND, Hollenberg NK. Flavanols for cardiovascular health: the science behind the sweetness. J Hypertens 23: 1453–1459, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Fisher ND, Hughes M, Gerhard-Herman M, Hollenberg NK. Flavanol-rich cocoa induces nitric-oxide-dependent vasodilation in healthy humans. J Hypertens 21: 2281–2286, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Gomez-Guzman M, Jimenez R, Sanchez M, Zarzuelo MJ, Galindo P, Quintela AM, Lopez-Sepulveda R, Romero M, Tamargo J, Vargas F, Perez-Vizcaino F, Duarte J. Epicatechin lowers blood pressure, restores endothelial function, and decreases oxidative stress and endothelin-1 and NADPH oxidase activity in DOCA-salt hypertension. Free Radic Biol Med 52: 70–79, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Hollenberg NK, Martinez G, McCullough M, Meinking T, Passan D, Preston M, Rivera A, Taplin D, Vicaria-Clement M. Aging, acculturation, salt intake, and hypertension in the Kuna of Panama. Hypertension 29: 171–176, 1997. [DOI] [PubMed] [Google Scholar]
- 17.Hollenberg NK, Rivera A, Meinking T, Martinez G, McCullough M, Passan D, Preston M, Taplin D, Vicaria-Clement M. Age, renal perfusion and function in island-dwelling indigenous Kuna Amerinds of Panama. Nephron 82: 131–138, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Huttemann M, Lee I, Malek MH. (-)-Epicatechin maintains endurance training adaptation in mice after 14 days of detraining. FASEB J 26: 1413–1422, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang M, Wei Q, Wang J, Du Q, Yu J, Zhang L, Dong Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene 25: 4056–4066, 2006. [DOI] [PubMed] [Google Scholar]
- 20.Jiang M, Yi X, Hsu S, Wang CY, Dong Z. Role of p53 in cisplatin-induced tubular cell apoptosis: dependence on p53 transcriptional activity. Am J Physiol Renal Physiol 287: F1140–F1147, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Kim YK, Kim HJ, Kwon CH, Kim JH, Woo JS, Jung JS, Kim JM. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J Appl Toxicol 25: 374–382, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Kruidering M, Maasdam DH, Prins FA, de Heer E, Mulder GJ, Nagelkerke JF. Evaluation of nephrotoxicity in vitro using a suspension of highly purified porcine proximal tubular cells and characterization of the cells in primary culture. Exp Nephrol 2: 324–344, 1994. [PubMed] [Google Scholar]
- 23.Kruidering M, Van de Water B, de Heer E, Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther 280: 638–649, 1997. [PubMed] [Google Scholar]
- 24.Lee RH, Song JM, Park MY, Kang SK, Kim YK, Jung JS. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells. Biochem Pharmacol 62: 1013–1023, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516–8525, 2003. [DOI] [PubMed] [Google Scholar]
- 26.Megyesi J, Udvarhelyi N, Safirstein RL, Price PM. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol Renal Fluid Electrolyte Physiol 271: F1211–F1216, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Mukhopadhyay P, Horvath B, Zsengeller Z, Zielonka J, Tanchian G, Holovac E, Kechrid M, Patel V, Stillman IE, Parikh SM, Joseph J, Kalyanaraman B, Pacher P. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic Biol Med 52: 497–506, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nogueira L, Ramirez-Sanchez I, Perkins GA, Murphy A, Taub PR, Ceballos G, Villarreal FJ, Hogan MC, Malek MH. (-)-Epicatechin enhances fatigue resistance and oxidative capacity in mouse muscle. J Physiol 589: 4615–4631, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26: 3203–3213, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F. (–)-Epicatechin activation of endothelial cell endothelial nitric oxide synthase, nitric oxide, and related signaling pathways. Hypertension 55: 1398–1405, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F. (–)-Epicatechin induces calcium- and translocation-independent eNOS activation in arterial endothelial cells. Am J Physiol Cell Physiol 300: C880–C887, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, Sies H, Kwik-Uribe C, Schmitz HH, Kelm M. (-)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci USA 103: 1024–1029, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22: 7265–7279, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Tanabe K, Lanaspa MA, Kitagawa W, Rivard CJ, Miyazaki M, Klawitter J, Schreiner GF, Saleem MA, Mathieson PW, Makino H, Johnson RJ, Nakagawa T. Nicorandil as a novel therapy for advanced diabetic nephropathy in the eNOS-deficient mouse. Am J Physiol Renal Physiol 302: F1151–F1160, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taub PR, Ramirez-Sanchez I, Ciaraldi TP, Perkins G, Murphy AN, Naviaux R, Hogan M, Maisel AS, Henry RR, Ceballos G, Villarreal F. Alterations in skeletal muscle indicators of mitochondrial structure and biogenesis in patients with type 2 diabetes and heart failure: effects of epicatechin rich cocoa. Clin Transl Sci 5: 43–47, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17: 3–8, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4: 307–320, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe N, Zmijewski JW, Takabe W, Umezu-Goto M, Le Goffe C, Sekine A, Landar A, Watanabe A, Aoki J, Arai H, Kodama T, Murphy MP, Kalyanaraman R, Darley-Usmar VM, Noguchi N. Activation of mitogen-activated protein kinases by lysophosphatidylcholine-induced mitochondrial reactive oxygen species generation in endothelial cells. Am J Pathol 168: 1737–1748, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Q, Dong G, Franklin J, Dong Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int 72: 53–62, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Yamazaki KG, Romero-Perez D, Barraza-Hidalgo M, Cruz M, Rivas M, Cortez-Gomez B, Ceballos G, Villarreal F. Short- and long-term effects of (–)-epicatechin on myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 295: H761–H767, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamazaki KG, Taub PR, Barraza-Hidalgo M, Rivas MM, Zambon AC, Ceballos G, Villarreal FJ. Effects of (–)-epicatechin on myocardial infarct size and left ventricular remodeling after permanent coronary occlusion. J Am Coll Cardiol 55: 2869–2876, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res 12: 5817–5825, 2006. [DOI] [PubMed] [Google Scholar]