

Abstract
Much remains to be discovered about the etiology of heart valve disease and the molecular level mechanisms that drive it. The MAPK/ERK pathway influences calcification in many cell types and has been linked to the expression of a contractile phenotype in valvular interstitial cells (VICs). However, a direct correlation between MAPK/ERK pathway activity and VIC calcification has not been previously described. Thus the role of the MAPK pathway in the calcification of VIC cultures was investigated by measuring ERK activation in both calcifying and noncalcifying VIC environments and then, conversely, analyzing the effects of ERK pathway inhibition on VIC calcification and phenotype. Prolonged elevation of phosphorylated ERK-1/2 was found in calcifying VIC cultures, whereas directly blocking phosphorylation of ERK-1/2 resulted in a dramatic decrease in nodule number, nodule size, and total calcified area. Application of the ERK pathway inhibitor was also associated with a dramatic decrease in apoptosis, which may have contributed to the decreased nodule formation obtained via ERK inhibition. Real-time PCR analysis revealed that calcified samples exhibited significantly elevated expression of several myofibroblastic and osteoblastic markers, while ERK inhibition substantially reduced the expression of these markers, often to levels comparable to the noncalcifying control. These data suggest that the MAPK pathway plays an important role in regulating the phenotype and calcification of VICs, wherein sustained pathway activation is associated with increased VIC calcification. These findings may be used to further elucidate the mechanisms of valvular disease and identify potential treatment targets.
Keywords: heart valve, signaling pathways, extracellular matrix, mineralization
although calcification is the most common problem associated with aortic heart valve disease (21, 30), the molecular level events that underlie this process are not yet clearly understood. The process of calcification is believed to be a highly programmed sequence of different intracellular signaling events, which influence many fundamental cellular functions, such as proliferation, migration, gene transcription, and apoptosis (30, 39). Valvular interstitial cells (VICs) are the most prominent cell type in the native valve leaflet, and these cells play a major role in the process of valve calcification (2, 8).
Currently, there exists no FDA-approved treatment to halt the progression of valvular calcification. This is due, in part, to our incomplete understanding of the etiology and mechanisms involved in valvular disease. Thus far, factors such as cytokines (17, 24, 50), mechanics (31, 32, 51), and the extracellular matrix (ECM) environment (9, 14, 29) have been shown to influence or regulate valve calcification. Numerous studies have also documented the roles of atherogenic risk factors and genetic predisposition in valve calcification, as reviewed in Refs. 30 and 37, leading to the development of promising in vitro and in vivo experimental models for studying the etiology of valvular disease (37). The differentiation of VICs to an osteoblast-like phenotype is also believed to significantly contribute to valve calcification (24, 27, 28, 39), and several studies have confirmed the involvement of the osteogenic Runx2/Cbfa1 and Wnt/Lrp5 signaling pathways in this disease process (3, 30, 37, 38, 46). However, despite these tremendous advances in our understanding of valve calcification over the last 10–15 years, much remains to be learned about the relationship between calcification and intracellular events.
The mitogen-activated protein kinase (MAPK) pathway is responsible for conveying information about the extracellular environment to the cell nucleus and is known to play a critical role in osteoblast differentiation and mineralization (9, 10). MAPKs become activated upon cellular recognition of various growth factors or upon cell adhesion to ECM proteins (11, 53), and then their signaling diverges into three separate downstream pathways: 1) extracellular-regulated kinases 1 and 2 (ERK-1/2), which are the subject of the present investigation; 2) c-Jun NH2-terminal kinases, which regulate cytokine expression; and 3) p38, which primarily affects apoptosis. The MAPK/ERK pathway is involved in a wide range of cellular functions, including transcription, proliferation, migration, survival, differentiation (43, 44), and, as noted earlier, calcification.
ERK has been shown to positively regulate calcification in various osteoblast and osteoblast precursor cell types (9, 10, 15, 20, 44), primarily via activation of the Runx2/CBFa-1 (core binding factor-α1) transcription factor (9). Inhibition of ERK in these systems has the effect of attenuating osteoblastic differentiation and mineralization. With a few exceptions (23, 36), most research also points toward a positive correlation between ERK activation and calcification in cells that are not destined to calcify as part of their normal phenotype (i.e., vascular cells). Consistent with the actions of ERK in osteoblasts, several groups have found that ERK activation tends to positively regulate calcification and osteoblastic differentiation in vascular smooth muscle cell cultures (6, 43, 47). Far less is known regarding the relationship between ERK and heart valve calcification. Serotonin-induced valvular disease has been correlated with upregulation of ERK phosphorylation and signaling (52). Further motivation for characterizing the ERK/calcification relationship in valves is provided by a recent study that examined ERK-1/2 expression in explanted human valves exhibiting end-stage calcific degeneration (1). Immunohistological staining of these explanted valves revealed high levels of phosphorylated ERK in calcified samples, while the presence of phosphorylated ERK was undetectable in healthy control valves. This same investigation named stimulation of ERK signaling as a reason for the ineffectiveness of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors (statins) at slowing end-stage valvular calcification (1).
As noted above, recent end-point clinical findings support the hypothesis that valve calcification is linked to ERK activity (1). However, although a few publications have suggested an involvement of ERK in VIC fibrosis (4, 45, 52), the relationship between ERK activation and VIC dysfunction or calcification remains quite unexplored. In the present study, we describe our work toward achieving a better understanding of the molecular level mechanisms that regulate valve calcification by exploring the role of the MAPK/ERK pathway in VIC dysfunction. Through these investigations, we eventually hope to also identify treatments that prevent or inhibit calcification.
MATERIALS AND METHODS
All chemicals and cell culture solutions were obtained from Sigma-Aldrich (St. Louis, MO), unless noted otherwise.
VIC isolation and culture.
VICs were isolated from porcine aortic valve leaflets (Hormel, Austin, MN) by collagenase digestion, as previously described (18), and cultured in growth medium (15% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in medium 199) at 37°C, 5% CO2 for two to four passages.
