

TNF-α is one of the major inflammatory cytokines that plays an important function in inflammation and is increased in patients with cardiovascular disease. TNF-α primarily targets vasculature and upregulates adhesion molecules such as ICAM-1. Our study demonstrated that N-acetyl-Ser-Asp-Lys-Pro diminished TNF-α-induced ICAM-1 expression mainly through inhibition of NF-κB pathway.
Keywords: Ac-SDKP, endothelial cells, inflammation, NF-κB, TNF-α
Abstract
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a naturally occurring tetrapeptide that prevents inflammation and fibrosis in hypertension and other cardiovascular diseases. We previously showed that, in angiotensin II-induced hypertension, Ac-SDKP decreased the activation of nuclear transcription factor NF-κB, whereas, in experimental autoimmune myocarditis and hypertension animal models, it also reduced the expression of endothelial leukocyte adhesion molecule ICAM-1. However, the mechanisms by which Ac-SDKP downregulated ICAM-1 expression are still unclear. TNF-α is a proinflammatory cytokine that induces ICAM-1 expression in various cell types via TNF receptor 1 and activation of the classical NF-κB pathway. We hypothesized that in endothelial cells Ac-SDKP suppresses TNF-α-induced ICAM-1 expression by decreasing IKK phosphorylation that as a consequence leads to a decrease of IκB phosphorylation and NF-κB activation. To test this hypothesis, human coronary artery endothelial cells were treated with Ac-SDKP and then stimulated with TNF-α. We found that TNF-α-induced ICAM-1 expression was significantly decreased by Ac-SDKP in a dose-dependent manner. Ac-SDKP also decreased TNF-α-induced NF-κB translocation from cytosol to nucleus, as assessed by electrophoretic mobility shift assay, which correlated with a decrease in IκB phosphorylation. In addition, we found that Ac-SDKP decreased TNF-α-induced IKK phosphorylation and IKK-β expression. However, Ac-SDKP had no effect on TNF-α-induced phosphorylation of p38 MAP kinase or ERK. Thus we conclude that Ac-SDKP inhibition of TNF-α activation of canonical, i.e., IKK-β-dependent, NF-κB pathway and subsequent decrease in ICAM-1 expression is achieved via inhibition of IKK-β.
NEW & NOTEWORTHY
TNF-α is one of the major inflammatory cytokines that plays an important function in inflammation and is increased in patients with cardiovascular disease. TNF-α primarily targets vasculature and upregulates adhesion molecules such as ICAM-1. Our study demonstrated that N-acetyl-Ser-Asp-Lys-Pro diminished TNF-α-induced ICAM-1 expression mainly through inhibition of NF-κB pathway.
inflammation plays a key role in the initiation, development, and progression of cardiovascular diseases, such as hypertension and atherosclerosis (38). One of the critical steps in the inflammatory response is the recruitment of leukocytes to the inflammatory lesion sites (20). This is achieved through processes of leukocyte adhesion and transendothelial migration in which cell adhesion molecules expressed on endothelial cells, such as ICAM-1, play an important role. These molecules bind to different ligands expressed on circulating leukocytes and are upregulated in response to proinflammatory stimuli, such as TNF-α. The increase in endothelial ICAM-1 expression in response to inflammatory stimuli was shown to be in temporal and spatial association with the accumulation of inflammatory cells within the affected tissue (1). Therefore, factors affecting endothelial adhesion molecule expression are important in regulating inflammatory processes.
N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP) is a naturally occurring tetrapeptide with anti-inflammatory and antifibrotic properties (32). Ac-SDKP is hydrolyzed mainly by angiotensin-converting enzyme (ACE), and it contributes to the anti-inflammatory and antifibrotic effects of ACE inhibitors. It has been shown that Ac-SDKP exerts antifibrotic effects by binding to Ac-SDKP receptors in rat cardiac fibroblasts (43). We previously reported that Ac-SDKP decreased cardiac and renal inflammatory cell infiltration in various models of hypertension, as well as in the model of heart failure after myocardial infarction (27, 28, 33). In addition, Lin et al. (14) demonstrated that Ac-SDKP prevented macrophage infiltration in the aorta in rats with angiotensin II (Ang II)-induced hypertension, and it suppressed ICAM-1 mRNA that is often used as a marker of endothelial cell inflammatory response (15). Furthermore, Nakagawa et al. (22) showed that Ac-SDKP reduced ICAM-1 protein expression in experimental autoimmune myocarditis (EAM) (22). Altogether, these results indicate that, under inflammatory conditions, Ac-SDKP affects ICAM-1 expression. However, the molecular mechanisms involved in Ac-SDKP-mediated anti-inflammatory effects on endothelial cells, including ICAM-1 downregulation, are still largely unclear.
TNF-α is one of the major inflammatory cytokines produced by a number of cells, including vascular endothelial cells (31), that plays an important function in inflammation (35). TNF-α-induced ICAM-1 expression in the endothelium is achieved through the activation of so-called “canonical” or “classical” NF-κB signaling pathway that depends on IKK-β and IKK-γ and induces the transcription of genes that regulate inflammation and cell survival (37). TNF-α initiates ligand-dependent oligomerization of the TNF-α receptor 1 (TNFR1) and recruitment of TNFR-associated factors (TRAFs) to the cytosolic TNFR1 domain (9, 42). These factors activate IKK-β and IKK-γ subunits of IKK complex by phosphorylation and conformational change. Activated (phosphorylated) IKK-β in turn phosphorylates IκB inhibitor on two specific serine residues, and this causes IκB degradation and the release of NF-κB. Released NF-κB is translocated into the nucleus, where it binds to specific promoter regions of target genes (19, 24). TNF-α signaling was also shown to be transduced via p-38 and ERK MAP kinase pathways (41).
