

Abstract
The extracellular calcium-sensing receptor CaSR is expressed in blood vessels where its role is not completely understood. In this study, we tested the hypothesis that the CaSR expressed in vascular smooth muscle cells (VSMC) is directly involved in regulation of blood pressure and blood vessel tone. Mice with targeted CaSR gene ablation from vascular smooth muscle cells (VSMC) were generated by breeding exon 7 LoxP-CaSR mice with animals in which Cre recombinase is driven by a SM22α promoter (SM22α-Cre). Wire myography performed on Cre-negative [wild-type (WT)] and Cre-positive SM22αCaSRΔflox/Δflox [knockout (KO)] mice showed an endothelium-independent reduction in aorta and mesenteric artery contractility of KO compared with WT mice in response to KCl and to phenylephrine. Increasing extracellular calcium ion (Ca2+) concentrations (1–5 mM) evoked contraction in WT but only relaxation in KO aortas. Accordingly, diastolic and mean arterial blood pressures of KO animals were significantly reduced compared with WT, as measured by both tail cuff and radiotelemetry. This hypotension was mostly pronounced during the animals' active phase and was not rescued by either nitric oxide-synthase inhibition with nitro-l-arginine methyl ester or by a high-salt-supplemented diet. KO animals also exhibited cardiac remodeling, bradycardia, and reduced spontaneous activity in isolated hearts and cardiomyocyte-like cells. Our findings demonstrate a role for CaSR in the cardiovascular system and suggest that physiologically relevant changes in extracellular Ca2+ concentrations could contribute to setting blood vessel tone levels and heart rate by directly acting on the cardiovascular CaSR.
Keywords: calcium-sensing receptor, CaSR, vascular smooth muscle cells, blood pressure regulation, G protein-coupled receptor, blood vessel tone regulation
the extracellular calcium -sensing receptor CaSR was the first G protein-coupled receptor identified that has an ion, Ca2+, as its primary physiological agonist. The CaSR was initially found in the parathyroid glands, where it acts as a controller of free ionized extracellular Ca2+ concentration ([Ca2+]o) in the blood via the regulation of parathyroid hormone (PTH) secretion (6). Systemic administration of pharmacological modulators of the CaSR has been shown to affect blood pressure in various animal models (50). However, it is unknown whether these effects are due to systemic or local actions of the CaSR. Indeed, the CaSR expression is found in the vasculature (7, 54, 59), but a definitive role for this receptor in blood pressure regulation has never been firmly established. In the vascular endothelium, CaSR activation is thought to stimulate nitric oxide (NO) production (34, 59) and/or endothelium-derived hyperpolarization (EDH) (54). In rat subcutaneous small arteries, Ca2+o evokes a biphasic response with contraction followed by dilatation in a concentration-dependent manner (40). Furthermore, activation of the endothelial CaSR in mouse aortic segments induces endothelium-dependent and -independent relaxations (34). Alteration in CaSR expression or function in vascular smooth muscle cells (VSMC) has been linked to pathological conditions such as microvascular complications during diabetes (34), vascular calcification (1), and idiopathic pulmonary arterial hypertension (55). Allosteric CaSR activators, termed calcimimetics, have been on the market since 2004 for the treatment of hyperparathyroidism secondary to kidney failure (3). In patients with chronic kidney disease (60), and in a rat model of surgically induced chronic kidney disease (39), calcimimetic treatment is associated with antihypertensive effects. However, because calcimimetics are phenylalkylamine derivatives, their ability to reduce blood pressure has been ascribed to potential off-target effects on L-type Ca2+ channels (37, 52). Given the importance of the CaSR in many physiological processes (43), the aim of this study was to determine whether the CaSR in VSMC contributes directly to blood pressure regulation. For this purpose, we generated animals with targeted CaSR ablation from smooth muscle cells by breeding mice with LoxP sites flanking exon seven of the CaSR (13) with mice in which Cre recombinase expression is transcriptionally regulated by the SM22α promoter (30). This approach allowed us to study animals with targeted deletion of the CaSR from VSMC throughout the vascular system, visceral smooth muscle, and cardiac cells but not skeletal muscle cells (30, 56).
MATERIALS AND METHODS
Chemicals and reagents.
All chemicals and reagents were obtained from Sigma-Aldrich (Poole, Dorset, UK) and Life Technologies (Paisley, Strathclyde, UK) unless otherwise stated.
Animals.
Commercially available SM22α-Cre+ mice (Jackson Immunoresearch Laboratories, Bar Harbor, ME) were crossed with exon7-LoxP CaSR sites (13). The CaSR-LoxP strain was generated from C57BL/6 × SVJ129 backcrossed with C57BL/6 for eight generations. CaSR-LoxP × SM22α-Cre mice were inbred for at least three generations before being used for experiments. Genotyping for CaSR-LoxP sites and Cre was performed as described elsewhere (13, 25, 56). All animal work was conducted according to UK legislation and all procedures were independently reviewed and approved by the UK Home Office (Home Office Project License 30/3007) and conformed to the guidelines from directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Euthanasia for tissue collection was performed by cervical dislocation according to UK legislation [A(SP)A 1986, Schedule 1].
Genotyping.
DNA from ear biopsies was extracted using DirectPCR Ear DNA Extraction System (Viagen, Los Angeles, CA) with proteinase K (Bioline, London, UK) according to the manufacturer's instructions. Genotype was determined using the SM22α promoter and Cre coding region primers from (25) and the LoxP primers already described (13). Primer sequences were as follows: SM22-Pro: CAGACACCGAAGCTACTCTCCTTCC; SM22-Cre: CGCATAACCAGTGAAACAGCATTGC; CaSR fl up: GTGACGGAAAACATA CTGC; and CaSR fl low: CGAGTACAGGCTTTGATGC. BioTaq Taq-polymerase (Bioline) was used for polymerase chain reaction (PCR). Final concentrations per reaction were as follows: 100 nM per primer, 1 mM dNTPs (Bioline), 4 mM MgCl2, 2 U polymerase, and 1.6% ear lysate. PCR conditions were as follows: 5-min initial denaturation at 95°C, 40 cycles of 30 s 95°C, 60 s 50°C (Cre) or 47°C (LoxP), 120 s 72°C, and a final elongation of 10 min at 4°C. Amplification products for Cre were visualized using agarose gel electrophoresis with an expected amplicon size of 500 bp in case of at least one Cre-positive allele. Amplification products for LoxP were visualized using polyacrylamide gel electrophoresis with an expected amplicon size of 167 bp for floxed alleles and 133 bp for nonfloxed alleles.
Real-time reverse transcription PCR.
Real-time reverse transcription PCR (RT-qPCR) was performed on endothelium-denuded aortas as described previously (22). Gene expression was calculated based on the 2−ΔΔCt method (33), normalized to the expression of the housekeeping gene β-actin. Samples with Ct values ≥31 for Pecam-1 were considered to be devoid of endothelial contamination. Primers were as follows: calcium-sensing receptor exon 6–7 (CaSR6-7), forward: GTGGTGAGACAGATGCGAGT and reverse: GCCAGGAACTCAATCTCCTT; β-actin, forward: TCCTAGCACCATGAAGATCA and reverse: CCACCGATCCACACAGAGTA; and Pecam-1, forward: AGACATGGAATACCAGTGCAGAG and reverse: ACAGGATGGAAATCACAACTTCAT.
Western blot.
CaSR immunoblot of microsomal membrane fractions from endothelium-denuded aortas or lysate from HEK-293 cells stably expressing the human CaSR (positive control) was performed as described previously (53) using an antibody directed against the amino terminus of the CaSR (Anaspec, Freemont, CA). Expression of α-smooth muscle actin was detected using a mouse monoclonal antibody.
Intracellular calcium imaging.
The thoracic aorta was dissected and cleaned in cold DMEM-F12 supplemented with 1% penicillin/streptomycin, cut open for endothelium removal, and cut into 1- to 2-mm segments, subsequently incubated for 30 min at 37 °C in 5% CO2 in an enzymatic digestion solution containing DMEM-F12 supplemented with 1 mg/ml papain, 1 mg/ml collagenase from Clostridium histolyticum, 1 mg/ml fatty acid free BSA, 1 mg/ml DTT, and 0.5 mg/ml soybean trypsin inhibitor. The cell suspension was seeded onto glass coverslips coated with poly-d-lysine and allowed to settle for 30 min at 37 °C in 5% CO2, after which cells were incubated for 30 min with extracellular solution (135 mM NaCl, 10 mM glucose, 5 mM KCl, 5 mM HEPES, 1.2 mM MgCl2, and 1 mM CaCl2, pH 7.4) containing 1.4 μM fura-2 AM, dissolved in DMSO. Intracellular calcium (Ca2+i) recordings were performed as described previously (46).
Wire myography.