Unless otherwise specified, VICs used in all experiments were seeded at a density of 50,000 cells/cm2 onto 24-well or 96-well plates. During the execution of experiments, the VICs were cultured in low-serum medium (1% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, in medium 199), and the medium was changed each day until day 5.
Culture substrate coatings.
Tissue culture polystyrene (TCPS) plates (24-well or 96-well) were coated with type I collagen (Coll) (Inamed Biomaterials, Fremont, CA; 2 μg/cm2), fibronectin (FN, 5 μg/cm2), fibrin (FB, 1.5 μg/cm2), or left untreated (TCPS). For the FB coating, plates were first incubated overnight at 4°C in fibrinogen (1 mg/ml), followed by three washes with 0.05% Tween 20 in phosphate-buffered saline (PBS) and 1 h incubation with thrombin (0.6 mg/ml) at 37°C (12). All coatings were prepared in 50 mM bicarbonate coating buffer, pH 8.5, and rinsed three times with PBS before cell seeding. The amounts of adsorbed proteins were measured on separate plates using the bicinchoninic acid protein assay (Pierce, Rockford, IL) to verify adsorption of protein coatings.
ERK-1/2 activation assay.
ERK-1/2 activity was assayed using an ELISA-based Dual Detect CELISA Assay Kit for ERK-1/2 (Thr202/Tyr204)/(Thr185/Tyr187) (Millipore, Danvers, MA), according to the manufacturer's instructions. VICs were cultured on 96-well plates that were coated with the different ECM components mentioned above or left uncoated (TCPS). At days 1 and 5, cells were fixed in 4% formaldehyde in Tris-buffered saline (TBS). Cells were then washed with TBST wash buffer (TBS with 0.05% Tween-20), quenched with 1% H2O2 in TBST, and blocked using 3% BSA in TBST. Primary antibodies against total ERK (monoclonal mouse anti-ERK, clone MK12) and phosphorylated ERK-1/2 [monoclonal rabbit anti-phospho-ERK-1/2 (Thr202/Tyr204)/(Thr185/Tyr187), clone AW39R] were diluted in blocking buffer and incubated with the samples overnight at 4°C. Plates were washed and incubated for 1 h with the corresponding secondary antibodies for total ERK and phospho-ERK-1/2: horseradish peroxidase (HRP)-labeled goat anti-mouse IgG and AP-labeled goat anti-rabbit IgG, respectively, then developed in HRP and AP chromogenic substrate solutions, respectively, for 30 min. Fluorescence was read at 550/590 nm (excitation/emission) to detect the HRP signal, and 360/460 nm to detect the AP signal (Synergy HT plate reader, Bio-Tek Instruments, Winooski, VT). The HRP readings represent the amount of total ERK in the cells, whereas the AP readings represent the amount of phosphorylated ERK-1/2 (Thr202/Tyr204)/(Thr185/Tyr187) in the cells. ERK-1/2 activation was expressed as the amount of phosphorylated ERK-1/2 relative to total ERK.
MEK-1/2 inhibition.
VICs on the aforementioned coatings (Coll, FN, FB, and uncoated TCPS) were treated with U-0126 [1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene; Calbiochem, San Diego, CA]. U-0126 specifically inhibits MEK-1/2, thus inhibiting activation of ERK-1/2 (7). At various time points during VIC culture, media was supplemented with U-0126 (5 μM) or left untreated. These studies were repeated using an alternative MEK inhibitor, PD-98059 (2′-amino-3′-methoxyflavone; 5 μM; Calbiochem) to confirm the MAPK specificity of these inhibition experiments. For conditions that received multiple days of inhibitor treatment, the U-0126 or PD-98059 was applied every other day until day 5, starting on day 1. In a separate set of experiments, U-0126 was applied at only one time point during the 5-day culture: 30 min, 1 h, or 1 day following cell seeding.
Quantification of nodule number and size.
After 5 days of culture in the presence or absence of U-0126 or PD-98059, VIC cultures were stained with Alizarin Red S (ARS) to facilitate quantification of calcified nodules, as ARS stains mineralized deposits red. Cultures were fixed with 10% neutral buffered formalin, stored at 4°C overnight, and stained with a 2% solution of ARS in PBS. Positively stained nodules were manually counted under a microscope (Olympus IX51 with Hamamatsu 285 digital camera and Simple PCI digital imaging software; Compix, Imaging Systems, Cranberry Township, PA). Nodule size was measured using ImageJ software (National Institutes of Health; http://rsb.info.nih.gov/ij/), and photomicrographs were captured under ×40 and ×100 magnifications.
Quantification of cell number.
At time points of 1, 3, and 5 days, VICs were lysed with radioimmunoprecipitation assay buffer [1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 1 mM iodoacetamide, 140 mM NaCl, 10 mM Tris·HCl, (pH 8.0)]. The amount of DNA in sample lysates was measured via the Quanti-iT PicoGreen assay (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions.
Migration assay.
Migration was assayed via a modified fence method (25), wherein VICs were seeded within 2 mm2 removable silicone wells, grown to confluency, and then allowed to migrate following the detachment of silicone isolators (defined as day 0). Grid-patterned transparencies were attached underneath plates containing VIC cultures to track cell movement over time. Photomicrographs were taken of the leading edge of cell migration under ×40 magnification (Olympus IX51) every 24 h for 5 days. Net cell edge displacement was measured by overlaying time course images and then quantifying migration distance (NIH ImageJ) by measuring the advancement of the leading cell edge subtracted from the migration area recorded on day 0 within a single grid space.
Apoptosis assay.