The aim of this work was to evaluate the effect of Ac-SDKP on TNF-α-stimulated ICAM-1 expression. We hypothesized that, in human coronary artery endothelial cells (HCAECs), Ac-SDKP suppresses TNF-α-induced ICAM-1 expression by suppressing NF-κB activation via inhibition of IKK-β phosphorylation.
MATERIALS AND METHODS
Materials.
Endothelial cell growth medium-2 (EGM-2) and endothelial cell basal medium (EBM) were purchased from Lonza. Protease inhibitors and phosphatase inhibitors for Western blotting were purchased from Roche. Recombinant human TNF-α was purchased from R&D Systems. IKK inhibitor IMD-0354 (IMD) and the antibody raised against ICAM-1 were purchased from Santa Cruz Biotechnology. The antibodies against phospho-IKK, phospho-p38, total p38, phospho-IκB, IκB, phospho-ERK, total ERK, and GAPDH were from Cell Signaling Technology. Ac-SDKP was purchased from Bachem.
Cell culture and Ac-SDKP treatment.
HCAECs were purchased from Lonza and maintained in EGM-2 supplemented with 5% FBS. The cells were incubated at 37°C in 5% CO2-95% air. On the day before experiments, HCAECs were starved in EGM-2 medium containing 0.5% FBS, at 37°C for overnight, and then switched to serum-free EBM medium during the treatment. HCAECs were treated with vehicle (control), TNF-α (0.5 ng/ml), TNF-α plus Ac-SDKP (10 nM), or Ac-SDKP alone. Cells were preincubated with Ac-SDKP for 30 min before the addition of TNF-α. On the basis of our previous experience in performing time-course experiments, TNF-α-induced phospho-IκB reached the highest level at 15 min; therefore, this time point was chosen to stimulate cells with TNF-α to assess phospho-IKK, phospho-IκB, phospho-p38, and phospho-ERK. An ACE inhibitor captopril (10 μM) was added to all groups to prevent breakdown of Ac-SDKP by ACE that is expressed by endothelial cells because the previous study has shown that the addition of captopril to the cell culture media increased the peptide half-life (7).
Cell lysate preparation and Western blot analysis.
For cell lysate preparation, HCAECs were washed with cold PBS and lysed in lysis buffer (20 mM Tris·HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM β-glycerophosphate, 1% Triton X-100, 1 mM PMSF, protease, and phosphatase inhibitor cocktail) while being vortexed. Cell lysates were then incubated on ice for 30 min and centrifuged (14,000 g for 10 min at 4°C), and supernatants were transferred to a tube. Protein concentration was determined using the Bradford assay (Bio-Rad). Total protein was separated by electrophoresis on SDS-PAGE, and the proteins were electro-transferred onto PVDF membranes (Millipore). Membranes were then probed with primary antibodies against ICAM-1 (1:1,000), phospho-IκBα (1:1,000), total IκB (1:1,000), phospho-ERK1/2 (1:1,000), phospho-p38 (1:1,000), GAPDH (1:1,000), or phospho-IKK (1:1,000). Horseradish peroxidase (HRP)-conjugated anti-rabbit IgGs or anti-mouse IgGs (Cell Signaling Technology) were used to visualize proteins by a chemiluminescence reaction (GE Healthcare). The expression of proteins was normalized to GAPDH expression.
RT-PCR for IKK-β mRNA expression.
Total RNA was extracted from HCAECs using an RNeasy kit (Qiagen). RT-PCR was performed using a two-step protocol. RNA (0.2 μg) was reverse transcribed into cDNA using an Omniscript kit (Qiagen), and mRNA levels of IKK-β were determined by real-time PCR using ABI 7900. Gene expression was quantified and analyzed using the comparative cycle threshold (CT) method as described in the Applied Biosystems user bulletin. All data were normalized to GAPDH as an internal control. Primer sequences are designed by TIB MOLBIOL and are as follows: IKK-β forward 5′-CCCCGATAAGCCTGCCACT, reverse 5′-TTCCTCTTGGGCTCTTGAAGGATA; GAPDH forward 5′-GGGAAGGTGAAGGTCGGAGTC, reverse 5′-GGTCAATGAAGGGGTCATTGATG.
Extraction of cell nuclear fraction.
Extraction of nuclear fraction of HCAECs was performed by using Active Motif Nuclear extraction kit according to the manufacturer's protocol. Briefly, HCAECs were washed twice with ice-cold PBS containing phosphatase inhibitors and then detached and incubated in a hypotonic buffer. Cells were then centrifuged, and the supernatants containing cytosolic fraction were removed. The remaining cell pellets were resuspended in lysis buffer and incubated on ice on a rocking platform at 150 rpm for 30 min and then centrifuged. The supernatant was the nuclear fraction.
EMSA.
