

Abstract
Idiopathic pulmonary fibrosis (IPF) is an incurable complex genetic disorder that is associated with sequence changes in 7 genes (MUC5B, TERT, TERC, RTEL1, PARN, SFTPC, and SFTPA2) and with variants in at least 11 novel loci. We have previously found that 1) a common gain-of-function promoter variant in MUC5B rs35705950 is the strongest risk factor (genetic and otherwise), accounting for 30-35% of the risk of developing IPF, a disease that was previously considered idiopathic; 2) the MUC5B promoter variant can potentially be used to identify individuals with preclinical pulmonary fibrosis and is predictive of radiologic progression of preclinical pulmonary fibrosis; and 3) MUC5B may be involved in the pathogenesis of pulmonary fibrosis with MUC5B message and protein expressed in bronchiolo-alveolar epithelia of IPF and the characteristic IPF honeycomb cysts. Based on these considerations, we hypothesize that excessive production of MUC5B either enhances injury due to reduced mucociliary clearance or impedes repair consequent to disruption of normal regenerative mechanisms in the distal lung. In aggregate, these novel considerations should have broad impact, resulting in specific etiologic targets, early detection of disease, and novel biologic pathways for use in the design of future intervention, prevention, and mechanistic studies of IPF.
I. CLINICAL AND PATHOLOGICAL FEATURES OF IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis (IPF) affects 5 million worldwide, disproportionately affects men, is more prevalent with age, and is inexplicably increasing in prevalence (75, 142, 151). Nevertheless, IPF is likely underdiagnosed (72, 151, 165). Death occurs in up to 50% of affected individuals within 3–5 yr following the diagnosis (75, 92, 165). Recently pirfenidone (94) and nintedanib (170) have been shown to slow IPF progression; however, IPF remains a progressive disease and no treatment (short of lung transplantation) is known to impact the poor survival associated with this disease (75). While IPF takes years to develop, most patients with IPF are diagnosed in the advanced stage of disease when little can be done to influence survival. Earlier diagnosis of IPF will identify subjects with more salvageable lung, enable treatment of those with a lower burden of lung disease (121, 163), and may reveal novel molecular targets for intervention that are effective in the preclinical and/or early clinical stages of this progressive disease.
The increasing prevalence of IPF may be explained to some extent by our aging population (75, 151), a greater physician awareness of the disease, screening of first-degree family members affected by the disease, and earlier detection in asymptomatic individuals in which case disease is found incidentally after high-resolution computed tomographic scans (HRCT) for other suspected conditions; however, the observed increase in prevalence is not fully understood. In the United States, IPF has an estimated incidence of 20–30/100,000 individuals, and this number increases with each decade between 50 and 90 yr of age (151). For instance, in 70- to 79-yr-old individuals, the incidence has been reported as high as 75/100,000 (36, 64). However, several studies imply a much higher prevalence of IPF than previously thought even if all asymptomatic fibrotic cases did not progress to established IPF. In a chest HRCT study of almost 2,500 smokers undergoing screening for lung cancer, bilateral interstitial lung abnormalities were found in 8%, and these abnormalities were associated with reduced lung volumes (225). Similarly, chest CT abnormalities consistent with IPF were found in 1.8% of those older than 50 yr of age (72). Among asymptomatic members of families with two or more cases of pulmonary fibrosis [familial interstitial pneumonia (FIP)], 14% have interstitial abnormalities on CT scan and 35% have interstitial abnormalities on transbronchial biopsy (111). We have found that the MUC5B promoter variant rs35705950 that is consistently and strongly associated with IPF (13, 47, 66, 146, 156, 178, 191, 226, 236) is also predictive of those with preclinical forms of pulmonary fibrosis (OR = 6.3 per allele, 95% CI 3.1–12.7), suggesting that IPF risk variants may be helpful in identifying subjects with a higher risk of disease development.
While a number of exposures [metal and wood dust (7, 67, 68, 81, 136), viruses (118, 190, 200, 233), and drugs (43, 69, 139)] have been found to be associated with the development of IPF, the most important environmental risk factor for IPF is cigarette smoking. In addition to occupational exposures, such as asbestos (15, 124, 173), ever having smoked cigarettes remains a risk factor for the development of IPF even after smoking cessation (8), suggesting that the response induced by cigarette smoke-related lung injury may become self-sustaining (187). For FIP, we have shown that cigarette smoking is a strong risk factor for the development of disease (189; OR = 3.6; 95% CI = 1.3–9.8). This finding suggests that cigarette smoking contributes significantly even in a genetic model of pulmonary fibrosis.
The morphological hallmark of IPF on imaging and histology is usual interstitial pneumonia (UIP). On the chest radiograph, UIP is characterized by basal predominant fine reticulonodular abnormality, which may be present for many years before clinical presentation with IPF. However, these features are relatively nonspecific (129). HRCT scans with appropriate technique is critical for evaluation of IPF (130). The characteristic HRCT features are reticular abnormality, traction bronchiectasis and honeycombing, with basal and peripheral predominant distribution, and with absence of features (such as mosaic attenuation or centrilobular nodules) that would suggest an alternative diagnosis (166). A confident radiologic diagnosis of UIP, by an experienced radiologist, based on these clinical features, correctly predicts the histological diagnosis of UIP in over 90% of cases (41, 50, 73, 74, 129, 183). Radiologic honeycombing is the most important single predictor of a UIP diagnosis (73). If honeycombing is absent, but all other HRCT features are highly suggestive of UIP, the likelihood of UIP is more than 80% (29). Thus the radiologic diagnosis of UIP may sometimes be accepted even in the absence of honeycombing, particularly in older individuals in whom the pretest probability of IPF is already high (46). Importantly, the HRCT features of UIP secondary to collagen vascular disease or other causes, such as asbestosis or chronic hypersensitivity pneumonitis, are indistinguishable from those of IPF (198). Identification of an underlying cause for UIP therefore remains an important task for the pulmonary clinician.
The histological expression of IPF is UIP. IPF, either familial or sporadic, represents a lung-limited condition recognized by progressive scarring of the pulmonary interstitium that results from persistent collagen production by proliferating fibroblasts/myofibroblasts and regenerating alveolar epithelia. This represents a constellation of microscopic findings including fibrosis of the alveolar interstitium alternating with normal appearing lung, fibroblastic foci, and subpleural microscopic honeycombing (164, 208). Fibroblastic foci are distinct subepithelial interstitial collections of myofibroblasts that are capable of producing mucopolysaccaride and collagen. Fibroblastic foci are not necessarily profuse and are more likely to be found in the fibrotic interstitium adjacent to denser areas of fibrosis and in the subpleural regions of the lung (34). It has been put forth that fibroblastic foci represent the site of the initial response to a yet undefined injury to type 1 alveolar epithelial cells, and due to the likely temporally heterogeneous nature of the histological findings in UIP, the alveolar epithelial injury is further postulated to be recurrent over time leading to the development of numerous foci of injury and repair resulting in the progressive accumulation of extracellular matrix (179). Fibroblastic foci are further hypothesized to be the starting point of the uncontrolled fibroproliferative process that eventually encroaches on the normal lung; however, there is inadequate support for this assumption.
An additional and unique histological feature of histological UIP is microscopic honeycombing. This is distinct from radiologic honeycombing and is characterized by clusters of small cysts, usually in a subpleural location and in densely fibrotic areas. Honeycomb cysts are often filled with mucus and occasionally inflammatory cells. These microscopic cystic structures are often lined by both pseudostratified ciliated columnar epithelium similar to that which line the major airways and distal bronchioles (199) and alveolar epithelium consisting of type 2 alveolar epithelial cells. In some cases, honeycomb cysts are entirely lined by alveolar type 2 cells. It has been long thought that honeycomb cysts represent the end stage of IPF since these structures are found in densely fibrotic areas of IPF/UIP; however, the origin and implications of these cystic structures remain debatable. Alternatively, the cysts could be the result of aberrant attempts at regeneration of the terminal airways, possibly via activation of dormant developmental pathways (181) (Figure 1).