Apoptosis was measured using an ELISA-based HT TiterTACS Assay Kit (Trevigen, Gaithersburg, MD), which detects DNA fragmentation. At days 1 and 5, cells were fixed in 3.7% buffered formaldehyde solution for 7 min, washed with PBS, and postfixed in 100% methanol for 20 min. Following manufacturer's instructions, the cells were permeabilized with proteinase K, quenched with 2.5% H2O2 in methanol, and then incubated with the labeling reaction mix (TdT, Biotin-dNTP, unlabeled dNTP) to label breaks in DNA. Streptavidin-HRP and then TACS-Sapphire were added to the wells to detect apoptotic cells; the reaction was stopped with 2 N HCl, and absorbance was read at 450 nm.
Apoptosis inhibition and stimulation.
To better understand the role of apoptosis in VIC calcification, VIC cultures were treated with either 2–20 μM M50054 [2,2′-methylene-bis(1,3-cyclohexanedione); Calbiochem] or 0.2–2 μM methotrexate (Calbiochem) for 5 days and then analyzed for calcification via nodule counts. M50054 directly inhibits apoptosis via mechanisms that block caspase-3 activation (49), while methotrexate is commonly used as an apoptosis-inducing agent.
RNA isolation.
Total RNA was isolated using TRI Reagent (Molecular Research Center, Cincinnati, OH), according to the manufacturer's instructions. VICs were lysed with 200 μl TRI Reagent per well at 4°C with 50× protease inhibitor cocktail (BD Biosciences, San Jose, CA). The homogenate was stored at room temperature for 5 min to complete the dissociation of nucleoprotein complexes, at which point 0.15 ml chloroform per 600 μl TRI Reagent was added to the homogenate, followed by centrifugation at 13,000 g for 15 min. After centrifugation, RNA was precipitated from the upper aqueous phase by adding 0.3 ml isopropanol per 600 μl TRI Reagent to the tubes and then centrifuged at 13,000 g for 8 min. After this centrifugation step, the RNA pellet was washed with 75% ethanol and centrifuged at 8,000 g for 5 min. The RNA pellet was air dried and dissolved in 75 μl H2O at 60°C for 15 min. RNA samples were stored at −20°C until subsequent use.
Quantitative real-time PCR analysis.
Custom primers for various markers of cell contractility and osteogenic activity were obtained from Invitrogen (Carlsbad, CA) and are listed in Table 1. For cDNA construction, 250 ng of original RNA isolated from samples were reverse transcribed using iScript (Bio-Rad Laboratories, Hercules, CA) as per manufacturer's instructions. Samples were processed for real-time PCR analysis by combining 0.5 μl of the cDNA construction, 5 μM of primers, and SYBR Green SuperMix (Bio-Rad) in a 15-μl reaction, as specified in the manufacturer's protocol. For thermocycling, a standard protocol was used: PCR reactions were run over 40 cycles of denaturing at 95°C for 15 s and annealed at 60°C for 1 min; this was followed by a melting curve analysis for 80 cycles of 55°C + 0.5°C/cycle, 10 s per cycle, to further confirm the purity of the final PCR products, with each condition performed in triplicate (iCycler iQ Real-Time PCR Instrument, Bio-Rad). A standard comparative threshold cycle (or ΔΔCT) method was used to analyze the PCR data. The CT of all samples were first normalized to β-actin as an internal control, and then the ΔCT values for experimental samples were further normalized to the negative control (VICs on Coll, which represented a noncalcifying condition).
Table 1.
Primer sequences for real-time PCR
| RhoA | F-5′-ACC AGT TCC CAG AGG TGT ATG T-3′ |
| R-5′-TTG GGA CAG AAA TGC TTG ACT TC-3′ | |
| Alkaline phosphatase (ALP) | F-5′-ATG AGC TCA ACC GGA ACA A-3′ |
| R-5′-GTG CCC ATG GTC AAT CCT-3′ | |
| Core binding factor α1 (CBFa1) | F-5′-GAG GAA CCG TTT CAG CTT ACT G-3′ |
| R-5′-CGT TAA CCA ATG GCA CGA G-3′ | |
| Matrix metalloproteinase-13 (MMP-13) | F-5′-ACA AGG GAT CCA GTC TCT CTA TGG T-3′ |
| R-5′-TCC AGG GAT AAT GAA GGA TCA CA-3′ | |
| Osteocalcin (OCN) | F-5′-TCA ACC CCG ACT GCG ACG AG-3′ |
| R-5′-TTG GAG CAG CTG GGA TGA TGG-3′ | |
| Transforming growth factor-β (TGF-β1) | F-5′-CGA GCC AGA GGC GGA CTA C-3′ |
| R-5′-TTG GTT GCC GCT TTC CA-3′ | |
| β-Actin | F-5′-ATG GTG GGT ATG GGT CAG AA-3′ |
| R-5′-CGG AGC TCG TTG TAG AAG GT-3′ |
Statistics.
All experiments were performed a minimum of three separate times, with n ≥ 3. Data were compared using ANOVA with Tukey's honestly significant difference post hoc test. P values ≤ 0.05 were considered statistically significant. Data are presented as means ± SD.
RESULTS
Generation of calcifying and noncalcifying VIC cultures.
Consistent with the findings in Ref. 42, culture of VICs on FN or Coll was associated with significantly less calcification than that obtained with VIC cultures on FB or TCPS (P < 0.0001). Thus the use of Coll or FN as a culture substrate enabled the culture of relatively noncalcifying VICs, while culture on FB produced highly calcified samples, and culture on TCPS functioned as not only a standard control, but was also associated with a high amount of calcification (Fig. 1 and Ref. 42).
Fig. 1.
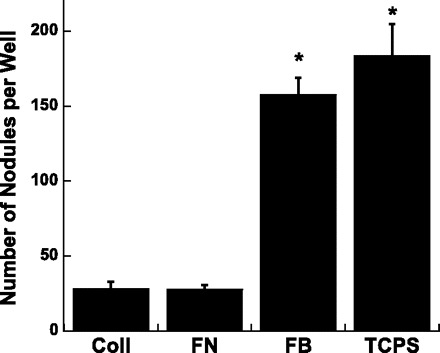
The number of calcified nodules formed by valvular interstitial cells (VICs) cultured on different substrate coatings for 5 days; nodules were identified and counted following Alizarin Red S staining. Coll, collagen type I; FN, fibronectin; FB, fibrin; TCPS, tissue culture polystyrene (uncoated control). Values are means ± SD. *P < 0.0001 compared with Coll or FN.