EMSA was performed by using EMSA gel shift kit (Panomics) according to the manufacturer's protocol. Oligonucleotides corresponding to NF-κB binding sequence (5′-AGTTGAGGGGACTTTCCCAGGC-3′, from Panomics) were used. HCAECs were seeded in a 100-mm culture dish and kept overnight in cell growth medium containing 0.5% FBS. Cells were then preincubated with Ac-SDKP (10 nM) for 30 min in the presence of captopril (10 μM) in serum-free EBM medium (Lonza). On the basis of our previous experience, TNF-α-induced NF-κB DNA binding activity was time dependent (data not shown) and reached the highest level after 1 h of stimulation. Therefore, we used this time point to challenge the cells with TNF-α (0.5 ng/ml for 1 h at 37°C). HCAEC's nuclear extracts (2 μg) were incubated with Poly D (I-C) at room temperature for 5 min. The nuclear extracts were then incubated with biotin-labeled probes at 15°C for 30 min. After electrophoresis on a 6% polyacrylamide gel, the samples on gel were transferred onto a presoaked Pall Biodyne B nylon membrane (VWR International). The membrane was cross linked using a UV crosslinker for 3 min and then developed by adding the blocking buffer and streptavidin-HRP conjugate. Optical density of the bands was compared as described previously (34).
Statistical analysis.
Results are expressed as means ± SE. Contrast statements in an analysis of variance routine were used to examine all pairwise comparisons. A Hochberg's method was used to adjust for multiple comparisons. A difference was considered significant if the adjusted P value was <0.05.
RESULTS
In endothelial cells Ac-SDKP suppresses TNF-α- stimulated ICAM-1 expression.
Effect of Ac-SDKP on TNF-α-stimulated ICAM-1 expression was examined by Western blotting. ICAM-1 expression was undetectable in untreated cells but was greatly increased by TNF-α treatment. Pretreating the cells with Ac-SDKP significantly decreased TNF-α-stimulated ICAM-1 expression in a concentration-dependent manner, without affecting the expression of housekeeping protein GAPDH. The strongest inhibition was achieved with a 10 nM dose (Fig. 1). When concentration of Ac-SDKP was increased to 100 nM, this inhibition was reversed (data not shown). Therefore, the dose of 10 nM Ac-SDKP was used for the rest of the experiments. These results indicate that Ac-SDKP suppresses TNF-α-induced ICAM-1 expression.
Fig. 1.
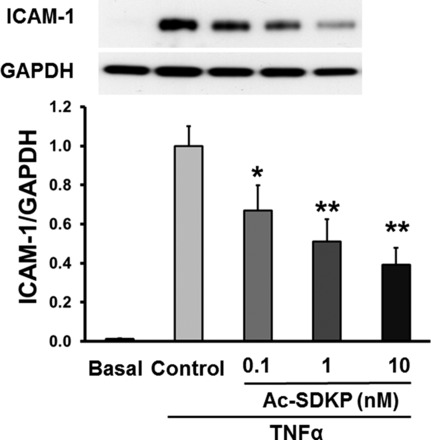
Effect of N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) on TNF-α-stimulated ICAM-1 expression. Human coronary artery endothelial cells (HCAECs) were pretreated with Ac-SDKP and then stimulated with TNF-α (0.5 ng/ml) for 6 h. Top: representative Western blots. Bottom: quantitative evaluation of ICAM-1 protein expression normalized to GAPDH, *P < 0.05; **P < 0.01, n = 7.
Ac-SDKP suppresses TNF-α-induced NF-κB activation through decreasing the phosphorylation of IKK and Iκβ.
TNF-α is a potent activator of NF-κB in endothelial cells (35). The TNF-α-induced activation of NF-κB requires activation of the enzymatic complex IKK that is followed by the phosphorylation of IκB protein (8). Therefore, we analyzed the effect of Ac-SDKP on TNF-α-induced IKK expression and phosphorylation. As shown in Fig. 2A, phosphorylated IKK was undetectable in untreated cells but greatly elevated by TNF-α treatment. Ac-SDKP significantly reduced IKK phosphorylation in TNF-α-stimulated cells. In addition, the data showed that neither TNF-α nor Ac-SDKP treatment affected the protein expression levels of the IKK-α subunit. However, IKK-β subunit protein expression levels were significantly elevated by TNF-α treatment, and this increase was significantly diminished by Ac-SDKP (Fig. 2B). Ac-SDKP treatment alone had no effect on either the basal level of phosphorylated IKK or nonphosphorylated IKK-β. These data indicate that Ac-SDKP inhibits TNF-α-induced IKK phosphorylation and IKK-β protein expression. However, IKK-β mRNA expression was not affected by Ac-SDKP (Fig. 2B).
Fig. 2.
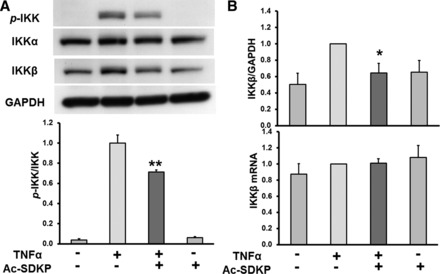
A: effect of Ac-SDKP on TNF-α-stimulated IKK phosphorylation (p-IKK). HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml) for 15 min. Top: representative Western blots. Bottom: quantitative evaluation of p-IKK expression normalized to total IKK, **P < 0.01, n = 7. B: effect of Ac-SDKP on TNF-α-stimulated IKK-β expression. HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml). Top: quantitative evaluation of IKK-β expression normalized to GAPDH, *P < 0.05, n = 7. Bottom: quantitative evaluation of IKK-β mRNA expression, n = 5.