Wire myography on mouse aortas and mesenteric arteries (MA) was performed as described elsewhere (5, 17, 19). In brief, thoracic aortas and second order MA of 4- to 6-mo-old mice were carefully dissected in cold buffer (118 mmol/l NaCl, 3.4 mmol/l KCl, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4, 25 mmol/l NaHCO3, 11 mmol/l glucose, and 1 mmol/l CaCl2, bubbled with 95% O2-5% CO2) and cut into 2-mm segments. For the study of vascular responses to the calcimimetic NPS R-568, the extracellular solution contained 1.5 mM CaCl2. The vessel rings were then carefully mounted in a wire myograph (Danish MyoTechnology, Aarhus, Denmark) using two gold-plated tungsten wires (diameter = 25 μm) for MA or stainless steel wires (diameter = 40 μm) for aortas. The vessels were let to equilibrate for 20 min at 37°C and then gradually stretched (up to 2 mN for mesenteric arteries and 5 mN for aorta). The plateau values after each stretch were used to calculate the lumen diameter that would give a target transmural pressure, which for MA was set as 70 mmHg and for aortas was set as 100 mmHg. After normalization, vessels were left to equilibrate for another 30 min and tested for viability using phenylephrine (PE) and acetylcholine (ACh). Where stated, the endothelium was removed by gentle rubbing of the vessel lumen with a clean human hair, or an air bubble was pushed through the lumen of the vessel. Lack of relaxation in response to 10 μM ACh was used to confirm the successful endothelial denudation. High KCl (40 mM) was used as an alternative, depolarizing, vasoconstrictor (11, 41, 51). When required, the vessels were pretreated with nitro-l-arginine methyl ester (l-NAME) for at least 20 min before the experiment, and concentration-response curves were obtained by cumulative addition of rising concentrations of the agonists [PE, ACh, CaCl2, and S-ntroso-N-acetylpenicillamine (SNAP)] directly to the bath. Solutions were changed every 20 min to avoid alterations of osmolarity. To calculate the percentage change in contractility of KO vessels compared with WT, each contraction data point was normalized to the average of individual maximal contraction values of WT vessels. For the dilatation experiments, vessels were first precontracted with PE, and then vasodilators were added to the bath. Zero percent relaxation is the level of tension before the vasorelaxant agonist was added, and 100% relaxation reflects the return of the tension to the basal level.
Tail-cuff and radiotelemetry measurements.
The blood pressure of male, age-matched animals (4–9 mo) was measured by tail cuff on awake, conscious animals during daytime using a two-channel CODA system (Kent Scientific, Torrington, CT), following the manufacturer's instructions and using 20 acclimatization and 10 measurement cycles.
Radiotelemetry was performed on 3-mo-old mice using implantable probes (4). Mice were anesthetized by subcutaneous injections with a combination of fluanisone (10 mg/kg body wt), fentanyl (0.3 mg/kg body wt), and midazolam (1 mg/kg body wt) and placed in dorsal recumbence on a thermostatically controlled heating platform for maintaining body temperature at 37.5°C. The body hair was liberally removed from the skin on the neck, and the skin was disinfected by isopropyl alcohol. A 1- to 1.5-cm-long midline incision through the skin on the neck was made, and the mandibular glands were carefully separated. The carotid artery along the left side of the trachea was carefully isolated with fine-tipped forceps from the surrounding tissues. The distal side of the carotid artery (just before the bifurcation) was occluded permanently with 6-0 nonabsorbable suture, and another suture was placed proximately for temporal occlusion of blood flow to allow for placement of the catheter. The additional suture was placed in the middle to hold the catheter in place after the cannulation of the artery. The artery was pierced using the 25-gauge bent needle just proximately the permanent ligation suture and the catheter was inserted into the vessel using Vessel Cannulation Forceps (Data Sciences International). The catheter was advanced into the artery until it reached the occlusion suture, the loose middle suture was tightened, and the occlusion suture was released. The catheter was then advanced further until the sensing region was positioned in the aortic arch. The occlusion suture was tightened and a subcutaneous pocket was made by inserting small surgical scissors. A HD-X11 radiotelemetry transmitter (Data Sciences International, New Brighton, MN) was then placed in the pocket. The terminal end of the positive lead was positioned with a small hemostat subcutaneously from the neck incision to the left caudal rib region (∼1 cm to the left of the xyphoid process). The negative lead terminal end was positioned with a small hemostat subcutaneously from the neck incision to the right pectoral muscle. The skin incision was closed using 6-0 nonabsorbable suture. A painkiller (0.2 ml/kg; Temgesic; Schering-Plough Europe) was injected subcutaneously at the end of operation. Mice were allowed to recover for 2 days before housing them in metabolic cages where they were kept on control diet for 4 days. The standard (control) diet was then changed to a high-salt diet (containing 4% NaCl) for a period of 5 days. For reasons of animal welfare, mice were then transferred back in normal housing cages and kept on standard diet for a washout period of 3 days, after which drinking water of the mice was supplemented with 0.5 g/l l-NAME.
Telemetry signals from the probes were recorded for the whole duration of the experiment in intervals of 5 min in quintuplicates resulting in 60 measurements per hour. Registration was performed with Dataquest A.R.T software 4.3. Analyses were performed with Ponemah analyses software 5.00 (all Data Sciences International) and Microsoft Excel 2010. Raw values were concentrated to hourly datapoints. For statistical analysis, 3-h intervals of the raw data were averaged for nighttime (active phase, 2000–2300) and daytime (resting phase, 800–1100). The standard diet vs. high-salt diet and standard diet vs. 0.5 g/l l-NAME diet phases were treated as two separate experiments because of the different housing conditions. For graphical representation only, traces were calculated as rolling average over 4 h.
Cardiac cine-MRI.
Cardiac cine-MRI scans of aged (14-mo-old) mice were acquired using a Bruker Biospec 94/20 9.4 Tesla small bore (20 cm) MRI spectrometer equipped with S116 high-performance gradient insert and Avance II electronics (Bruker, Ettlingen, Germany). Scans were acquired using a 72-mm transmit/receive quadrature polarized birdcage coil. Mice were kept under general anesthesia using 1–5% isoflurane in a 40% O2/air at 1.2 l/min during scanning. Animals were ECG, respiration, and temperature monitored using a Model 1025 monitoring and gating system (SA Instruments, Stony Brook, NY). The temperature of the animals was kept at 37°C using temperature-controlled bath feeding into the restraining scaffold. T2 weighted, respiratory and cardiac gated, fast-imaging with steady-state precession pro-cine scans were performed with a field of view of 32 × 32 mm, an echo time of 1.228 ms, a repetition time of 5 ms, a band width of 125 kHz, and slice thickness of 1–1.5 mm were collected from apex to the aortic arch using Paravision 5.0 (Bruker). Gating was performed using the Small Animal Monitor software and monitoring equipment according to the manufacturer's instructions (SA Instruments). Scans performed for each mouse included a Tri-axial pilot orientation scan, a central long-axis cine-scan, and short-axis cinescans covering subapical to suprabasal image plane. Scans were analyzed using the academic version of Segment 1.9 R2626 (http://segment.heiberg.se) (21) by one person blinded to the genotype of the animals. Functional parameters were assessed from multislice short-axes cine MR images by manual markup of left-ventricular endocardium and epicardium in the first timeframe of the slice showing the papillary muscles. The markup was then propagated in time and adjusted to fit the new timeframe. After the markup of the slice was completed, the markup for the whole cine-cycle was propagated apically and basally and contours were again adjusted to fit the respective scans. End diastole and systole were defined as the timeframe showing highest and lowest ventricular volume using the autodetection feature of the Segment software. For wall thickness measurements, the thicknesses of the second and third apical slice in end systole and diastole, measured with the respective tool in the Segment software, were averaged. Based on the long axis scans, hearts of KO animals showing full apical end-systolic closure were selected as remodeled subgroup.
Histomorphology and fibrosis staining.
Sections from paraffin-embedded hearts and aortas were stained with hematoxylin and eosin (H&E). To detect fibrosis in heart sections of aged mice, collagen deposition was visualized in 8-μm cryosections of paraformaldehyde-fixed hearts stained with picrosirius-red mixture. The 8-μm cryosections of paraformaldehyde-fixed (4% in PBS for 4 h) hearts of adult mice were thawed in PBS for 30 min and washed twice in distilled water for 2 min. Sections were then prestained with 1% fast green for 15 min, washed in 1% acetic acid for 15 s, and twice in distilled water for 2 min. Final staining was achieved by incubation in a solution of 0.1% fast green, 0.1% direct red, and 1.3% picric acid for 2:30 h. Sections were then washed twice in 1% acetic acid for 15 s and distilled water for 2 min and finally dehydrated in rising concentrations of ethanol, cleared in xylene, and mounted in DPX:Xylene 4:1, resulting in green staining for all tissues and red staining for fibrotic tissue. Fibrotic area was quantified using CellProfiler 2.1.1 (28) by one person blinded to the genotype of the animals. All epicardial and endocardial positive (red) staining was manually removed along with other image artifacts (e.g., tissue folding). An image analysis pipeline was run to separate different shades of red pixels (fibrosis positive) from background tissue (green). Cardiac fibrosis percentage was calculated by dividing the number of detected red pixels per 100 positively stained pixels (red + green).
Ex vivo measurement of baseline heart rate.
Rapidly dissected hearts were retrogradely perfused in Langendorff fashion. Baseline heart rates were measured using longitudinal force tonometry (31). Six-month-old male mice were killed by cervical dislocation. The thorax was rapidly opened and the heart, lungs, and thoracic aorta were removed to ice cold Krebs buffer (118 mmol/l NaCl, 3.4 mmol/l KCl, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4, 25 mmol/l NaHCO3, 11 mmol/l glucose, and 1 mmol/l CaCl2). The aorta was cannulated with a 20-gauge hypodermic needle filled with Krebs buffer and the tip of the needle was advanced to the aortic valve. The needle was attached to the perfusion apparatus and the heart perfused in Langendorff mode (31) at a constant flow rate of 3 ml/min (Gilson Minipuls 3 peristaltic pump; Gilson, Luton, UK) with warmed (37.5°C) Krebs buffer pregassed with O2-CO2 (95-5%). Coronary perfusion pressure was measured by means of a pressure transducer (MLT 844; ADInstruments, Chalgrove, UK) located immediately before the warming coil, where a Condon mercury manometer was also located to accommodate perfusion fluid during pressure changes. A clip was attached to the apex of the heart and linked via a thread and pulley to an isometric tension transducer (50-g sensitivity range; ADInstruments) for measuring contractile tension. A resting diastolic tension of 2 g was applied at the start of the experiment and readjusted periodically. The heart was surrounded by a heated jacket (37.5°C). Isometric cardiac contractions were recorded by means of a Powerlab 8/30, Chart 5, data acquisition system (ADInstruments), and heart rate was derived from the cardiac contractions signals.