FIGURE 1.

Traditional and hypothesized relationship between fibroblastic foci and honeycomb cysts. The traditional thinking is that honeycomb cysts are a consequence of extensive fibroproliferation throughout the lung parenchymal and represent a pathophysiological consequence of persistent fibroproliferation. Based on our findings and observations made by other investigators, we have hypothesized persistent bronchiolo-alveolar injury results in a failure to repair the airway epithelia and that independent of fibroproliferation, this inability to regenerate the distal airway epithelia results in honeycomb formation. We further hypothesize that overexpression of MUC5B by airway progenitor cells inhibits normal repair and accelerates the development of honeycomb cysts.
An additional prominent histological feature, though not specific to UIP, is type 2 alveolar epithelial cell hyperplasia. These cells function as the progenitors for alveolar type 1 cells, which under normal conditions represent 90% of the alveolar epithelial surface area (181). Additionally, type II alveolar epithelial cells are the source of pulmonary surfactant. Following an injury to the alveolus, reconstitution of alveolar structure by type II cell proliferation and differentiation to type I cells is considered to be essential for maintenance of the epithelial barrier and restoration of gas exchange. In IPF, however, type 2 alveolar epithelial cell hyperplasia persists replacing type 1 cells and likely contributes to collagen accumulation both by the release of fibroblast growth factors, and by transitioning to fibroblasts and eventually myofibroblasts (epithelial-mesenchymal transition) (6, 180, 229). Moreover, while the alveolar epithelial cells undergo apoptosis, the fibroblasts/myofibroblasts appear to be resistant to this process.
Over the past decade, consensus has been reached on the diagnosis of IPF (92, 165, 207, 208). It depends on clinical features (progressive and persistent dyspnea, disease localized to the lungs, and no clear etiology), an HRCT of the lung showing lower zone predominant reticular infiltrates, traction bronchiectasis, and subpleural honeycomb changes, or a surgical lung biopsy demonstrating UIP. It should be noted that microscopic honeycombing does not correlate with HRCT scan honeycombing which may represent dilated airways due to traction from the fibrotic process. Histological honeycombing is almost always present to some degree in UIP; however, this is not the case for HRCT scan honeycombing. If typical HRCT findings are seen, coupled with the expected clinical presentation, and there is no history of connective tissue disease (CTD) and serum autoantibodies are absent, the diagnosis of IPF is fairly secure. However, in up to 33% of IPF subjects when first evaluated, HRCT honeycombing is absent. Instead, the other features of IPF including bilateral peripheral subpleural lower zone predominate reticular infiltrates and traction bronchiectasis are seen. Even in this instance, surgical lung biopsy more often reveals UIP with microscopic honeycombing than NSIP.
Other fibrotic lung diseases have clinical and histological expressions that must be distinguished from IPF. IPF falls into a classification of interstitial lung diseases based on their histological appearance and known as the idiopathic interstitial pneumonias (IIP) (90, 93, 165, 207, 208). IPF/UIP is most common followed by the histological entity of nonspecific interstitial pneumonia (NSIP). NSIP can be idiopathic (iNSIP), but more often represents either a pulmonary manifestation of one of the known CTD or possibly a lung dominant expression of a CTD (LD-CTD). In LD-CTD patients, circulating autoantibodies are present, but clinical features necessary to establish a CTD diagnosis are lacking (48, 176). NSIP can also be one of the histological expressions of chronic hypersensitivity pneumonitis (HP) or a drug-induced lung disease (217). Compared with IPF, NSIP occurring with iNSIP, CTD, or LD-CTD occurs in younger subjects, is more often seen in women and has a much more benign prognosis (210). In pulmonary fibrosis, whether idiopathic, familial, due to chronic hypersensitivity pneumonitis, CVD, LD-CVD, or even asbestosis, either NSIP or UIP may be present.
While IPF is a distinct entity, there are significant variations in the clinical course. The expected course is progressive dyspnea with or without cough with death occurring within 3–5 yr from the consequences of hypoxemia. However, in up to 20% of IPF subjects there is prolonged survival (6–12 yr). In ∼5–10% of patients with IPF, acute decompensations occur, referred to as acute exacerbations (33, 186). Pathologically, acute exacerbations are characterized by the histological lesion of organizing diffuse alveolar damage (DAD), which is superimposed on the preexisting UIP. Patients with acute exacerbations experience a marked increase in their dyspnea over 3–7 days, become more hypoxic, and are increasingly difficult to oxygenate (123). Imaging of the chest shows new bilateral diffuse ground glass opacities, and over 50% of episodes of acute exacerbations are fatal in spite of high-dose glucocorticoids and broad-spectrum antibiotics. For those who survive, mortality within the next year is high. There is also an accelerated course in IPF with declines in lung function and death within 3 yr after diagnosis, but without acute exacerbations. In this group, during the initial evaluation, there is often more severe physiological impairment, i.e., forced vital capacity and total lung capacity <60% of predicted, diffusing capacity for carbon monoxide (DLCO) <35% of predicted, hypoxemia requiring supplemental oxygen and sometimes the presence of pulmonary hypertension. An accelerated course can be anticipated if at a 6-mo interval follow-up, physiological testing demonstrates 5–10% declines in the percentage of the physiological predicted values compared with the initial values (116). Other than the genotype (146, 147, 158), there are few, if any, validated biomarkers which predict outcome, particularly when the patient is first evaluated. For example, it has been shown that IPF subjects with the MUC5B promoter variant rs35705950 have improved survival compared with those without this variant (158) (Figure 2), suggesting that the MUC5B variant either identifies individuals earlier in the course of disease or represents a different pathophysiological subtype of IPF. While a number of biological markers have been shown to be prognostic for existing disease (21, 32, 39, 49, 52, 53, 56, 61, 71, 80, 87, 97, 108, 109, 122, 134, 150, 153, 158, 161, 169, 174, 195, 215, 219, 227, 235, 239), with the exception of the MUC5B promoter variant (72) and MMP7 (174), it is unclear if these biomarkers are useful for predicting preclinical/mild disease.
FIGURE 2.

Kaplan-Meier survival curves for patients with IPF by MUC5B promoter variant rs35705950 genotype for the INSPIRE cohort (A) and the Chicago cohort (B). In the INSPIRE cohort, the hazard ratios controlled for age, gender, FVC, DLCO, and MMP7. In the Chicago cohort, the hazard ratios controlled for age, gender, FVC, and DLCO. [Adapted from Peljto et al. (158), with permission from American Medical Association.]
In the past decade recent novel treatment trials for IPF have focused on inhibition of the fibroproliferative process with the primary end point being improvement or stabilization of the FVC percent predicted (94, 170). Secondary end points included quality of life and/or mortality. Multiple pathways that enhance fibrosis have been interrogated by novel therapies in human IPF and have been negative except for two recent trials testing pirfenidone (94) and nintedanib (170), which have both been shown to slow IPF progression. Pirfenidone, thought to interfere with the profibrotic molecule transforming growth factor (TGF)-β1 (94), and nintedanib, a triple-kinase inhibitor of fibroblast growth factors (170), have been approved for use in IPF. Although in neither trial was there mortality or quality of life benefits, there were impressive decreases in the rate of the FVC decline after 1 yr of treatment. While both pirfenidone and nintedanib slow the rate of disease progression, the long-term effects of these drugs on mortality are unknown; however, review of the trials suggest that pirfenidone and nintedanib may prove to reduce mortality (89).
Bilateral lung transplant remains an option for qualified IPF patients offering a 50% 3-yr survival post-transplant (143). However, a relatively small number of cases qualify or receive this potential life-saving procedure. Other adjunct treatments that may contribute to improve the outcome of IPF include use of oxygen to manage hypoxia, possibly sildenafil for pulmonary hypertension, nocturnal continuous positive airway pressure for obstructive sleep apnea, and the treatment of gastric acid reflux aspiration (31, 76, 132).