We next analyzed whether Ac-SDKP affected TNF-α-stimulated IκB phosphorylation. The addition of TNF-α to human endothelial cells caused significant increase in the protein levels of phosphorylated IκB with a concomitant decrease of total IκB protein levels. The levels of phosphorylated IκB were significantly decreased in cells treated with Ac-SDKP (Fig. 3). Phosphorylated IκB protein in non-TNF-α-treated and in cells treated with Ac-SDKP alone was not detectable. These data indicate that Ac-SDKP interferes with TNF-α-induced IκB phosphorylation.
Fig. 3.
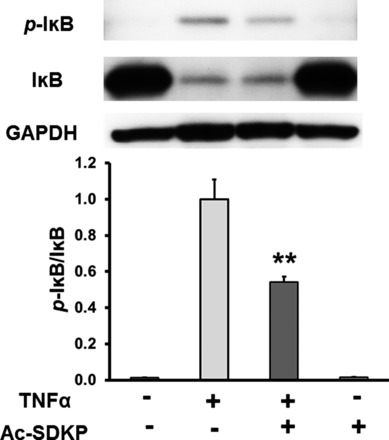
Effect of Ac-SDKP on TNF-α-stimulated IκB phosphorylation. HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml) for 15 min. Top: representative Western blots. Bottom: quantitative evaluation of p-IκB normalized to total IκB, **P < 0.01, n = 8.
Lastly, we tested whether Ac-SDKP-mediated decrease in TNF-α-induced IKK-β and IκB phosphorylation resulted in inhibited NF-κB activation in human endothelial cells. As shown in Fig. 4, TNF-α significantly increased formation of NF-κB-DNA complex. Conversely, pretreatment with Ac-SDKP significantly reduced TNF-α-stimulated NF-κB-DNA complex formation. Ac-SDKP alone had no effect. These results indicate that Ac-SDKP reduced TNF-α-induced NF-κB translocation from cytosol to nucleus binding to DNA in TNF-α-stimulated cells.
Fig. 4.
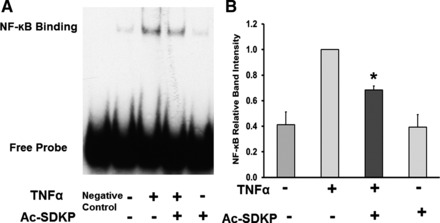
Effect of Ac-SDKP on TNF-α-stimulated NF-κB activation. HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml) for 1 h. A: representative NF-κB DNA binding. B: quantitative evaluation of NF-κB DNA binding. Negative control is the EMSA probe only without nuclear extract. *P < 0.05, n = 6.
TNF-α-stimulated ICAM-1 expression is mediated by IKK.
To confirm that TNF-α-stimulated ICAM-1 expression is mediated by IKK in our experimental settings, we used a selective IKK-β inhibitor, IMD (37). As shown in Fig. 5, IMD significantly inhibited TNF-α-induced IKK phosphorylation. In addition, it also reduced IKK-β expression, whereas IMD treatment alone had no effect on the basal phosphorylation of IKK and IKK-β. We further determined whether IMD is able to inhibit TNF-α-mediated ICAM-1 expression. Similar to the data shown in Fig. 1, TNF-α treatment dramatically increased ICAM-1 expression, which was significantly suppressed by IMD. IMD treatment alone had no effect on ICAM-1 expression (Fig. 6). These data indicate that phosphorylation of IKK plays a role in TNF-α-mediated ICAM-1 expression.
Fig. 5.
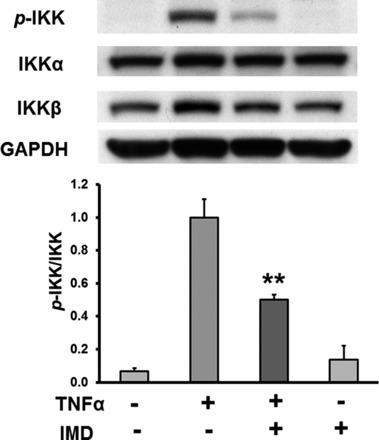
Effect of IKK-β inhibitor IMD-0354 (IMD) on TNF-α-stimulated p-IKK. HCAECs were pretreated with IMD (0.1 μM) and then stimulated with TNF-α (0.5 ng/ml) for 15 min. Top: representative Western blots. Bottom: quantitative evaluation of p-IKK expression normalized to total IKK, **P < 0.01, n = 6.
Fig. 6.
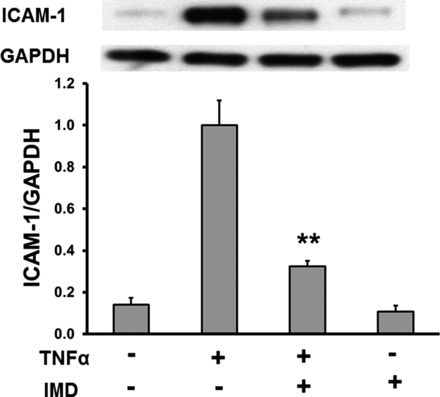
Effect of IKK-β inhibitor IMD on TNF-α-stimulated ICAM-1 expression. HCAECs were pretreated with IMD (0.1 μM) and then stimulated with TNF-α (0.5 ng/ml) for 6 h. Top: representative Western blots. Bottom: quantitative evaluation of ICAM-1 protein expression normalized to GAPDH, **P < 0.01, n = 7.