Spontaneous activity of pulmonary vein sleeve cells in lung slices.
Murine lung slices were prepared as described previously (44). In brief, 4- to 6-mo-old mice were killed by cervical dislocation and, after opening of the chest cavity, the trachea was cannulated and lungs were inflated by injecting ∼1.2 ml of low melting point agarose (37°C, 1.8% in sHBSS). The agarose was stiffened by applying cold sHBSS supplemented with 20 mM HEPES, pH 7.3 over the lungs. The stiffened lungs were cut into ∼180-μm thick slices using a Vf-300 microtome (Precisionary Instruments, Greenville, NC). Slices were kept in a humidified cell culture incubator with 5% CO2 in DMEM:F12, supplemented with 10% FBS. Slices were loaded in the dark with 20 μM Oregon-Green BAPTA-1 AM in the presence of 0.1% Pluronic F127 and 200 μM sulfobromophtalein in sHBSS at room temperature for 1 h, followed by deesterification for at least 30 min in the dark in sHBSS with 200 μM sulfobromophtalein. Slices were then mounted in a custom-built imaging chamber and imaged on a Nikon Eclipse Ti microscope, using a Nikon S Fluor ×40 oil immersion objective, with an excitation of 470 ± 15 nm. Emission was measured at 525 ± 25 nm. Images were acquired with a frame rate of 25 frames per second using OptoFluor software (Cairn Research, Faversham, UK). Image acquisition was performed for ∼20-s periods to record the frequency of the spontaneous activity. Image analysis was performed using ImageJ software (47).
Statistical analyses.
Statistical analyses were performed using GraphPad Prism 6.04 (GraphPad Software, La Jolla, CA). Statistical tests employed are stated with the respective results or in the figure legends. Where two curves were compared, asterisks above individual data points indicate point-by-point comparison by Holm-Sidak posttest of two-way ANOVA; plus-signs indicate difference of the fitted curves by extra sum-of-squares F-test and two-way ANOVA; n states the number of individual biological repeats (number of animals, cell batches, etc.).
RESULTS
Generation of SM22αCaSRΔflox/Δflox mice.
Breeding of SM22α-Cre+ mice with mice in which LoxP sites flanked exon 7 of the CaSR gene (LoxP-CaSR+/+) (13) yielded SM22αCaSRΔflox/Δflox mice (which were termed KO). Age- and sex-matched mice expressing only floxed CaSR alleles but no Cre-recombinase (SM22α-Cre−/−-LoxP-CaSR+/+) were used as controls for all experiments (termed WT). The resulting truncated Δexon7 product was shown to be nonfunctional (13), and these animals were viable and fertile (56). Representative genotyping is shown in Fig. 1A. Aortas of KO animals appeared histomorphologically normal and indistinguishable from those of WT mice of comparable ages (Fig. 1B), suggesting that CaSR deletion does not affect the histomorphology of blood vessels.
Fig. 1.
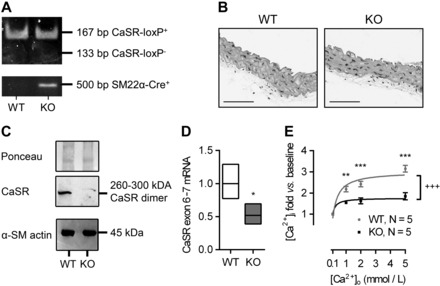
Characterization of the SM22αCaSRΔflox/Δflox mouse. A: typical genotyping of wild-type (WT) and knockout (KO) mice. B: representative histological sections of aortas from WT and KO animals (n = 4). Scale bars = 100 μm. C: Western blot analysis of calcium-sensing receptor (CaSR) expression with Ponceau staining as loading control (top and middle) and α-smooth muscle actin (α-SM actin; bottom) expression in endothelium-denuded and adventitia-removed, pooled aortas; n = 18 (WT) and 17 (KO) mice. D: relative mRNA expression of the full-length CaSR (exon 6–7 vs. WT) in endothelium-denuded, adventitia-removed aortas of WT and KO animals; mean (line) ± 1SD (box); n = 4. **P < 0.01, Student's t-test (performed on ΔΔCt values vs. WT). E: intracellular Ca2+ concentration ([Ca2+]i) in freshly isolated WT and KO VSMC exposed to increasing extracellular Ca2+ concentration ([Ca2+]o; 0.1–5 mmol/l), reported as fold changes from baseline values. Curves were fitted as hyperbolic. Data are means ± SE; n = 3 (WT) and 4–5 (KO). **P < 0.01. ***P < 0.001, two-way ANOVA with Holm-Sidak posttest. +++P < 0.001, extra sum-of-squares F-test for curve comparison.
Molecular and functional CaSR expression in the vasculature was found to be significantly reduced in KO compared with WT animals. Bona fide, CaSR-like immunoreactivity, corresponding to the molecular mass of the fully mature dimeric form of the receptor (53), was reduced in membrane fractions from endothelium-denuded KO aortas compared with that seen in WT, as shown by Western blot using a polyclonal antibody that preferentially detects the dimeric form of the CaSR. CaSR deletion from VSMC did not affect the expression levels of α-smooth muscle actin (Fig. 1C). RT-qPCR carried out in endothelium- and adventitia-denuded aortas from WT and KO animals showed that expression of the CaSR (exon 6–7) was significantly reduced in KO vessels compared with WT (Fig. 1D). Classically, CaSR activation is linked to phosphoinositide turnover and an increase in intracellular Ca2+ concentration ([Ca2+]i) (6). In fura-2 loaded VSMC isolated from WT and KO mouse aortas, increasing [Ca2+]o from 0.1 to 5 mmol/l evoked a concentration-dependent increase in [Ca2+]i in WT VSMC, which was significantly smaller in KO VSMC (Fig. 1E).
Ex vivo blood vessel contractility is impaired in SM22αCaSRΔflox/Δflox mice.
Previous studies have suggested a role for the CaSR in vascular tone regulation (50). To test this hypothesis directly, we performed ex vivo tension measurements on isolated aortas and MA from WT and KO mice. Rings of aortas and MA of KO animals showed a significantly smaller contraction in response to a depolarizing stimulus (i.e., high K+) compared with aortic rings from WT (Fig. 2, A and B). A comparable level of reduction in KO aortas and MA contractility was observed when PE (1 nmol/l-30 μmol/l), an agonist of the Gq/11 protein-coupled α1-adrenoceptor, was employed (Fig. 2, C and D). Except for the responses to PE, where potency was slightly but significantly reduced in KO aortas and MA compared with WT control, EC50 values in all other experiments remained unchanged (Table 1). In contrast, endothelium-dependent relaxation of PE-precontracted blood vessels in response to ACh was modestly, but significantly, enhanced in KO aortas (Fig. 2E), and reduced in KO MA (Fig. 2F).
Fig. 2.
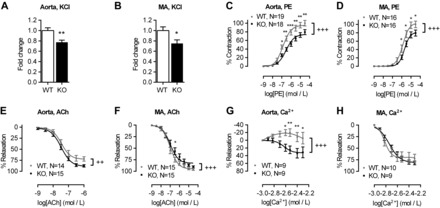
Blood vessel contractility of SM22αCaSRΔflox/Δflox mice is impaired. Aorta (WT, n = 33; KO, n = 30) and mesenteric artery (MA) responses to high K+ (KCl; 40 mmol/l; WT, n = 33; KO, n = 34; A and B), phenylephrine (PE; 1 nmol/l-30 μmol/l; C and D), acetylcholine (ACh, 1 nmol/l to 30 μmol/l, PE-preconstricted to ∼60% max; E and F), and Ca2+o (1–5 mmol/l, PE-preconstricted to ∼60% max; G and H). Results were normalized to the averaged maximum control response in WT. Curves were fitted as sigmoidal concentration-response (for PE and ACh) or 3rd-order polynomial (for Ca2+). Data are means ± SE. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test, or two-way ANOVA with Holm-Sidak posttest. ++P < 0.01, +++P < 0.001, extra sum-of-squares F-test for curve comparison.
Table 1.