II. GENETIC DETERMINANTS OF PULMONARY FIBROSIS
A. Risk of Pulmonary Fibrosis
Current evidence suggests that IPF results from a combination of multiple genetic and nongenetic risk factors, where the definition of nongenetic risk factors is broadly construed to include both exogenous (e.g., cigarette smoking and air pollution) and endogenous (e.g., systemic diseases and autoimmune conditions) elements that contribute to the overall risk of developing disease. While many nongenetic factors may also be under genetic control (e.g., autoimmune conditions), the concept is that complex diseases, which constitute most human illnesses, develop consequent to the combination of genetic factors and factors that are primarily nongenetic. The relative contribution of genetic versus nongenetic contributors differs across diseases and populations; within a disease like IPF, the relative contributions of each of these risks also differ among individuals. This complexity results in challenges for identification of genetic risk factors, and several different approaches have been used in combination to overcome these challenges for IPF.
Linkage analysis encompasses a group of statistical methods to examine the inheritance pattern of genetic variants within families to determine if there is a relationship between the genetic variants and the phenotype of interest. Linkage analyses rely on family data, which can be made up by a wide range of pedigree structures from extended pedigrees to affected sib pairs; the more distant the relationships between affected individuals, the more powerful the analysis. Linkage analysis is most powerful for rare, highly penetrant alleles, which likely account for a small proportion of pulmonary fibrosis.
Genetic association studies are the most commonly used study design to find disease genes in complex traits. Association studies can be based on a candidate region based on linkage analysis, for candidate genes identified in other ways, or in genome-wide association studies (GWAS). GWAS have historically been conducted using common variants, but are increasingly being explored in the context of whole exome or whole genome resequencing. The challenge for any GWAS is obtaining convincing statistical evidence for association with any given variant or group of variants in the face of many statistical tests. Moreover, association studies often result in common variants that are not particularly penetrant. Each of the above strategies has been applied to pulmonary fibrosis; here we give a brief overview of the evidence for pulmonary fibrosis genetic risk factors.
Clustering of pulmonary fibrosis in families first suggested a genetic component to disease risk, and several studies have identified both common and rare genetic variations that are linked and/or associated with disease (203). Cases of pulmonary fibrosis have been reported in closely related family members including monozygotic twins raised in different environments (12, 84, 185), in genetically related members of several families (10, 12, 70, 193), in consecutive generations in the same families (12, 65, 120), and in family members separated at an early age (193). While a single report suggests that familial interstitial pneumonia (FIP) is inherited as an autosomal recessive trait (212), other pedigrees demonstrate an autosomal dominant pattern of inheritance (1, 131, 193) perhaps with reduced penetrance (1, 10, 70, 84, 126, 140, 185, 193). We have reported over 100 families with 2 or more cases of IIP (a category of interstitial lung disease inclusive of IPF) and have found that FIP most closely follows an autosomal dominant inheritance pattern that involves all subtypes of IIP, is strongly associated with cigarette smoking (OR = 3.6; 95% confidence interval 1.3–9.8), and appears to be a complex genetic disorder (189), suggesting that the different histological types of idiopathic interstitial pneumonia may be related etiologically or even pathogenically. These findings highlight the complex nature of IPF and suggest that while a susceptibility gene may predispose to IIP, another event (a modifier gene, a medical condition, or a specific exposure) may result in a unique type of idiopathic interstitial pneumonia. Rare variants in several genes have been reported to segregate with familial interstitial fibrosis, including TERT (4, 211), TERC (4, 211), RTEL (30, 192), and PARN (192) in the telomerase pathway and surfactant protein genes SFTPC (119, 205, 213) and SFTPA2 (224). These studies have taken advantage of large families where available, but have also identified rare variants among those with no family history of disease. Reviewed in detail elsewhere (110), studies of these genes and specific variants implicate telomere maintenance (TERT, TERC, PARN and RTEL) and surfactant dysfunction (SFTPC, SFTPA2) in IPF pathogenesis.
Two large GWAS of patients (familial and sporadic) and controls have been conducted in pulmonary fibrosis (47, 146). Both studies confirmed the role of the promoter of the MUC5B gene in the risk of IPF (see below) and identified other common variants associated with risk. Noth et al. (146) reported association between pulmonary fibrosis and three polymorphisms in the TOLLIP gene, which is in the same region as the MUC5B gene. Our results demonstrate that common variants in both the TERT and TERC gene regions were associated with IPF (47), suggesting that both common and rare variants in the telomerase genes may be risk factors for disease (Figure 3). We also identified seven other loci associated with disease (4q22, FAM13A; 6p24, DSP; 10q24, OBFC1; 13q34, ATP11A; 19p13, DPP9; 7q22, 15q14-15). The pathways implicated by the genes identified in the GWAS confirmed the role of telomere maintenance (TERT, TERC, OBFC1) and host defense (MUC5B, ATP11A, TOLLIP), and identified barrier function (DSP, DPP9) as important for disease etiology. In aggregate, the genome-wide variants account for 30–35% of the risk of familial and sporadic pulmonary fibrosis, suggesting an important role for common genetic variation in both familial and sporadic disease. In fact, the risk of developing IPF for each of the significant genetic loci reported is equivalent for familial and sporadic forms of IPF (Figure 4), suggesting a similarity between these two types of IPF. This is important in several contexts, including efforts related to screening high-risk populations and to target therapeutics toward these common features that could potentially be applicable to broad groups of patients.
FIGURE 3.
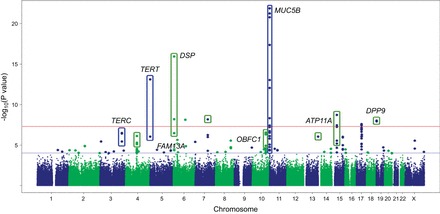
GWAS results at 439,828 SNPs with 1,616 cases and 4,683 controls under additive model. SNPs above red line were genome-wide significant at P < 5 × 10−8. These SNPs and SNPs between red and blue lines, corresponding to 5 × 10−8 < P value < 0.0001 were followed up in 876 cases and 1,890 controls. [Adapted from Fingerlin et al. (47), with permission from Nature Publishing Group.]
FIGURE 4.

The genetic basis of familial and sporadic idiopathic interstitial pneumonia is similar. A similar risk is conferred by common variants at each of the major IPF loci for both familial and sporadic forms of IPF. [Data derived from recent GWAS in Fingerlin et al. (47) and previously presented in Mathai et al. (128), with permission from Wolters Kluwer Health, Inc.]
The biological functions of the genes implicated so far suggest that increased injury may be a primary defect for a substantial proportion of those with pulmonary fibrosis, rather than, or in addition to, dysregulated repair mechanisms. The secondary nature of the chronic inflammation and remodeling may provide an additional explanation why many treatments aimed at decreasing inflammation have failed as therapeutics. The genetic findings are key for eventual identification of the causal variant(s) in each gene/region. As we gain a more comprehensive understanding of the mechanism of disease pathogenesis, the hope is that new therapeutics can be developed. As treatments that prolong life are identified, these genetic variants and other biomarkers may prove useful in personalizing approaches to treatment and identifying individuals who are at risk or at earlier stages of disease when the lung is more salvageable.
B. MUC5B Promoter Variant rs35705950
We discovered the MUC5B promoter variant rs35705950 as a risk variant for IPF using a positional cloning approach of linkage analysis in families followed by case-control association across the region of linkage (178). The linkage analysis was carried out among 88 families of European ancestry and identified a region of suggestive linkage on chromosome 11p15. Resequencing of the region in individuals of European ancestry identified polymorphisms that were then genotyped in familial and sporadic cases in addition to controls of European ancestry. The rs35705950 variant was the strongest and only independently associated variant in the region and has now been replicated in seven subsequent studies among individuals of European ancestry (13, 47, 66, 146, 191, 226, 236). Table 1 provides the risk allele carrier frequency, odds ratios, and confidence intervals for having one copy of the risk allele (54.9–67%) compared with having no copies for each of the published studies to date. The frequency of the risk allele (mean allele frequency) is ∼35% among European ancestry cases compared with ∼9% among European ancestry controls. The largest study (47) estimated that the odds of developing pulmonary fibrosis for those with one copy of the risk allele were 4.5 times (95% CI: 3.9, 5.2) the odds of those with no copies and that the odds for those with two copies are 20.2 times those with no copies (95% CI: 15.2–27.0). Strikingly, the risk associated with rs35705950 is very similar between familial and sporadic disease (178), indicating that the variant contributes substantially to both presentations of IPF. To date, the MUC5B promoter variant rs35705950 is the strongest known risk factor (genetic and otherwise) for the development of IPF. And while we and others have examined the association between rs35705950 and a variety of lung diseases [asbestosis, sarcoidosis, scleroderma associated interstitial lung disease, chronic obstructive pulmonary disease (COPD), asthma, and lung cancer] (13, 157, 191), the MUC5B promoter variant is only associated with IPF (13, 47, 66, 146, 178, 191, 226, 236) and idiopathic nonspecific interstitial pneumonia (66).
