

Abstract
Autophagy is a cellular process of “self-eating.” During autophagy, a portion of cytoplasm is enveloped in double membrane-bound structures called autophagosomes, which undergo maturation and fusion with lysosomes for degradation. At the core of the molecular machinery of autophagy is a specific family of genes or proteins called Atg. Originally identified in yeast, Atg orthologs are now being discovered in mammalian cells and have been shown to play critical roles in autophagy. Traditionally, autophagy is recognized as a cellular response to nutrient deprivation or starvation whereby cells digest cytoplasmic organelles and macromolecules to recycle nutrients for self-support. However, studies during the last few years have indicated that autophagy is a general cellular response to stress. Interestingly, depending on experimental conditions, especially stress levels, autophagy can directly induce cell death or act as a mechanism of cell survival. In this review, we discuss the molecular machinery, regulation, and function of autophagy. In addition, we analyze the recent findings of autophagy in renal systems and its possible role in renal pathophysiology.
Keywords: apoptosis, Atg, cytoprotection, mammalian target of rapamycin, phosphatidylinositol 3-kinase
during normal cell growth and development, a well-controlled balance is needed between the biosynthesis and catabolism of macromolecules. This balance is preserved by diverse regulatory mechanisms that respond to the environmental circumstances and provoke specific signals into systematic growth and developmental responses. In this continuous process, there is an invariable production of compounds that can be toxic to the cells. It has been recognized that appropriate degradation of irreversibly damaged proteins is vital to cellular viability and function (121). Often, damaged proteins develop abnormal intermolecular interactions to form aggregates that, when trapped inside other cellular components, are cytotoxic. In eukaryotic cells, the major systems responsible for protein degradation are the ubiquitin-proteasome pathway and the lysosomal system. The proteasomal pathway is restricted to the cytosol and nucleus and mainly involved in the degradation of short-lived proteins. In contrast, the lysosomal system comprises vesicular organelles with various hydrolases including proteases for substance degradation. It is generally believed that lysosomes are responsible for the degradation of long-lived proteins. The major channel that delivers cytoplasmic constituents including proteins to lysosomes for degradation is autophagy (55).
An Evolving View of Autophagy
Autophagy, a term derived from the Greek, means self (auto)-eating (phagy). In a cell, three types of autophagy have been recognized: macroautophagy, microautophagy, and chaperone-mediated autophagy (Fig. 1). Macroautophagy starts with the de novo formation of a cup-shaped isolation double membrane (also called phagophore or preautophagosome) that engulfs a portion of cytoplasm. The isolation membrane then encloses to form a mature vesicle, i.e., autophagosome, that subsequently fuses with a lysosome, leading to the degradation of intra-autophagosomal components by lysosomal hydrolases. In contrast, microautophagy involves the engulfment of cytoplasm instantly at the lysosomal membrane by invagination, protrusion, and separation. Chaperone-mediated autophagy, on the other hand, is a process of direct transport of unfolded proteins via the lysosomal chaperonin hsc70 and LAMP-2A, a lysosomal membrane receptor. Of these three types, macroautophagy is most prevalent and most frequently referred to as “autophagy,” the focus of our discussion in this review.
Fig. 1.
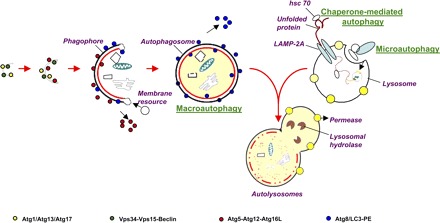
The 3 forms of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy starts with the de novo formation of a cup-shaped isolation double membrane that engulfs a portion of cytoplasm. Microautophagy involves the engulfment of cytoplasm instantly at the lysosomal membrane by invagination, protrusion, and separation. Chaperone-mediated autophagy is a process of direct transport of unfolded proteins via the lysosomal chaperonin hsc70 and LAMP-2A. All forms of autophagy subsequently lead to the degradation of intra-autophagosomal components by lysosomal hydrolases. PE, phosphatidylethanolamine.
For many years, autophagy was considered to be primarily involved in cellular architectural changes that prevail during differentiation and development. However, in the late 1960s, autophagy was found to be enhanced in various organs including newborn kidney (29). This and other related observations led to the belief that breakdown of cytoplasmic components by cellular autophagy could be functionally related to gluconeogenesis (106). Sybers et al. (125) further proposed that cellular injury stimulates the endoplasmic reticulum (ER) to enclose the damaged components, permitting localized lysosomal digestion without causing injury to the entire cell. Later, autophagy was shown to be particularly selective in removing cytoplasmic components from the cytoplasm and to play an important role in intracellular turnover (107).
Autophagy is constitutive in all eukaryotic cell types containing a lysosomal compartment. This basal autophagy contributes to long-lived protein degradation as well as organelle turnover and occurs in parallel with the proteasomal pathway to control the quality or health of the cytoplasm. Autophagy can also be induced as a cellular reaction to various physiological challenges including nutrient starvation, cell growth control, antiaging, intracellular protein and organelle turnover, development, eradication of microorganisms, cell death, tumor suppression, and inherent immunity (80). While a role for autophagy in cell survival is well recognized, cell death from excessive cellular consumption or autophagy has also been suggested (9, 30). To complicate these circumstances further, cytotoxic events generally induce autophagy, making it hard to conclude whether autophagy is a death mechanism or a failed attempt at cellular defense (79). Although autophagy may engulf cytoplasm nonselectively, a selective or specific sequestration of organelles has been observed and proposed to play a role in certain diseases including cancer, cardiomyopathy, renal disorders, neurodegenerative disorders, and viral, bacterial, and parasitic infections (17, 23, 68, 81, 90, 94, 98, 145).
Core Molecular Machinery of Autophagy
The voyage into the understanding of the molecular mechanism of autophagy began with the identification of the autophagy-related (Atg) genes in yeast (95–97). To date, over 30 Atg genes have been cloned. Notably, mammalian Atg orthologs have been identified, showing high conservation with yeast genes (54). Atgs constitute the core molecular machinery of autophagy, which is involved in the induction of the isolation membrane, engulfment of cytoplasm, formation of the autophagic vesicles, and fusion with lysosome. A summary of Atg proteins and their functions in autophagy is provided in Table 1.
Table 1.