EC50 values from wire myography experiments
| Genotype |
||
|---|---|---|
| Treatment | WT | KO |
| Aorta | ||
| PE | −6.871 ± 0.083 (n = 19) | −6.534 ± 0.093* (n = 18) |
| PE + l-NAME | −7.450 ± 0.106 (n = 6) | −7.281 ± 0.145ns (n = 6) |
| PE-E | −7.491 ± 0.164 (n = 9) | −7.353 ± 0.303ns (n = 8) |
| ACh | −7.469 ± 0.095 (n = 14) | −7.475 ± 0.067ns (n = 15) |
| ACh + l-NAME | −6.150 ± 0.142 (n = 4) | −6.146 ± 0.263ns (n = 5) |
| SNAP | −6.892 ± 0.167 (n = 5) | −6.886 ± 0.279ns (n = 5) |
| Calcium | NA | −2.636 ± 0.081 (n = 9) |
| MA | ||
| PE | −5.875 ± 0.080 (n = 16) | −5.702 ± 0.070* (n = 16) |
| PE + l-NAME | −6.047 ± 0.070 (n = 11) | −5.928 ± 0.095ns (n = 9) |
| PE-E | −6.352 ± 0.111 (n = 10) | −6.263 ± 0.120ns (n = 11) |
| ACh | −7.073 ± 0.056 (n = 15) | −6.898 ± 0.072ns (n = 15) |
| ACh + l-NAME | −6.374 ± 0.161 (n = 9) | −6.148 ± 0.182ns (n = 7) |
| SNAP | −6.717 ± 0.134 (n = 6) | −6.646 ± 0.071ns (n = 6) |
| Calcium | −2.832 ± 0.045 (n = 9) | −2.782 ± 0.046ns (n = 10) |
Values presented as log(EC50) in mol/l ± SE. EC50 values from wire myography experiments presented in Figs. 2 and 4 (see figures for experimental conditions), which were fitted with sigmoidal concentration-response curves, with exception of extracellular Ca2+ concentration-response curves for wild-type (WT) aortas that could not be fitted with a sigmoidal concentration-response curve.
KO, knockout; MA, mesenteric arteries; PE, phenylephrine; PE-E, PE-endothelium denuded; l-NAME, nitro-l-arginine methyl ester; snap, S-ntroso-N-acetylpenicillamine.
P < 0.05, extra sum-of-squares F-test; ns, not significant.
In line with this reduction in contractility in response to contractile stimuli, we observed a reduction in Ca2+i signaling in VSMC isolated from KO mice compared with WT cells in the presence of depolarizing concentrations of K+ (60 mmol/l KCl, KO vs. WT: 3.81 ± 0.62 vs. 1.90 ± 0.53 fold vs. baseline; n = 5; P < 0.05, Student's t-test).
To investigate the effects of the physiological CaSR agonist Ca2+ on the vascular tone, we precontracted aortas and MA from WT and KO animals with PE to ∼60% of maximal tone and then cumulatively added rising concentrations of Ca2+o (from 1 to 5 mmol/l). Aortas of WT animals displayed significantly different response to Ca2+o than aortas from KO animals, showing a moderate contraction to Ca2+o that reached 19.9 ± 7.8% at 2.5 mmol/l Ca2+o (P < 0.05, one sample t-test vs. baseline), while aortas from KO animals showed only relaxation that reached 33.4 ± 11.8% in the presence of 4 mmol/l Ca2+o (P < 0.05, one sample t-test vs. baseline; Fig. 2G). In contrast, MA displayed only relaxation to rising [Ca2+]o that was of comparable magnitude in vessels from WT and KO animals (Fig. 2H). To determine CaSR specificity of the responses observed in the aortas, aortic rings were preconstricted with PE to ∼ 60% of maximal tone and then treated with increasing concentrations of the calcimimetic NPS R-568 (10 nM-10 μM). NPS R-568 evoked contraction followed by relaxation in WT aortas while KO aortas were insensitive to low concentrations of calcimimetic (10 nM to 300 nM) and only responded with vasodilation at concentrations >1 μM NPS R-568 (Fig. 3A). After removal of the endothelium, aortas from WT mice still showed a contractile response to rising concentrations of NPS R-568 but did not show any relaxation even at very high levels of the calcimimetic. Endothelium-denuded aortas from KO animals were completely insensitive to NPS R-568 (Fig. 3B).
Fig. 3.
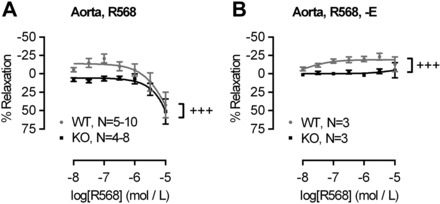
Pharmacological activation of the CaSR in WT and KO blood vessels. Responses to NPS R-568 (10 nmol/l to 10 μmol/l) in intact (A) or endothelium-denuded (B) aortas. Curves were fitted as sigmoidal concentration response. Data are means ± SE. +++P < 0.001, two-way ANOVA.
Inhibition of nitric oxide synthase (NOS) with l-NAME (100 μmol/l; Fig. 4, A and B) or endothelial denudation (Fig. 4, C and D) did not abolish the reduction in contractility in KO aortas and MA compared with WT. These results indicate that the impaired vascular contractility observed in KO animals was not due to an increase in endothelium-mediated relaxation. Abrogation of NO signaling with l-NAME led to ACh-induced contraction (16, 27, 42) in both WT and KO aortas, which again was significantly smaller in aortas from KO animals compared with WT (Fig. 4E, negative relaxation = contraction). In MA, l-NAME abrogated the differences in the relaxation response to ACh between KO and WT (Fig. 4F). Endothelium-independent relaxation following treatment with the NO donor SNAP (1 nmol/l - 30 μmol/l) showed no difference in ACh sensitivity between WT and KO endothelium-denuded aortas and MA (Fig. 4, G and H). EC50 values of all agonists in these series of experiments were unchanged between WT and KO animals (Table 1).
Fig. 4.
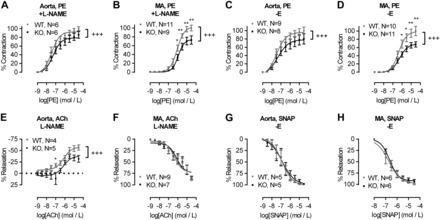
Role of nitric oxide (NO) in endothelium-dependent relaxation in WT and KO blood vessels. Aorta and MA responses to PE (1 nmol/l to 30 μmol/l) in the presence of the NO synthase inhibitor nitro-l-arginine methyl ester (l-NAME; 100 μmol/l; A and B) or endothelium denuded (-E; C and D). E and F: aorta and MA responses to ACh (1 nmol/l to 30 μmol/l) in the presence of l-NAME (100 μmol/l). G and H: relaxation of endothelium-denuded (-E) aortas and MA from WT and KO mice to increasing concentrations of the NO donor S-ntroso-N-acetylpenicillamine (SNAP). Curves were fitted as sigmoidal concentration-response. Data are means ± SE. *P < 0.05, **P < 0.01, two-way ANOVA with Holm-Sidak posttest. +++P < 0.001, extra sum-of-squares F-test for curve comparison.
SM22αCaSRΔflox/Δflox mice are hypotensive.
The impaired vascular contractility of KO mice suggested an effect of VSMC-CaSR deletion on blood pressure in vivo, and indeed, tail-cuff measurements in adult male mice showed that KO animals exhibited a reduction in blood pressure [∼5% systolic, 9% diastolic, and 7% mean arterial pressure (MAP) compared with WT; Fig. 5, A–C]. Longitudinal radiotelemetry experiments confirmed this reduction in systolic and diastolic blood pressure and MAP. Notably, this reduction was only apparent during nighttime, when the animals were active, but not during daytime when the animals were resting. Short-term l-NAME treatment (38) increased blood pressure in animals of both genotypes but did not abolish the hypotension of KO relative to WT mice (Fig. 5, D–F, and Table 2). Pulse height was increased in KO animals compared with WT mice (Fig. 5G), particularly following l-NAME treatment, owing to the bigger difference in diastolic than systolic pressures between the genotypes. Short-term dietary NaCl supplementation yielded no changes in systolic or diastolic blood pressure in either WT or KO animals (Table 2).
Fig. 5.
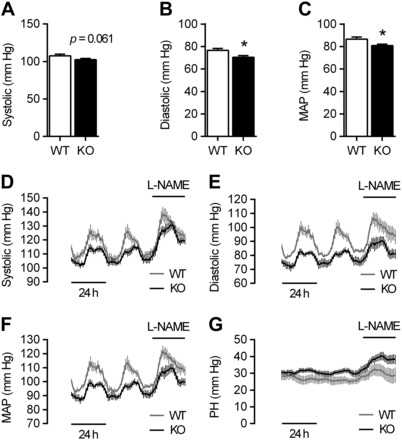
Tail-cuff and radiotelemetry blood pressure measurements of WT and KO animals. A–C: tail-cuff measurements of systolic (A), diastolic (B), and mean arterial pressures (MAP; C) of WT and KO mice. Data are means ± SE; n = 20 (WT) and 35 (KO). *P < 0.05, Student's t-test. D-G: longitudinal radiotelemetry measurements of systolic (D) and diastolic blood pressure (E), MAP (F), and pulse height (PH; G) of WT and KO mice in the presence or absence of l-NAME treatment. Data are means ± SE; n = 5 (WT and KO).
Table 2.