Ac-SDKP does not inhibit TNF-α-induced MAPK activation.
TNF-α-stimulated recruitment of TRAF protein complex to the intracellular domain of TNF-α receptor can activate MAPK pathways as well (18). Therefore, we analyzed the effect of Ac-SDKP on TNF-α-induced ERK and p38 MAP kinase activation. The results in Fig. 7 indicate that TNF-α significantly increased p38 phosphorylation; however, Ac-SDKP failed to inhibit TNF-α-induced activation of this kinase. ERK phosphorylation was also examined, but we found that TNF-α failed to activate ERK under our experimental conditions (Fig. 8).
Fig. 7.
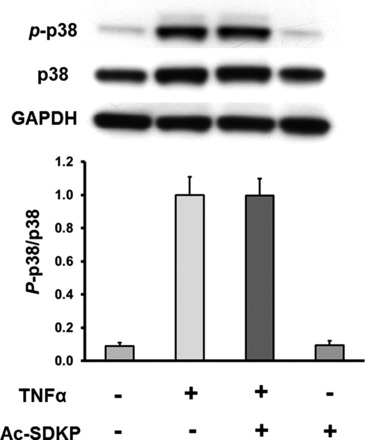
Effect of Ac-SDKP on TNF-α-stimulated p38 phosphorylation (p-p38). HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml) for 15 min. Top: representative Western blots. Bottom: quantitative evaluation of p-p38 normalized to total p38, n = 4.
Fig. 8.
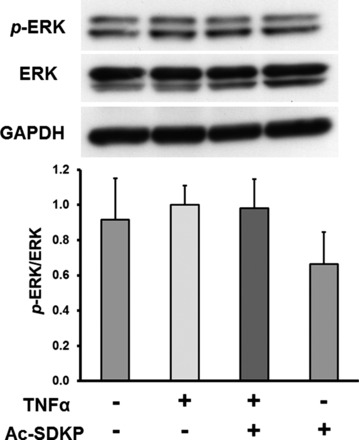
Effect of Ac-SDKP on TNF-α-stimulated ERK phosphorylation (p-ERK). HCAECs were pretreated with Ac-SDKP (10 nM) and then stimulated with TNF-α (0.5 ng/ml) for 15 min. Top: representative Western blots. Bottom: quantitative evaluation of p-ERK normalized to total ERK, n = 4.
DISCUSSION
Intercellular adhesion molecule ICAM-1 expressed in endothelial cells plays an important role in vascular inflammatory responses in which it regulates leukocyte transendothelial migration to the inflammation sites (10). Previous studies from our group have found that Ac-SDKP reduced cardiac ICAM-1 expression in animals with experimental autoimmune myocarditis and Ang II-induced hypertension (14, 22). In this study, we found that in HCAECs Ac-SDKP-inhibited TNF-α-stimulated 1) ICAM-1 expression in a dose-dependent manner, 2) NF-κB cytosol to nuclear translocation, 3) IκB and IKK phosphorylation, and 4) IKK-β expression. We also found that that these inhibitory effects of Ac-SDKP were not p38 or ERK dependent. Therefore, the present study provides the first in vitro evidence that Ac-SDKP suppresses TNF-α-stimulated NF-κB-dependent ICAM-1 expression by acting upstream from IKK complex and by causing a decrease in IKK and IκB phosphorylation.
Ac-SDKP is an endogenous bioactive tetrapeptide released from thymosin-β4 by prolyl-oligopeptidase (2, 7). Both clinical and experimental studies have shown that Ac-SDKP is associated with inflammation. Ac-SDKP is normally present in nanomolar concentrations in human plasma and circulating mononuclear cells (43), and the level of Ac-SDKP in pericardial fluid was altered in the patients with tuberculous pericarditis (25). In addition, a number of studies, including ours, have demonstrated that Ac-SDKP prevents or reverses inflammation in various animal models. Ac-SDKP reduced cardiac inflammation in hypertension and in postmyocardial infarction heart failure (26, 40). Ac-SDKP also prevented cardiac inflammation and dysfunction in rats with galectin-3-induced myocarditis (16). In animal models of T cell-mediated EAM, Nakagawa et al. (22) found that Ac-SDKP prevented cardiac inflammation and reduced cardiac ICAM-1 protein expression. In addition, a previous report showed that Ac-SDKP suppressed aortic ICAM-1 mRNA expression in Ang II-induced hypertension (14). Together, these data indicate that one of the modes of Ac-SDKP anti-inflammatory effects is through the inhibition of ICAM-1 expression that consequently decreases inflammatory cell transendothelial migration and accumulation at lesion sites. However, the molecular mechanisms by which Ac-SDKP downregulates ICAM-1 expression are unclear.