Activation of intracellular signaling pathways.
Having generated VIC samples that contained either little or extensive calcification, ERK-1/2 activity was measured at various time points following cell seeding (30 min, 1 h, 1 day, and 5 days) to investigate how these cell populations differed from each other on a molecular level. As shown in Fig. 2, the correlation observed between nodule numbers and ERK-1/2 activation was time point dependent. Namely, at later time points, most notably at 24 h following cell seeding, the more calcified conditions (FB, TCPS) had significantly greater ERK-1/2 activity than the less-calcified conditions (Coll, FN). In fact, at this time point, ERK phosphorylation on the minimally calcifying conditions was not even detectable. On day 5, ERK activity on FB was still elevated compared with Coll and FN conditions, suggesting that sustained, later term activation of ERK signaling was associated with VIC calcification. ERK activation at earlier time points (30 min, 1 h) did not correlate with calcification, as ERK-1/2 activity was high on all conditions except TCPS.
Fig. 2.
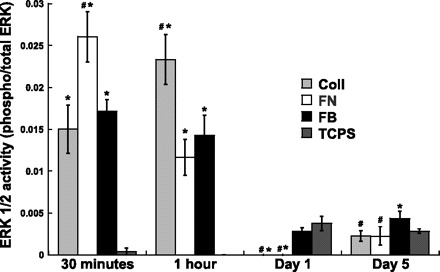
Extracellular-regulated kinase 1 and 2 (ERK-1/2) activity of VICs, as expressed by the ratio of phosphorylated ERK-1/2 (Thr202/Tyr204)/(Thr185/Tyr187) to total ERK. VICs were cultured on substrates known to promote different levels of nodule formation, and ERK-1/2 activity was quantified at multiple time points following cell seeding. Values are means ± SD; n = 4 samples per condition. #P < 0.05 compared with FB at the same time point. *P < 0.05 compared with TCPS at the same time point.
Inhibition of ERK-1/2 activation affects VIC calcification.
As seen in representative photomicrographs of VIC cultures (Fig. 3), blocking the ERK pathway via the application of U-0126 led to a significant decrease in the number of nodules formed in all ECM-coated conditions. These qualitative observations were supported by quantitative data describing nodule number, size, and total calcified area per well (Fig. 4). Treatment with U-0126 decreased both nodule number (Fig. 4A) and average nodule size (Fig. 4B) relative to untreated controls for each substrate. Total calcified area, which was obtained by summing the area of all individual nodules in each well, also significantly decreased upon administration of U-0126 (Fig. 4C). Overall, blocking ERK-1/2 activation via administration of U-0126 resulted in consistent inhibition of nodule formation across all culture conditions. Figures 3 and 4 demonstrate that similar calcification-inhibition effects were achieved via application of an alternative MEK inhibitor, PD-98059. The potent inhibition of nodule formation by two separate MEK inhibitors suggests that these effects were MAPK specific and not due to potential off-target effects of the inhibitors. Only the U-0126 inhibitor was further explored in subsequent experiments.
Fig. 3.
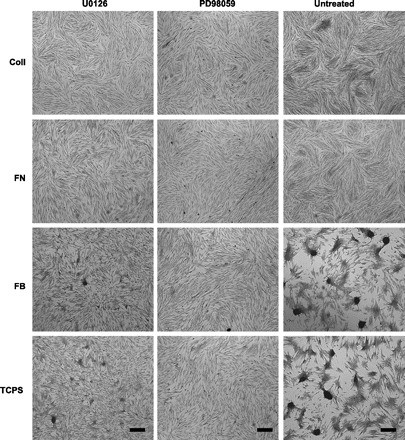
Montage of photomicrographs showing nodule formation in day 5 VIC cultures that received treatment with U-0126 or PD-98059 or remained untreated. In environments that usually support nodule formation (FB, TCPS), nodule formation was significantly attenuated via the administration of U-0126 or PD-98059. ×40 Magnification; scale bar represents 100 μm.
Fig. 4.
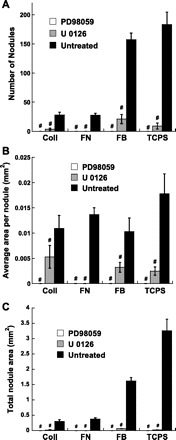
Characterization of nodule formation in day 5 VIC cultures following inhibition of the mitogen-activated protein kinase (MAPK)/ERK pathway via application of U-0126 or PD-98059. A: total nodule number per well. B: average size of nodules. C: total nodule area per well. Values are means ± SD; n = 9 samples per condition. #P < 0.0001 compared with corresponding untreated condition.
Nodule formation was also assessed following U-0126 application at varying time points. Specifically, U-0126 was applied once to VIC cultures at 30 min, 1 h, or 1 day after cell seeding, replaced by fresh culture medium after 12 h, and then nodule counts on these samples were performed at the standard day 5 end point. As demonstrated in Fig. 5, nodule formation was significantly reduced only when ERK-1/2 activation was inhibited at the 24-h time point. Consistent with the trends identified in Fig. 2, blocking ERK-1/2 activation at early time points (30 min, 1 h) did not impact VIC culture calcification.
Fig. 5.
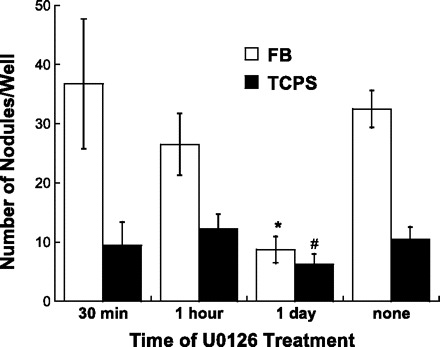
Quantification of nodule number in day 5 VIC cultures following a one-time application of U-0126 at 30 min, 1 h, or 1 day after cell seeding. Values are means ± SD; n = 4 samples per condition. *P < 0.001 compared with untreated FB. #P < 0.05 compared with untreated TCPS.
