

We show for the first time preserved arterial baroreflex control of sympathetic nerve activity in type 2 diabetes patients with selective impairment in cardiac baroreflex control that appears related to obesity. These findings lend important insight into blood pressure regulation in a patient population prone to developing hypertension.
Keywords: muscle sympathetic nerve activity, obesity, arterial blood pressure, insulin resistance, modified oxford
Abstract
Despite greater blood pressure reactivity to acute cardiovascular stressors and a higher prevalence of hypertension in type 2 diabetes (T2D) patients, limited information is available regarding arterial baroreflex (ABR) control in T2D. We hypothesized that ABR control of muscle sympathetic nerve activity (MSNA) and heart rate (HR) are attenuated in T2D patients. Seventeen T2D patients (50 ± 2 yr; 31 ± 1 kg/m2), 9 weight-matched controls (WM-CON, 46 ± 2 yr; 32 ± 2 kg/m2) and 10 lean controls (Lean-CON, 49 ± 3 yr; 23 ± 1 kg/m2), underwent bolus infusions of sodium nitroprusside (100 μg) followed 60 s later by phenylephrine (150 μg) and weighted linear regression performed. No group differences in overall sympathetic baroreflex gain were observed (T2D: −2.5 ± 0.3 vs. WM-CON: −2.6 ± 0.2 vs. Lean-CON: −2.7 ± 0.4 arbitrary units·beat·mmHg−1, P > 0.05) or in sympathetic baroreflex gain when derived separately during blood pressure (BP) falls (nitroprusside) and BP rises (phenylephrine). In contrast, overall cardiac baroreflex gain was reduced in T2D patients compared with Lean-CON (T2D: 8.2 ± 1.5 vs. Lean-CON: 15.6 ± 2.9 ms·mmHg−1, P < 0.05) and also tended to be reduced in WM-CON (9.3 ± 1.9 ms·mmHg−1) compared with Lean-CON (P = 0.059). Likewise, during BP rises, cardiac baroreflex gain was reduced in T2D patients and weight-matched controls compared with lean controls (P < 0.05), whereas no group differences were found during BP falls (P > 0.05). Sympathetic and cardiac ABR gains were comparable between normotensive and hypertensive T2D patients (P > 0.05). These findings suggest preserved ABR control of MSNA in T2D patients compared with both obese and lean age-matched counterparts, with a selective impairment in ABR HR control in T2D that may be related to obesity.
NEW & NOTEWORTHY
We show for the first time preserved arterial baroreflex control of sympathetic nerve activity in type 2 diabetes patients with selective impairment in cardiac baroreflex control that appears related to obesity. These findings lend important insight into blood pressure regulation in a patient population prone to developing hypertension.
the arterial baroreflex (ABR) plays an important role in the beat-to-beat regulation of arterial blood pressure (BP) by rapidly adjusting cardiac output and peripheral vascular resistance via alterations in parasympathetic and sympathetic nerve activity. Type 2 diabetes (T2D) has been characterized by exaggerated increases in BP during acute stressors (39–41), and T2D patients are prone to developing hypertension (50). However, whether impairments in ABR function may contribute to alterations in BP regulation in T2D remains unclear.
Animal studies have provided evidence suggesting that T2D may negatively impact ABR function. In obese Zucker rats, reductions in cardiovagal baroreflex sensitivity have been reported (8), and as insulin resistance progresses in adult obese Zucker rats, they become hypertensive and exhibit further impairment in ABR function including a blunted baroreflex control of sympathetic nerve activity (29, 48). To our knowledge, no studies have examined sympathetic baroreflex control in T2D patients. Moreover, although cardiovagal baroreflex sensitivity has previously been examined in T2D, studies have yielded mixed results (17, 20, 47) and, of note, previous investigations have mainly used closed loop spontaneous baroreflex measures. The latter is an important point to consider in that spontaneous methods to estimate baroreflex sensitivity (i.e., gain) only determine the gain around the operating point of the baroreflex stimulus-response curve, which may be different from the gain determined when “opening” the ABR loop and driving BP over a broader range pharmacologically (14, 44). Open loop methods more fully characterize the maximal gain of the baroreflex. Also, a separate analysis of ABR function during falls and rises in BP can provide important information (51). Indeed, previous work has demonstrated differences in ABR function in humans when examining falls vs. rises in BP separately that would have otherwise been missed when examining the overall baroreflex gain alone (18, 26, 51). Thus it is clear that a comprehensive assessment of ABR function is warranted in patients with T2D.
Given this background, we tested the hypothesis that T2D patients exhibit reductions in sympathetic and cardiac baroreflex gain compared with age, weight, and sex-matched control subjects. Furthermore, since body weight-matched controls for the T2D patients were obese, and obesity has been shown to alter ABR function (3, 6, 21–23), we also recruited an age-matched lean control subject group for comparison. Lastly, due to the prevalence of hypertension among patients with T2D (4, 50), and given that hypertension can also negatively impact ABR function (7, 12, 35), we compared those with and without diagnosed hypertension within the T2D patient group. Muscle sympathetic nerve activity (MSNA), heart rate (HR), and BP were continuously measured during pharmacologically induced decreases and increases in BP (i.e., modified Oxford) to drive BP over a broad range and to also examine ABR responsiveness to falls and rises in BP separately. In addition, estimates of spontaneous baroreflex sensitivity derived from resting oscillations in BP were also examined to provide a more complete assessment of ABR function in T2D patients.
METHODS
All experimental procedures and protocols conformed to the Declaration of Helsinki and were approved by the University of Missouri Health Sciences Institutional Review Board. Each subject received a verbal and written explanation of the study objectives, measurement techniques, and risks and benefits associated with the investigation before providing written informed consent. Before participation, each subject completed a medical health history questionnaire and a 12-h fasting blood chemistry screening including a lipid panel, metabolic panel, glucose, insulin, and HbA1C measurements.
Subjects.
A total of 36 subjects participated: 17 T2D patients (duration of disease: 12 ± 2 yr); 9 age-, sex-, and body weight-matched healthy controls (WM-CON); and 10 age- and sex-matched lean controls (Lean-CON). Seven of the T2D patients had a clinical diagnosis of hypertension in their medical record and were taking antihypertensive medications including ACE inhibitors (n = 5), angiotensin II receptor blockers (n = 1), and diuretics (n = 5). During recruitment we excluded any patients taking medications directly influencing the sympathetic nervous system (e.g., clonidine). Primary subject characteristics are provided in Table 1.
Table 1.