Table 1.
Summary of studies conducted in NHWs investigating the relationship between the MUC5B promoter variant rs35705950 and IPF
| Location | IPF (%SNP) | Controls (%SNP) | Odds Ratio GT Versus GG | Odds Ratio TT Versus GG | P Value | Reference Nos. |
|---|---|---|---|---|---|---|
| United States | 367 (60.8%) | 802 (20.7%) | 5.7 (4.3–7.5) | 9.6 (4.7–19.4) | 8.9 × 10−41 | 236 |
| United Kingdom | 110 (67.0%) | 416 (19.6%) | 6.6 (4.1–10.7) | 11.8 (4.3–33.7) | 2.0 × 10−17 | 191 |
| France | 142 (65.5%) | 1383 (20.2%) | 6.4 (4.5–9.0) | 19.1 (9.0–36.1) | 9.0 × 10−29 | 13 |
| United States | 2,492 (63.1%) | 1890 (19.6%) | 4.5 (3.9–5.2) | 20.2 (15.2–27.0) | 7.2 × 10−95 | 47 |
| United States | 324 (57.2%) | 702 (21.3%) | 2.4 (2.1–2.8) overall | 2.4 × 10−50 | 146 | |
| United States | 84 (55.9%) | 689 (21.8%) | 3.2 (2.2–4.6) overall | 1.2 × 10−10 | 226 | |
| Germany | 71 (54.9%) | 35 (8.6%) | 11.1 (3.3–37.0) overall | <0.001 | 66 | |
IPF, idiopathic pulmonary fibrosis; SNP, single nucleotide promoter.
While initial studies of rs35705950 are among those of European ancestry, recent studies have examined the evidence for association with pulmonary fibrosis in other race or ethnic groups (66, 156, 223). The frequency of the T allele at rs35705950 is 11, 8, and 1% among European, South Asian, and East Asian populations, respectively, and was observed only once among 504 individuals (1,008 alleles) from Africa (1,000 genomes) (54). Even though rs35705950 is rare among Asian and African ancestries, the evidence so far indicates that the risk associated with the variant among Asians (66, 223) and Mexicans (156) is similar to the risk observed among European ancestries (Table 2). This similarity is important as the MUC5B promoter variant rs35705950 is considered a target for early diagnosis and intervention (72), and rs35705950 is considered for prognostic potential (158).
Table 2.
Summary of studies conducted in Asians and Hispanics investigating the relationship between the MUC5B promoter variant rs35705950 and IPF
| Location | IPF (%SNP) | Controls (%SNP) | Odds Ratio GT Versus GG | Odds Ratio TT Versus GG | P Value | Reference Nos. |
|---|---|---|---|---|---|---|
| Mexico | 82 (50.0%) | 108 (12.5%) | 7.90 (3.63–18.31) overall | 3.8 × 10−09 | 156 | |
| Korea | 228 (2.2%) | 102 (0.0%) | Unable to calculate | 156 | ||
| China | 165 (6.7%) | 1013 (1.6%) | 4.33 (1.99–9.42) overall | 0.001 | 223 | |
| Japan | 44 (6.8%) | 310 (1.6%) | 4.34 (1.02–18.49) overall | 0.03 | 66 | |
IPF, idiopathic pulmonary fibrosis; SNP, single nucleotide promoter.
The elevated risk associated with rs35705950 is substantially higher than for most other common risk variants for complex disease, with the exception of the human leukocyte antigen (HLA) region for some autoimmune diseases such as type I diabetes and systemic lupus erythematosus (SLE), which have odds ratios greater than 10 (42) and 4 (167), respectively, for specific HLA alleles. Other mucin polymorphisms that are associated with gastric and colon cancer have much lower risk estimates than those estimated for rs35705950. For example, a meta-analysis of studies of rs4072037 in the MUC1 gene estimate the OR for the rare allele to be 0.58 (95% CI: 0.44, 0.75) among European ancestries and 0.60 (95% CI: 0.54, 0.66) among Asian ancestries under a dominant model for the rare allele (40), corresponding to an OR of ∼1.7 for the common allele. Similarly, the strongest known association with type 2 diabetes is with rs7903146 in the TC7FL2 gene; the OR for risk is estimated to be 1.49 (95% CI: 1.34, 1.66) among a Danish sample and 1.45 among a West African sample (95% CI: 1.19, 1.77) (58).
C. Rare Versus Common IPF Risk Alleles
Like other complex diseases, both rare and common variants contribute to risk of pulmonary fibrosis, with rare variants typically having larger effects than the more common variants. However, in IPF the strongest known risk factor for pulmonary fibrosis, the MUC5B promoter variant rs35705950, has a larger effect on disease risk than anticipated by most models of the relationship between disease allele frequency and impact on risk, given the relatively common frequency of the risk allele (Figure 5). The rare variants identified in genes like TERT are associated with a large increased risk of disease compared with common variants, but by virtue of their rare frequency, individually account for a small proportion of disease. The more common variants generally have lesser effects, but because they are so common, have the potential to explain more of the population burden of disease. It is important to note that given the low incidence of disease, even the substantial relative increases in risk associated with any of the common variants still results in a low absolute risk for IPF. The impacts of rs35705950 and other common risk factors have similar impacts among both familial and sporadic cases, indicating that studies of either group may have important implications for the other.
FIGURE 5.
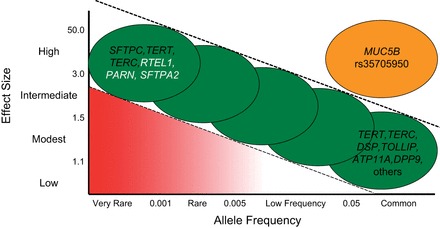
Relationship between allele frequency and penetrance of the risk allele, with examples of genes with risk alleles for IPF. Genes in black font have variants reported for both familial and sporadic IPF, and genes in white font have variants reported only for familial IPF. [Adapted from Figure 1 in Antonarakis et al. (2), with permission from Nature Publishing Group.]
D. Mucin Gene Expression and Disease
The risk allele at rs35705950 is a gain-of-function variant associated with increased expression of MUC5B in unaffected subjects (178) and among subjects with IPF (Helling et al., unpublished data). The MUC5B promoter variant rs35705950 is located in a highly conserved, regulatory region 3 kb upstream from the transcription start site of the MUC5B gene. Our results indicate that the variant allele is associated with increased MUC5B mRNA expression in lung tissue from unaffected individuals and patients with IPF (Figure 6A; Refs. 177, 178). Moreover, we have found that enhanced expression of MUC5B is localized to the distal airway, respiratory bronchiole, and the honeycomb cyst (Figure 6, B and C; Ref. 177), and that the MUC5B promoter variant rs35705950 enhances expression of MUC5B in the bronchiolo-alveolar epithelia (141). These findings suggest that dysregulation of MUC5B, especially in the peripheral airspace, is a consequence of the MUC5B promoter variant and that overexpression of MUC5B in this region of the lung may be involved in the pathogenesis of IPF.
FIGURE 6.