Atg proteins in autophagy
| Gene | Function in Autophagy | Cellular Localization | Reference | Other Names | Entrez Gene ID | Molecular Mass, kDa |
|---|---|---|---|---|---|---|
| Atg1 | Protein serine/threonine kinase forming complex with Atg13p and Atg17 during PAS formation | Cytoplasm | 75 | AUT3, APG1 | 852695 | 10 |
| Atg2 | Peripheral membrane protein required for vesicle formation and cycling with Atg9 between preautophagosomal structure and mitochondria | Cytoplasm | 137 | SPO72, APG2 | 855479 | 17 |
| Atg3 | E2-like enzyme involved in Atg8-PE conjugation | Cytoplasm | 140 | AUT1, APG3 | 855741 | 35 |
| Atg4 | Conserved cysteine protease for Atg8 cleavage and processing and attachment of autophagosomes to microtubules | Cytoplasm | 62 | AUT2, APG4 | 855498 | 55 |
| Atg5 | Conserved protein undergoing conjugation with Atg12, leading to oligomerization with Atg16p to form a complex involved in Atg8 lipidation | Cytoplasm, autophagic vacuole | 39 | APG5 | 855954 | 33 |
| Atg6 | Subunit of PI3-kinase complexes I and II that are essential for autophagy | Membrane fraction | 20 | VPS30, Beclin1, APG6 | 855983 | 63 |
| Atg7 | Mediating conjugation of Atg12p with Atg5p and Atg8p with PE | Cytosol and membrane fraction (mainly mitochondria) | 142 | CVT2, APG7 | 856576 | 71 |
| Atg8 | Conserved protein that undergoes COOH-terminal conjugation to PE; Atg8p-PE is a component of autophagosome vesicles | Cytosolic to autophagic vacuolar membrane | 128 | APG8, LC3 | 852200 | LC3 I: 18; LC3 II: 16 |
| Atg9 | Transmembrane protein involved in formation of autophagic vesicles; cycles between preautophagosomal structure and other cytosolic punctate structures; forms complex with Atg23p and Atg27p | Preautophagosomal membrane structure | 114 | APG9, AUT9 | 851406 | 115 |
| Atg10 | Conserved E2-like conjugating enzyme that mediates formation of Atg12p-Atg5p conjugate, a critical step in autophagy | Unclear | 111 | APG10 | 850684 | 20 |
| Atg11 | Adapter protein that directs receptor-bound cargo and recruits other proteins to preautophagosomal structure | Preautophagosomal membrane structure | 85 | CVT9 | 856162 | 135 |
| Atg12 | Conserved protein that undergoes conjugation with Atg5p, leading to oligomerization with Atg16p and formation of a complex involved in Atg8p lipidation | Membrane fraction | 39 | APG12 | 852518 | 21 |
| Atg13 | Regulatory subunit of Atg1 signaling complex; stimulates Atg1 kinase activity; required for vesicle formation during autophagy and involved in Atg9p, Atg23p, and Atg27p cycling | Extrinsic to membrane | 114 | APG13 | 856315 | 83 |
| Atg14 | Targets PI3-kinase complex I (with Vps34p, Vps15p, and Vps30p) and other proteins to preautophagosomal structure | Preautophagosomal membrane structure | 92 | APG14, CVT12 | 852425 | 40 |
| Atg15 | Lipase required for intravacuolar lysis of autophagosomes; targeted to intravacuolar vesicles during autophagy via multivesicular body pathway | Vacuolar lumen | 31 | AUT5, CVT17 | 850432 | 58 |
| Atg16 | Conserved protein that interacts with Atg12-Atg5 conjugates to form Atg12-Atg5-Atg16 multimers in preautophagosomal structure | Preautophagosomal membrane structure | 74 | SAP18, APG16 | 855194 | 17 |
| Atg17 | Scaffold protein responsible for preautophagosomal structure organization; forming Atg1-Atg13-Atg17 complex; stimulates Atg1 kinase activity | Preautophagosomal membrane structure | 45 | APG17 | 851142 | 49 |
| Atg18 | Phosphoinositide binding protein required for vesicle formation in autophagy | Cytosol | 58 | AUT10, CVT18 | 850577 | 55 |
| Atg19 | Receptor protein for cytoplasm-to-vacuole targeting pathway | Cytosol, preautophagosomal membrane structure | 21 | CVT19 | 854072 | 48 |
| Atg20 | Contains a Phox homology domain that binds PI3P | Endosome, preautophagosomal membrane structure | 147 | CVT20 | 851445 | 72 |
| Atg21 | Similar to Atg18 | Cytosol | 122 | HSV1 | 856004 | 55 |
| Atg22 | Vacuolar integral membrane protein required for efflux of amino acids during autophagic body breakdown; null mutation causes a gradual loss of viability during starvation | Integral to vacuolar membrane | 144 | AUT4 | 850319 | 59 |
| Atg23 | Cycles between PAS and non-PAS locations; forms a complex with Atg9p and Atg27p | Preautophagosomal membrane structure | 134 | CVT23 | 851154 | 51 |
| Atg24 | Contains PI-binding domain and forms complexes with Atg20 | Cytosol, preautophagosomal membrane structure | 5 | CVT | 853416 | 49 |
| Atg27 | Type I membrane protein involved in autophagy and involved in membrane delivery to preautophagosomal structure; binds PI3P | Membrane, preautophagosomal membrane structure | 146 | ETF1 | 853261 | 30 |
| Atg29 | Autophagy-specific protein that is required for recruitment of other Atg proteins (mainly Atg17) to preautophagosomal structure | Cytosol, preautophagosomal membrane structure | 49 | NA | 855937 | 25 |
PE, phosphatidylethanolamine; PAS, preautophagosome; PI, phosphatidylinositol; PI3-kinase, phosphatidylinositol 3-kinase; PI3P, phosphatidylinositol 3-phosphate.
The formation of autophagosomes is reliant upon several Atgs that are part of two novel conjugation systems required for initiation and elongation of the isolation membrane (96) (Fig. 2). The first conjugating event involves the conjugation of a single lipid molecule, phosphatidylethanolamine, to the COOH terminus of Atg8/LC3. In this conjugation event, Atg7 and Atg3 act as E1- and E2-homologous enzymes, respectively, in the ubiquitin pathway. For this conjugation to occur, Atg8/LC3 is initially cleaved by Atg4, a cysteine protease (41). The cleaved/activated Atg8/LC3 (LC3-I) is then activated by Atg7 and further transferred to Atg3. Atg3 conjugates Atg8/LC3 to phosphatidylethanolamine to form LC3-II, a lipidated form associated with autophagic membranes or vesicles (53) (Fig. 2A). Of note, redistribution of LC3 from the cytosol to autophagic vesicles and formation of LC3-II are commonly used as a marker for autophagy (83). The second conjugation event of autophagy involves the covalent attachment of the COOH-terminal glycine of Atg12 to Atg5 through an internal lysine residue (84). In this conjugation, Atg7 and Atg10 act as E1- and E2-homologous enzymes, respectively. Atg7 hydrolyzes ATP and activates Atg12 through the formation of a thioester bond. The activated Atg12 is then transferred to Atg5 via Atg10. The Atg5-Atg12 complex associates noncovalently to coiled-coil protein Atg16, which oligomerizes to form large units that are required for targeting the Atg8/LC3 into the limiting membrane and elongation of the sequestering membrane (61) (Fig. 2B). As these two conjugation events of autophagy are interdependent, Atg8/LC3 conjugation depends on the Atg5-Atg12-Atg16 complex activity. Consequently, LC3 conjugation or LC3-II formation is not detectable in Atg5-null cells and animals (133).
Fig. 2.
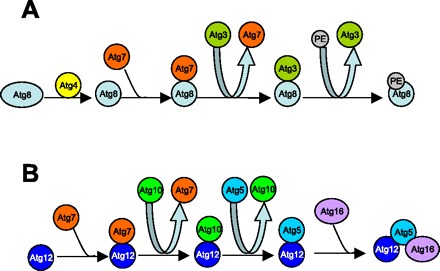
Two essential ubiquitin-like conjugation systems in autophagy. A: the first conjugating system includes Atg8, -4, -7, and -3 and PE. This system conjugates a single PE molecule to the COOH terminus of Atg8/LC3. B: the second conjugation system includes Atg12, -7, -10, -5, and 16. This conjugation leads to the covalent attachment of the COOH-terminal glycine of Atg12 to Atg5 through an internal lysine residue.
Besides the conjugation events, autophagy is also regulated by two unique kinase complexes (Fig. 3). The first kinase complex includes an autophagy-specific phosphatidylinositol 3-kinase (PI3-kinase), Vps34 (homolog to human class III PI3-kinase). The involvement of PI3-kinases in autophagy was initially suggested by the observation that autophagosome formation was inhibited by pharmacological inhibitors of PI3-kinases including 3-methyladenine, wortmannin, and LY-294002 (14, 118). As a PI3-kinase, Vps34 phosphorylates phosphatidylinositol (PI) to produce phosphatidylinositol 3-phosphate (PI3P), a docking lipid that promotes protein complex formation, membrane enclosing, and the consequent sequestration of cytoplasmic components in autophagic vacuoles (82, 105) (Fig. 3A). Importantly, Vps34 is associated with and probably regulated by Vps15 and Vps30/Atg6/beclin-1, which plays an important role in focusing the two conjugation events to the emerging autophagic units (50). Therefore, the PI3-kinase complex not only controls the nucleation of autophagic vesicles but also guides the conjugation events. Notably, beclin-1 in this complex has several interacting partners including Bcl-2, UVRAG, Ambra, and Bif-1 or Endophilin B1 (35, 103, 126). These molecular interactions are critical to the regulation of autophagy.
Fig. 3.
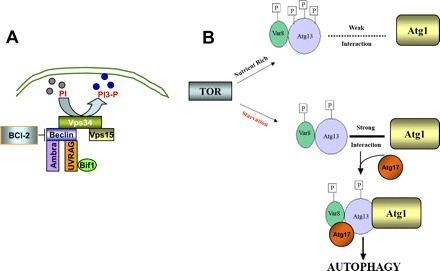
Two essential kinase systems in autophagy. A: class III phosphatidylinositol 3-kinase (PI3-kinase) complex. Vps34, a class III PI3-kinase, phosphorylates phosphatidylinositol (PI) to produce phosphatidylinositol 3-phosphate (PI3P), a docking lipid that promotes protein complex formation, membrane enclosing, and the consequent sequestration of cytoplasmic components in autophagic vacuoles. Vps34 forms complexes with and is regulated by beclin-1 and Vps15. Beclin-1 further interacts with and is regulated by Bcl-2, Ambra, and UVRAG. B: serine/threonine protein kinase complex. Regulation of this complex is centered on the interaction between Atg13 and Atg1, a serine/threonine protein kinase. In the presence of nutrients and growth factors, target of rapamycin (TOR) kinase is active and phosphorylates Atg13, preventing its association with Atg1. During starvation, TOR is not active, resulting in the activation of phosphatases and partial dephosphorylation of Atg13. Dephosphorylated Atg13 associates tightly with and activates Atg1, leading to the recruitment of Atg17 and the formation of the autophagic serine/threonine kinase complex.