Main subject characteristics
| Lean-CON | WM-CON | T2D | P Value | |
|---|---|---|---|---|
| Sex, male/female | 6/4 | 3/6 | 9/8 | |
| Age, yr | 49 ± 3 | 46 ± 2 | 50 ± 2 | 0.540 |
| Weight, kg | 70 ± 3 | 86 ± 5* | 87 ± 4* | 0.011 |
| BMI, kg/m2 | 23 ± 1 | 32 ± 2* | 31 ± 1* | <0.001 |
| Waist circumference, cm | 84 ± 4 | 106 ± 5* | 101 ± 3* | <0.001 |
| Waist-to-hip ratio | 0.85 ± 0.04 | 0.96 ± 0.04 | 0.94 ± 0.02 | 0.065 |
| Glucose, mg/dl | 87 ± 3 | 90 ± 4 | 152 ± 11*† | <0.001 |
| HbA1c, % | 5.4 ± 0.1 | 5.5 ± 0.1 | 7.6 ± 0.3*† | <0.001 |
| HbA1c, mmol/mol | 36 ± 1.1 | 37 ± 1.1 | 60 ± 3.3*† | <0.001 |
| Insulin, μIU/ml | 4.6 ± 0.5 | 7.3 ± 0.8 | 9.9 ± 1.1* | 0.004 |
| HOMA-IR | 1.0 ± 0.1 | 1.7 ± 0.3 | 3.6 ± 0.5*† | <0.001 |
| Triglycerides, mg/dl | 116 ± 20 | 104 ± 12 | 144 ± 16 | 0.231 |
| HDL, mg/dl | 61 ± 7 | 52 ± 7 | 39 ± 3* | 0.007 |
| Anti-hyperglycemic medications, n | ||||
| Biguanide | 0 | 0 | 13 | |
| Sulfonylurea | 0 | 0 | 9 | |
| DPP-4 inhibitor | 0 | 0 | 1 | |
| Insulin | 0 | 0 | 2 |
Values are means ± SE.
T2D, type 2 diabetes patients; Lean-CON, lean control; WM-CON, weight-matched control; BMI, body mass index; HOMA-IR, homeostatic model assessment of insulin resistance.
P < 0.05 vs. Lean-CON;
P < 0.05 vs. WM-CON.
Cardiovascular and metabolic measurements.
HR was continuously monitored using a standard lead II surface electrocardiogram (ECG, Quinton Q710 Foremost Equipment, Rochester, NY). BP was measured on a beat-to-beat basis using servo-controlled finger photoplethysmography (Finometer, Finapres Medical Systems, Amsterdam, The Netherlands) placed on the middle finger of the left hand and supported on a bedside table positioned at the level of the heart. The changes in arterial BP measured by photoplethysmography have been shown to provide an accurate estimate of directly measured intra-arterial BP (24, 49). In addition, an automated sphygmomanometer (Welch Allyn, Skaneateles Falls, NY) recorded resting BP every minute by auscultation of the brachial artery of the right arm to validate absolute BP measurements from the Finometer (10, 57). Respiratory movements were monitored using a strain-gauge pneumobelt placed in a stable position around the abdomen (Pneumotrace; UFI, Morro Bay, CA) to avoid the influence of any large respiratory excursions on cardiovascular variables.
Multiunit postganglionic MSNA was recorded using standard microneurographic techniques, as previously described (16, 56, 57). Briefly, a tungsten microelectrode was placed into the peroneal nerve near the left fibular head. Signals were amplified, filtered (bandwidth: 0.7–2.0 kHz), rectified, and integrated (0.1-s time constant) to obtain mean voltage neurograms (Nerve Traffic analyzer, model 662c-3; University of Iowa Bioengineering, Iowa City, IA). MSNA was identified by the presence of spontaneous bursts with characteristic pulse synchronicity and by its responsiveness to end-expiratory breath holds but not to arousal or skin stimulation. MSNA signals were obtained in all WM controls, but we were unable to attain quality MSNA signals in one lean control, four T2D patients (3 normotensive and 1 hypertensive), and one additional hypertensive T2D patient was highly sensitive to the procedure so it was stopped. All neural cardiovascular data was acquired at a frequency of 1,000 Hz using Chart version 5.2 (Powerlab, ADInstruments, Bella Vista, NSW, Australia) and stored for later analysis.
Experimental protocols.
On the experimental day, T2D patients were instructed to refrain from medication use and all subjects arrived at the laboratory between 7:00 AM and 9:00 AM following an overnight fast. Subjects were also requested to abstain from caffeinated beverages the morning of the study and strenuous physical activity and alcohol for at least 24 h before experimental sessions. All experiments were performed in a dimly-lit room at an ambient room temperature of 22–24°C. Upon arrival, subjects were positioned supine and a venous catheter was inserted into the antecubital or a hand vein of the right arm for blood sampling and drug infusion. Next, subjects were instrumented for HR, BP, respiratory movements, and MSNA. Once all signals were acquired, data were collected for at least a 10-min baseline period to determine resting neural cardiovascular values. ABR control of HR and MSNA was then tested using the modified Oxford technique. Briefly, a bolus injection of sodium nitroprusside (100 μg) was followed 60 s later by a bolus infusion of phenylephrine (150 μg) to decrease and increase BP by ∼15 mmHg, respectively (46, 51, 52). A minimum of three trials of the modified Oxford technique was performed in all subjects. Trials were separated by a minimum of 15 min.
Data analysis.
Resting values for HR, BP, and MSNA were calculated as mean values over the initial resting baseline period. Resting MSNA was quantified as burst frequency (bursts/min) and burst incidence (bursts/100 heartbeats). In addition, for ABR analysis, MSNA burst height was quantified and expressed as a percentage of the average of the three largest bursts during the baseline period [assigned a mean value of 100 arbitrary units (AU)]. If no MSNA burst was detected for a particular cardiac cycle, a value of zero was assigned to that cardiac cycle (i.e., total MSNA). MSNA was analyzed using a custom LabVIEW program (15, 16).
Sympathetic baroreflex sensitivity.
To determine sympathetic baroreflex sensitivity during the modified Oxford, MSNA was averaged over 3-mmHg diastolic BP ranges (bins), and a weighted linear regression analysis between MSNA and diastolic BP was performed. MSNA within each pressure bin was calculated as total MSNA (total height of all MSNA bursts relative to the number of cardiac cycles) and expressed as AU/beat. MSNA during the modified Oxford was also expressed as burst incidence (bursts/100 heartbeats). Diastolic BP was used for this analysis because changes in MSNA correlate most closely with changes in diastolic BP (53). Each modified Oxford trial was analyzed in three segments. First, overall arterial baroreflex gain was determined from the point at which diastolic BP started to decrease following infusion of sodium nitroprusside until the diastolic BP peak following phenylephrine infusion, thus encompassing the MSNA response to both a decrease and an increase in BP. The MSNA responses to acute hypotension and hypertension were also separately examined using the segments from the onset of the diastolic BP fall after nitroprusside infusion until the nadir of the BP change and from the lowest diastolic BP after phenylephrine administration until the peak rise in BP, respectively. Since poor linear regression modeling could increase the chances of committing a type II error, a minimum R value of 0.5 was used for accepting the linear regressions. In this regard, linear regressions could not be constructed for one T2D patient due to the lack of data points, and two T2D patients did not reach R ≥ 0.5 for BP rises (phenylephrine), and one lean control did not reach R ≥ 0.5 for BP falls (sodium nitroprusside). All baroreflex trials were evaluated separately and the multiple trials from each subject were averaged together to obtain a mean response for that subject.