Physiological parameters from longitudinal radiotelemetry recordings
| Control |
Treated |
|||
|---|---|---|---|---|
| WT Control | KO Control | WT l-NAME | KO l-NAME | |
| Nighttime | ||||
| Systolic, mmHg | 127.3 ± 4.9 | 112.1 ± 2.6* | 138.7 ± 5.8 | 126.7 ± 3.1++## |
| Diastolic, mmHg | 101.4 ± 4.9 | 81.3 ± 2.2** | 106.7 ± 4.4 | 88.9 ± 3.9**+++ |
| MAP, mmHg | 114.6 ± 4.7 | 97.3 ± 2.4* | 121.8 ± 4.4 | 107.1 ± 3.4*+++# |
| PH, mmHg | 25.8 ± 2.5 | 30.8 ± 1.5 | 32.0 ± 3.4 | 37.8 ± 1.9+# |
| HR, beats/min | 621.1 ± 19.7 | 515.7 ± 16.1** | 489.3 ± 29.6 | 409.1 ± 11.4*+++### |
| dp/dt, mmHg/s | 1,934.9 ± 162.3 | 2,437.2 ± 244.3 | 1,884.5 ± 186.8 | 2,436.9 ± 236.2+ |
| Daytime | ||||
| Systolic, mmHg | 108.6 ± 2.3 | 106.6 ± 2.3 | 123.9 ± 3.7 | 118.9 ± 2.5### |
| Diastolic, mmHg | 82.7 ± 1.5 | 76.8 ± 3.4 | 95.1 ± 5.6 | 80.3 ± 3.6*+ |
| MAP, mmHg | 96.0 ± 1.60 | 92.01 ± 2.8 | 109.1 ± 4.1 | 98.9 ± 3.0+## |
| PH, mmHg | 26.0 ± 2.0 | 29.9 ± 1.9 | 28.8 ± 4.4 | 38.6 ± 3.0+ |
| HR, beats/min | 482.8 ± 23.9 | 459.7 ± 20.0 | 450.4 ± 23.5 | 372.2 ± 23.5+# |
| dp/dt, mmHg/s | 2,008.4 ± 95.1 | 2,399.2 ± 259.7 | 1,919.6 ± 279.8 | 2,647.9 ± 261.3+ |
| Control |
Treated |
|||
|---|---|---|---|---|
| WT Control | KO Control | WT High-Salt | KO High-Salt | |
| Nighttime | ||||
| Systolic, mmHg | 133.5 ± 1.8 | 126.6 ± 4.4 | 133.1 ± 4.5 | 123.5 ± 3.3+ |
| Diastolic, mmHg | 104.1 ± 1.6 | 92.4 ± 3.8** | 103.8 ± 1.0 | 90.5 ± 3.3**+++ |
| MAP, mmHg | 119.0 ± 1.3 | 109.7 ± 4.2* | 118.1 ± 2.1 | 107.1 ± 3.2*++ |
| PH, mmHg | 29.4 ± 2.0 | 34.2 ± 1.6 | 29.3 ± 3.9 | 33.0 ± 1.1 |
| HR, beats/min | 638.9 ± 2.3 | 545.3 ± 22.8** | 610.5 ± 19.8 | 515.2 ± 24.6**+++ |
| dp/dt, mmHg/s | 2,412.1 ± 109.4 | 2,628.5 ± 300.3 | 2,041.8 ± 104.6 | 2,307.4 ± 231.4 |
| Daytime | ||||
| Systolic, mmHg | 116.04 ± 3.2 | 119.4 ± 3.9 | 114.0 ± 2.6 | 113.6 ± 1.9 |
| Diastolic, mmHg | 87.15 ± 1.6 | 84.7 ± 4.1 | 85.6 ± 1.4 | 78.6 ± 2.3 |
| MAP, mmHg | 101.65 ± 2.4 | 102.0 ± 4.1 | 99.7 ± 1.5 | 95.9 ± 1.9 |
| PH, mmHg | 28.88 ± 1.8 | 34.7 ± 2.3 | 28.5 ± 2.5 | 35.0 ± 1.9+ |
| HR, beats/min | 489.0 ± 13.6 | 457.7 ± 32.8 | 469.8 ± 14.0 | 382.9 ± 9.1*++# |
| dp/dt, mmHg/s | 2,181.7 ± 96.1 | 2,451.4 ± 245.8 | 2,197.5 ± 127.3 | 2,293.6 ± 186.3 |
Data are means ± SE; n = 5. Physiological parameters from longitudinal radiotelemetry recordings (cf. Figs. 5 and 6) from WT and KO mice kept on control drinking water vs. drinking water supplemented with nitro-l-arginine methyl ester (l-NAME; 0.5 g/l) and on control diet vs. a high-salt diet (4% NaCl supplemented). MAP, mean arterial pressure; PH, pulse height; HR, heart rate. *+#P < 0.05, **++##P < 0.01, ***+++###P < 0.001, two-way ANOVA with Holm-Sidak posttest; *WT vs. KO (posttest); +WT vs. KO (ANOVA); #control vs. l-NAME/high salt (ANOVA).
SM22αCaSRΔflox/Δflox mice are bradycardic.
In addition to the impaired vascular reactivity that is associated with hypotension in the SM22αCaSRΔflox/Δflox mice, radiotelemetry experiments also revealed that heart rates of these animals were ∼5% (daytime) to 17% (nighttime) below those of WT mice (Fig. 6A and Table 2). Furthermore, during nighttime, the positive first derivative of the blood pressure curve (dp/dt), an indirect measure of inotropy (36), was significantly higher in the KO compared with WT mice. Short-term l-NAME treatment, while decreasing heart rate in mice of both genotypes, did not affect the difference in heart rates between WT and KO animals nor did it influence dp/dt in these animals (Fig. 6B and Table 2). A high-salt diet led to a small reduction in heart rate of WT and KO animals during daytime and did not affect dp/dt (Table 2).
Fig. 6.
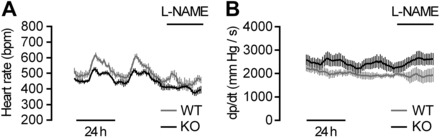
Longitudinal radiotelemetry measurements of heart rate (A) and the positive first derivative of the blood pressure curve (dp/dt; B) of WT and KO mice in the presence or absence of l-NAME treatment. Data are means ± SE; n = 5 (WT and KO).
SM22αCaSRΔflox/Δflox mice exhibit cardiac remodeling.
The observed bradycardia presented by the KO mice could be accounted for, at least in part, by direct CaSR ablation from cardiomyocytes as the SM22α promotor has been shown to direct Cre expression transiently to the heart (30). This consideration, together with recent evidence suggesting that the CaSR plays a role in electromechanical coupling of cardiomyocytes (48), led us to investigate more thoroughly the cardiac phenotype of SM22αCaSRΔflox/Δflox mice. While there were no differences between hearts from WT and KO animals by superficial observation or any histomorphological changes seen by H&E staining (Fig. 7A), KO animals showed a reduction in fibrosis compared with age-matched WT controls [Fig. 7B; 0.93 ± 0.08 vs. 2.09 ± 0.43% fibrotic area, n = 9 (KO), 10 (WT), P < 0.05, Student's t-test with Welch's correction].
Fig. 7.
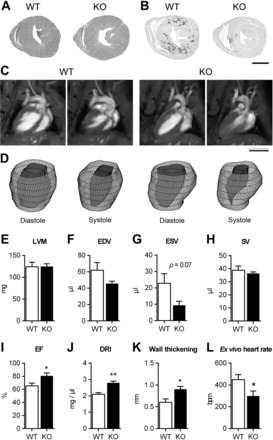
Cardiac phenotype of WT and KO animals. Hematoxylin and eosin staining (A) and picrosirius red staining (B) of heart sections showing reduced occurrence of fibrosis in KO hearts compared with WT. Representative long-axis MRI scans (C) and 3D reconstructions (D) of left ventricles of WT and remodeled KO mice (5 out of 11 investigated) in end diastole and end systole. Left ventricular mass (LVM; E), left ventricular end diastolic (EDV; F), end systolic (ESV; G), and stroke volume (SV; H), ejection fraction (EF; I), diastolic remodeling index (DRI; J), and wall thickening (end-systolic minus end-diastolic wall thickness; K) of remodeled hearts. Data are means ± SE; n = 5. **P < 0.01, *P < 0.05, Student's t-test. L: base intrinsic heart rate [beats per min (bpm)] of ex vivo retrograde perfused hearts. Data are means ± SE; n = 9 (WT) and 10 (KO). *P < 0.05, Student's t-test.
Cardiac cine-MRI showed that the long axes of WT and KO hearts of aged animals were comparable between genotypes in end-diastole but presented with a visible difference in end-systole in hearts from 5 out of 11 investigated KO animals. In these hearts, complete closure of the apical left ventricular cavity (2nd apical short-axis MRI slice) was seen, while it was still visible at the end-systole of the hearts of WT mice (Fig. 7C; Supplemental Video S1; Supplemental Material is available online at the journal website). These functional differences can be easily observed in morphological 3D reconstructions of the left ventricle endocardium (Fig. 7D; Supplemental Videos S2 and S3). These five hearts were thus termed “remodeled” and analyzed as a separate group. MRI analysis showed that left ventricular mass (Fig. 7E), left ventricular end-diastolic volume (Fig. 7F), end end-diastolic volume (Fig. 7G), and stroke volume (Fig. 7H) were unaltered between WT and remodeled KO hearts. An observed decrease in end-systolic volume did not reach statistical significance. Ejection fraction (Fig. 7I), end-diastolic remodeling index (Fig. 7J, size of the ventricle vs. the end-diastolic volume), and wall thickening (Fig. 7K) were increased in remodeled hearts from KO mice compared with WT hearts.
To test more directly the hypothesis that direct CaSR ablation from cardiomyocytes was responsible for the observed bradycardia in the SM22αCaSRΔflox/Δflox mouse, we also investigated the base frequency of nonpaced hearts from WT and KO mice ex vivo in the absence of autonomic regulation in a retrogradely perfused Langendorff preparation (31). Isolated hearts from KO animals showed a significantly reduced basal heart rate compared with that measured in the hearts of WT animals (Fig. 7L), suggesting that CaSR deletion from the heart has a direct impact on chronotropy. Pacemaker activity also occurs in the pulmonary veins, which contain cardiomyocyte-like pulmonary vein sleeve cells (PVCs). PVCs employ the same electrochemical coupling machinery as atrial cardiomyocytes (14), can act as pacemaker cells (12, 26, 57) and can be easily studied in ex vivo lung slice preparations as they are present in the murine lung. In PVCs from KO animals, there was a reduced rate of spontaneous activity, measured as changes in Ca2+i compared with WT mice (KO vs WT: 0.58 ± 0.10 vs 1.00 ± 0.09 fold; n = 6; P < 0.05, Student's t-test), suggesting that the bradycardia is an intrinsic feature of the KO animals.