Impact of ERK pathway inhibition on cell number, migration, and apoptosis.
The MAPK/ERK pathway can affect many cell processes that may contribute to calcification (24); thus the impact of ERK inhibition on VIC proliferation, migration, and apoptosis was examined. Since the purpose of these experiments was to investigate how U-0126 lowered nodule number, these studies focused on conditions in which nodules were consistently formed, namely FB and TCPS. As shown in Fig. 6, administration of U-0126 decreased the number of cells on both TCPS and FB surfaces. Meanwhile, Fig. 7 indicates that the decreased calcification levels observed upon U-0126 treatment were not due to changes in migration. Migration of VICs treated with U-0126 was statistically similar to migration of untreated VICs at all time points up to 5 days. Lastly, a strong correlation was observed between inhibition of ERK-1/2 activation (via U-0126) and apoptosis. Specifically, by day 5, cultures that received U-0126 had drastically reduced levels of apoptosis (Fig. 8).
Fig. 6.
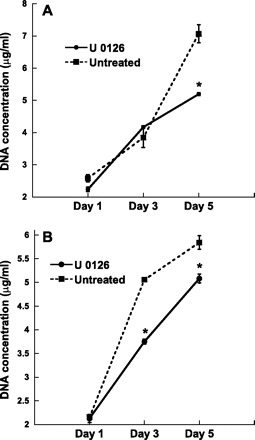
Change in VIC cell number over time when cultured on TCPS (A) or FB (B) in the presence or absence of U-0126. Values are means ± SD; n = 9 samples per condition. *P < 0.001 compared with untreated condition.
Fig. 7.
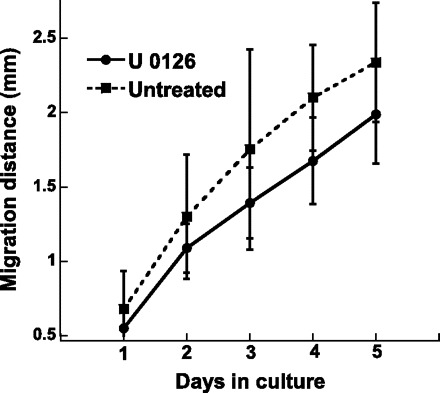
Cumulative migration distance of VICs cultured on TCPS, with or without administration of U-0126 for a total of 5 days. Values are means ± SD; n = 12 samples per condition.
Fig. 8.
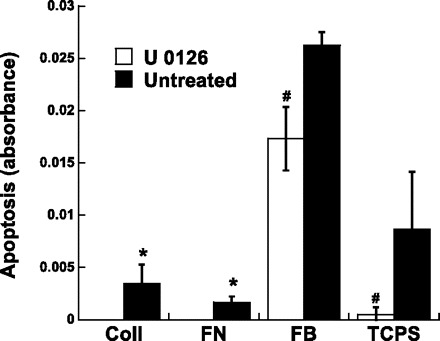
Apoptosis of VICs cultured on each of the four different substrates and treated with U-0126 or left untreated 5 days. Values are means ± SD; n = 3 samples per condition. #P < 0.05 compared with paired untreated condition. *P < 0.05 compared with untreated TCPS or untreated FB.
Because of the dramatic decrease in apoptosis levels in U-0126-treated cultures, the role of apoptosis in VIC culture calcification was further explored via the application of a direct apoptosis inhibitor (M50054) to calcifying cultures (FB, TCPS) or an apoptosis inducer (methotrexate) to noncalcifying cultures (Coll, FN). As shown in Fig. 9, direct inhibition of apoptosis was indeed sufficient to significantly decrease nodule formation on both TCPS and FB in a dose-dependent manner. Meanwhile, induction of apoptosis on Coll and FN did not promote the formation of nodules on those surfaces (not shown).
Fig. 9.
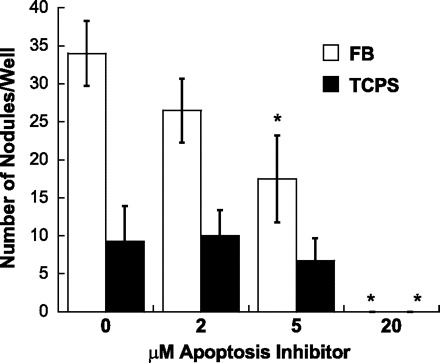
Impact of direct apoptosis inhibition (via application of M50054) on nodule formation in day 5 VIC cultures. Values are means ± SD; n = 4 samples per condition. *P < 0.05 compared with corresponding untreated condition.
Impact of ERK pathway inhibition on cell phenotype.
The expression of various phenotypic markers (Table 1) was quantified via real-time PCR to examine whether VIC differentiation to a contractile or osteoblast-like phenotype was related to calcification trends. Calcifying VICs often express markers related to a myofibroblast phenotype [i.e., α-smooth muscle actin (α-SMA), RhoA, matrix metalloproteinase-13 (MMP-13)] and/or markers related to osteogenic differentiation [i.e., alkaline phosphatase (ALP), osteocalcin (OCN), CBFa-1] (24, 31, 34, 35, 39, 42, 48). Expression of transforming growth factor-β (TGF-β1) can be associated with either one of the dysfunctional VIC phenotypes (myofibroblast or osteoblast). All data were normalized to β-actin and then to a noncalcifying control.