For spontaneous sympathetic baroreflex sensitivity, MSNA was averaged over 3-mmHg diastolic BP bins during the baseline period, and the relationship between the spontaneous changes in MSNA and diastolic BP was assessed using a weighted linear regression analysis. In addition to total MSNA, burst incidence (percentage of hearbeats associated with a burst of MSNA) was also calculated for spontaneous sympathetic baroreflex sensitivity (27).
Cardiac baroreflex gain.
Cardiac baroreflex gain was estimated from the relationship between systolic BP and R-R interval following nitroprusside and phenylephrine infusion. We calculated overall cardiac baroreflex gain, as well as gains to BP falls and rises separately using similar parameters to identify segments as described for sympathetic baroreflex measures above, with the only difference being the use of systolic BP. For systolic BP falls, data were assessed beginning at the start of the systolic BP fall nearest to the bolus injection of nitroprusside, and concluded at the nadir. For systolic BP rises, data were assessed beginning at the systolic BP rise nearest to the bolus injection of phenylephrine and concluded at the highest systolic BP peak prior to systolic BP and R-R interval becoming discordant. Overall cardiac baroreflex gain was determined from the point at which systolic BP started to decrease following infusion of sodium nitroprusside until the systolic BP peak following phenylephrine infusion. Piecewise linear regression was applied to derive cardiac baroreflex gains, as previously described (52). Briefly, this analysis defines “breakpoints” within the data, and as such provides a method to objectively eliminate the nonlinear threshold and saturation portions of the curve, which are commonly defined by subjective visual inspection alone. This analysis required at least five data points to define the presence of threshold and/or saturation (52). While this approach can be used for determination of cardiac baroreflex gains, it cannot be applied for sympathetic baroreflex gains because of the more limited beat-to-beat data since not all cardiac cycles are associated with a burst of MSNA. Threshold and saturation regions were excluded when detected, and the slope of the linear portion of the systolic BP and R-R interval relationship was determined and defined as cardiac baroreflex gain. Schwartz's Bayesian information criterion (BIC) and Akaike information criterion (AIC) were used to statistically determine the preference for piecewise linear regressions (52). A likelihood ratio test with an α of 0.05 was used when discrepancies between BIC and AIC were observed.
Spontaneous cardiac baroreflex sensitivity was estimated during the baseline period using the sequence technique (Nevrokard software, Izola, Slovenia). Briefly, sequences of three or more consecutive beats where systolic BP and R-R interval change in the same direction were identified as baroreflex sequences. A linear regression was applied to each individual sequence, and an overall average was calculated for a measure of spontaneous cardiac baroreflex sensitivity. Only sequences where R2 was >0.85 were accepted. Gains were determined for all sequences combined and also separately for up (increase systolic BP: increase R-R interval] and down (decrease systolic BP: decrease R-R interval) sequences (28).
HR variability.
Time domain HR variability was determined by the standard deviation of the normal-to-normal intervals (SDNN) and the root-mean-square of successive differences in R-R interval (RMSSD). SDNN is considered an estimate of overall HR variability, and RMSSD is an estimate of short-term components of HR variability, which is primarily mediated by parasympathetic nerve activity (1). Power spectral analysis using fast Fourier transformation was also performed in the HF range (0.15–0.4), which is also considered to predominantly represent parasympathetic tone (1, 34).
Blood processing.
For blood work analyses, samples were sent to a commercial laboratory (Boyce & Bynum Pathology Laboratories, Columbia, MO) for analysis of glucose, lipids, and HbA1C measures. Insulin was measured via an EIA assay (ALPCO, Salem, NH). Insulin resistance was calculated using the homeostatic model assessment of insulin resistance (HOMA-IR): HOMA-IR = (glucose × insulin)/22.5.
Statistical analysis.
All data are reported as mean ± SE. Statistical comparisons of physiological variables between T2D patients and weight-matched controls, and between normotensive and hypertensive T2D patients, were made using t-tests. Comparisons between T2D patients, weight-matched controls and lean controls were made using one-way ANOVA. When group differences were found, one-way analysis of covariance (ANCOVA) was performed with body mass index (BMI) as a covariate to further determine whether BMI (the main difference between control groups) was a mediating factor. Pearson product-moment correlations were used to test for associations between BMI, waist circumference, waist-to-hip ratio, and HbA1C and sympathetic and cardiac ABR gains. Fisher least significance difference post hoc analyses were made when significant differences were detected. Data were analyzed using SigmaPlot 13 (Systat Software) and statistical significance was set at P < 0.05.
RESULTS
Subject characteristics.
As expected, body weight and BMI were similar among weight-matched controls and T2D patients but were lower in lean controls (P < 0.05) (Table 1). T2D patients had significantly elevated plasma glucose and HbA1C compared with weight-matched and lean controls (P < 0.05) (Table 1). Insulin was also greater in T2D patients; however, this only reached statistical significance compared with lean controls, and not weight-matched controls (P = 0.12; Table 1). Nevertheless, HOMA-IR was significantly higher in T2D patients compared with weight-matched and lean controls (P < 0.05). Separate t-tests indicated significantly greater plasma insulin and HOMA-IR in weight-matched controls compared with lean controls (both P < 0.05). No differences in resting systolic, diastolic, or mean BP were found among groups (P > 0.05; Table 2). Resting HR was significantly higher in T2D patients and weight-matched controls compared with lean controls (P < 0.05; Table 2). Interestingly, resting MSNA was not different between any of the groups (P > 0.05; Table 2; Fig. 1), whereas RMSSD and SDNN, time domain measures indicative of parasympathetic nerve activity, were significantly reduced in T2D patients compared with lean controls (P < 0.05) with a trend for similar reductions in weight-matched controls (Table 2).
Table 2.
Resting cardiovascular and MSNA measures
| Lean-CON | WM-CON | T2D | P Value | |
|---|---|---|---|---|
| Cardiovascular variables | ||||
| Heart rate, beats/min | 58 ± 3 | 69 ± 1* | 66 ± 2* | 0.004 |
| Systolic BP, mmHg | 114 ± 3 | 118 ± 6 | 125 ± 3 | 0.082 |
| Diastolic BP, mmHg | 73 ± 2 | 79 ± 3 | 78 ± 2 | 0.235 |
| Mean BP, mmHg | 87 ± 2 | 92 ± 4 | 94 ± 2 | 0.167 |
| MSNA | (n = 9) | (n = 9) | (n = 12) | |
| Burst frequency, bursts/min | 22 ± 4 | 25 ± 5 | 29 ± 3 | 0.556 |
| Burst incidence, bursts/100 heartbeats | 39 ± 7 | 37 ± 8 | 44 ± 3 | 0.645 |
| Heart rate variability | ||||
| SDNN, ms | 69 ± 10 | 49 ± 6 | 43 ± 6* | 0.047 |
| RMSSD, ms | 60 ± 14 | 39 ± 9 | 29 ± 5* | 0.041 |
| HF power, ms2 | 1420 ± 677 | 652 ± 259 | 363 ± 114 | 0.110 |
Values are means ± SE.