The second kinase complex of autophagy is centered on Atg1 and Atg13 (75). In the classical model of autophagy induced by nutrient starvation, Atg13 is one of the downstream effectors of target of rapamycin (TOR)2 kinase (48). In the presence of nutrients including growth factors, TOR2 kinase is active and phosphorylates Atg13, preventing its association with Atg1, a serine/threonine protein kinase. Under this condition, Atg1 contributes to the formation of lysosomal hydrolase carrier vesicles, but not autophagosomes (3). During starvation, TOR2 kinase is inhibited, resulting in the activation of phosphatases and partial dephosphorylation of Atg13. Dephosphorylated Atg13 associates tightly with and activates Atg1, leading to the recruitment of Atg17 and the formation of the second kinase complex of autophagy. The Atg1-Atg13-Atg17 complex participates in elongation of the isolation membrane and progression toward a complete autophagosome structure (51) (Fig. 3B).
Once the autophagosome is fused at each side, it gets ported and blended with a neighboring lysosome, forming an autolysosome. In yeast, components of the SNARE machinery are required for autophagosome porting and blending with the lysosome (28). The inner membrane of the autophagosomes and the sequestered components of the cytoplasm are then degraded by lysosomal hydrolases. Once the macromolecules are degraded inside the lysosome, the monomeric units are exported to the cytosol for recycling (55).
Regulation of Autophagy
The complex molecular machinery of autophagy suggests that its regulation can be extremely complicated and may involve multiple signaling inputs. Indeed, many of the major signaling pathways have been shown to regulate autophagy under various conditions. Of note, these signaling pathways may cross talk and regulate at different levels in the autophagic cascade, including induction and expansion of the isolation membrane, enclosure of the isolation membrane to form autophagosome, and fusion with lysosome.
TOR kinase.
TOR kinase is a principal regulator of autophagy that shuts off autophagy in cells growing in the presence of nutrients including amino acids and growth factors. Being autophagy inhibitory, TOR activity prevents the formation of Atg complexes including the Atg1-Atg13-Atg17 serine/threonine protein kinase complex and the Vps34-Atg6-Vps15 lipid kinase complex (51, 82, 124). It also interferes with the two ubiquitin-like conjugation systems of autophagy. As a result, induction and expansion of the isolation membrane is abrogated. Conversely, TOR is inactivated when a cell is nutrient deprived, unleashing the autophagic cascade. Therefore, inhibition of TOR has been suggested to be obligatory for the induction of autophagy (2). As a result, TOR may provide a central platform for autophagic signaling pathways and their integration (Fig. 4), although it remains to be determined whether TOR inhibition is a universal mechanism for autophagy under other types of cellular stress.
Fig. 4.
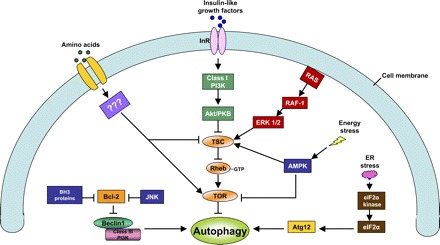
Major regulatory mechanisms of autophagy. The major signaling pathways of autophagy regulation under conditions of nutrient (amino acids, growth factors) deprivation, endoplasmic reticulum (ER) stress, energetic stress or depletion (see detailed discussion in text) are depicted. The pathways may cross talk and regulate autophagy at different levels, including induction and expansion of the isolation membrane, enclosure of the isolation membrane to form autophagosome, and fusion with lysosome. PI3K, PI3-kinase; AMPK, AMP-dependent protein kinase; TSC, tuberous sclerosis complex; eIF-2α, eukaryotic initiation factor-2α; InR, insulin receptor; RAS, rat sarcoma (a protein encoded by rat sarcoma virus oncogene).
Class I PI3-kinase/Akt.
Class I PI3-kinase/Akt regulates autophagy mainly through the modulation of TOR activity. Briefly, PI3-kinase phosphorylates phosphatidylinositol (4,5)P2 to produce phosphatidylinositol (3,4,5)P3, which activates phosphoinositide-dependent kinase-1 (PDK-1). PDK-1 can phosphorylate and activate Akt. Akt, also called protein kinase B, can further phosphorylate and inactivate tuberous sclerosis complex (TSC). TSC is a GTPase-activating protein complex, and one of its downstream targets is Rheb. When TSC is inactivated by Akt, the GTPase activity of Rheb is blocked, leading to a GTP-bound form of Rheb, which is the major activator of TOR (42). Therefore, by activating TOR, class I PI3-kinase/Akt suppresses autophagy. Importantly, class I PI3-kinase /Akt is the critical link between growth factor signaling and TOR. Growth factors such as insulin-like growth factor bind to their tyrosine kinase receptors, leading to the activation of PI3-kinase/Akt and TOR, resulting in the inhibition of autophagy (Fig. 4).
Amino acid signaling.
It has been recognized for decades that amino acids, the major end products of autophagic degradation, are also crucial regulators or inhibitors of autophagy (89). This provides a feedback mechanism to control the level of autophagy to maintain cellular homeostasis. Of note, not all but only a few amino acids are involved in autophagy regulation (76, 88, 119). Alanine, leucine, glutamine, and phenylalanine seem particularly effective (12, 87, 119). These amino acids have their own recognition sites at the cell surface and are believed to initiate signal transduction to suppress autophagy (46). A major mechanism for amino acid-mediated inhibition of autophagy is TOR activation. In 1995, Blommaart et al. (15) showed that in isolated rat hepatocytes the inhibitory effects of amino acids on autophagy are prevented by rapamycin, the TOR inhibitor. A role for TOR in amino acid-mediated autophagy inhibition was also demonstrated later in yeast by Noda and Ohsumi (91). The mechanism whereby amino acids activate TOR remains unclear, but it seems to be independent of class I PI3-kinase/Akt.
Bcl-2 family proteins.
The interaction between Bcl-2 and beclin-1 suggested that autophagy could be subjected to regulation by Bcl-2 family proteins (67). In 2005, Pattingre et al. (103) further demonstrated the inhibitory effects of Bcl-2 on autophagy in both yeast and mammalian cells. The inhibitory effects of Bcl-2 were shown to depend on its physical interaction with beclin-1 (103). The antiautophagy activities have now been shown not only for Bcl-2 but also for its homologs including Bcl-XL, Bcl-w, and Mcl-1 (32, 70, 104). Binding and sequestration of beclin-1 by these proteins is expected to disturb the formation of the class III PI3-kinase (Vps34-Vps15-beclin-1) complex that is critical to the induction of autophagy, and as a result autophagy is inhibited. Conversely, BH3-only proteins can bind Bcl-2/Bcl-XL to allow beclin-1 to induce autophagy (70, 151). It is noteworthy that, in addition to beclin-1, Bcl-2 family proteins may also regulate autophagy by other mechanisms including Ca2+ homeostasis (40).
AMP-dependent protein kinase.
AMP-dependent protein kinase (AMPK) is sensitive to the cytosolic AMP-to-ATP ratio and responds to metabolic stresses affecting this ratio, such as glucose deprivation, hypoxia, ischemia, heat shock, or oxidative stress (47). Not surprisingly, AMPK is involved in autophagy induction under these conditions. In yeast, autophagy induction during metabolic stress requires AMPK (138). However, the involvement of AMPK in mammalian autophagy is controversial. Nevertheless, recent studies by Meijer and Codogno (77) and Meley et al. (78) have further clarified the role of AMPK in autophagy in metabolically stressed mammalian cells. A major mechanism for AMPK to induce autophagy is suppression of TOR signaling. AMPK can directly inhibit TOR via phosphorylation. In addition, AMPK can activate TSC, leading to the inhibition of TOR.
Eukaryotic initiation factor-2α.
In response to cellular stress, eukaryotic initiation factor-2α (eIF-2α) is phosphorylated and regulated by a family of evolutionarily conserved serine/threonine kinases. In 2002, Talloczy et al. (127) showed that eIF-2α phosphorylation by GCN2 (the yeast eIF-2α kinase) is essential for starvation-induced autophagy in yeast. A role for eIF-2α phosphorylation was also shown to be important to autophagy during viral infection in murine embryonic fibroblasts. More recently, eIF-2α has been implicated in autophagy under ER stress. For example, Kouroku et al. (57) demonstrated that ER stress-associated autophagy can be prevented by eIF-2α mutations that block its phosphorylation. ER stress-associated autophagy is also attenuated by dominant-negative inhibition of protein kinase regulated by RNA (PKR)-like ER kinase (PERK), the protein kinase responsible for eIF-2α phosphorylation (57). It was suggested that eIF-2α phosphorylation/activation may lead to selective translation of specific transcription factors, resulting in the expression of Atg12 and formation of the Atg5-Atg12-Atg16 complex that is critical to autophagy induction (57).