Figure 10 reveals that trends in calcification were closely matched by changes in the expression of myofibroblastic and osteoblastic markers. In Fig. 10A, it is seen that culture of VICs in a calcifying environment (in this case, culture on TCPS) was associated with strong upregulation of MMP-13, ALP, CBFa-1, OCN, and TGF-β1 on day 5 relative to the negative control environment. Mimicking the results obtained for nodule number, addition of U-0126 to VICs on TCPS significantly decreased the expression of MMP-13, ALP, CBFa-1, and TGF-β1 relative to untreated TCPS. In the cases of CBFa-1 and TGF-β1, U-0126 treatment reduced the expression to levels that were almost identical to those found for the negative control.
Fig. 10.
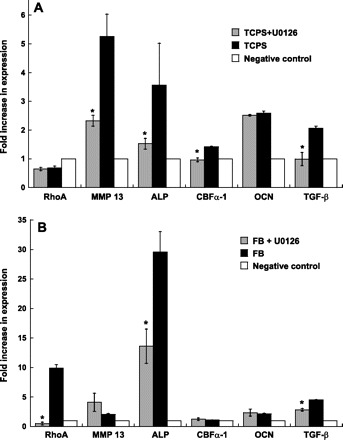
Phenotypic characterization (via real-time PCR) of VICs cultured on TCPS (A) or FB (B) in the presence or absence of U-0126. Values are means ± SD; n = 3 samples per condition. MMP-13, matrix metalloproteinase-13; ALP, alkaline phosphatase; CBFa1, core binding factor-α1; OCN, osteocalcin; TGF-β, transforming growth factor-β. *P < 0.05 compared with untreated.
VICs on FB (Fig. 10B) did not follow the aforementioned trends quite as cleanly as those on TCPS, but did still display evidence of myofibroblast/osteoblast activity that was decreased upon addition of U-0126. Specifically, culture of VICs on FB led to an increase in the expression of RhoA, MMP-13, ALP, OCN, and TGF-β1 relative to the negative control. Addition of U-0126 to VICs on FB then decreased the expression of RhoA, ALP, and TGF-β1 relative to the untreated FB condition.
DISCUSSION
The characteristics of explanted diseased valves have been described in recent years: they contain cells that exhibit markers of myofibroblast or osteoblast-like phenotypes, they are rich in inflammatory cytokines and osteogenic growth factors, and they possess grossly altered ECM arrangement (14, 16, 19, 26, 27, 30, 39). However, because most of this information comes from end-point analyses of diseased valves, the molecular mechanisms and signaling pathways that underlie these changes have not been fully examined. Several studies have implicated the Wnt/Lrp5/β-catenin and Runx2/Cbfa1 pathways, which are involved in osteogenesis, as positive regulators of valve calcification (3, 28, 30, 38, 46). However, it is unlikely that the complex process of valve calcification can be fully explained by analysis solely of these two pathways.
Thus this study has focused on better understanding the role of the MAPK/ERK pathway in VIC calcification. The MAPK/ERK pathway regulates various cellular functions, such as proliferation, migration, transcription, and apoptosis (41). As noted earlier, ERK is also an important regulator of mineralization processes in many different tissues (6, 13, 22, 43, 44, 52) and has recently been linked to the presence of calcification in explanted human valves (1). The first step in this study was the creation of culture environments, which represented different levels of VIC calcification, as investigating the differences between a calcifying VIC culture and a noncalcifying VIC culture first requires that each of those conditions be generated. Pulling from our laboratory's previous work in this area (29, 42), noncalcifying conditions were created via culture of VICs on either Coll or FN coatings, whereas calcifying conditions were obtained via culture of VICs on either unmodified TCPS or FB coatings. Because the calcifying vs. noncalcifying conditions created for this work were achieved via adsorption of different protein coatings to the culture substrates, it is likely that integrin-initiated events underlie many of the observations made herein. Regardless of specific integrin roles, however, the results in this work still clearly demonstrate that VIC calcification is strongly impacted by ERK pathway activity. Measurement of ERK-1/2 activation (Fig. 2) supported the hypothesis that differences in VIC calcification were associated with changes in ERK-1/2 activation, although with conditions: while ERK-1/2 activation levels at day 1 and 5 time points did correlate with calcification trends, ERK-1/2 activation levels at short-term time points did not. The importance of ERK-1/2 activation at the longer term time points was further emphasized by an ERK inhibition experiment, which demonstrated that ERK pathway inhibition was successful in reducing nodule formation only when performed at one of the later time points.
In the present study, blocking ERK-1/2 activation (via U-0126 or PD-98059) caused a universal and dramatic decrease in the number of calcific nodules. The decrease in calcific nodule number occurred concurrently with a decrease in average nodule size, resulting in a greatly diminished total calcified area for all conditions. As noted earlier, ERK pathway activity has been associated with increased mineralization in various vascular cells (6, 13, 22, 43, 44, 52). Thus the results of the present study are highly consistent with published findings related to MAPK and the calcification of several vascular cell types. However, one recent publication did find an inverse relationship between MAPK and α-SMA, a myofibroblast marker often associated with increased VIC calcification. In that study, blocking ERK in VIC cultures was associated with increased amounts of α-SMA (5). However, it is challenging to draw direct conclusions about nodule formation or calcification solely from α-SMA expression. VICs frequently express α-SMA without forming nodules or calcifying (4, 29, 42). Moreover, VIC dysfunction is much more closely tied to α-SMA organization (i.e., whether it is arranged in stress fibers) than the level of α-SMA expression; this is also the reason that α-SMA was not included in the PCR-based phenotype panel in the present study. Thus, despite a previous report that ERK inhibition stimulated production of a marker that may be related to increased VIC calcification, the extensive nodule characterization and phenotypic analysis in the present study strongly points toward ERK inhibition as a potent method to decrease nodule formation in VIC cultures.