DISCUSSION
Vascular phenotype.
SM22αCaSRΔflox/Δflox mice show changes in their cardiovascular system: the main findings of this study are that SM22αCaSRΔflox/Δflox mice exhibit impaired vasoconstriction, systemic hypotension, and bradycardia.
Ex vivo tension measurements of aortas and mesenteric arteries from WT and KO mice demonstrated impaired contractile responses to high K+ (a depolarizing stimulus) and impaired maximum contractility in response to PE (GPCR-mediated). In addition, the potency of PE to contract KO blood vessels was also impaired. These findings indicate that the CaSR sensitizes the response of blood vessels to contractile stimuli. Importantly, no histological difference was found between blood vessels from WT and KO mice, indicating that changes in vessel morphology were not responsible for the observed changes in blood vessel contractility. Furthermore, the reduction in maximum contractility was independent of inhibition of NOS or endothelial denudation while vascular relaxation in response to an exogenous NO donor remained unchanged. In addition, inhibition of NOS signaling can unmask the effects of endothelium-derived contracting factors released from the endothelium upon stimulation that in the absence of NO can cause pronounced vasoconstriction (16, 27, 42). This contractile effect of ACh in the presence of l-NAME was reduced in KO compared with WT aortas, providing yet another indication that the impaired contractility of blood vessels lacking the VSMC CaSR is independent of the contractile stimulus.
Despite the fact that the sensitivity to an NO donor was comparable in WT and KO vessels, the endothelium-dependent relaxation was differentially affected in MA and aortas. ACh evoked enhanced relaxation in KO aortas, which may be explained by the aforementioned reduced contractile component of the ACh response. KO MA, in contrast, exhibited a small but significant suppression of the ACh-mediated relaxation that was absent when l-NAME was added. These findings suggest a reduced endothelium-derived NO signaling in MA, as induced by endothelial stimulation (e.g., ACh), while the sensitivity of VSMC to NO itself was unaffected in KO animals.
In line with our results of genetic ablation, Loot et al. (34) have recently found that pharmacological inhibition of the CaSR attenuated vascular contraction in response to PE and high K+. The congruence of these results of pharmacological CaSR inhibition in WT mice and CaSR ablation in genetically altered mice provides another line of evidence that the VSMC-CaSR is indeed directly responsible for the modulation of vascular tone, rather than possible off-target effects of calcilytic drugs (35) or unexpected side-effects of the genetic ablation. This contractility-enhancing effect of the CaSR is not limited to VSMC, since, using the same animal model as the one we have used for the current studies, we have recently reported that airways of KO mice showed a reduction in contractility in response to the muscarinic ACh receptor agonist methacholine compared with WT mice (56), similarly to the observed reduction in blood vessel contractility in response to PE in this study. Finally, the observed effects in SM22αCaSRΔflox/Δflox mice were unlikely to be due to impaired SM22α function, as constitutive ablation of SM22α was shown to lead to increased MA vasoconstriction in response to an α1-adrenoceptor agonist (58). In the current study, the CaSR agonist Ca2+o induced vasorelaxation in WT and KO MA and KO aortas, likely due to stimulation of the endothelial CaSR (34, 54, 59). However, in WT aortas, we observed [Ca2+]o-dependent vasoconstriction. These effects of increasing [Ca2+]o inducing vasoconstriction in WT aortas are consistent with previous findings by Ohanian et al. (40), who reported vasoconstriction in response to physiological [Ca2+]o in rat subcutaneous small arteries. These authors suggested that the contractile effect was not CaSR-mediated. Similarly, Loot et al. (34) have reported vascular contraction in response to rising concentrations of Ca2+o in endothelium-denuded or l-NAME-treated mouse aortic rings. Here, we have demonstrated that the contracting component of the Ca2+o response was lost in KO aortas. Similarly, airways of WT mice have been shown to react with robust contraction to rising concentrations of the CaSR agonists Ca2+o and spermine and this contractile response was completely lost in airways from animals lacking the CaSR in smooth muscle cells (56). Moreover, results obtained by treating PE-preconstricted aortas with the calcimimetic NPS R-568 mimicked the results observed by using Ca2+o to modulate vascular tone. The calcimimetic-mediated contraction in WT aortas was endothelium independent, further corroborating a role for the VSMC CaSR in vessel contractility. Together, these results clearly support the idea that the CaSR mediates Ca2+o-induced contractions in smooth muscle cells. Cultured VSMC of KO animals reacted with impaired Ca2+i mobilization to stimulation with both Ca2+o and K+, indicating Ca2+i-signaling as one mechanism of action by which the CaSR affects blood vessel tone. Together, these results demonstrate that the observed impaired vascular contractility in blood vessels of SM22αCaSRΔflox/Δflox mice, as shown by reduced responses to primary contractile stimuli, is an effect, which is independent of the endothelium and dependent on the VSMC CaSR.
Tail-cuff and radiotelemetry measurements confirmed the hypotensive phenotype of SM22αCaSRΔflox/Δflox animals, which was predicted from the ex vivo vessel tone studies. In both circumstances, we observed a significant reduction in diastolic blood pressure and MAP. This reduction was more apparent during the animals' active than resting phase, suggesting that the modulating effect of the CaSR on blood pressure is dependent on sympathetic activity, again supporting the idea that the effects of the CaSR on blood vessel tone and blood pressure are dependent on the presence of other contractile, or activating, stimuli. A contribution of the cardiac phenotype of the animals, i.e., the reduction in heart rate, to the observed hypotension in SM22αCaSRΔflox/Δflox animals is likely, although the stronger reduction in diastolic than systolic blood pressure points to peripheral resistance being the principal cause of the hypotension in KO animals.
Acute l-NAME treatment did not abolish this difference in WT and KO mice diastolic blood pressure, mirroring the aforementioned ex vivo results on isolated blood vessels, where NOS inhibition or endothelial denudation did not abolish the impaired vascular contractility either. This rules out the possibility that, at least in the short term, the difference in blood pressure between WT and KO mice might be NO driven. Short-term exposure to a high-salt-supplemented diet has been shown to rescue the loss of angiotensin-converting enzyme-induced hypotension in mice of a similar background (9). However, the lack of any change in blood pressure in KO animals kept on a high-salt diet here suggests that the kidney is not likely to be involved in causing the observed hypotension in our strain.
Pulse height was increased in KO animals following l-NAME treatment. While elevated pulse height is recognized as being a feature of increased arterial stiffening, reduced blood vessel compliance is also accompanied by isolated systolic hypertension in humans and animals (15, 18), which clearly is not the case in KO animals. Indeed, the increased pulse height can be attributed solely to the greater reduction in diastolic than systolic blood pressure in KO animals. In KO animals, l-NAME treatment induced a larger increase in systolic blood pressure (and, therefore, pulse height) than diastolic blood pressure. It is known that the importance of endothelium-dependent NO signaling declines with the reduction of arterial size and plays a major role in aorta and other big arteries, while endothelium-dependent hyperpolarization plays a major role in smaller resistance arteries (49). Therefore, the observed increase in systolic blood pressure in KO mice could be ascribed to NOS inhibition having a greater effect in the large conduit arteries, than in resistance arteries (45). Furthermore, l-NAME administration in the drinking water led to a reduction in heart rate of KO animals, which was comparable to that observed in WT animals. These results suggest the existence of an intact baroreflex response to the increase in peripheral resistance, or at least that the response of the cardiovascular system to systemic NO synthesis inhibition, is not dependent on the VSMC-CaSR.
Collectively, the observations described herein support the hypothesis that the loss of the CaSR from VSMC leads to a reduction of vascular tone, manifesting in vivo as a reduction in diastolic blood pressure, which is dependent on the physical activity of the mice. The presence of both a cardiac as well as vascular phenotype in these animals suggests that the observed hypotensive phenotype is likely to be accounted for by a combination of CaSR ablation from both cardiac and vascular systems and further studies are required to dissect these effects in greater detail using cell-specific gene ablation studies in which Cre recombinase is driven by alternative promoters.
It has long been established that the CaSR can translate even minute changes in [Ca2+]o into intracellular responses (6). These changes in extracellular [Ca2+]o can also be elicited by extrusion of [Ca2+]i; indeed, excitation and subsequent extrusion of [Ca2+]i into the intracellular space have been conclusively demonstrated to activate the CaSR on neighboring cells and elicit intracellular responses in a paracrine fashion, acting in a way as a “third messenger” (10, 23, 24, 29). On the basis of our observations in this study and the aforementioned considerations, we therefore propose a hypothesis of how the VSMC-CaSR and endothelial-CaSR could directly contribute to the maintenance of blood vessel tone (Fig. 8). A contractile stimulus, e.g., via depolarization or α1-adrenoceptor agonists, leads to an increase in [Ca2+]i in VSMC. Restoration of normal resting Ca2+i levels is achieved in part via extrusion of Ca2+i into the interstitium, where it is feasible that such localized increase in Ca2+o would activate the VSMC-CaSR in an auto-/paracrine fashion, thus causing potentiation of contraction. Loss of such a mechanism in the SM22αCaSRΔflox/Δflox mice could account for the observed reduction in VSMC contractility, thus leading to the observed in vivo hypotension. This role of the CaSR as modulator of VSMC contractility is further supported by the fact that, conversely, increased expression of the CaSR and an associated increase in [Ca2+]i have been reported in pulmonary arterial smooth muscle cells from patients with pulmonary arterial hypertension and surrogate animal models (55), which is alleviated by pharmacological CaSR inhibition (20).