Mitogen-activated protein kinases.
Mitogen-activated protein kinases (MAPKs), including ERK1/2, p38, and JNK, are a family of serine/threonine protein kinases involved in a wide range of cellular responses. In various models of autophagy, MAPK activation has been documented (26, 115). During nutrient starvation both ERK1 and ERK2 are activated, and inhibition of ERK1/2 abrogates starvation-induced autophagy (105). Consistently, amino acids have been shown to suppress autophagy by inhibiting ERK1/2 in human colon cancer cells (102). A role for JNK in autophagy induction has also been suggested. For example, the recent work by Wei et al. (138a) demonstrated a critical role for JNK-mediated phosphorylation of Bcl-2 in autophagy during cellular starvation. They showed that JNK1, but not JNK2, is activated during starvation stress and phosphorylates Bcl-2 at multiple sites, leading to its dissociation from beclin-1 and autophagy induction. Similar observations were shown by Pattingre et al. (101) in ceramide-induced autophagy. JNK signaling may also directly induce beclin-1 transcription in response to ceramide treatment (64). For autophagy regulation by p38, vom Dahl and colleagues (136) conducted a series of studies showing that p38 plays a key role in cell swelling-induced autophagy. It is important to recognize that the role and regulation of individual MAPKs in autophagy are very complicated and can vary from one experimental model to another.
It is important to note that the current knowledge of autophagy regulation is mainly derived from research characterizing the autophagic response to nutrient deprivation or starvation. Whether the regulatory mechanisms are central to autophagy under other stress conditions remains to be determined. It is likely that specific stimuli may trigger unique signaling pathways that are then integrated with the core regulatory mechanisms to induce autophagy.
Autophagy in Cell Survival
A role for autophagy in cell survival was initially recognized in experimental models of nutrient depletion or starvation. This is not surprising, as autophagy breaks down the engulfed cytoplasmic constituents including proteins and organelles, resulting in the production and recycling of nutrients to sustain the intermediary metabolisms and biosynthetic pathways that are essential to cell viability (117). This is clearly shown by autophagic degradation of proteins, resulting in the generation of amino acids. Under normal circumstances, cells maintain amino acid levels by new synthesis or proteasomal degradation of short-lived proteins rather than autophagy. However, during an extended period of starvation, de novo synthesis and proteasomal degradation does not occur because of the lack of substrates and energy and the vital amino acids are produced by autophagy (79). In fact, immediately after birth neonates use autophagy as an important source of oxidizable substrates during sudden interruptions of maternal nutrient supply. Kuma et al. (60) demonstrated that autophagy is upregulated during early neonatal periods in the liver, heart, lung, diaphragm, pancreas, and muscle. The autophagic degradation of proteins in the liver produces amino acids, which are converted to glucose and utilized by brain and erythrocytes as a metabolic energy source. It is also understood that autophagy generates amino acids to supply the tricarboxylic cycle as well as substrates for new protein synthesis that are necessary for cell survival during starvation (69, 79). In autophagy-deficient cells, insufficient generation of starvation-induced proteins such as argininosuccinate synthetase, heat shock protein, and Ape1 were documented, leading to cell death in response to starvation (132). Consistently, during the neonatal period Atg5 knockout mice are depleted of amino acids and show declined cardiac ATP production, which is accompanied by myocardial injury and animal death (60). Autophagy is also important to the maintenance of cellular energy homeostasis and bioenergetics during growth factor deprivation. For example, in BAX-BAK knockout mice, autophagy can maintain cell survival during IL-3 deprivation for a remarkable period of several weeks (69). Conversely, inhibition of autophagy by silencing autophagy genes or pharmacological inhibitors ultimately leads to cell death, further confirming a critical role for autophagy in cell viability (16).
In addition to the well-documented role of autophagy in cell survival during nutrient and growth factor deprivation, autophagy has been shown to be protective against cell death during stress by many recent studies. Ferraro and Cecconi (34) showed that autophagy can postpone the beginning of the apoptotic program or slow down apoptosis, and, when it fails to do so, apoptosis takes over the fate of the cell. In this and other related studies, pharmacological inhibitors of autophagy such as 3-methyladenine, bafilomycin, monesin, and chloroquine increase apoptosis in various cell types. Of note, while 3-methyladenine blocks the formation of autophagosomes, bafilomycin, monesin, and chloroquine inhibit the fusion of autophagosome with lysosome or lysosomal degradative activity (19, 141). As a result, treatment with bafilomycin, monesin, or chloroquine increases autophagic vacuoles, which can be shown by LC3-GFP punctate accumulation in the cytoplasm and also by increased LC3-II signal in immunoblot analysis. The death-promoting effects of pharmacological inhibitors of autophagy suggest that autophagy is cytoprotective in a variety of cell injury models. Moreover, the protective role of autophagy has been supported by studies using Atg-deficient cells or animals and also by studies using small interfering RNA (siRNA) to knock down critical Atg genes. With these approaches, inhibition of autophagy accelerates starvation-induced apoptosis as well as mitochondrial membrane potential loss and release of cytochrome c (16). In camptothecin-treated cells, autophagy interrupts apoptosis and its inhibition triggers apoptosis by mitochondrial depolarization and caspase 9 activation (1). More recently, Apel et al. (7) showed that irradiation induces autophagy and autophagy-related genes in therapy-resistant cancer cells. Importantly, siRNA knockdown of the induced Atgs can sensitize the cells to radiotherapy. Autophagy is also induced in normal kidney cells during nephrotoxicity (100, 104, 143). As discussed below, blockade of autophagy can exacerbate cell death in these experimental models, further supporting a survival role for autophagy in malignant as well as normal cells.
In vivo in mice, loss of Atg7 in the liver results in obvious alterations in a series of stress-related proteins, followed by the occurrence of ER and oxidative stresses and tissue injury (73). Consistently, Kim et al. (52) showed that hepatocytes lose both Atg7 and Atg6/beclin-1 by calpain-2 activation during anoxia-reoxygenation or liver ischemia-reperfusion, leading to inhibition of autophagy. This autophagy blockade is associated with the onset of mitochondrial permeability transition and ultimately cell death by necrosis and apoptosis (52). In the heart, ischemia induces autophagy through an AMPK-dependent mechanism, whereas subsequent reperfusion stimulates autophagy through an Atg6/beclin-1-dependent, AMPK-independent mechanism. Interestingly, it was concluded that autophagy may be protective during ischemia; however, it may be detrimental during reperfusion (72).
Together, these in vitro and in vivo studies indicate that autophagy may well be a cellular response to stress that, depending on the experimental conditions, can be an important protective mechanism for cell survival. Obviously, autophagy can maintain the viability of starved cells by temporally digesting portions of cytoplasm to supply vital substrates and energy. However, it is much less clear how autophagy protects against cell death under other stress conditions. In this regard, it has been suggested that autophagy may protect by eliminating damaged and potentially dangerous organelles, such as leaky mitochondria (37). Nevertheless, this possibility remains to be carefully tested and evaluated, and may not be the sole or even major mechanism for cell survival via autophagy.
Autophagic Cell Death
While a cytoprotective role for autophagy has been demonstrated in a variety of experimental models, autophagy has also been suggested to induce or participate in cell death under certain circumstances. As a matter of fact, cell death resulting from autophagy has been classified as type II cell death, in parallel to apoptotic (type I) and necrotic (type III) cell death (18, 36). Autophagic cell death was originally suggested by observations from apoptosis-defective or -blocked cells. A notable example was shown by Shimizu et al. (120) using Bax and Bak double-knockout (Bax−/− Bak−/−) mouse embryonic fibroblasts. In these cells, apoptosis is blocked because of the loss of Bax and Bak, two proapoptotic Bcl-2 family proteins that are essential to mitochondrial outer membrane permeabilization, and consequent release of apoptogenic factors including cytochrome c. When these cells are injured by classical apoptotic insults, they do not die by apoptosis like wild-type cells. However, in response to apoptotic insults, the Bax−/− Bak−/− cells do not show long-term viability or clonogenic ability, either. Instead, they undergo massive autophagy followed by ultimate cell death. Remarkably, autophagy seems to contribute to the delayed nonapoptotic cell death, because the latter is abrogated when autophagy is blocked (120). Consistently, Yu et al. (150) showed that when the progression of apoptosis is blocked by peptide caspase inhibitors, cells die with autophagic features. Importantly, the cell death can be prevented by autophagy inhibitors and siRNA knockdown of autophagy genes including Atg6/beclin-1. More recently, lipopolysaccharide-treated U937 monocytoid cells were shown to undergo autophagic cell death that was Atg6/beclin-1 dependent (139). Neuronal cells are predominantly vulnerable to developing autophagic stress in aging, because of an increased requirement for autophagic clearance of damaged constituents, and decreased degradative and biosynthetic reserves (135). In pancreatic cancer cells, Ozpolat et al. (99) found that protein kinase C (PKC)-δ constitutively suppresses autophagy through the induction of tissue transglutaminase (TG)2 and inhibition of PKC-δ/TG2 signaling leads to significant autophagy and Atg6/beclin-1-dependent cell death. In the case of cancer therapy, Aoki et al. (6) suggested that curcumin can restrain the growth of malignant gliomas in vitro and in vivo through the induction of autophagy and autophagic cell death.