BP, blood pressure; MSNA, muscle sympathetic nerve activity; SDNN, standard deviation of the normal-to-normal intervals; RMSSD, root-mean-square of successive differences in R-R interval; HF, high frequency.
P < 0.05 vs. Lean-CON.
Fig. 1.
Original recordings of muscle sympathetic nerve activity (MSNA) and electrocardiogram (ECG) in 3 type 2 diabetes patients (T2D), 3 weight-matched controls (WM-CON), and 3 lean controls (Lean-CON). BMI, body mass index.
Sympathetic ABR gain.
Individual and mean summary data for sympathetic ABR gains derived during the modified Oxford are presented in Fig. 2A. Overall sympathetic ABR gain for total MSNA was not different between T2D patients and weight-matched controls (P > 0.05). Similar results were found for overall sympathetic ABR gain using MSNA burst incidence (T2D: −3.3 ± 0.3 vs. WM-CON: −3.2 ± −0.2 bursts·100 heartbeats−1·mmHg−1, P > 0.05). Likewise, no significant group differences were detected in sympathetic ABR gain during BP falls (nitroprusside) or BP rises (phenylephrine; Fig. 2A). Furthermore, when including lean controls, no differences in sympathetic ABR gain were seen among the three groups (P > 0.05; Fig. 3). These results were similar when using total MSNA or MSNA burst incidence (data not shown) as the dependent variable (P > 0.05). Mean weighted linear regression R values were comparable among groups for overall sympathetic ABR gains (T2D: −0.88 ± 0.02, WM-CON: −0.91 ± 0.01, Lean-CON: −0.83 ± 0.05), BP falls (T2D: −0.79 ± 0.04, WM-CON: −0.77 ± 0.03, Lean-CON: −0.75 ± 0.03), and BP rises (T2D: −0.76 ± 0.07, WM-CON: −0.76 ± 0.03, Lean-CON: −0.78 ± 0.06).The changes in diastolic BP following drug infusions were comparable among groups (sodium nitroprusside: T2D, −17 ± 1; WM-CON, −18 ± 1; Lean-CON, −18 ± 1 mmHg; P > 0.05; phenylephrine: T2D, +17 ± 1; WM-CON, +21 ± 2; Lean-CON, +18 ± 2 mmHg; P > 0.05). No significant correlations were noted between sympathetic ABR gain and BMI, waist circumference, waist-to-hip ratio, or HbA1C (all P > 0.05).
Fig. 2.
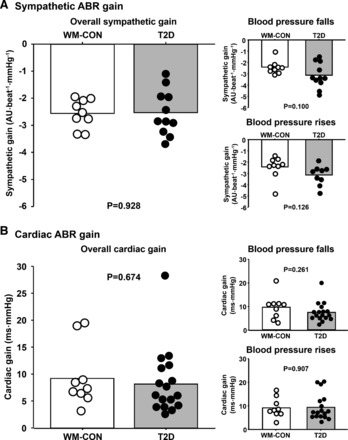
Individual and mean summary data for overall sympathetic arterial baroreflex (ABR) gain (A) and cardiac ABR gain (B) with separate analyses of sympathetic and cardiac ABR gain during falls and rises in BP following bolus intravenous infusion of nitroprusside and phenylephrine, respectively, in type 2 diabetes patients (T2D) and weight-matched controls (WM-COM). AU, arbitrary units.
Fig. 3.
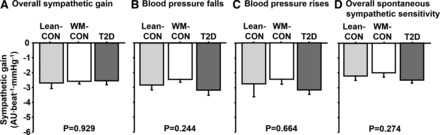
Overall sympathetic ABR gain (A), sympathetic ABR gain during falls (B), and sympathetic ABR gain during rises in BP (C), and spontaneous sympathetic baroreflex sensitivity (D) in lean controls (Lean-CON), weight-matched controls (WM-CON), and type 2 diabetes patients (T2D).
For spontaneous measures of sympathetic ABR sensitivity, no differences were observed between any of the groups for the control of total MSNA (P > 0.05, Fig. 3D). Likewise, spontaneous sympathetic ABR sensitivity as determined by MSNA burst incidence was also similar among groups (T2D, −4.3 ± 0.5; WM-CON, −3.2 ± 0.3; Lean-CON, −4.0 ± 0.5 bursts·100 heartbeats−1·mmHg−1, P > 0.05). Variations in diastolic BP (BP ranges) during the baseline period were not different among groups (T2D, 22 ± 3; WM-CON, 25 ± 3; Lean-CON, 22 ± 1 mmHg, P > 0.05).
Cardiac ABR gain.
Individual and mean summary data for cardiac ABR gains derived during the modified Oxford are presented in Fig. 2B. Overall cardiac ABR gain was not different between T2D patients and weight-matched controls (P > 0.05). Similarly, no significant group differences were detected in cardiac ABR gain during BP falls (nitroprusside) or BP rises (phenylephrine). However, overall cardiac ABR gain was significantly reduced in T2D patients compared with lean controls (P < 0.05) and also tended to be reduced in weight-matched controls compared with lean controls (P = 0.059, Fig. 4A). Likewise, during BP rises, cardiac ABR gain was reduced in weight-matched controls and T2D patients compared with lean controls (P < 0.05), whereas no group differences were found during BP falls (P > 0.05; Fig. 4, B and C). When adjusting for BMI (ANCOVA), no significant differences were found among groups for overall cardiac gain and cardiac gains during BP falls and rises (P > 0.05), suggesting BMI is a mediating factor. Mean regression R values were comparable among groups for overall cardiac ABR gains (T2D: −0.84 ± 0.03, WM-CON: −0.82 ± 0.02, Lean-CON: −0.84 ± 0.02), BP falls (T2D: −0.91 ± 0.02, WM-CON: −0.89 ± 0.03, Lean-CON: −0.88 ± 0.02), and BP rises (T2D: −0.90 ± 0.02, WM-CON: −0.90 ± 0.02, Lean-CON: −0.89 ± 0.03). Changes in systolic BP following drug infusion appeared to be greatest, although not statistically significant, in T2D patients and lowest in lean controls for both BP falls and rises (BP falls: T2D, −43 ± 4; WM-CON, −38 ± 4; Lean-CON, −32 ± 4 mmHg; BP rises: T2D, +52 ± 6; WM-CON, +46 ± 2; Lean-CON, +37 ± 5 mmHg). A marginal association between overall cardiac ABR gain and BMI was observed (R = −0.3, P = 0.076). Interestingly, a stronger association was observed between cardiac ABR gain and HbA1C (R = −0.426, P < 0.01). No significant correlations were noted between overall cardiac ABR gain and waist circumference (R = −0.2) or waist/hip ratio (R = 0.14) (both P > 0.05).
Fig. 4.