Fig. 8.
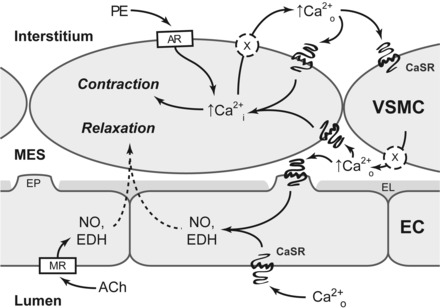
Hypothetical mechanism for the CaSR mediated auto-/paracrine amplification of contraction in vascular smooth muscle cells (VSMC). In VSMC, adrenoceptor (AR) agonists (e.g., PE) increase [Ca2+i], causing blood vessel constriction. Ca2+i is then extruded from the cell into the interstitium. Locally accumulating Ca2+o activates the CaSR on the same and neighboring VSMC, thus amplifying and synchronizing VSMC contractility. Ca2+i is also extruded into the myo-endothelial space (MES) where it can activate the CaSR on endothelial cell (EC) projections (EP), which penetrate the internal elastic lamina (EL). Activation of the endothelial CaSR leads to VSMC relaxation via a mechanism likely involving NO synthesis and endothelium-derived hyperpolarizations (EDH), in a fashion similar to dilating agents like acetylcholine (ACh) acting on muscarinic receptors (MR).
Cardiac phenotype.
CaSR ablation from SM22α-positive tissues also leads to its deletion from the developing heart (30), which could account for the aforementioned bradycardia in KO mice. Therefore, direct loss of the CaSR from the heart could impact negatively on pacemaker frequency or intracellular coupling. Previously, it has been demonstrated in ventricular cardiomyocytes that CaSR activation leads to increased Ca2+i signaling and increased cell shortening culminating in a positive inotropic effect and modification of their electromechanical coupling (48). Isolated ex vivo hearts recapitulated the reduction in heart rate observed by radiotelemetry in vivo. Indeed, isolated PVC from KO mice, which show similar electrophysiological hallmarks of cardiac pacemaker cells (12, 14, 26, 57), also exhibited reduced lower spontaneous activity than cells isolated from WT mice, results suggesting that the observed bradycardia in KO mice could be ascribed to decreased pacemaker activity. The observed cardiac remodeling, i.e., stronger mode of contraction, together with the increase in dp/dt, could thus be interpreted as compensatory response to the bradycardic and hypotensive phenotype. The reduction in fibrosis suggests that KO animals might be protected against the development of age-related cardiac fibrosis (2), maybe via a beneficial effect of their reduced heart rate (8) but could also be a direct consequence of CaSR deletion from the heart, as recently indicated in a rat model of cardiac hypertrophy (32). Further studies will be necessary to completely elucidate the role of the cardiac CaSR.
Conclusions.
Taken together, our studies highlight a physiological role for the CaSR in the cardiovascular system. In VSMC, CaSR-mediated regulation of contraction contributes to vessel contractility. Loss of this mechanism, likely together with direct cardiac effects, could account for the observed hypotension in SM22ACaSRΔflox/Δflox mice. The net balance between the effects of the CaSR in smooth muscle (pro-contractile) and the endothelium (pro-relaxing) determines the influence of the vascular CaSR on vascular tone regulation. Furthermore, KO mice exhibit both in vivo and ex vivo bradycardia, linking the CaSR to direct modulation of cardiac function. The results of our present study thus implicate the CaSR directly in the local regulation of the cardiovascular system.
GRANTS
This work was supported by the European Union though a Marie Curie Initial Training Network (“Multifaceted CaSR,” 264663, to D. Riccardi, P. J. Kemp, and S. A. Price), Amgen, (to D. Riccardi), the Novo Nordisk Foundation (to V. Matchkov), the US Department of Veterans Affairs Merit Review Grant (1I01BX001960 to W. Chang), and the Danish Medical Research Council (to R. A. Fenton).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.S., P.L.Y., W.C., K.J.B., D.H.E., and D.R. conception and design of research; M.S., P.L.Y., I.L.-F., T.S.D., S.C.B., P.J.E., A.A., J.G., K.R., V.V.M., R.A.F., W.C., A.S., D.T.W., S.A.P., and D.R. performed experiments; M.S., P.L.Y., I.L.-F., S.C.B., P.J.E., A.A., K.R., V.V.M., R.A.F., M.K., and P.J.K. analyzed data; M.S., P.L.Y., V.V.M., R.A.F., M.K., K.J.B., D.T.W., S.A.P., and P.J.K. interpreted results of experiments; M.S. and P.L.Y. prepared figures; M.S. drafted manuscript; M.S., P.L.Y., S.C.B., and D.R. edited and revised manuscript; M.S., P.L.Y., I.L.-F., T.S.D., S.C.B., P.J.E., A.A., J.G., K.R., V.V.M., R.A.F., W.C., M.K., A.S., K.J.B., D.T.W., S.A.P., D.H.E., P.J.K., and D.R. approved final version of manuscript.
Supplementary Material
REFERENCES
- 1.Alam MU, Kirton JP, Wilkinson FL, Towers E, Sinha S, Rouhi M, Vizard TN, Sage AP, Martin D, Ward DT, Alexander MY, Riccardi D, Canfield AE. Calcification is associated with loss of functional calcium-sensing receptor in vascular smooth muscle cells. Cardiovasc Res 81: 260–268, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis 2: 158–173, 2011. [PMC free article] [PubMed] [Google Scholar]
- 3.Block GA, Martin KJ, de Francisco AL, Turner SA, Avram MM, Suranyi MG, Hercz G, Cunningham J, Abu-Alfa AK, Messa P, Coyne DW, Locatelli F, Cohen RM, Evenepoel P, Moe SM, Fournier A, Braun J, McCary LC, Zani VJ, Olson KA, Drueke TB, Goodman WG. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 350: 1516–1525, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Boedtkjer E, Praetorius J, Matchkov VV, Stankevicius E, Mogensen S, Fuchtbauer AC, Simonsen U, Fuchtbauer EM, Aalkjaer C. Disruption of Na+,HCO3− cotransporter NBCn1 (slc4a7) inhibits NO-mediated vasorelaxation, smooth muscle Ca2+ sensitivity, and hypertension development in mice. Circulation 124: 1819–1829, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Broegger T, Jacobsen JC, Secher Dam V, Boedtkjer DM, Kold-Petersen H, Pedersen FS, Aalkjaer C, Matchkov VV. Bestrophin is important for the rhythmic but not the tonic contraction in rat mesenteric small arteries. Cardiovasc Res 91: 685–693, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 366: 575–580, 1993. [DOI] [PubMed] [Google Scholar]
- 7.Bukoski RD, Bian K, Wang Y, Mupanomunda M. Perivascular sensory nerve Ca2+ receptor and Ca2+-induced relaxation of isolated arteries. Hypertension 30: 1431–1439, 1997. [DOI] [PubMed] [Google Scholar]
- 8.Busseuil D, Shi Y, Mecteau M, Brand G, Gillis MA, Thorin E, Asselin C, Romeo P, Leung TK, Latour JG, Des Rosiers C, Bouly M, Rheaume E, Tardif JC. Heart rate reduction by ivabradine reduces diastolic dysfunction and cardiac fibrosis. Cardiology 117: 234–242, 2010. [DOI] [PubMed] [Google Scholar]
- 9.Carlson SH, Oparil S, Chen YF, Wyss JM. Blood pressure and NaCl-sensitive hypertension are influenced by angiotensin-converting enzyme gene expression in transgenic mice. Hypertension 39: 214–218, 2002. [DOI] [PubMed] [Google Scholar]
- 10.Caroppo R, Gerbino A, Fistetto G, Colella M, Debellis L, Hofer AM, Curci S. Extracellular calcium acts as a “third messenger” to regulate enzyme and alkaline secretion. J Cell Biol 166: 111–119, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cawley T, Geraghty J, Osborne H, Docherty JR. Effects of portal hypertension on responsiveness of rat mesenteric artery and aorta. Br J Pharmacol 114: 791–796, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang SL, Chen YC, Yeh YH, Lin YK, Wu TJ, Lin CI, Chen SA, Chen YJ. Heart failure enhanced pulmonary vein arrhythmogenesis and dysregulated sodium and calcium homeostasis with increased calcium sparks. J Cardiovasc Electrophysiol 22: 1378–1386, 2011. [DOI] [PubMed] [Google Scholar]
- 13.Chang W, Tu C, Chen TH, Bikle D, Shoback D. The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal 1: ra1, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeRuiter MC, Gittenberger-De Groot AC, Wenink AC, Poelmann RE, Mentink MM. In normal development pulmonary veins are connected to the sinus venosus segment in the left atrium. Anat Rec 243: 84–92, 1995. [DOI] [PubMed] [Google Scholar]
- 15.Essalihi R, Dao HH, Yamaguchi N, Moreau P. A new model of isolated systolic hypertension induced by chronic warfarin and vitamin K1 treatment. Am J Hypertens 16: 103–110, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Feletou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol 164: 894–912, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez-Rodriguez S, Edwards DH, Newton B, Griffith TM. Attenuated store-operated Ca2+ entry underpins the dual inhibition of nitric oxide and EDHF-type relaxations by iodinated contrast media. Cardiovasc Res 84: 470–478, 2009. [DOI] [PubMed] [Google Scholar]