In vascular endothelial cells, TNF-α-induced ICAM-1 expression is carried out through TNFR1 receptor and is mediated by the canonical NF-κB pathway (17, 21, 42). TNF-α-induced endothelial cell inflammatory responses have been associated with various cardiovascular diseases. In particular, long-term responses involving gene upregulation and protein synthesis of TNF-α-dependent molecules such as ICAM-1 affect endothelial cell function and interaction with immune cells (17). Therefore, we hypothesized that in HCAECs Ac-SDKP suppression of TNF-α-induced ICAM-1 expression is achieved through inhibition of NF-κB pathway. We first demonstrated that TNF-α increased ICAM-1 expression in HCAECs, which is in agreement with the previous studies performed on human umbilical vein endothelial cells (21). We then tested whether Ac-SDKP affected TNF-α-stimulated ICAM-1 expression, and we showed that treatment with Ac-SDKP indeed inhibited TNF-α-mediated ICAM-1 expression in a dose-dependent manner. To further dissect the molecular mechanisms involved in the decreased ICAM-1 expression, we analyzed the effect of Ac-SDKP on different components of canonical NF-κB pathway.
We demonstrated that Ac-SDKP inhibited TNF-α-induced cytosol to nuclear translocation of NF-κB that would result in a decreased amount of nuclear NF-κB available to bind to DNA and to initiate the transcription of TNF-α-dependent genes (3). These data are in agreement with our previous in vivo study showing that Ac-SDKP decreased NF-κB activation in Ang II-induced hypertension (6). A key step in NF-κB activation involves phosphorylation of IκB. Therefore, we tested whether Ac-SDKP affects TNF-α-induced IκB phosphorylation. Indeed, we found that Ac-SDKP significantly inhibited TNF-α-stimulated IκB phosphorylation. These data implicated that the inhibitory effect of Ac-SDKP on TNF-α-induced NF-κB pathway may be exerted on the molecules upstream from IκB such as IKK complex.
It is well established that activation of the IKK that phosphorylates IκB is critical in the activation of NF-κB. The IKK complex consists of two catalytic subunits, IKK-α and IKK-β, and a noncatalytic adaptor subunit, IKK-γ (9). Gene-depletion studies have demonstrated that IKK-β, but not IKK-α, plays an essential role in NF-κB activation mediated by TNF-α (12, 13, 29, 39). Stimulation by TNF-α induces trimerization of TNFR1 that leads to the recruitment of various adaptor proteins, ubiquitin ligases, and kinases to the cytosolic TNFR1 domain that phosphorylate and activate IKK-β (9). The exact mechanism of this phosphorylation remains unclear, but it could be mediated by both upstream kinases and transautophosphorylation (4). Our data show that in nonstimulated cells IKK phosphorylation was very low, but it dramatically increased in response to TNF-α. Treatment with TNF-α also significantly increased IKK-β protein levels but not IKK-α, which is in agreement with the established role of IKK-β, but not IKK-α, in IKK activation by TNF-α (4). Pretreatment with Ac-SDKP decreased TNF-α-stimulated IKK phosphorylation and IKK-β protein levels, indicating that the inhibitory effects of Ac-SDKP on TNF-α-stimulated IκB phosphorylation are mediated by a decreased activity of IKK complex. We observed a similar pattern of IKK phosphorylation and IKK-β expression when we used IMD (a specific IKK-β inhibitor that inhibits ATP attachment to IKK-β) (11) to downregulate NF-κB pathway and ICAM-1 expression in response to TNF-α. Therefore, the observed decrease in total IKK phosphorylation in cells treated with Ac-SDKP may be due mainly to decreased IKK-β phosphorylation. There are many IKK kinase candidates implicated in IKK-β phosphorylation, such as TAK1 (36) and MAP kinase kinases (23), and it is possible that some of these kinases may be involved in Ac-SDKP-mediated inhibition of NF-κB signaling pathway. On the other hand, the observed decrease in TNF-α-dependent IKK-β expression in response to IMD was less than the decrease observed in response to Ac-SDKP, indicating that, in addition to altering IKK-β phosphorylation, Ac-SDKP may also affect the levels of nonphosphorylated IKK-β. This effect may involve Ac-SDKP inhibition of IKK-β protein and/or mRNA expression, potentiation of IKK-β degradation, and/or Ac-SDKP inhibition of phosphatases such as PP2A (5) and PP2C-β (30) implicated in IKK-β dephosphorylation. Together our data indicate that Ac-SDKP downregulation of NF-κB pathway and ICAM-1 expression in response to TNF-α is achieved through the inhibition of IKK-β kinase. However, the complexity of the signaling pathway upstream of the IKK-β introduces numerous molecules as possible Ac-SDKP targets. Therefore, further studies are needed to dissect this question.
In addition to activating NF-κB pathway, TNF-α stimulation of TNFR1 and subsequent recruitment of protein complex to TNFR1 cytoplasmic domain activates MAP kinase pathway as well (18). These pathways involve signaling through ERK and p38 MAP kinases. Our data show that TNF-α dramatically increased p38 phosphorylation but did not affect ERK phosphorylation. Ac-SDKP had no effect on TNF-α-stimulated p38 phosphorylation. These data suggest that the inhibitory effect of Ac-SDKP is mainly through inhibition of NF-κB pathway and is independent of p38 MAP kinase or ERK. However, it is unclear at which point Ac-SDKP interacts with NF-κB pathway. Because TNF-α-mediated p38 phosphorylation is not affected by Ac-SDKP, it is not likely that Ac-SDKP directly interacts with the TNF-α receptor or associated proteins (TRAFs).