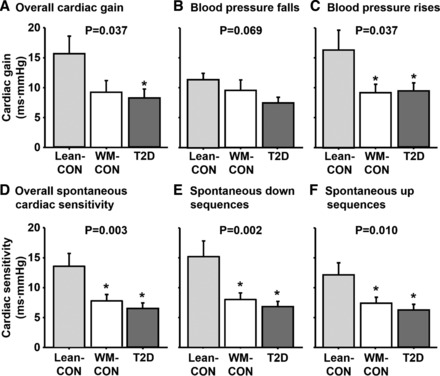
A–C overall cardiac ABR gain (A), cardiac ABR gain during falls (B), and rises (C) in BP in lean controls (Lean-CON), weight-matched controls (WM-CON), and type 2 diabetes patients (T2D). D–F: spontaneous cardiac ABR sensitivity for all sequences (D), down sequences (E), and up sequences (F) in Lean-CON, WM-CON, and T2D patients. *P < 0.05 vs. Lean-CON.
Spontaneous cardiac ABR sensitivity was significantly reduced in T2D patients and weight-matched controls compared with lean controls (P < 0.05), as well as cardiac ABR sensitivity derived during BP falls and BP rises separately (both P < 0.05 vs. Lean-CON; Fig. 4, D–F). Unlike the modified Oxford, the group differences observed in spontaneous cardiac ABR sensitivity persisted when adjusting for BMI (P < 0.05). Interestingly, the number of sequences detected were significantly lower among lean controls compared with T2D patients and weight-matched controls for up sequences (T2D, 42 ± 5; WM-CON, 44 ± 5; Lean-CON, 25 ± 6, P < 0.05). Likewise, lean controls had fewer down sequences, but this was only significant compared with weight-matched controls (T2D, 42 ± 4; WM-CON, 52 ± 5; Lean-CON, 30 ± 7, P < 0.05). Variations in systolic BP (BP ranges) during the baseline period were not different among groups (T2D, 35 ± 2; WM-CON, 44 ± 5; Lean-CON, 40 ± 5 mmHg, P > 0.05).
Sympathetic and cardiac ABR gain in normotensive vs. hypertensive T2D patients.
A comparison of resting metabolic and neural cardiovascular variables for T2D patients with and without hypertension is provided in Table 3. No differences were noted between groups for any resting variable (P > 0.05). The similar systolic, diastolic, and mean BP values between normotensive (NTN) and hypertensive (HTN) T2D patients is a consequence of the T2D patients being well-controlled with BP lowering medications [ACE inhibitors (n = 5), Angiotensin II receptor blockers (n = 1), and diuretics (n = 5)]. Overall sympathetic ABR gain, as well as gains during BP falls and rises during the modified Oxford, were similar between T2D patients with and without hypertension (P > 0.05) (Fig. 5, A–C). For spontaneous measures of sympathetic ABR sensitivity, no differences were also observed between groups for either the control of total MSNA (NTN-T2D, −2.6 ± 0.1 vs. HTN-T2D, −2.4 ± 0.4 AU·beat−1·mmHg−1, P > 0.05) or burst incidence (NTN-T2D, −4.7 ± 0.4 vs. HTN-T2D, −4.7 ± 0.1 bursts·100 heartbeats−1·mmHg−1, P > 0.05).
Table 3.
Normotensive and hypertensive T2D subject characteristics
| NTN-T2D | HTN-T2D | P Value | |
|---|---|---|---|
| Sex, male/female | 5/5 | 4/3 | |
| Age, yr | 47 ± 3 | 54 ± 2 | 0.096 |
| Weight, kg | 86 ± 5 | 89 ± 5 | 0.706 |
| BMI, kg/m2 | 31 ± 2 | 31 ± 1 | 0.838 |
| Glucose, mg/dl | 152 ± 17 | 151 ± 15 | 0.978 |
| HbA1c, % | 7.9 ± 0.4 | 7.2 ± 0.4 | 0.249 |
| HbA1c, mmol/mol | 63 ± 4.5 | 55 ± 4.6 | 0.263 |
| Insulin, μIU/ml | 10.2 ± 1.4 | 9.4 ± 2 | 0.720 |
| HOMA-IR | 3.8 ± 0.7 | 3.3 ± 0.6 | 0.623 |
| Triglycerides, mg/dl | 151 ± 26 | 134 ± 14 | 0.638 |
| HDL, mg/dl | 39 ± 4 | 39 ± 3 | 0.994 |
| Cardiovascular variables | |||
| Heart rate, beats/min | 67 ± 2 | 64 ± 4 | 0.486 |
| Systolic BP, mmHg | 124 ± 3 | 128 ± 5 | 0.484 |
| Diastolic BP, mmHg | 77 ± 2 | 79 ± 3 | 0.462 |
| Mean BP, mmHg | 92 ± 2 | 95 ± 3 | 0.438 |
| MSNA | (n = 7) | (n = 5) | |
| Burst frequency, bursts/min | 31 ± 3 | 25 ± 5 | 0.348 |
| Burst incidence, bursts/100 heartbeats | 47 ± 4 | 40 ± 6 | 0.287 |
Values are means ± SE.
NTN, normotensive; HTN, hypertensive.
Fig. 5.
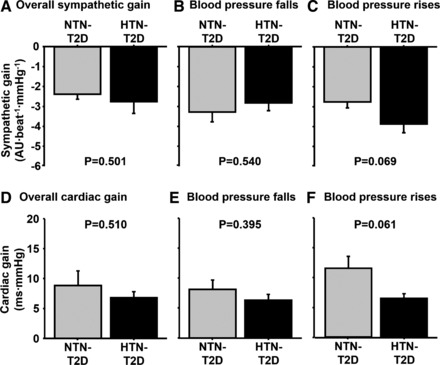
A comparison of sympathetic ABR gains (A–C) and cardiac ABR gains (D–F) derived during the modified Oxford trials between normotensive (NTN-T2D) and hypertensive type 2 diabetes patients (HTN-T2D).
For cardiac ABR gain during the modified Oxford, no differences between T2D patients with and without hypertension were seen for overall gain, and for BP falls and rises (P > 0.05; Fig. 5, D–F). Similarly, no group differences were seen for any measure of spontaneous cardiac ABR sensitivity (P > 0.05).
DISCUSSION
The primary novel findings of the present study are twofold: 1) ABR control of MSNA is not reduced in the presence of T2D, and 2) ABR control of HR is impaired similarly among T2D patients and obese subjects compared with healthy lean subjects of the same age. These findings indicate a preserved ABR control of MSNA in T2D patients with a selective impairment in ABR control of HR in T2D that may be related to obesity.