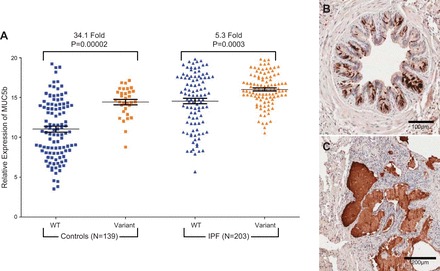
A: MUC5B expression in IPF (n = 203) and unaffected subjects (n = 139) stratified by MUC5B promoter variant (rs35705950) genotype. MUC5B is produced in the bronchiolo-alveolar epithelia (B) and honeycomb cysts (C) of patients with IPF. [Adapted from Seibold et al. (178).]
However, the MUC5B promoter variant rs35705950 does not fully explain the observed differential expression of MUC5B between cases and controls (178, 232). Polymorphisms in the proximal promoter region within 1 kb of the transcription start site of MUC5B are also associated with expression changes in MUC5B (88). Among Japanese, three of these variants (rs17228946, rs17235346, and rs17235353) associated with increased expression were associated with the lung disease panbronchiolitis (88). Moreover, the 4-kb promoter region of the MUC5B gene has 3 CpG islands and other regulatory sites, suggesting that expression of MUC5B may be a function of genetic and nongenetic factors. Ongoing studies are examining epigenetic and environmental influences such as cigarette smoking on MUC5B expression and whether these effects are modified by rs35705950.
Dysregulation of mucin expression is commonly observed in gastric and colorectal cancer (86, 154, 168, 194, 197, 220, 221). The pathways implicated by these studies and animal models are strongly suggestive of alterations in host defense and barrier function as drivers of disease risk. For example, the mouse knockout of MUC2 upregulates the TLR pathway (17), and knockout of MUC4 is associated with increased colonization of Helicobacter pylori (149). Erosion of barrier function due to alterations in MUC1 and MUC4 are important for metastasis in gastric (MUC1) (197), ovarian (MUC4) (160), and lung cancer (MUC4) (160). There are few examples of specific sequence variants in mucin genes that are convincingly associated with either disease or expression differences, however. One exception is the SNP noted above in MUC1 that is associated with gastric cancer (rs4072037); the exonic SNP is associated with alternative splicing of the MUC1 gene (144). This same SNP may be associated with serous borderline tumors, mucinous borderline, and invasive tumors of the ovary and endometroid tumors (228).
E. Evolutionary Selection
The magnitude of increased risk associated with the risk allele of rs35705950, which occurs at a baseline frequency of ∼9% of the European-ancestry population, is striking. Because pulmonary fibrosis typically has an onset well after reproductive age, the T allele at rs35705950, which increases MUC5B expression, is not expected to be subject to negative selection due to increasing risk for pulmonary fibrosis. Recent work demonstrating the necessity of MUC5B for mucociliary clearance, infection control, and immune regulation in mice indicates that increased MUC5B may provide a selective advantage with improved host defense, hence resulting in positive selective pressure on the T allele at rs35705950. In fact, among patients with IPF, the MUC5B promoter variant is associated with an improved prognosis (158) and a lower bacterial burden (137), suggesting a relationship between the variant allele and improved host defense. In light of these observations and the fact that rs35705950 is rare among East Asians and Africans, investigations of the evidence for selection at rs35705950 are underway; the genome-wide data available allow studies of the strength of evidence of selection at the MUC5B locus as well as other loci. Comparison of these data with those in other populations can provide estimates of the age and genealogy of the risk variant that may shed light on important selective events relevant to the genesis of idiopathic pulmonary fibrosis.
III. HOMEOSTATIC AND PATHOBIOLOGICAL FUNCTIONS OF LUNG MUCINS
As a crucial mechanism of homoeostatic innate defense, mucociliary clearance requires the coordinated functions of its two components: mucus and cilia. Mucus is comprised of water, salts, cells, cellular debris, and other macromolecules that are held together in a mesh formed by polymeric mucin glycoproteins (45, 206). Mucins are released by nonciliated secretory cells, club cells in the distal lung (11, 44, 204). In the extracellular space, the mucus gel they form is positioned above an epithelial layer that is also densely populated with ciliated cells. The interactions between mucus and cilia are tightly regulated by factors that include the quantities and qualities of mucins produced and the maintenance of a cilia structures and motility. Indeed, dysregulated mucin production, aberrant cilia assembly, and poor control of the salt and water contents in the mucus layer are factors that are known to impact mucociliary clearance function and lead to disease development and exacerbation. The regulation and control of mucin and cilia functions in health and disease are presented below.
A. Mucin Genes
MUC5AC and MUC5B, and their orthologs (“Muc” in mice) are expressed throughout the upper and lower respiratory tract. In human upper airways, MUC5AC is found in the nasopharynx, while MUC5B is found in nasal and oral gland secretions. In tracheobronchial conducting airways, both mucins are expressed by surface and submucosal gland mucous cells, but their expression ordinarily favors higher relative expression of MUC5AC by surface epithelia and MUC5B by the glands. In bronchioles (small airways that lack cartilage or glands), very little MUC5AC is present, and MUC5B predominates. In mice, which lack cartilaginous or gland-rich airways distal to the trachea, in the baseline state surface epithelial cell expression of Muc5b predominates (45, 175, 234).
In diseased conditions in humans, or in animal models of lung disease, mucin expression and localization change dramatically. In human airways, MUC5AC is induced in both submucosal glands and surface epithelia (99, 152, 230). While this finding with MUC5AC is quite consistent, the effects of disease states on MUC5B vary substantially. The variable expression of MUC5B in human disease is best studied in bronchial biopsies, which favor detection of transcript and protein levels in surface epithelia. In human asthma, whereas MUC5AC expression in bronchial biopsies increases significantly and consistently, MUC5B expression decreases (152). Importantly, this decrease is driven primarily by allergic status: patients with the highest levels of allergic inflammation display MUC5B expression levels that are lower than healthy controls and less severely allergic patients (230). Perhaps not surprisingly, the mechanisms that regulate the control and expression of MUC5AC/Muc5ac are characterized more substantially than MUC5B/Muc5b.
Data demonstrating MUC5AC/Muc5ac expression mechanisms have been extensively studied in the context of asthma. The type 2 inflammatory cytokine IL-13 induces MUC5AC/Muc5ac expression in human asthma and in mouse models (114). This process has been linked to the signal transduction and activator of transcription (STAT)-6 pathway (113). However, the lack of a cis STAT-6 DNA binding motif in the MUC5AC/Muc5ac promoter regions suggests that its role is indirect (234). Rather, signaling pathways that involve control through IL-13 and the epidermal growth factor receptor and Notch, ultimately leading to the inactivation of forkhead box A2 (FOXA2) and activation of SAM pointed domain-containing Ets transcription factor (SPDEF) in IL-13, have been characterized extensively (22, 23, 107, 155, 196, 222). Other pathways downstream of inflammation, such as mitogen-activated protein kinases (MAPKs), nuclear factor κB (NF-κB), and hypoxia inducible factor-1 (HIF-1) have also been described (24, 25, 234). The roles of these pathways in MUC5AC/Muc5ac regulation have been recently reviewed elsewhere (24, 25, 234).
Given its more complex patterns of expression, much less is known about MUC5B/Muc5b regulation. MUC5B/Muc5b is abundantly expressed in naive airways from early life time points onward (175). However, whereas IL-13 potently stimulates MUC5AC expression, IL-13 negatively affects MUC5B levels in human asthma (152, 230). This is supported further by data in differentiated human airway epithelial cell cultures, where IL-13 directly downregulates MUC5B expression (152, 237). Levels of MUC5B expression by surface epithelia also decrease in early stages of COPD (19, 79). However, MUC5B increases later in COPD (19, 98), and also in cystic fibrosis (CF) (37, 99) and IPF (177, 178) consistent with a lack of an IL-13 driven component in these diseases. In contrast to what is observed in humans, mice show little negative control of Muc5b expression by IL-13. Muc5b increases two- to threefold in inflammation driven by allergen sensitization/challenge or transgenic overexpression of IL-13 (114, 234). However, in a chronic cigarette smoke exposure model of COPD, a transient downregulation followed by recovery of Muc5b expression occurs (Evans, unpublished observation). Taken together, these data suggest that the complex control of MUC5B/Muc5b expression involves multiple levels of regulatory machinery. Indeed, the MUC5B/Muc5b promoter contains both core elements that are highly conserved including the 3-kb single nucleotide promoter variant rs35705950 that is present in primates and controls MUC5B levels by >30-fold in the lungs (Figure 6A). Whether this site is actively bound by transcription factors or comprises an added layer of epigenetic control is yet to be determined. Evidence for epigenetic control of MUC5B has been shown in studies using carcinoma cell lines from various tissues (216), but the role of the SNP in controlling MUC5B expression in these cell lines and in nonpulmonary organs in vivo is not yet known.