In summary, in the present study, we demonstrated that Ac-SDKP inhibits TNF-α-induced ICAM-1 expression in HCAECs by targeting NF-κB signaling pathway through the inhibition of IκB kinase (Fig. 9). These data add insight into the molecular mechanism of our previous in vivo evidence that Ac-SDKP downregulated ICAM-1, and this may contribute to further investigation on Ac-SDKP anti-inflammatory activity and cardiovascular protective effect, as well as to the development of Ac-SDKP-based therapies for treating inflammatory diseases.
Fig. 9.
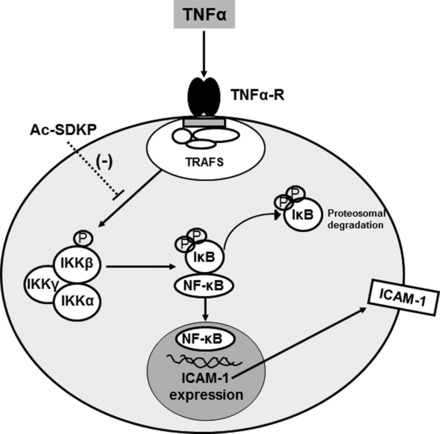
Schematic representation of postulated mechanisms of Ac-SDKP-mediated downregulation of TNF-α-induced NF-κB signaling cascade in HCAECs. TNF-α binds to TNF-α receptor (TNF-α-R) and leads to serine phosphorylation of IKK-β. Phosphorylated and activated IKK-β phosphorylates IκB, which then causes NF-κB to be released from the IκB-NF-κB complex and to be translocated to the nucleus, where it binds to the ICAM-1 promoter and initiates the transcription of ICAM-1 gene. Ac-SDKP inhibits TNF-α-induced phosphorylation of IKK-β and subsequently the phosphorylation of IκB, leading to the inhibitions of ICAM-1 expression. TRAFs, TNF-α receptor-associated factors.
Limitation.
We demonstrate that the inhibitory effect of Ac-SDKP on TNF-α-induced ICAM-1 expression is mainly through inhibition of the NF-κB pathway, and that is independent of p38 MAP kinase or ERK. We cannot exclude the possibility that other signaling pathways also contribute to this inhibitory effect of Ac-SDKP, and therefore further investigations are warranted. In addition, TNF-α is one of the stimuli that induce ICAM-1 expression. We have not explored whether Ac-SDKP modulates other cytokines that mediate ICAM-1 expression because this was not the focus of our current study.
Perspective.
TNF-α is one of the major inflammatory cytokines produced by a number of cells, including vascular endothelial cells, that plays an important function in inflammation. Patients with cardiovascular disease have increased expression and/or plasma concentrations of inflammatory markers, such as TNF-α, which primarily target vascular endothelial cells and upregulate specific adhesion molecules such as ICAM-1, contributing to the pathophysiology of cardiovascular disease. Our study demonstrated that Ac-SDKP diminished TNF-α-induced ICAM-1 expression mainly through inhibition of NF-κB pathway in HCAECs. These data, along with our previous in vivo data, support the possibility that Ac-SDKP may be beneficial for the treatment of cardiovascular diseases with inflammatory and fibrotic components.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.Z., X.-P.Y., P.N., E.L.P., and O.A.C. conception and design of research; L.Z. performed experiments; L.Z., X.-P.Y., B.J., N.-E.R., P.N., E.L.P., and O.A.C. analyzed data; L.Z., X.-P.Y., B.J., N.-E.R., P.H., P.N., and O.A.C. interpreted results of experiments; L.Z. and B.J. prepared figures; L.Z. and B.J. drafted manuscript; L.Z., X.-P.Y., B.J., N.-E.R., P.H., P.N., E.L.P., and O.A.C. edited and revised manuscript; L.Z., X.-P.Y., B.J., N.-E.R., P.H., P.N., E.L.P., and O.A.C. approved final version of manuscript.