- 18.Franklin SS, Gustin WT, Wong ND, Larson MG, Weber MA, Kannel WB, Levy D. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 96: 308–315, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Garland CJ, Yarova PL, Jimenez-Altayo F, Dora KA. Vascular hyperpolarization to beta-adrenoceptor agonists evokes spreading dilatation in rat isolated mesenteric arteries. Br J Pharmacol 164: 913–921, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo Q, Huang JA, Yamamura A, Yamamura H, Zimnicka AM, Fernandez R, Yuan JX. Inhibition of the Ca(2+)-sensing receptor rescues pulmonary hypertension in rats and mice. Hypertens Res 37: 116–124, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heiberg E, Sjogren J, Ugander M, Carlsson M, Engblom H, Arheden H. Design and validation of segment–freely available software for cardiovascular image analysis. BMC Med Imaging 10: 1, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hobaus J, Hummel DM, Thiem U, Fetahu IS, Aggarwal A, Mullauer L, Heller G, Egger G, Mesteri I, Baumgartner-Parzer S, Kallay E. Increased copy-number and not DNA hypomethylation causes overexpression of the candidate proto-oncogene CYP24A1 in colorectal cancer. Int J Cancer 133: 1380–1388, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofer AM, Curci S, Doble MA, Brown EM, Soybel DI. Intercellular communication mediated by the extracellular calcium-sensing receptor. Nat Cell Biol 2: 392–398, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Hofer AM, Gerbino A, Caroppo R, Curci S. The extracellular calcium-sensing receptor and cell-cell signaling in epithelia. Cell Calcium 35: 297–306, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci USA 99: 7142–7147, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honjo H, Boyett MR, Niwa R, Inada S, Yamamoto M, Mitsui K, Horiuchi T, Shibata N, Kamiya K, Kodama I. Pacing-induced spontaneous activity in myocardial sleeves of pulmonary veins after treatment with ryanodine. Circulation 107: 1937–1943, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Kojda G, Laursen JB, Ramasamy S, Kent JD, Kurz S, Burchfield J, Shesely EG, Harrison DG. Protein expression, vascular reactivity and soluble guanylate cyclase activity in mice lacking the endothelial cell nitric oxide synthase: contributions of NOS isoforms to blood pressure and heart rate control. Cardiovasc Res 42: 206–213, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42: 71–75, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Lembrechts R, Brouns I, Schnorbusch K, Pintelon I, Kemp PJ, Timmermans JP, Riccardi D, Adriaensen D. Functional expression of the multimodal extracellular calcium-sensing receptor in pulmonary neuroendocrine cells. J Cell Sci 126: 4490–4501, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis 41: 179–184, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Liao R, Podesser BK, Lim CC. The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol 303: H156–H167, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu L, Wang C, Sun D, Jiang S, Li H, Zhang W, Zhao Y, Xi Y, Shi S, Lu F, Tian Y, Xu C, Wang L. Calhex ameliorates cardiac hypertrophy by inhibiting cellular autophagy in vivo and in vitro. Cell Physiol Biochem 36: 1597–1612, 2015. [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[-delta delta C(T)] method. Methods 25: 402–408, 2001. [DOI] [PubMed] [Google Scholar]
- 34.Loot AE, Pierson I, Syzonenko T, Elgheznawy A, Randriamboavonjy V, Zivkovic A, Stark H, Fleming I. Ca2+ sensing receptor cleavage by calpain partially accounts for altered vascular reactivity in mice fed a high fat diet. J Cardiovasc Pharmacol 61: 528–535, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Marquis RW, Lago AM, Callahan JF, Rahman A, Dong X, Stroup GB, Hoffman S, Gowen M, DelMar EG, Van Wagenen BC, Logan S, Shimizu S, Fox J, Nemeth EF, Roethke T, Smith BR, Ward KW, Bhatnagar P. Antagonists of the calcium receptor. 2. Amino alcohol-based parathyroid hormone secretagogues. J Med Chem 52: 6599–6605, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Morimont P, Lambermont B, Desaive T, Janssen N, Chase G, D'Orio V. Arterial dP/dtmax accurately reflects left ventricular contractility during shock when adequate vascular filling is achieved. BMC Cardiovasc Disord 12: 13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakagawa K, Parekh N, Koleganova N, Ritz E, Schaefer F, Schmitt CP. Acute cardiovascular effects of the calcimimetic R-568 and its enantiomer S-568 in rats. Pediatr Nephrol 24: 1385–1389, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Newaz M, Blanton A, Fidelis P, Oyekan A. NAD(P)H oxidase/nitric oxide interactions in peroxisome proliferator activated receptor (PPAR)alpha-mediated cardiovascular effects. Mutat Res 579: 163–171, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Odenwald T, Nakagawa K, Hadtstein C, Roesch F, Gohlke P, Ritz E, Schaefer F, Schmitt CP. Acute blood pressure effects and chronic hypotensive action of calcimimetics in uremic rats. J Am Soc Nephrol 17: 655–662, 2006. [DOI] [PubMed] [Google Scholar]
- 40.Ohanian J, Gatfield KM, Ward DT, Ohanian V. Evidence for a functional calcium-sensing receptor that modulates myogenic tone in rat subcutaneous small arteries. Am J Physiol Heart Circ Physiol 288: H1756–H1762, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Plane F, Wiley KE, Jeremy JY, Cohen RA, Garland CJ. Evidence that different mechanisms underlie smooth muscle relaxation to nitric oxide and nitric oxide donors in the rabbit isolated carotid artery. Br J Pharmacol 123: 1351–1358, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med 18: 286–290, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: conception, development, adult physiology, and disease. Annu Rev Physiol 74: 271–297, 2012. [DOI] [PubMed] [Google Scholar]
- 44.Rietdorf K, Bootman MD, Sanderson MJ. Spontaneous, pro-arrhythmic calcium signals disrupt electrical pacing in mouse pulmonary vein sleeve cells. PLoS One 9: e88649, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruiz A, Lopez RM, Perez T, Castillo C, Castillo EF. The effects of NG-nitro-l-arginine methyl ester on systolic pressure, diastolic pressure and pulse pressure according to the initial level of blood pressure. Fundam Clin Pharmacol 22: 45–52, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Rushton DJ, Mattis VB, Svendsen CN, Allen ND, Kemp PJ. Stimulation of GABA-induced Ca2+ influx enhances maturation of human induced pluripotent stem cell-derived neurons. PLoS One 8: e81031, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schreckenberg R, Dyukova E, Sitdikova G, Abdallah Y, Schluter KD. Mechanisms by which calcium receptor stimulation modifies electromechanical coupling in isolated ventricular cardiomyocytes. Pflügers Arch 467: 379–388, 2015. [DOI] [PubMed] [Google Scholar]
- 49.Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M, Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol 28: 703–711, 1996. [DOI] [PubMed] [Google Scholar]
- 50.Smajilovic S, Yano S, Jabbari R, Tfelt-Hansen J. The calcium-sensing receptor and calcimimetics in blood pressure modulation. Br J Pharmacol 164: 884–893, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sward K, Albinsson S, Rippe C. Arterial dysfunction but maintained systemic blood pressure in cavin-1-deficient mice. PLoS One 9: e92428, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thakore P, Ho WS. Vascular actions of calcimimetics: role of Ca(2)(+) -sensing receptors versus Ca(2)(+) influx through L-type Ca(2)(+) channels. Br J Pharmacol 162: 749–762, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward DT, Brown EM, Harris HW. Disulfide bonds in the extracellular calcium-polyvalent cation-sensing receptor correlate with dimer formation and its response to divalent cations in vitro. J Biol Chem 273: 14476–14483, 1998. [DOI] [PubMed] [Google Scholar]
- 54.Weston AH, Absi M, Ward DT, Ohanian J, Dodd RH, Dauban P, Petrel C, Ruat M, Edwards G. Evidence in favor of a calcium-sensing receptor in arterial endothelial cells: studies with calindol and Calhex 231. Circ Res 97: 391–398, 2005. [DOI] [PubMed] [Google Scholar]
- 55.Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca(2+)-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yarova PL, Stewart AL, Sathish V, Britt RD Jr, Thompson MA, AP PL, Freeman M, Aravamudan B, Kita H, Brennan SC, Schepelmann M, Davies T, Yung S, Cholisoh Z, Kidd EJ, Ford WR, Broadley KJ, Rietdorf K, Chang W, Bin Khayat ME, Ward DT, Corrigan CJ, JP TW, Kemp PJ, Pabelick CM, Prakash YS, Riccardi D. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci Transl Med 7: 284ra260, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu MC, Huang CF, Chang CM, Chen YC, Lin CI, Chen SA. Diverse cell morphology and intracellular calcium dynamics in pulmonary vein cardiomyocytes. Heart Vessels 26: 101–110, 2011. [DOI] [PubMed] [Google Scholar]
- 58.Zeidan A, Sward K, Nordstrom I, Ekblad E, Zhang JC, Parmacek MS, Hellstrand P. Ablation of SM22alpha decreases contractility and actin contents of mouse vascular smooth muscle. FEBS Lett 562: 141–146, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Ziegelstein RC, Xiong Y, He C, Hu Q. Expression of a functional extracellular calcium-sensing receptor in human aortic endothelial cells. Biochem Biophys Res Commun 342: 153–163, 2006. [DOI] [PubMed] [Google Scholar]
- 60.Zitt E, Woess E, Mayer G, Lhotta K. Effect of cinacalcet on renal electrolyte handling and systemic arterial blood pressure in kidney transplant patients with persistent hyperparathyroidism. Transplantation 92: 883–889, 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.