Analysis of VIC cell number, migration, apoptosis, and phenotype after U-0126 treatment revealed that the nodule-inhibiting actions of U-0126 were at least partly attributable to changes in apoptosis and VIC phenotype. Formation of dystrophic nodules in VIC cultures is associated with substantial increases in apoptosis, and apoptosis was significantly decreased in U-0126-treated cultures. Application of a direct apoptosis inhibitor to VICs cultured in procalcific environments (FB or TCPS) resulted in a significant decrease in nodule number, thereby demonstrating that a certain level of apoptosis was necessary for the formation of nodules in these cultures. The hypothesis that apoptosis inhibition preceded the reduction of VIC nodule formation is further supported by the results in Ref. 33, which demonstrated that increased apoptosis precedes calcification in smooth muscle cell cultures, and that these apoptotic bodies may act as nucleating structures for calcium crystal formation. Moreover, an apoptosis assay performed by our group at 1 day postseeding (before nodule formation; data not shown) revealed that apoptosis on the minimally calcifying surfaces was lower than that found on TCPS and FB, which also supports the conclusion that inhibition of apoptosis can lead to decreased nodule formation. However, our findings do somewhat contradict those documented by Jian et al. (16), who found that apoptosis inhibition reduced calcification, but not the formation of nodules. It is possible that these differences arose from the different source of the VICs, timing of inhibitor application, type of inhibitor used, and timing of end-point nodule analysis.
The decreased cell number with U-0126 administration was not due to an increase in necrotic cell death (data not shown), nor an increase in apoptosis (Fig. 8 shows a decrease in apoptosis), and was thus likely due to decreased cell proliferation. VIC differentiation to a myofibroblast phenotype is often associated with an increase in proliferation (24), and cell proliferation is considered a hallmark feature of aortic valve disease (37). Although the images in Fig. 3 suggest a sparser cell population on untreated TCPS and FB relative to U-0126-treated conditions, Fig. 6 provides quantitative evidence against this observation. A high density of viable cells is found in the nodules and precalcific “ridges” formed in VIC cultures (42, 50), and the three-dimensional nature of these cell-rich nodules can lead to the appearance of low culture confluency, despite high cell numbers. Overall, the present findings that calcified cultures were associated with both increased apoptosis and increased proliferation are consistent with many previous studies (28, 37, 40). Ultimately, however, it remains to be discovered whether the reduction of VIC proliferation, such as that achieved upon treatment with U-0126, can directly influence the ability of VICs to form nodules in these cultures. The highly calcifying, untreated TCPS condition had a similar number of cells at day 5 as the minimally calcifying Coll and FN substrates (not shown). Thus, while the proliferation data help us to better understand the impact of ERK pathway inhibition on VIC behavior, it is not yet clear how to directly relate this information to nodule formation.
Lastly, ERK pathway inhibition exhibited a powerful effect on VIC phenotype. Numerous myofibroblastic and osteoblastic markers were elevated in calcifying VIC cultures, but treatment of those cultures with U-0126 rendered them more phenotypically similar to the noncalcifying VIC condition. Thus, even though VICs were placed in an environment that normally promotes nodule formation and emergence of osteoblast-like and myofibroblastic VIC phenotypes, inhibition of ERK activation was able to preserve the more quiescent, noncalcifying VIC phenotype.
Conclusions.
Development of therapies for the prevention and treatment of calcific valvular disease would be greatly facilitated by better understanding valvular disease on a molecular level. Our current work clearly demonstrates that the MAPK/ERK pathway is involved in the process of VIC calcification and that inhibition of this pathway can dramatically reduce calcification and help VICs to maintain a relatively quiescent phenotype. Of course, the ERK pathway does not act in isolation, as many other pathways intersect with or run parallel to ERK (10). Nevertheless, it is hoped that these findings may be applied to elucidating the etiology of valve calcification, identifying targets for blocking calcification, and designing biomaterial environments for the creation of engineered valve tissue.
GRANTS
Funding for this work was provided by a National Science Foundation Career Award (CBET-0547374) to K. S. Masters and a Herb Fellowship to X. Gu.
REFERENCES
- 1.Anger T, El-Chafchak J, Habib A, Stumpf C, Weyand M, Daniel WG, Hombach V, Hoeher M, Garlichs CD. Statins stimulate RGS-regulated ERK-1/2 activation in human calcified and stenotic aortic valves. Exp Mol Pathol 85: 101–111, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Butcher JT, Nerem RM. Porcine aortic valve interstitial cells in three-dimensional culture: comparison of phenotype with aortic smooth muscle cells. J Heart Valve Dis 13: 478–485, 2004. [PubMed] [Google Scholar]
- 3.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. J Am Coll Cardiol 47: 1707–1712, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cushing MC, Liao JT, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor-beta1 interactions with matrix molecules. Matrix Biol 24: 428–437, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Cushing MC, Mariner PD, Liao JT, Sims EA, Anseth KS. Fibroblast growth factor represses Smad-mediated myofibroblast activation in aortic valvular interstitial cells. FASEB J 22: 1769–1777, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding HT, Wang CG, Zhang TL, Wang K. Fibronectin enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells via ERK pathway. J Cell Biochem 99: 1343–1352, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273: 18623–18632, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan TC, Pandit A. Living artificial heart valve alternatives: a review. Eur Cell Mater 6: 28–45, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Franceschi RT, Ge C, Xiao G, Roca H, Jiang D. Transcriptional regulation of osteoblasts. Ann N Y Acad Sci 1116: 196–207, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol 176: 709–718, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giancotti FG, Tarone G. Positional control of cell fate through joint integrin/receptor protein kinase signaling. Annu Rev Cell Dev Biol 19: 173–206, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Harpel PC, Gordon BR, Parker TS. Plasmin catalyzes binding of lipoprotein (a) to immobilized fibrinogen and fibrin. Proc Natl Acad Sci USA 86: 3847–3851, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, Itoh K. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res 17: 1785–1794, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Hinton RB Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, and Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res 98: 1431–1438, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem 275: 9645–9652, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 75: 457–465, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Jian B, Xu J, Connolly J, Savani RC, Narula N, Liang B, Levy RJ. Serotonin mechanisms in heart valve disease I: serotonin-induced up-regulation of transforming growth factor-beta1 via G-protein signal transduction in aortic valve interstitial cells. Am J Pathol 161: 2111–2121, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson CM, Hanson MN, Helgeson SC. Porcine cardiac valvular subendothelial cells in culture: cell isolation and growth characteristics. J Mol Cell Cardiol 19: 1185–1193, 1987. [DOI] [PubMed] [Google Scholar]