T2D has previously been characterized by exaggerated increases in arterial BP during acute cardiovascular stressors (39–41). Given the importance of the ABR in buffering elevations in BP during acute stress, we reasoned that the presence of T2D would impair ABR function, including baroreflex control of MSNA. However, contrary to our hypothesis, ABR control of MSNA was unaffected by the presence of T2D. Indeed, we found no differences in any indexes of sympathetic baroreflex gain between T2D patients and either weight-matched obese controls or lean controls. This was surprising given reports of impaired ABR control of SNA in human studies of insulin resistance and obesity (21, 22), and in obese animal models (11, 29, 48), although findings in animals appear to be mixed (54). Nonetheless, we were unable to confirm the previous obesity studies and even found no changes in ABR-MSNA gain in obese-T2D patients. While we cannot completely explain these findings, several factors warrant discussion. First, previous studies that reported impaired ABR control of MSNA in obese humans used steady-state drug infusions (22) to assess the baroreflex rather than rapid bolus infusions as in the current study, which limits the comparison of results. Also, previous studies that have reported impaired ABR control of MSNA in obese humans used subjects with markedly greater BMI (41 ± 1 kg/m2) compared with the obese weight-matched controls as well as the T2D patients in the present study (Table 1) (22). This is important considering that greater visceral adiposity has also been associated with higher resting MSNA (2, 30). Perhaps the effect of obesity on ABR control of MSNA in the present study may have been minimized by recruitment of T2D patients and weight-matched controls on the lower end of the obesity spectrum. Also, for the T2D patients, blood glucose control and medication usage may have impacted our finding of preserved ABR-MSNA control. Indeed, despite significantly elevated fasting glucose values, all T2D patients in the present study were taking antihyperglycemic medications (Table 1), similar to most of the clinical T2D population. The potential impact of antihyperglycemic medications, alone or in combination, on ABR function remains unclear and warrants future consideration. Nevertheless, the results of the present study suggest preservation of ABR control of MSNA in treated T2D patients.
In contrast to ABR-MSNA control, overall cardiac ABR gain was significantly reduced in T2D patients compared with lean controls. Likewise, during BP rises, cardiac ABR gain was significantly reduced in T2D patients as well as weight-matched controls compared with lean controls. Since the T2D patients and weight-matched controls were obese, and obesity has been shown to reduce cardiac ABR function (3, 6), these findings were not surprising. Indeed, we reasoned that ABR control of HR would be impaired in both T2D patients and weight-matched controls; however, we hypothesized that the presence of T2D would further impair ABR control of HR. Yet, the magnitude of impairment in ABR control of HR that was observed was overall quite similar between T2D patients and weight-matched controls. These data suggest that the impairment may be a result of the coexistence of obesity, which is supported by a lack of group differences in cardiac ABR gain when taking BMI into account using a covariate analysis. Although reductions in cardiac baroreflex gain with obesity are consistent with previous reports (3, 6), it is not entirely clear why obesity would selectively impair ABR control of HR in the present study, but not impair ABR control of MSNA. It is likely that reductions in cardio-vagal tone/activity contribute to impaired ABR HR control with obesity. The importance of vagal efferent activity in ABR control of HR is well documented and impairments in vagal activity to the heart have been reported in obesity (31, 45). We also observed lower indexes of cardio-vagal tone in T2D patients and obese controls compared with lean controls (Table 2). These data suggest that in the course of metabolic disease progression in obese subjects parasympathetic regulation deficits may appear before the onset of sympathetic impairments. However, alterations in parasympathetic activity may not be the only reason for selective cardiac baroreflex impairment with obesity. Indeed, there is a growing body of research suggesting that descending neural inputs from hypothalamic regions, as well as inputs from brainstem regions, can influence ABR control of HR and MSNA independently (19, 36, 38, 42). Likewise, the hypothalamus has been implicated in both metabolic and autonomic nervous system disturbances in diet-induced animal models of obesity and in human obesity (43, 55, 58). Given previous work suggesting preservation of the afferent arm of the ABR in obese animals (29), impairments in vagal efferent activity or central nuclei may likely be involved and warrant future consideration.
T2D is often associated with hypertension (4, 50), which has previously been shown to impair ABR function (7, 12, 35). For this reason, we compared ABR function in T2D patients with and without hypertension testing the hypothesis that hypertensive T2D patients would exhibit further reductions in cardiac and sympathetic ABR gain compared with normotensive T2D patients. However, no differences in ABR control of HR or MSNA were found between these groups (Fig. 5). It is worth noting that all hypertensive T2D patients were currently on a blood pressure lowering treatment regimen (≥ 1 antihypertensive medication) and were well-controlled as evidenced by comparable resting BP to normotensive T2D patients and healthy controls. Thus our findings are only related to well-treated hypertensive T2D patients and additional studies are needed to determine whether untreated hypertension among T2D patients further influences ABR control of HR or MSNA.
Perspectives
Alterations in the control of sympathetic and parasympathetic neural activity, and ultimately impairment in control of BP, have been associated with an increased risk for cardiovascular disease and mortality (9, 13, 37). Previous studies have demonstrated that patients with T2D have exaggerated increases in both MSNA and BP to acute stressors (27, 39–41). These data are clinically important given that greater cardiovascular reactivity increases the risk for myocardial infarction and stroke among T2D patients (5, 32, 33, 59). Moreover, such data are consistent with the observation that T2D patients often develop hypertension (4, 50). Because the ABR plays a vital role in the regulation of BP, we hypothesized that baroreflex dysfunction would be present in patients with T2D. Although we found a blunting of cardiac baroreflex sensitivity in T2D patients compared with age-matched lean controls, a similar impairment was present in obese non-T2D subjects suggesting that T2D did not cause further ABR dysfunction than obesity alone. Moreover, ABR control of MSNA was not different in T2D patients or obese subjects compared with lean controls. These findings suggest that an impairment in ABR sympathetic control may not contribute to BP dysregulation in patients with T2D. Nevertheless, additional viewpoints should be considered. These might include whether alterations in the transduction of MSNA to changes in vascular conductance contribute to BP dysregulation in patients with T2D. Indeed, previous findings have suggested greater α-adrenergic responsiveness among patients with T2D (25). Furthermore, additional studies are needed to determine whether an impairment in ABR control of MSNA manifests with further progression of metabolic disease or with higher levels of obesity in humans with T2D.
In summary, the results from the present study demonstrate that ABR control of MSNA is not reduced in the presence of T2D and that ABR control of HR is impaired similarly among T2D patients and obese subjects compared with age-matched lean controls. Taken together, these findings suggest a maintained ABR control of MSNA in T2D patients with a selective impairment in ABR control of HR in T2D that may be a result of the co-existence of obesity.
GRANTS
This research was supported by an American Heart Association Grant in Aid (20160072 to P. J. Fadel) and by an ACSM Foundation Research Grant from the American College of Sports Medicine Foundation (to S. W. Holwerda). The results of the study do not constitute endorsement by ACSM.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.W.H., C.N.Y., and P.J.F. conception and design of research; S.W.H., L.C.V., R.M.R., K.C., C.N.Y., and P.J.F. performed experiments; S.W.H., L.C.V., and C.N.Y. analyzed data; S.W.H., L.C.V., C.N.Y., and P.J.F. interpreted results of experiments; S.W.H., L.C.V., and C.N.Y. prepared figures; S.W.H. and P.J.F. drafted manuscript; S.W.H., L.C.V., C.N.Y., and P.J.F. edited and revised manuscript; S.W.H., L.C.V., R.M.R., K.C., C.N.Y., and P.J.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Charla Jay for assistance with screening blood draws and patient recruitment. We also thank Joseph W. LeMaster, Uzma Khan, James P. Fisher, and Adam Alter for assistance during the initiation of these studies. This research was submitted in partial fulfilment of the requirements for a Ph.D. for S. W. Holwerda.