B. Mucin Structures
Whereas the molecular control of mucin expression of MUC5AC/Muc5ac and MUC5B/Muc5b have been studied most extensively, the biochemical features of these mucins at the glycoprotein level have been described but not extensively tested at functional levels. In part, this is because of their extraordinarily large sizes (in the mega-Dalton range) and the characteristics as thick viscoelastic polymers. Both MUC5AC/Muc5ac and MUC5B/Muc5b contain disulfide-rich NH2 and COOH termini that form disulfide linked polymers (Figure 7). These give rise to polymers that can vary in assemblies of 2 to >20 oligomers. Disruption of these with thiol reducing agents such as dithiothreitol or N-acetyl cysteine reduces their sizes and increase mucociliary transport.
FIGURE 7.
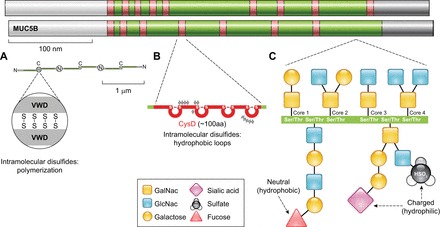
Polymeric mucin structures. MUC5AC and MUC5B apoprotein backbones are comprised by NH2-terminal (white) and COOH-terminal (gray) cysteine-rich domains that are conserved in polymeric mucins and von Willebrand factor. The central portions of MUC5AC and MUC5B are comprised of a mucin glycosylation domain (green) that is rich in serines and threonines that are sites of O-linked glycan attachments. The glycosylation domain is interrupted by additional cysteine-rich CysD domains (red). A: the COOH-terminal von Willebrand-like domain (VWD) has a cysteine-knot structure that is the site of disulfide dimerization in the endoplasmic reticulum; each dimer is ∼1 μm in length. In the Golgi, dimers further assemble at their NH2 termini via covalent disulfide bonds and other noncovalent interactions to form extensive polymeric structures that are 10s to 100s of microns long. B: in cysteine-rich CysD domains, intramolecular disulfide bonds also assemble. CysDs contain hydrophobic amino acids and form loops that may promote mucin mesh network alignment. C: in the mucin glycosylation domain, glycan linkages occur on the hydroxylated ends of serine and threonine residues through the initial attachment of N-acetylgalactosamine (GalNac). This initial O-glycosylation step is followed by galactose and/or N-acetylglucosamine (GlcNac) attachments in specific core linkages. These can be elaborated with additional galactose and GlcNac moieties and be modified further by the attachment of fucose, sialic acid, and sulfate end groups. These terminal groups carry charges and structural characteristics that mediate mucin biophysical properties and interactions with host-derived and foreign materials.
Glycosylation is another property that lends to mucin size and charge heterogeneity (35, 45, 206). Glycosylation is controlled by numerous enzymes that include core machinery that introduce O-linked N-acetylgalactosamines to serine and threonine amino acids, followed by Core-linkage and terminal glycosylation machinery that result in very large and diverse carbohydrate structures. Terminal glycans include N-acetylglucosamine, galactose, fucose, and sialic acid, which vary in size, polarity, and charge density, which can also be affected by sulfation. Importantly, terminal glycosylation of mucins utilizes in many cases the same machinery that determines blood antigen types (e.g., Lewis and H antigens). In human asthma, the presence of H antigen secretor status (fucosyltransferase 2-mediated α1,2 fucosyaltion) has been linked to heightened risk for severe asthma exacerbation and hospitalization (78). This could relate either to changes in host defense or physical obstruction, but these have not been determined.
C. Secreted Polymeric Mucin Functions
The pathological obstructive effects of secreted mucus reflect aberrant control of its homeostatic regulation. Mucus can either be tightly adherent to or loosely associated with underlying epithelia. In the colon, tight association is responsible for keeping the intestinal flora and underlying tissues separated. In the lungs, loosely associated mucus is responsible for trapping inhaled particles, including bacteria, while still allowing for transport out of the airways by ciliary and cough driven forces, followed by elimination by expectoration or swallowing (45, 206).
These clearance mechanisms also remove endogenous debris, including dying epithelial cells and leukocytes. While the means of transport of these is somewhat intuitive, recent data also show that in addition to physical elimination, mucin glycans also interact with carbohydrate interacting lectins that are present on leukocyte surfaces in a highly selective fashion. Sialic acid residues that are present in α2,3 linkages on the surface of MUC5B/Muc5b bind to sialic acid binding immunoglobuluin-like lectin receptor (Siglec)-8, which is present on human eosinophils, and Siglec-F, which is present on mouse eosinophils and resident alveolar macrophages (9, 135). These interactions have recently been shown to mediate apoptosis in human and mouse eosinophils (100), but the effects of Muc5b sialoside-Siglec-F signaling in alveolar macrophages are not yet determined. Interactions between siglecs, as well as other glycan-selective immunoreceptors such as dectins, have only recently been identified. Their interactions have opened a new field for studying how first and second lines of innate host defense are linked.
D. Pathological Effects of Mucins
When mucus is overproduced, excessively secreted, and/or overly viscous and adherent, materials that need to be eliminated accumulate and drive disease exacerbations. This is particularly prominent in cystic fibrosis, but adherent mucus plugs are also seen in severe asthma (77, 115, 182), COPD (91), and IPF (178). Mucus accumulation results in plugging that is linked to airflow obstruction and can lead to fatal asphyxiation, but may also compromise underlying cell function. In a mouse model of asthma, genetic deletion of Muc5ac resulted in protection from mucus plugging and airway hyperreactivity acutely (231). Under chronic conditions, mucus accumulation can also lead to pathogen and leukocyte accumulation, which result in impaired immune defense and chronic tissue injury.
E. Cilia
Cilia are organelles that protrude from plasma membranes, with two types of cilia: motile and nonmotile. Motile cilia are present in specialized cells that are involved in fluid and particle transport. Nonmotile cells are present in almost all cells, which typically have a single primary cilium whose functions include sensory signal transduction. Both are constructed from tubulin-formed axonemes that also contain structural anchors such as dyneins and nexin. In motile cilia, these form a ring of nine microtubule doublets with a central pair of singlet microtubules (9+2) axonemes, but in nonmotile cilia, the central pairs are absent, resulting in outer ring only (9+0) axonemes (16).
Motile cilia are present in specialized cells involved in particle transport. In the lungs, ciliated cells are abundant throughout the conducting airways and are typically present in roughly equal numbers compared with nonciliated/secretory cells. Ciliated airway epithelial cells are easily identified by their apical surfaces with numerous motile cilia in clusters of 100–300 projecting into the airway lumen space and surface mucus. Although the ciliated and mucus layers in the lumen were once considered to be separate entities, so-called “sol” and “gel” phases, recent studies demonstrate that this separation is not so distinct. Due to the presence of highly glycosylated membrane mucins (MUC1, MUC4, and MUC16) along and between cilia, the periciliary layer (PCL) forms a grafted brush that allows for the free movement of cilia. The overlying, viscous layer of mucus that serves to transport adsorbed particles and pathogens is positioned atop the grafted brush, thus creating what is now referred to as a gel-on-brush structure (18). Both the membrane mucins lining the cilia surfaces and the mucus gel itself are highly glycosylated and hydrated. Charged sugars on the mucins participate with ions such as sodium, calcium, chloride, and bicarbonate to maintain an osmotically balanced hydrogel. Dysfunction of these components could lead to a loss of hydration, which causes cilia to collapse as water is removed from the PCL. For example, excess osmoles in mucus pulls water from the PCL, reducing its thickness via collapse of cilia (63). This results in defective mucus transport, particle/debris accumulation, and impaired repair processes following injury (Figure 8).