It is unclear how autophagy triggers cell death. However, several possible mechanisms have been proposed. The first possibility can be called the autophagic stress hypothesis, which emphasizes that excessive autophagy provokes an imbalance between autophagic stimulation and the cellular capability to complete autophagic degradation and restore essential cellular components. This scenario culminates in a specific form of stress, i.e., autophagic stress, followed by cell death (24, 25). The second mechanism may be called the “wrong eating” hypothesis, which indicates that autophagy may mistakenly digest vital organelles or proteins that are essential to cell viability. Finally, the third mechanism is the “atrophy” hypothesis, which emphasizes that excessive autophagy may deplete the cells of too much cytoplasm, resulting in atrophic cell death. In this case, cells die as a result of starvation and self-digestion, and whether autophagy per se induces cell death can be questioned.
It remains controversial as to whether autophagic cell death is pathophysiologically relevant or just an observation from artificial experimental models (59). This question was raised when autophagic cell death was first revealed in studies using apoptosis-defective models. Currently, the most hotly debated issue regarding autophagic cell death is probably in the area of tumorigenesis. There is a consensus that knockout of key autophagic genes (and therefore suppression of autophagy) is associated with tumor development, suggesting that autophagy is a bona fide mechanism of tumor suppression. For example, Atg6/beclin-1 knockout mice have a clearly increased frequency in spontaneous lymphoma and mammary neoplasia (113). The tumor-suppressing effects of autophagy seem readily explainable by the activation of autophagic cell death: when autophagy is defective, cancer cell growth is unchecked by autophagic cell death, resulting in tumorigenesis. However, this straightforward explanation has been seriously challenged recently (71). Instead, the emerging idea is that autophagy is cytoprotective in tumors. By limiting tumor cell necrosis and inflammation, autophagy contributes to the maintenance of genomic stability (71). In addition, autophagy may directly inhibit cell growth in tumors by degrading organelles or proteins that are essential to cell cycle regulation such as cyclin E and Rb (56, 66).
Autophagic Survival and Autophagic Cell Death: Where Is the Connection?
It is clear from studies during the last few years that autophagy is not just a response to starvation but a more general response to cell stress. Moreover, it has been recognized that autophagy occurs early during cell stress and, when the stress persists, cells die (Fig. 5). As discussed above, autophagy may be a mechanism for cell survival but may also induce cell death. Autophagy and its signaling seem to be integrated with that of cell death. Clearly, many signaling pathways activated by cell stress also play roles in the regulation of both autophagy and cell death. These include TOR, PI3-kinase, Akt, and MAPK, to name a few (see Regulation of Autophagy above). Even at the level of core machineries, autophagy and cell death share key molecules, which may determine the fate of the cells, to live or to die.
Fig. 5.
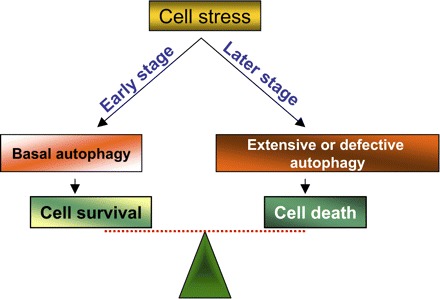
Roles of autophagy in cell death and survival. At the early stage of cell stress, autophagy is induced and is cytoprotective. When cell stress is too severe at late stage, excessive autophagy may trigger cell injury and death.
Atg6/beclin-1.
Atg6 is a key autophagy protein involved in the formation of PI3-kinase complex that controls the nucleation of autophagic vesicles. Interestingly, beclin-1, the mammalian ortholog of Atg6, was originally identified by yeast two-hybrid screening as a protein that interacts with Bcl-2 (67), a well-recognized apoptosis regulator that antagonizes apoptosis by interacting and neutralizing proapoptotic molecules (148). Later studies have pinpointed the molecular interaction via a Bcl-2 homology 3 (BH3) domain in beclin-1 (70, 93). The beclin-1/Bcl-2 interaction suggests that these two molecules may provide a cross talk channel between autophagy and apoptosis. Indeed, by sequestering Bcl-2, beclin-1 has been suggested to induce apoptosis or sensitize cells to apoptosis (93). On the other hand, Bcl-2 and its homolog Bcl-XL can suppress autophagy by sequestering beclin-1 (63). It is noteworthy that Bcl-2 and Bcl-XL may also have additional mechanisms to antagonize autophagy. For example, Bcl-2 expressed in the ER can attenuate autophagy by blocking Ca2+ rise in the ER and agonist-induced Ca2+ fluxes (40). Thus the beclin-1-Bcl-2/Bcl-XL axis may be one of the key rheostats that govern the cellular response to stress: autophagy or apoptosis.
Atg5.
Atg5 is another protein at the crossroad of autophagy and apoptosis. During autophagy, Atg5 forms a protein conjugation complex with Atg12 and Atg16, which is essential for elongation of the isolation membrane. However, Yousefi et al. (149) showed that Atg5 is proteolytically cleaved by calpains, releasing a 24-kDa fragment. Notably, the truncated Atg5 then translocates into mitochondria and binds Bcl-XL, leading to the release of cytochrome c, caspase activation, and apoptosis (149). In addition, Atg5 may trigger cell death by physically interacting with Fas Associated via Death Domain (FADD) by its COOH terminus (112). These observations, although requiring confirmation by studies in other model systems, suggest that Atg5 can be a critical switch between autophagy and cell death.
p53.
The tumor suppressor p53 has a well-recognized role in apoptosis induction. It may induce apoptosis indirectly via transcription of proapoptotic genes or directly by activating mitochondrial permeabilization molecules including Bax (43, 131). Interestingly, recent studies have also suggested a role for p53 in autophagy regulation. However, it remains controversial as to whether p53 signaling stimulates or inhibits autophagy. In 2005, Feng et al. (33) showed that transient activation of p53 by cellular stress can attenuate TOR signaling, leading to autophagy. Crighton et al. (27) further identified damage-regulated autophagy modulator (DRAM) as a transcriptional target of p53 that, upon induction, activates autophagy. In line with these observations, the involvement of p53 in autophagy has been confirmed in other experimental models (4, 22, 43, 104, 129). In contrast, Tasdemir et al. (130) showed recently that autophagy is dramatically induced when p53 is deleted or inhibited. In this study, p53 was shown to be degraded during autophagy and maintenance of p53 levels prevented autophagy. Morselli et al. (86) further suggested that cytosolic, and not nuclear, p53 inhibits autophagy. While these findings require further studies to confirm, they suggest that physiological levels of p53 may actually repress autophagy. Thus, depending on the experimental conditions, p53 may have both pro- and antiautophagy effects. Despite the recognition of the roles played by p53 in both autophagy and apoptosis, little is known about the mechanism that governs the dual roles and the potential switch between them.
Regulation of Autophagy in Renal Pathophysiology
The research on autophagy in renal physiology has been scarce but has spun over three decades. In 1975, Pfeifer and Scheller (109) suggested an important role for cellular autophagy in the turnover of cytoplasmic constituents in rat kidneys. Interestingly, autophagy was shown to occur in a diurnal fashion, minimum during the night and maximum during the day. After that, Pfeifer along with Guder (108) studied the stimulation of autophagy by parathyroid hormone and cAMP in isolated tubular fragments from the rat kidney cortex. These studies showed that the increase in the number of autophagic vacuoles is proportional to the production of ammonia and glucose, suggesting a role for autophagy in gluconeogenesis in kidneys. Further studies by Pfeifer and Warmuth-Metz (110) demonstrated the inhibitory effects of insulin on autophagy in renal proximal tubular cells. Inhibition of autophagy was also demonstrated in renal proximal tubules during uninephrectomy and diabetes, providing an anticatabolic mechanism for renal hypertrophy under these experimental conditions (11, 44). In 1983, Berkenstam et al. (13) isolated autophagic vacuoles from kidney cortex, demonstrating enriched lysosomal enzymes and high proteolytic activity of the organelles. In addition to proximal tubules, autophagy has been implicated in the physiology of other segments or cells of the kidneys. In the thick ascending limb, functional unloading resulted in an increased sequestration of cytoplasmic components into autophagic vacuoles within a short time interval (10). Autophagy has also been identified in podocytes during cellular differentiation and recovery from damage caused by experimental puromycin aminonucleoside-induced nephrosis (8). Interestingly, electron microscopy revealed two types of autophagy in podocytes in renal biopsy specimens. While type I autophagy fails to transform into autophagosomes, type II forms complete autophagic vacuoles and apparently plays a significant role in the clearance of proteins and lipids (116).