REFERENCES
- 1.Anonymous. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology Circulation 93: 1043–1065, 1996. [PubMed] [Google Scholar]
- 2.Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation 106: 2533–2536, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez GE, Davy BM, Ballard TP, Beske SD, Davy KP. Weight loss increases cardiovagal baroreflex function in obese young and older men. Am J Physiol Endocrinol Metab 289: E665–E669, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Arauz-Pacheco C, Parrott MA, Raskin P. Treatment of hypertension in adults with diabetes. Diabetes Care 26, Suppl 1: S80–82, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Barrett-Connor EL, Cohn BA, Wingard DL, Edelstein SL. Why is diabetes mellitus a stronger risk factor for fatal ischemic heart disease in women than in men? The Rancho Bernardo Study. JAMA 265: 627–631, 1991. [PubMed] [Google Scholar]
- 6.Beske SD, Alvarez GE, Ballard TP, Davy KP. Reduced cardiovagal baroreflex gain in visceral obesity: implications for the metabolic syndrome. Am J Physiol Heart Circ Physiol 282: H630–H635, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Bristow JD, Gribbin B, Honour AJ, Pickering TG, Sleight P. Diminished baroreflex sensitivity in high blood pressure and ageing man. J Physiol 202: 45P–46P, 1969. [PubMed] [Google Scholar]
- 8.Bunag RD, Barringer DL. Obese Zucker rats, though still normotensive, already have impaired chronotropic baroreflexes. Clinical Exp Hypertens A 10, Suppl 1: 257–262, 1988. [DOI] [PubMed] [Google Scholar]
- 9.Charkoudian N, Rabbitts JA. Sympathetic neural mechanisms in human cardiovascular health and disease. Mayo Clin Proc 84: 822–830, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Credeur DP, Holwerda SW, Boyle LJ, Vianna LC, Jensen AK, Fadel PJ. Effect of aging on carotid baroreflex control of blood pressure and leg vascular conductance in women. Am J Physiol Heart Circ Physiol 306: H1417–H1425, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis G. Baroreflex and somato-reflex control of blood pressure, heart rate and renal sympathetic nerve activity in the obese Zucker rat. Exp Physiol 96: 623–634, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Eckberg DL. Carotid baroreflex function in young men with borderline blood pressure elevation. Circulation 59: 632–636, 1979. [DOI] [PubMed] [Google Scholar]
- 13.Eckberg DL, Drabinsky M, Braunwald E. Defective cardiac parasympathetic control in patients with heart disease. N Engl J Med 285: 877–883, 1971. [DOI] [PubMed] [Google Scholar]
- 14.Fadel PJ, Raven PB. Human investigations into the arterial and cardiopulmonary baroreflexes during exercise. Exp Physiol 97: 39–50, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fairfax ST, Holwerda SW, Credeur DP, Zuidema MY, Medley JH, Dyke PC 2nd, Wray DW, Davis MJ, Fadel PJ. The role of alpha-adrenergic receptors in mediating beat-by-beat sympathetic vascular transduction in the forearm of resting man. J Physiol 591: 3637–3649, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fairfax ST, Padilla J, Vianna LC, Davis MJ, Fadel PJ. Spontaneous bursts of muscle sympathetic nerve activity decrease leg vascular conductance in resting humans. Am J Physiol Heart Circ Physiol 304: H759–H766, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figueroa A, Baynard T, Fernhall B, Carhart R, Kanaley JA. Endurance training improves postexercise cardiac autonomic modulation in obese women with and without type 2 diabetes. Eur J Appl Physiol 100: 437–444, 2007. [DOI] [PubMed] [Google Scholar]
- 18.Fisher JP, Kim A, Young CN, Fadel PJ. Carotid baroreflex control of arterial blood pressure at rest and during dynamic exercise in aging humans. Am J Physiol Regul Integr Comp Physiol 299: R1241–R1247, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fontes MA, Tagawa T, Polson JW, Cavanagh SJ, Dampney RA. Descending pathways mediating cardiovascular response from dorsomedial hypothalamic nucleus. Am J Physiol Heart Circ Physiol 280: H2891–H2901, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Frattola A, Parati G, Gamba P, Paleari F, Mauri G, Di Rienzo M, Castiglioni P, Mancia G. Time and frequency domain estimates of spontaneous baroreflex sensitivity provide early detection of autonomic dysfunction in diabetes mellitus. Diabetologia 40: 1470–1475, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Grassi G, Dell'Oro R, Facchini A, Quarti Trevano F, Bolla GB, Mancia G. Effect of central and peripheral body fat distribution on sympathetic and baroreflex function in obese normotensives. J Hypertens 22: 2363–2369, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Grassi G, Seravalle G, Cattaneo BM, Bolla GB, Lanfranchi A, Colombo M, Giannattasio C, Brunani A, Cavagnini F, Mancia G. Sympathetic activation in obese normotensive subjects. Hypertension 25: 560–563, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Grassi G, Seravalle G, Dell'Oro R, Turri C, Bolla GB, Mancia G. Adrenergic and reflex abnormalities in obesity-related hypertension. Hypertension 36: 538–542, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Harms MP, Wesseling KH, Pott F, Jenstrup M, Van Goudoever J, Secher NH, Van Lieshout JJ. Continuous stroke volume monitoring by modelling flow from non-invasive measurement of arterial pressure in humans under orthostatic stress. Clin Sci (Lond) 97: 291–301, 1999. [PubMed] [Google Scholar]
- 25.Hogikyan RV, Galecki AT, Halter JB, Supiano MA. Heightened norepinephrine-mediated vasoconstriction in type 2 diabetes. Metabolism 48: 1536–1541, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Holwerda SW, Fulton D, Eubank WL, Keller DM. Carotid baroreflex responsiveness is impaired in normotensive African American men. Am J Physiol Heart Circ Physiol 301: H1639–H1645, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holwerda SW, Restaino RM, Manrique C, Lastra G, Fisher JP, Fadel PJ. Augmented pressor and sympathetic responses to skeletal muscle metaboreflex activation in type 2 diabetes patients. Am J Physiol Heart Circ Physiol 310: H300–H309, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holwerda SW, Reynolds LJ, Restaino RM, Credeur DP, Leidy HJ, Thyfault JP, Fadel PJ. The influence of reduced insulin sensitivity via short-term reductions in physical activity on cardiac baroreflex sensitivity during acute hyperglycemia. J Appl Physiol (1985) 119: 1383–1392, 2015. [DOI] [PubMed] [Google Scholar]