- 19.Kaden JJ, Kilic R, Sarikoc A, Hagl S, Lang S, Hoffmann U, Brueckmann M, Borggrefe M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med 16: 869–872, 2005. [PubMed] [Google Scholar]
- 20.Khatiwala CB, Peyton SR, Metzke M, Putnam AJ. The regulation of osteogenesis by ECM rigidity in MC3T3–E1 cells requires MAPK activation. J Cell Physiol 211: 661–672, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Knight RL, Wilcox HE, Korossis SA, Fisher J, Ingham E. The use of acellular matrices for the tissue engineering of cardiac valves. Proc Inst Mech Eng [H] 222: 129–143, 2008. [DOI] [PubMed] [Google Scholar]
- 22.Kono SJ, Oshima Y, Hoshi K, Bonewald LF, Oda H, Nakamura K, Kawaguchi H, Tanaka S. Erk pathways negatively regulate matrix mineralization. Bone 40: 68–74, 2007. [DOI] [PubMed] [Google Scholar]
- 23.Liao XB, Zhou XM, Li JM, Yang JF, Tan ZP, Hu ZW, Liu W, Lu Y, Yuan LQ. Taurine inhibits osteoblastic differentiation of vascular smooth muscle cells via the ERK pathway. Amino Acids 34: 525–530, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol 171: 1407–1418, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mann BK, West JL. Cell adhesion peptides alter smooth muscle cell adhesion, proliferation, migration, and matrix protein synthesis on modified surfaces and in polymer scaffolds. J Biomed Mater Res 60: 86–93, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Mohler ER 3rd. Mechanisms of aortic valve calcification. Am J Cardiol 94: 1396–1402, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Mohler ER 3rd, Chawla MK, Chang AW, Vyavahare N, Levy RJ, Graham L, Gannon FH. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis 8: 254–260, 1999. [PubMed] [Google Scholar]
- 28.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation 103: 1522–1528, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arterioscler Thromb Vasc Biol 29: 246–253, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more). Arterioscler Thromb Vasc Biol 26: 1721–1728, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Otto CM. Calcification of bicuspid aortic valves. Heart 88: 321–322, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pho M, Lee W, Watt DR, Laschinger C, Simmons CA, McCulloch CA. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol 294: H1767–H1778, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res 87: 1055–1062, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis 13: 841–847, 2004. [PubMed] [Google Scholar]
- 35.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104: 2525–2532, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Radcliff K, Tang TB, Lim J, Zhang Z, Abedin M, Demer LL, Tintut Y. Insulin-like growth factor-I regulates proliferation and osteoblastic differentiation of calcifying vascular cells via extracellular signal-regulated protein kinase and phosphatidylinositol 3-kinase pathways. Circ Res 96: 398–400, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Rajamannan NM. Calcific aortic stenosis: lessons learned from experimental and clinical studies. Arterioscler Thromb Vasc Biol 29: 162–168, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation 112: I229–I234, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 107: 2181–2184, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, Singh RJ, Stone NJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation 105: 2660–2665, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren J, Fang CX. Small guanine nucleotide-binding protein Rho and myocardial function. Acta Pharmacol Sin 26: 279–285, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez KJ, Masters KS. Regulation of valvular interstitial cell calcification by components of the extracellular matrix. J Biomed Mater Res. In Press. [DOI] [PMC free article] [PubMed]
- 43.Roy J, Kazi M, Hedin U, Thyberg J. Phenotypic modulation of arterial smooth muscle cells is associated with prolonged activation of ERK1/2. Differentiation 67: 50–58, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Salasznyk RM, Klees RF, Hughlock MK, Plopper GE. ERK signaling pathways regulate the osteogenic differentiation of human mesenchymal stem cells on collagen I and vitronectin. Cell Commun Adhes 11: 137–153, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Setola V, Dukat M, Glennon RA, Roth BL. Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Mol Pharmacol 68: 20–33, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest 115: 1210–1220, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simmons CA, Nikolovski J, Thornton AJ, Matlis S, Mooney DJ. Mechanical stimulation and mitogen-activated protein kinase signaling independently regulate osteogenic differentiation and mineralization by calcifying vascular cells. J Biomech 37: 1531–1541, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. Int J Biochem Cell Biol 35: 113–118, 2003. [DOI] [PubMed] [Google Scholar]
- 49.Tsuda T, Ohmori Y, Muramatsu H, Hosaka Y, Takiguchi K, Saitoh F, Kato K, Nakayama K, Nakamura N, Nagata S, Mochizuki H. Inhibitory effect of M50054, a novel inhibitor of apoptosis, on anti-Fas-antibody-induced hepatitis and chemotherapy-induced alopecia. Eur J Pharmacol 433: 37–45, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res 95: 253–260, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Willems IE, Havenith MG, Smits JF, Daemen MJ. Structural alterations in heart valves during left ventricular pressure overload in the rat. Lab Invest 71: 127–133, 1994. [PubMed] [Google Scholar]
- 52.Xu J, Jian B, Chu R, Lu Z, Li Q, Dunlop J, Rosenzweig-Lipson S, McGonigle P, Levy RJ, Liang B. Serotonin mechanisms in heart valve disease II: the 5-HT2 receptor and its signaling pathway in aortic valve interstitial cells. Am J Pathol 161: 2209–2218, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yee KL, Weaver VM, Hammer DA. Integrin-mediated signalling through the MAP-kinase pathway. IET Syst Biol 2: 8–15, 2008. [DOI] [PubMed] [Google Scholar]