The role of autophagy in renal pathology was not recognized until very recently. In 2007, Chien et al. (23) demonstrated the induction of autophagic genes (beclin-1 and LC3) during renal ischemia-reperfusion in rats, suggesting the first evidence of autophagy in this pathological condition. Although the role of autophagy was not determined, beclin-1 and LC3 expression was detected along with apoptosis in injured renal tubules. Interestingly, both autophagy and apoptosis were suppressed by the expression of Bcl-XL and Bcl-2 (23). In 2008, Suzuki et al. (123) further demonstrated the formation of autophagosome-like structures in renal tubular cells during ischemia-reperfusion in mice and, notably, in transplanted kidneys in humans. In cultured renal tubular HK2 cells, autophagy inhibitors could suppress H2O2-induced cell death, suggesting that autophagy might contribute to cell death under the condition (123). Very recently, Gozuacik et al. (38) showed autophagy in renal tubular cells following tunicamycin-induced ER stress in mice. In vitro results from cell cultures further suggested that autophagy may serve as a second cell-killing mechanism that acts in concert with apoptosis during ER stress (38).
In contrast, a protective role of autophagy has been suggested in other settings of renal pathology. In a mouse model of cisplatin-induced nephrotoxicity, our recent work (104) demonstrated a time-dependent increase of autophagic vacuoles by electron microscopy in renal tubular cells, accompanied by LC3-II accumulation. In cultured renal proximal tubular cells, cisplatin induced autophagy within hours, before apoptosis. Importantly, inhibition of autophagy by pharmacological inhibitors and beclin-1 knockdown increased apoptosis during cisplatin treatment, suggesting a protective role for autophagy in cisplatin-induced renal injury and nephrotoxicity (104). Similarly, Yang et al. (143) demonstrated cisplatin-induced autophagy in cultured renal tubular cells and suggested a cytoprotective or prosurvival role for autophagy in this experimental model. Autophagy was also shown in renal tubular cells during cyclosporine A-induced nephrotoxicity. Notably, under this condition autophagy was induced by ER stress and apparently acted as a protective mechanism against cell death (100).
Concluding Remarks
Autophagy is a conserved catabolic pathway that occurs in all eukaryotic cells. By engulfing and degrading portions of cytoplasm, autophagy reprocesses substances for biosynthetic or metabolic needs. Remarkable progress has been made recently in the understanding of the core machinery of autophagy and its regulation. Importantly, it is now recognized that autophagy is a general cellular response to stress, not limited to nutrient deprivation or starvation. Depending on the experimental conditions, autophagy may protect against cell death; however, excessive autophagy may also lead to cell injury and death. It remains largely unknown how autophagy protects or kills a cell. Despite recent intense research on autophagy in other organ systems, very limited is known about the role and regulation of autophagy in renal pathophysiology. Nevertheless, emerging evidence has demonstrated autophagy in renal disease conditions including renal ischemia-reperfusion and nephrotoxicity. The role of autophagy in the development of renal pathology has been suggested, but whether it is protective or detrimental remains unclear and may depend on the experimental condition. Further studies should determine the role of autophagy in renal diseases in in vivo animal models. In addition, the mechanisms of autophagy induction and regulation in renal pathophysiology should be investigated. A thorough mechanistic understanding of autophagy may provide novel strategies for the prevention and treatment of related renal diseases.
REFERENCES
- 1.AbedinMJAbedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 14: 500–510, 2007. [DOI] [PubMed] [Google Scholar]
- 2.AbeliovichHAbeliovich H, Dunn WA Jr, Kim J, Klionsky DJ. Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 151: 1025–1034, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AbeliovichHAbeliovich H, Zhang C, Dunn WA Jr, Shokat KM, Klionsky DJ. Chemical genetic analysis of Apg1 reveals a non-kinase role in the induction of autophagy. Mol Biol Cell 14: 477–490, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.AbidaWMAbida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res 68: 352–357, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AnoYAno Y, Hattori T, Oku M, Mukaiyama H, Baba M, Ohsumi Y, Kato N, Sakai Y. A sorting nexin PpAtg24 regulates vacuolar membrane dynamics during pexophagy via binding to phosphatidylinositol-3-phosphate. Mol Biol Cell 16: 446–457, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.AokiHAoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol 72: 29–39, 2007. [DOI] [PubMed] [Google Scholar]
- 7.ApelAApel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 68: 1485–1494, 2008. [DOI] [PubMed] [Google Scholar]
- 8.AsanumaKAsanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, Kominami E, Tomino Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 17: 1165–1167, 2003. [DOI] [PubMed] [Google Scholar]
- 9.BaehreckeEHBaehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6: 505–510, 2005. [DOI] [PubMed] [Google Scholar]
- 10.BahroMBahro M, Gertig G, Pfeifer U. Short-term stimulation of cellular autophagy by furosemide in the thick ascending limb of Henle's loop in the rat kidney. Cell Tissue Res 253: 625–629, 1988. [DOI] [PubMed] [Google Scholar]
- 11.Barbosa Junior AdeABarbosa Junior Ade A, Zhou H, Hultenschmidt D, Totovic V, Jurilj N, Pfeifer U. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch B Cell Pathol Incl Mol Pathol 61: 359–366, 1992. [DOI] [PubMed] [Google Scholar]
- 12.BergaminiEBergamini E, Bombara M, Del Roso A, Gori Z, Masiello P, Masini M, Pollera M, Vittorini S. The regulation of liver protein degradation by aminoacids in vivo. Effects of glutamine and leucine. Arch Physiol Biochem 103: 512–515, 1995. [DOI] [PubMed] [Google Scholar]
- 13.BerkenstamABerkenstam A, Ahlberg J, Glaumann H. Isolation and characterization of autophagic vacuoles from rat kidney cortex. Virchows Arch B Cell Pathol Incl Mol Pathol 44: 275–286, 1983. [DOI] [PubMed] [Google Scholar]
- 14.BlommaartEFBlommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243: 240–246, 1997. [DOI] [PubMed] [Google Scholar]
- 15.BlommaartEFBlommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 270: 2320–2326, 1995. [DOI] [PubMed] [Google Scholar]
- 16.BoyaPBoya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25: 1025–1040, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.BurschWBursch W, Ellinger A. Autophagy—a basic mechanism and a potential role for neurodegeneration. Folia Neuropathol 43: 297–310, 2005. [PubMed] [Google Scholar]
- 18.BurschWBursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann NY Acad Sci 926: 1–12, 2000. [DOI] [PubMed] [Google Scholar]
- 19.ButlerDButler D, Brown QB, Chin DJ, Batey L, Karim S, Mutneja MS, Karanian DA, Bahr BA. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res 8: 227–237, 2005. [DOI] [PubMed] [Google Scholar]
- 20.CaoYCao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 17: 839–849, 2007. [DOI] [PubMed] [Google Scholar]
- 21.ChangCYChang CY, Huang WP. Atg19 mediates a dual interaction cargo sorting mechanism in selective autophagy. Mol Biol Cell 18: 919–929, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.ChengYCheng Y, Qiu F, Tashiro S, Onodera S, Ikejima T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem Biophys Res Commun 376: 483–488, 2008. [DOI] [PubMed] [Google Scholar]
- 23.ChienCTChien CT, Shyue SK, Lai MK. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 84: 1183–1190, 2007. [DOI] [PubMed] [Google Scholar]
- 24.ChuCTChu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 65: 423–432, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ChuCTChu CT. Eaten alive: autophagy and neuronal cell death after hypoxia-ischemia. Am J Pathol 172: 284–287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.CorcelleECorcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fenichel P, Hofman P, Poujeol P, Mograbi B. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 3: 57–59, 2007. [DOI] [PubMed] [Google Scholar]
- 27.CrightonDCrighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126: 121–134, 2006. [DOI] [PubMed] [Google Scholar]
- 28.DarsowTDarsow T, Rieder SE, Emr SD. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J Cell Biol 138: 517–529, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De DuveCDe Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 28: 435–492, 1966. [DOI] [PubMed] [Google Scholar]
- 30.DebnathJDebnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy 1: 66–74, 2005. [DOI] [PubMed] [Google Scholar]
- 31.EppleUDEpple UD, Suriapranata I, Eskelinen EL, Thumm M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol 183: 5942–5955, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ErlichSErlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3: 561–568, 2007. [DOI] [PubMed] [Google Scholar]