FIGURE 8.
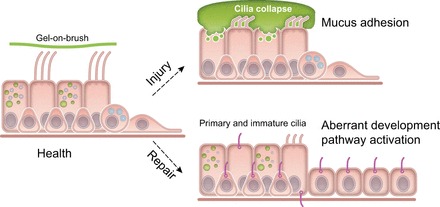
Regulation of motile and nonmotile cilia in the lungs. Under conditions of health, multiciliated cells are present in conducting airways. These specialized epithelial cells maintain the normal structure and function of apically localized motile cilia clusters through a balance of electrolyte and osmolyte homeostasis. In addition, membrane tethered mucins present along cilia adsorb water and maintain a stable “grafted brush” structure that supports an overlying gel. Normal repair programs following injury activate progenitor basal-like cells to differentiate fully to secretory and ciliated cells. Primary nonmotile cilia precede motile cilia in this normal repair process. Under conditions where mucins are overproduced or hypersecreted (such as the MUC5B promoter variant rs35705950), or when electrolyte homeostasis is disrupted (e.g., through excessive Na+ absorption), cilia collapse and aggregates of mucus adhere to airway surfaces, potentially worsening injury. Under injurious conditions, resulting from excess mucus or other forms of lung injury, aberrant repair could lead to abnormal activation of progenitor basal-like cells, resulting in partially differentiated ciliated cells that retain primary cilia, have poorly developed apical motile cilia, and developmental programs that are aberrantly activated.
Disease states that can result in cilia dysfunction include conditions of impaired ion transport such as cystic fibrosis, structural abnormalities of cilia components in primary ciliary dyskinesia (PCD) (101), and in diseases in which the polymeric mucins MUC5B and MUC5AC are overproduced (e.g., asthma) (133, 148). The recent discovery that the MUC5B promoter variant rs35705950 causes excessive MUC5B production in small airways suggests that this could result in impaired cilia-mediated clearance in the small airways. As a result of defective ciliary function, clearance is impaired, leading to chronic airway infection and inflammation, bronchiectasis, and distal lung remodeling occur.
In addition to motile cilia, primary cilia (highly conserved, nonmotile, solitary structures) are present in almost all nondividing cells of the airway and transduce essential signaling information from the extracellular milieu. A large number of ciliopathies (20, 51) as well as polycystic kidney disease (PKD) (38) are associated with defects in the primary cilium. The primary cilium is critical for multiple developmental pathways (55), and their dysregulation is prominent in IPF lung (181). Furthermore, it has also been shown that primary ciliogenesis precedes motile cilia during development and that primary cilia are transiently present in adult mouse airway during repair after respiratory virus injury (82). These lines of evidence support the notion that primary cilia play a role in cellular programming during repair following injury (Figure 8).
Tissue damage can itself perpetuate the activation of inflammatory cascades that if persistent can also chronically evoke injury-repair processes. While tissue repair may ordinarily result in resolution of injury and a return to homeostasis, chronic injury and/or aberrant repair can result in remodeling of tissues to pathological states. In diseased lungs, remodeling is seen as chronic mucous cell metaplasia/hyperplasia, small airway and alveolar fibrosis, and emphysematous alveolar destruction. These processes have been shown to be associated with endoplasmic reticulum (ER) stress or hypoxia in intracellular compartments, culminating in the release of proinflammatory and tissue growth factors such as TGF-α and -β and destructive proteases such as matrix metalloproteinases and cathepsins into the extracellular environments. Thus the long-term results of chronic mucus hypersecretion can result in enhanced lung injury.
IV. CLINICAL RELEVANCE OF MUC5B TO PULMONARY FIBROSIS
MUC5B is a validated target in IPF. As noted above, we have found that 1) a common gain-of-function (Figure 6A) promoter variant in MUC5B rs35705950 is the strongest risk factor (genetic and otherwise), accounting for 30–35% of the risk of developing IPF (178) (confirmed in 9 independent studies) (13, 66, 146, 156, 178, 191, 226, 236), including our GWAS [OR for T (minor) allele = 4.51; 95% CI = 3.91–5.21; P = 7.21 × 10−95 (47)]; 2) rs35705950 can potentially be used to identify individuals with preclinical pulmonary fibrosis (72) and is predictive of radiologic progression of preclinical pulmonary fibrosis (3); 3) IPF subjects with the MUC5B promoter variant rs35705950 have improved survival compared with those without this variant (158) (Figure 2); and 4) MUC5B may be involved in the pathogenesis of pulmonary fibrosis. Specifically, MUC5B message and protein are expressed in bronchiolo-alveolar epithelia of IPF (141, 177) (Figure 6, B and C) and the characteristic IPF honeycomb cysts (177, 178). Reflecting on these advances, it is reasonable to consider whether the MUC5B promoter variant rs35705950 could be used to assist in the diagnosis or clinical management of patients with IPF.
The MUC5B promoter variant rs35705950 may prove helpful in identifying asymptomatic cases of early IPF. For both the familial and sporadic forms IPF, it is generally accepted that pulmonary fibrosis can be present a number of years before symptoms become apparent, suggesting that the diagnosis of IPF can be made before the lung has been extensively and irreversibly damaged. While most patients with IPF are diagnosed when the disease is advanced, an earlier diagnosis of IPF will detect patients with a lower burden of lung disease (111, 121, 163), more salvageable lung, and may reveal novel molecular targets for intervention that are only effective in early stages of IPF. Preclinical pulmonary fibrosis (PrePF) is an emerging clinical phenotype that is characterized by specific identifiable chest CT abnormalities (subpleural reticular changes, honeycombing, traction bronchiectasis, centrilobular nodules, and ground glass) (163). Preclinical pulmonary fibrosis has been reported more frequently among smokers and in families with two or more cases of pulmonary fibrosis (163, 189), and we have found that the MUC5B promoter variant rs35705950 is predictive of those with PrePF (OR = 6.3 per allele; 95% CI 3.1–12.7; Ref. 72). Others have found that among asymptomatic first-degree family members of FIP, 14% have interstitial changes on CT scan and 35% have interstitial abnormalities on transbronchial biopsy (111). Moreover, we have recently found that rs35705950 is predictive of radiologic progression of PrePF (OR = 2.8 per allele; 95% CI 1.8–4.4; Ref. 3) which was associated with a greater decline in FVC (P = 0.0001) and an increased risk of death (HR = 3.7; 95% CI 1.3, 10.7; P = 0.02). Interstitial lung abnormalities on chest CT scan, a radiologic feature of PrePF, has recently been shown to be associated with a greater risk of all-cause mortality in four independent cohorts (162), suggesting that in addition to having radiologic features of IPF, PrePF is likely a harbinger of IPF. While the MUC5B promoter variant is predictive of PrePF, rs35705950 is present in ∼19% of the population (178), and IPF occurs infrequently (<0.1%; Refs. 151, 165). Thus additional biomarkers are needed to identify individuals with PrePF within at-risk populations. In aggregate, these findings suggest that 1) IPF is underdiagnosed (72, 111); 2) PrePF is prevalent in an identifiable at-risk population, such as asymptomatic family members of FIP families or even sporadic IPF (72, 111); 3) ∼75% of the cases of PrePF are progressive (3); 4) radiologic progression of preclinical is associated with increased morbidity and mortality (3, 162); and 5) the MUC5B variant rs35705950 in combination with peripheral blood biomarkers and radiologic screening should prove useful in identifying those with PrePF. Thus we have put forward a model that uses the MUC5B promoter variant rs35705950 to identify a population that is at risk of both Pre-PF and IPF (Figure 9). We postulate that the development of IPF takes many years and that the MUC5B promoter variant can be used to identify individuals early in their path of disease development. While strategies have not been developed for primary and secondary prevention of IPF, identification of both an at-risk population and those with PrePF facilitates intervention prior to the development of advanced lung fibrosis with the goal of preventing the persistent and extensive fibroproliferative response that destroys and displaces normal lung tissue.