- 29.Huber DA, Schreihofer AM. Attenuated baroreflex control of sympathetic nerve activity in obese Zucker rats by central mechanisms. J Physiol 588: 1515–1525, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones PP, Davy KP, Alexander S, Seals DR. Age-related increase in muscle sympathetic nerve activity is associated with abdominal adiposity. Am J Physiol Endocrinol Metab 272: E976–E980, 1997. [DOI] [PubMed] [Google Scholar]
- 31.Karason K, Molgaard H, Wikstrand J, Sjostrom L. Heart rate variability in obesity and the effect of weight loss. Am J Cardiol 83: 1242–1247, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Kissela BM, Khoury J, Kleindorfer D, Woo D, Schneider A, Alwell K, Miller R, Ewing I, Moomaw CJ, Szaflarski JP, Gebel J, Shukla R, Broderick JP. Epidemiology of ischemic stroke in patients with diabetes: the greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care 28: 355–359, 2005. [DOI] [PubMed] [Google Scholar]
- 33.Koskinen P, Manttari M, Manninen V, Huttunen JK, Heinonen OP, Frick MH. Coronary heart disease incidence in NIDDM patients in the Helsinki Heart Study. Diabetes Care 15: 820–825, 1992. [DOI] [PubMed] [Google Scholar]
- 34.Malliani A, Pagani M, Lombardi F, Cerutti S. Cardiovascular neural regulation explored in the frequency domain. Circulation 84: 482–492, 1991. [DOI] [PubMed] [Google Scholar]
- 35.Matsukawa T, Gotoh E, Hasegawa O, Shionoiri H, Tochikubo O, Ishii M. Reduced baroreflex changes in muscle sympathetic nerve activity during blood pressure elevation in essential hypertension. J Hypertens 9: 537–542, 1991. [DOI] [PubMed] [Google Scholar]
- 36.McDowall LM, Horiuchi J, Killinger S, Dampney RA. Modulation of the baroreceptor reflex by the dorsomedial hypothalamic nucleus and perifornical area. Am J Physiol Regul Integr Comp Physiol 290: R1020–R1026, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Osculati G, Grassi G, Giannattasio C, Seravalle G, Valagussa F, Zanchetti A, Mancia G. Early alterations of the baroreceptor control of heart rate in patients with acute myocardial infarction. Circulation 81: 939–948, 1990. [DOI] [PubMed] [Google Scholar]
- 38.Patel KP, Schmid PG. Role of paraventricular nucleus (PVH) in baroreflex-mediated changes in lumbar sympathetic nerve activity and heart rate. J Auton Nerv Syst 22: 211–219, 1988. [DOI] [PubMed] [Google Scholar]
- 39.Petrofsky J, Prowse M, Remigio W, Raju C, Salcedo S, Sirichotiratana M, Madani P, Chamala RR, Puckett E, Wong M, Fajita M, Kaur R, Moore S, Pereira A, Katikaneni S, Regula K, Elavarthy P, Kumar U, Raju L, Gadagoju A. The use of an isometric handgrip test to show autonomic damage in people with diabetes. Diabetes Tech Ther 11: 361–368, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Petrofsky JS, Lee S. The impact of rosiglitazone on cardiovascular responses and endurance during isometric exercise in patients with Type 2 diabetes. Med Sci Monitor 12: CR21–26, 2006. [PubMed] [Google Scholar]
- 41.Petrofsky JS, Stewart B, Patterson C, Cole M, Al Malty A, Lee S. Cardiovascular responses and endurance during isometric exercise in patients with Type 2 diabetes compared with control subjects. Med Sci Monitor 11: CR470–477, 2005. [PubMed] [Google Scholar]
- 42.Polson JW, Dampney RA, Boscan P, Pickering AE, Paton JF. Differential baroreflex control of sympathetic drive by angiotensin II in the nucleus tractus solitarii. Am J Physiol Regul Integr Comp Physiol 293: R1954–R1960, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci USA 108: 2939–2944, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raven PB, Fadel PJ, Ogoh S. Arterial baroreflex resetting during exercise: a current perspective. Exp Physiol 91: 37–49, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Rossi RC, Vanderlei LC, Goncalves AC, Vanderlei FM, Bernardo AF, Yamada KM, da Silva NT, de Abreu LC. Impact of obesity on autonomic modulation, heart rate and blood pressure in obese young people. Auton Neurosci 193: 138–141, 2015. [DOI] [PubMed] [Google Scholar]
- 46.Rudas L, Crossman AA, Morillo CA, Halliwill JR, Tahvanainen KU, Kuusela TA, Eckberg DL. Human sympathetic and vagal baroreflex responses to sequential nitroprusside and phenylephrine. Am J Physiol Heart Circ Physiol 276: H1691–H1698, 1999. [DOI] [PubMed] [Google Scholar]
- 47.Ruiz J, Monbaron D, Parati G, Perret S, Haesler E, Danzeisen C, Hayoz D. Diabetic neuropathy is a more important determinant of baroreflex sensitivity than carotid elasticity in type 2 diabetes. Hypertension 46: 162–167, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Schreihofer AM, Mandel DA, Mobley SC, Stepp DW. Impairment of sympathetic baroreceptor reflexes in obese Zucker rats. Am J Physiol Heart Circ Physiol 293: H2543–H2549, 2007. [DOI] [PubMed] [Google Scholar]
- 49.Shi X, Gallagher KM, Smith SA, Bryant KH, Raven PB. Diminished forearm vasomotor response to central hypervolemic loading in aerobically fit individuals. Med Sci Sports Exerc 28: 1388–1395, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 37: 1053–1059, 2001. [DOI] [PubMed] [Google Scholar]
- 51.Studinger P, Goldstein R, Taylor JA. Age- and fitness-related alterations in vascular sympathetic control. J Physiol 587: 2049–2057, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Studinger P, Goldstein R, Taylor JA. Mechanical and neural contributions to hysteresis in the cardiac vagal limb of the arterial baroreflex. J Physiol 583: 1041–1048, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sundlof G, Wallin BG. Human muscle nerve sympathetic activity at rest. Relationship to blood pressure and age. J Physiol 274: 621–637, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki H, Nishizawa M, Ichikawa M, Kumagai K, Ryuzaki M, Kumagai H, Saruta T, Ikeda H. Basal sympathetic nerve activity is enhanced with augmentation of baroreceptor reflex in Wistar fatty rats: a model of obesity-induced NIDDM. J Hypertens 17: 959–964, 1999. [DOI] [PubMed] [Google Scholar]
- 55.Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschop MH, Schwartz MW. Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122: 153–162, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev 59: 919–957, 1979. [DOI] [PubMed] [Google Scholar]
- 57.Vianna LC, Hart EC, Fairfax ST, Charkoudian N, Joyner MJ, Fadel PJ. Influence of age and sex on the pressor response following a spontaneous burst of muscle sympathetic nerve activity. Am J Physiol Heart Circ Physiol 302: H2419–H2427, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61–73, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao W, Katzmarzyk PT, Horswell R, Wang Y, Johnson J, Hu G. Sex differences in the risk of stroke and HbA(1c) among diabetic patients. Diabetologia 57: 918–926, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]