- 33.FengZFeng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102: 8204–8209, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.FerraroEFerraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys 462: 210–219, 2007. [DOI] [PubMed] [Google Scholar]
- 35.FimiaGMFimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125, 2007. [DOI] [PubMed] [Google Scholar]
- 36.GalluzziLGalluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, Kroemer G. Life, death and burial: multifaceted impact of autophagy. Biochem Soc Trans 36: 786–790, 2008. [DOI] [PubMed] [Google Scholar]
- 37.GalluzziLGalluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med 8: 78–91, 2008. [DOI] [PubMed] [Google Scholar]
- 38.GozuacikDGozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 15: 1875–1886, 2008. [DOI] [PubMed] [Google Scholar]
- 39.HanadaTHanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282: 37298–37302, 2007. [DOI] [PubMed] [Google Scholar]
- 40.Hoyer-HansenMHoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 25: 193–205, 2007. [DOI] [PubMed] [Google Scholar]
- 41.IchimuraYIchimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature 408: 488–492, 2000. [DOI] [PubMed] [Google Scholar]
- 42.InokiKInoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 37: 19–24, 2005. [DOI] [PubMed] [Google Scholar]
- 43.JiangMJiang M, Dong Z. Regulation and pathological role of p53 in cisplatin nephrotoxicity. J Pharmacol Exp Ther 327: 300–307, 2008. [DOI] [PubMed] [Google Scholar]
- 44.JuriljNJurilj N, Pfeifer U. Inhibition of cellular autophagy in kidney tubular cells stimulated to grow by unilateral nephrectomy. Virchows Arch B Cell Pathol Incl Mol Pathol 59: 32–37, 1990. [DOI] [PubMed] [Google Scholar]
- 45.KabeyaYKabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16: 2544–2553, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.KadowakiMKadowaki M, Karim MR, Carpi A, Miotto G. Nutrient control of macroautophagy in mammalian cells. Mol Aspects Med 27: 426–443, 2006. [DOI] [PubMed] [Google Scholar]
- 47.KahnBBKahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005. [DOI] [PubMed] [Google Scholar]
- 48.KamadaYKamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150: 1507–1513, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.KawamataTKawamata T, Kamada Y, Suzuki K, Kuboshima N, Akimatsu H, Ota S, Ohsumi M, Ohsumi Y. Characterization of a novel autophagy-specific gene, ATG29. Biochem Biophys Res Commun 338: 1884–1889, 2005. [DOI] [PubMed] [Google Scholar]
- 50.KiharaAKihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae J Cell Biol 152: 519–530, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.KimJKim J, Huang WP, Stromhaug PE, Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J Biol Chem 277: 763–773, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.KimJSKim JS, Nitta T, Mohuczy D, O'Malley KA, Moldawer LL, Dunn WA Jr, Behrns KE. Impaired autophagy: a mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology 47: 1725–1736, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.KirisakoTKirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 147: 435–446, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.KlionskyDJKlionsky DJ, Cregg JM, Dunn WA Jr, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell 5: 539–545, 2003. [DOI] [PubMed] [Google Scholar]
- 55.KlionskyDJKlionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 290: 1717–1721, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.KoneriKKoneri K, Goi T, Hirono Y, Katayama K, Yamaguchi A. Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res 27: 1453–1457, 2007. [PubMed] [Google Scholar]
- 57.KourokuYKouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14: 230–239, 2007. [DOI] [PubMed] [Google Scholar]
- 58.KrickRKrick R, Tolstrup J, Appelles A, Henke S, Thumm M. The relevance of the phosphatidylinositolphosphate-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS Lett 580: 4632–4638, 2006. [DOI] [PubMed] [Google Scholar]
- 59.KroemerGKroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9: 1004–1010, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.KumaAKuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. [DOI] [PubMed] [Google Scholar]
- 61.KumaAKuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277: 18619–18625, 2002. [DOI] [PubMed] [Google Scholar]
- 62.LangTLang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J 17: 3597–3607, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.LevineBLevine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4: 600–606, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LiDDLi DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, Huang WL, Zeng YX, Zhu XF. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 28: 886–898, 2008. [DOI] [PubMed] [Google Scholar]
- 66.LiangXHLiang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676, 1999. [DOI] [PubMed] [Google Scholar]
- 67.LiangXHLiang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72: 8586–8596, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.LockshinRALockshin RA, Zakeri Z. Apoptosis, autophagy, more. Int J Biochem Cell Biol 36: 2405–2419, 2004. [DOI] [PubMed] [Google Scholar]
- 69.LumJJLum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248, 2005. [DOI] [PubMed] [Google Scholar]
- 70.MaiuriMCMaiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J 26: 2527–2539, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MathewRMathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 21: 1367–1381, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.MatsuiYMatsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100: 914–922, 2007. [DOI] [PubMed] [Google Scholar]
- 73.MatsumotoNMatsumoto N, Ezaki J, Komatsu M, Takahashi K, Mineki R, Taka H, Kikkawa M, Fujimura T, Takeda-Ezaki M, Ueno T, Tanaka K, Kominami E. Comprehensive proteomics analysis of autophagy-deficient mouse liver. Biochem Biophys Res Commun 368: 643–649, 2008. [DOI] [PubMed] [Google Scholar]
- 74.MatsushitaMMatsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 282: 6763–6772, 2007. [DOI] [PubMed] [Google Scholar]
- 75.MatsuuraAMatsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae Gene 192: 245–250, 1997. [DOI] [PubMed] [Google Scholar]
- 76.MayMEMay ME, Buse MG. Effects of branched-chain amino acids on protein turnover. Diabetes Metab Rev 5: 227–245, 1989. [DOI] [PubMed] [Google Scholar]
- 77.MeijerAJMeijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy 3: 238–240, 2007. [DOI] [PubMed] [Google Scholar]
- 78.MeleyDMeley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem 281: 34870–34879, 2006. [DOI] [PubMed] [Google Scholar]
- 79.MizushimaNMizushima N. Autophagy: process and function. Genes Dev 21: 2861–2873, 2007. [DOI] [PubMed] [Google Scholar]
- 80.MizushimaNMizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ, 12 Suppl 2: 1535–1541, 2005. [DOI] [PubMed] [Google Scholar]
- 81.MizushimaNMizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.MizushimaNMizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 152: 657–668, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MizushimaNMizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 3: 542–545, 2007. [DOI] [PubMed] [Google Scholar]
- 84.MizushimaNMizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 35: 553–561, 2003. [DOI] [PubMed] [Google Scholar]
- 85.MonastyrskaIMonastyrska I, Shintani T, Klionsky DJ, Reggiori F. Atg11 directs autophagosome cargoes to the PAS along actin cables. Autophagy 2: 119–121, 2006. [DOI] [PubMed] [Google Scholar]
- 86.MorselliEMorselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, Vicencio JM, Soussi T, Kroemer G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 7: 3056–3061, 2008. [DOI] [PubMed] [Google Scholar]
- 87.MortimoreGEMortimore GE, Khurana KK, Miotto G. Amino acid control of proteolysis in perfused livers of synchronously fed rats. Mechanism and specificity of alanine co-regulation. J Biol Chem 266: 1021–1028, 1991. [PubMed] [Google Scholar]
- 88.MortimoreGEMortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr 7: 539–564, 1987. [DOI] [PubMed] [Google Scholar]
- 89.MortimoreGEMortimore GE, Schworer CM. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 270: 174–176, 1977. [DOI] [PubMed] [Google Scholar]
- 90.NakaiANakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624, 2007. [DOI] [PubMed] [Google Scholar]
- 91.NodaTNoda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966, 1998. [DOI] [PubMed] [Google Scholar]
- 92.ObaraKObara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes—Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae Mol Biol Cell 17: 1527–1539, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.ObersteinAOberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem 282: 13123–13132, 2007. [DOI] [PubMed] [Google Scholar]
- 94.Ogier-DenisEOgier-Denis E, Codogno P. Autophagy: a barrier or an adaptive response to cancer. Biochim Biophys Acta 1603: 113–128, 2003. [DOI] [PubMed] [Google Scholar]
- 95.OhsumiYOhsumi Y. [Autophagy in yeast, bulk protein degradation in the vacuole.] Seikagaku 69: 39–44, 1997. [PubMed] [Google Scholar]