REFERENCES
- 1.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood 84: 2068–2101, 1994. [PubMed] [Google Scholar]
- 2.Cavasin MA. Therapeutic potential of thymosin-beta4 and its derivative N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in cardiac healing after infarction. Am J Cardiovasc Drugs 6: 305–311, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J 9: 899–909, 1995. [PubMed] [Google Scholar]
- 4.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284: 309–313, 1999. [DOI] [PubMed] [Google Scholar]
- 5.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388: 548–554, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez GE, Rhaleb NE, Nakagawa P, Liao TD, Liu Y, Leung P, Dai X, Yang XP, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline reduces cardiac collagen cross-linking and inflammation in angiotensin II-induced hypertensive rats. Clin Sci (Lond) 126: 85–94, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grillon C, Lenfant M, Wdzieczak-Bakala J. Optimization of cell culture conditions for the evaluation of the biological activities of the tetrapeptide N-acetyl-ser-asp-lys-pro, a natural hemoregulatory factor. Growth Factors 9: 133–138, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Harikumar KB, Sung B, Pandey MK, Guha S, Krishnan S, Aggarwal BB. Escin, a pentacyclic triterpene, chemosensitizes human tumor cells through inhibition of nuclear factor-kappaB signaling pathway. Mol Pharmacol 77: 818–827, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2: a000158, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep 61: 22–32, 2009. [DOI] [PubMed] [Google Scholar]
- 11.Lennikov A, Hiraoka M, Abe A, Ohno S, Fujikawa T, Itai A, Ohguro H. IkappaB kinase-beta inhibitor IMD-0354 beneficially suppresses retinal vascular permeability in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci 55: 6365–6373, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Li Q, Estepa G, Memet S, Israel A, Verma IM. Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev 14: 1729–1733, 2000. [PMC free article] [PubMed] [Google Scholar]
- 13.Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med 189: 1839–1845, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CX, Rhaleb NE, Yang XP, Liao TD, D'Ambrosio MA, Carretero OA. Prevention of aortic fibrosis by N-acetyl-seryl-aspartyl-lysyl-proline in angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 295: H1253–H1261, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JM, Garcia-Alvarez MC, Bignon J, Kusinski M, Kuzdak K, Riches A, Wdzieczak-Bakala J. Overexpression of the natural tetrapeptide acetyl-N-ser-asp-lys-pro derived from thymosin beta4 in neoplastic diseases. Ann NY Acad Sci 1194: 53–59, 2010. [DOI] [PubMed] [Google Scholar]
- 16.Liu YH, D'Ambrosio M, Liao TD, Peng H, Rhaleb NE, Sharma U, Andre S, Gabius HJ, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol 296: H404–H412, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol 70: 317–325, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Mo SJ, Son EW, Lee SR, Lee SM, Shin DH, Pyo S. CML-1 inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in endothelial cells through inhibition of IkBalpha kinase. J Ethnopharmacol 109: 78–86, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol 24: 327–334, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Myers CL, Wertheimer SJ, Schembri-King J, Parks T, Wallace RW. Induction of ICAM-1 by TNF-α, IL-1-β, and LPS in human endothelial cells after downregulation of PKC. Am J Physiol 263: C767–C772, 1992. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa P, Liu Y, Liao TD, Chen X, Gonzalez GE, Bobbitt KR, Smolarek D, Peterson EL, Kedl R, Yang XP, Rhaleb NE, Carretero OA. Treatment with N-acetyl-seryl-aspartyl-lysyl-proline prevents experimental autoimmune myocarditis in rats. Am J Physiol Heart Circ Physiol 303: H1114–H1127, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano H, Shindo M, Sakon S, Nishinaka S, Mihara M, Yagita H, Okumura K. Differential regulation of IkappaB kinase alpha and beta by two upstream kinases, NF-kappaB-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci USA 95: 3537–3542, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Napetschnig J, Wu H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys 42: 443–468, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ntsekhe M, Matthews K, Wolske J, Badri M, Wilkinson KA, Wilkinson RJ, Sturrock ED, Mayosi BM. Scientific letter: Ac-SDKP (N-acetyl-seryl-aspartyl-lysyl-proline) and Galectin-3 levels in tuberculous pericardial effusion: implications for pathogenesis and prevention of pericardial constriction. Heart 98: 1326–1328, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb NE. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension 49: 695–703, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb NE. Antifibrotic effects of N-acetyl-seryl-aspartyl-Lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension 37: 794–800, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE. Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation 112: 2436–2445, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prajapati S, Gaynor RB. Regulation of Ikappa B kinase (IKK)gamma/NEMO function by IKKbeta-mediated phosphorylation. J Biol Chem 277: 24331–24339, 2002. [DOI] [PubMed] [Google Scholar]
- 30.Prajapati S, Verma U, Yamamoto Y, Kwak YT, Gaynor RB. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem 279: 1739–1746, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Ranta V, Orpana A, Carpen O, Turpeinen U, Ylikorkala O, Viinikka L. Human vascular endothelial cells produce tumor necrosis factor-alpha in response to proinflammatory cytokine stimulation. Crit Care Med 27: 2184–2187, 1999. [DOI] [PubMed] [Google Scholar]
- 32.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb NE. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens 22: 593–603, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhaleb NE, Peng H, Yang XP, Liu YH, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation 103: 3136–3141, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhaleb NE, Pokharel S, Sharma UC, Peng H, Peterson E, Harding P, Yang XP, Carretero OA. N-acetyl-Ser-Asp-Lys-Pro inhibits interleukin-1beta-mediated matrix metalloproteinase activation in cardiac fibroblasts. Pflügers Arch 465: 1487–1495, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci 13: 5094–5107, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Silverman N, Zhou R, Erlich RL, Hunter M, Bernstein E, Schneider D, Maniatis T. Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J Biol Chem 278: 48928–48934, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y, Nagai R. Novel IkB kinase inhibitors for treatment of nuclear factor-kB-related diseases. Expert Opin Investig Drugs 20: 395–405, 2011. [DOI] [PubMed] [Google Scholar]
- 38.Tracy RP. Inflammation in cardiovascular disease: cart, horse, or both? Circulation 97: 2000–2002, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Yang F, Tang E, Guan K, Wang CY. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 170: 5630–5635, 2003. [DOI] [PubMed] [Google Scholar]
- 40.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension 43: 229–236, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 15: 11–18, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Z, Connell MC, MacEwan DJ. TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal 19: 1238–1248, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Zhuo JL, Carretero OA, Peng H, Li XC, Regoli D, Neugebauer W, Rhaleb NE. Characterization and localization of Ac-SDKP receptor binding sites using 125I-labeled Hpp-Aca-SDKP in rat cardiac fibroblasts. Am J Physiol Heart Circ Physiol 292: H984–H993, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]