- 96.OhsumiYOhsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2: 211–216, 2001. [DOI] [PubMed] [Google Scholar]
- 97.OhsumiYOhsumi Y, Ohsumi M, Baba M. [Autophagy in yeast.] Tanpakushitsu Kakusan Koso 38: 46–52, 1993. [PubMed] [Google Scholar]
- 98.OrvedahlAOrvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ 2008. [DOI] [PMC free article] [PubMed]
- 99.OzpolatBOzpolat B, Akar U, Mehta K, Lopez-Berestein G. PKC delta and tissue transglutaminase are novel inhibitors of autophagy in pancreatic cancer cells. Autophagy 3: 480–483, 2007. [DOI] [PubMed] [Google Scholar]
- 100.PalletNPallet N, Bouvier N, Legendre C, Gilleron J, Codogno P, Beaune P, Thervet E, Anglicheau D. Autophagy protects renal tubular cells against cyclosporine toxicity. Autophagy 4: 783–791, 2008. [DOI] [PubMed] [Google Scholar]
- 101.PattingreSPattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem 2008. [DOI] [PMC free article] [PubMed]
- 102.PattingreSPattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J Biol Chem 278: 16667–16674, 2003. [DOI] [PubMed] [Google Scholar]
- 103.PattingreSPattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939, 2005. [DOI] [PubMed] [Google Scholar]
- 104.Periyasamy-ThandavanSPeriyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 74: 631–640, 2008. [DOI] [PubMed] [Google Scholar]
- 105.PetiotAPetiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275: 992–998, 2000. [DOI] [PubMed] [Google Scholar]
- 106.PfeiferUPfeifer U. [Cellular autophagy: glycogen segregation in early stages of a partial liver atrophy.] Virchows Arch B Cell Pathol 5: 242–253, 1970. [PubMed] [Google Scholar]
- 107.PfeiferUPfeifer U. Inhibition by insulin of the formation of autophagic vacuoles in rat liver. A morphometric approach to the kinetics of intracellular degradation by autophagy. J Cell Biol 78: 152–167, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.PfeiferUPfeifer U, Guder WG. Stimulation of cellular autophagy by parathyroid hormone and cyclic adenosine 3′,5′:monophosphate in isolated tubular fragments from the rat's kidney cortex. Virchows Arch B Cell Pathol 19: 51–67, 1975. [DOI] [PubMed] [Google Scholar]
- 109.PfeiferUPfeifer U, Scheller H. A morphometric study of cellular autophagy including diurnal variations in kidney tubules of normal rats. J Cell Biol 64: 608–621, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.PfeiferUPfeifer U, Warmuth-Metz M. Inhibition by insulin of cellular autophagy in proximal tubular cells of rat kidney. Am J Physiol Endocrinol Metab 244: E109–E114, 1983. [DOI] [PubMed] [Google Scholar]
- 111.PhillipsARPhillips AR, Suttangkakul A, Vierstra RD. The ATG12-conjugating enzyme ATG10 is essential for autophagic vesicle formation in Arabidopsis thaliana Genetics 178: 1339–1353, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.PyoJOPyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280: 20722–20729, 2005. [DOI] [PubMed] [Google Scholar]
- 113.QuXQu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112: 1809–1820, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.ReggioriFReggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell 6: 79–90, 2004. [DOI] [PubMed] [Google Scholar]
- 115.RouxPPRoux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.SatoSSato S, Kitamura H, Adachi A, Sasaki Y, Ghazizadeh M. Two types of autophagy in the podocytes in renal biopsy specimens: ultrastructural study. J Submicrosc Cytol Pathol 38: 167–174, 2006. [PubMed] [Google Scholar]
- 117.Scherz-ShouvalRScherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17: 422–427, 2007. [DOI] [PubMed] [Google Scholar]
- 118.SeglenPOSeglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79: 1889–1892, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.SeglenPOSeglen PO, Gordon PB, Poli A. Amino acid inhibition of the autophagic/lysosomal pathway of protein degradation in isolated rat hepatocytes. Biochim Biophys Acta 630: 103–118, 1980. [DOI] [PubMed] [Google Scholar]
- 120.ShimizuSShimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6: 1221–1228, 2004. [DOI] [PubMed] [Google Scholar]
- 121.SquierTCSquier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol 36: 1539–1550, 2001. [DOI] [PubMed] [Google Scholar]
- 122.StromhaugPEStromhaug PE, Reggiori F, Guan J, Wang CW, Klionsky DJ. Atg21 is a phosphoinositide binding protein required for efficient lipidation and localization of Atg8 during uptake of aminopeptidase I by selective autophagy. Mol Biol Cell 15: 3553–3566, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.SuzukiCSuzuki C, Isaka Y, Takabatake Y, Tanaka H, Koike M, Shibata M, Uchiyama Y, Takahara S, Imai E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem Biophys Res Commun 368: 100–106, 2008. [DOI] [PubMed] [Google Scholar]
- 124.SuzukiKSuzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J 20: 5971–5981, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.SybersHDSybers HD, Ingwall J, DeLuca M. Autophagy in cardiac myocytes. Recent Adv Stud Card Struct Metab 12: 453–463, 1976. [PubMed] [Google Scholar]
- 126.TakahashiYTakahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9: 1142–1151, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.TalloczyZTalloczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99: 190–195, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.TanidaITanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36: 2503–2518, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.TasdemirETasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy 4: 810–814, 2008. [DOI] [PubMed] [Google Scholar]
- 130.TasdemirETasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10: 676–687, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.TesniereATesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 15: 3–12, 2008. [DOI] [PubMed] [Google Scholar]
- 132.TsukadaMTsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae FEBS Lett 333: 169–174, 1993. [DOI] [PubMed] [Google Scholar]
- 133.TsukamotoSTsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science 321: 117–120, 2008. [DOI] [PubMed] [Google Scholar]
- 134.TuckerKATucker KA, Reggiori F, Dunn WA Jr, Klionsky DJ. Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J Biol Chem 278: 48445–48452, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.UchiyamaYUchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol 64: 233–246, 2001. [DOI] [PubMed] [Google Scholar]
- 136.vom DahlSvom Dahl S, Dombrowski F, Schmitt M, Schliess F, Pfeifer U, Haussinger D. Cell hydration controls autophagosome formation in rat liver in a microtubule-dependent way downstream from p38MAPK activation. Biochem J 354: 31–36, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.WangCWWang CW, Kim J, Huang WP, Abeliovich H, Stromhaug PE, Dunn WA Jr, Klionsky DJ. Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J Biol Chem 276: 30442–30451, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.WangZWang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol 21: 5742–5752, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138a.WeiYWei Y, Sinha S, Levine B. Dual role of JNK-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4: 949–951, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.XuYXu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem 281: 19179–19187, 2006. [DOI] [PubMed] [Google Scholar]
- 140.YamadaYYamada Y, Suzuki NN, Hanada T, Ichimura Y, Kumeta H, Fujioka Y, Ohsumi Y, Inagaki F. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem 282: 8036–8043, 2007. [DOI] [PubMed] [Google Scholar]
- 141.YamamotoAYamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23: 33–42, 1998. [DOI] [PubMed] [Google Scholar]
- 142.Yamazaki-SatoHYamazaki-Sato H, Tanida I, Ueno T, Kominami E. The carboxyl terminal 17 amino acids within Apg7 are essential for Apg8 lipidation, but not for Apg12 conjugation. FEBS Lett 551: 71–77, 2003. [DOI] [PubMed] [Google Scholar]
- 143.YangCYang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 294: F777–F787, 2008. [DOI] [PubMed] [Google Scholar]
- 144.YangZYang Z, Huang J, Geng J, Nair U, Klionsky DJ. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol Biol Cell 17: 5094–5104, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.YenWLYen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology (Bethesda) 23: 248–262, 2008. [DOI] [PubMed] [Google Scholar]
- 146.YenWLYen WL, Legakis JE, Nair U, Klionsky DJ. Atg27 is required for autophagy-dependent cycling of Atg9. Mol Biol Cell 18: 581–593, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.YorimitsuTYorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell 16: 1593–1605, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.YouleRJYoule RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59, 2008. [DOI] [PubMed] [Google Scholar]
- 149.YousefiSYousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8: 1124–1132, 2006. [DOI] [PubMed] [Google Scholar]
- 150.YuLYu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304: 1500–1502, 2004. [DOI] [PubMed] [Google Scholar]
- 151.ZhangHZhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]