FIGURE 9.

Indicates that the “at-risk” population and the population with preclinical pulmonary fibrosis is large (19% with the MUC5B promoter variant and 1.8% of individuals = 50 years of age, respectively), IPF is diagnosed in a small population with established end-stage disease (111, 163), and preclinical pulmonary fibrosis can be identified using the MUC5B variant rs35705950. In addition, recent findings (3) indicate that preclinical pulmonary fibrosis (detected via chest CT scan) is associated with a poor prognosis, suggesting that preclinical pulmonary fibrosis may be a precursor of IPF.
The MUC5B promoter variant rs35705950 and other genotypes may prove helpful in managing patients with IPF. While IPF is a distinct entity, there are substantial variations in the clinical course of this disease. IPF is a progressive lung disease with death occurring on average within 3–5 yr of diagnosis. Adverse prognosticators include age, cigarette smoking history, and features of more extensive lung disease (finger clubbing, lung volumes, radiologic features, and oxygen desaturation) (96). However, some patients (5–10%) have acute exacerbations (33, 186), and up to 20% have a prolonged survival. While we have shown that IPF subjects with the MUC5B promoter variant rs35705950 have improved survival compared with those without this variant (158) (Figure 2), others have found that both a TOLLIP allele (146) and a TLR3 allele (147) also provide prognostic information among individuals with IPF. These findings suggest that IPF subtypes defined by genetic variants may prove helpful in prognosticating among individuals with IPF, and further consideration should be given to inclusion of these prognostic factors in trials designed to test the utility of novel therapeutic agents.
V. PROPOSED MODELS OF MUC5B-INDUCED PULMONARY FIBROSIS
While it is indisputable that the MUC5B promoter variant rs35705950 is a validated gain-of-function etiologic target in PrePF and IPF, the mechanisms that link enhanced production of MUC5B to the development of pulmonary fibrosis are not obvious or even partially understood. However, accumulating evidence suggests that the distal airway epithelium is the likely site of MUC5B overexpression and may at least localize the investigative efforts to understand the fibroproliferative effects of MUC5B in the IPF lung. Given the above considerations and the emerging understanding of the pathogenesis of IPF, there are at least three (and undoubtedly many more) competing hypotheses that link enhanced production of MUC5B in the bronchiolo-alveolar region to the development of pulmonary fibrosis.
One line of reasoning focuses on the relationship between overexpression of MUC5B, the involvement of the terminal airway, microscopic honeycomb cyst formation, and repair/regeneration of the distal airspace in IPF (159, 164, 181, 208). The MUC5B promoter variant rs35705950 leads to enhanced expression of MUC5B in the bronchiolo-alveolar epithelia (Figure 6, B and C; Ref. 141), and several histopathological analyses (159, 177, 178) have confirmed involvement of MUC5B in the pathogenesis of IPF in that region of the lung. Microscopic honeycomb cysts contain abundant MUC5B, and our recent observations highlight the relationship between MUC5B overexpression and microscopic honeycomb cyst formation (Figure 10) (232). Accumulating evidence suggests that the distal airway epithelium is the principal origin of alveolar stem cells. Bronchiolo-alveolar epithelial cells give rise to alveolar type I (ATI) and alveolar type II (ATII) cells in bleomycin-induced pulmonary fibrosis in mice (171), and it has recently been demonstrated in mice that distal airway stem cells expressing Krt5 proliferate into interbronchial regions of alveolar ablation following H1N1 infection and assemble into alveoli-like structures (112). Furthermore, activated rare lineage-negative, Krt5+ epithelial stem/progenitor cells proliferate and migrate widely to occupy heavily injured areas in the distal lung following H1N1 infection and bleomycin challenge (214). Persistent Notch signaling after H1N1 injury leads to parenchymal cysts, indicative of abortive regeneration, analogous to current concepts regarding honeycomb cyst formation in the IPF lung. In H1N1 lung injury, it has been proposed that honeycomb cysts represent daughter structures derived from the aberrant differentiation of pulmonary progenitors expressing KRT5/Krt5 (112, 159, 171, 172, 214).
FIGURE 10.
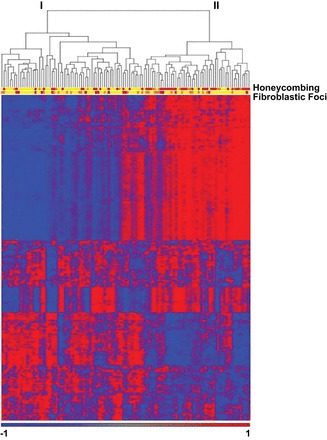
Gene expression profiling identifies two subtypes of IPF. mRNA profiles from 119 IPF lungs were subject to hierarchical clustering based on the expression of 472 transcripts that are differentially expressed at 5% FDR and with greater than 2-fold change in IPF compared with control lung. The most prominent feature of the heatmap is the group of 51 subjects (43%; subject group II) with relatively high expression compared with 68 subjects (57%; subject group I) of a large set of transcripts (transcript clusters A and B) and low expression of another set of transcripts (transcript cluster C). Transcript cluster B contains 80 unique transcripts upregulated in group II compared with group I IPF that include a number of genes that have been previously shown to be upregulated in IPF/UIP, namely, osteopontin, MMP1, MMP7, PLUNC, MUC5B, collagen COL17A1, and keratins 5, 6C, 15, and 17. Cluster C contains 71 unique transcripts that are downregulated in group II compared with group I IPF with a few previously implicated in IPF [advanced glycosylation end product-specific receptor (AGER)] or other chronic lung diseases [hedgehog interacting protein (HHIP)] and many novel IPF candidate genes. Functional enrichment analysis, using Fisher exact test, of the 121 unique transcripts in cluster A showed it to be strongly enriched in transcripts associated with the cilium genes (Benjamini corrected P value 3.7 × 10−11) and their structural components (axoneme, 3.9 × 10−11; dynein, 9.4 × 10−7). This cluster also contains a number of genes with unknown function. Decreased expression is represented by blue color while red indicates increased expression. [Adapted from Yang et al. (232), with permission from BMJ Publishing Group Ltd.]
In IPF, we have found that KRT5+ cells can be found within the fibrotic interstitium, and KRT5+ cells are in close association with MUC5B-rich honeycomb cysts (177). Cells lining honeycomb cysts in IPF appear to manifest intermediate features of ciliated and distal epithelial phenotypes, suggesting aberrant differentiation (177, 232). We have recently found that lung tissue samples from ∼40% of patients with IPF are highly enriched for transcripts of cilium genes, MUC5B, and MMP7, and this molecular phenotype is associated with microscopic honeycombing (Figure 10 and Ref. 232). In patient samples, bronchiolized epithelium expresses SOX2 and MUC5B but not SPDEF, consistent with abnormal programming of distal airway epithelia in IPF (159). Moreover, airway epithelium overlying fibroblastic foci contains p63-positive basal cells that are capable of undergoing epithelial to mesencyhmal transition (EMT) (85). Importantly, a striking number of cells in honeycomb cysts and bronchiolized epithelium express β-catenin (28), a gene in the Wnt developmental pathway with an established role in IPF (28, 60, 102, 103, 181, 218). The strong association of MUC5B expressing cells with KRT5+ cells lining honeycomb cysts suggests a role for MUC5B expression in the establishment or maintenance of honeycomb cysts by KRT5+ cells, and points to a role for the bronchiolo-alveolar epithelium as a potential site of initiation of the fibroproliferative response with involvement of MUC5B and stem/progenitor cells.
Based on these considerations, we hypothesize that excessive production of MUC5B by stem cells that attempt to regenerate injured bronchiolar and alveolar epithelium disrupt normal developmental pathways and highjack the normal reparative mechanisms in the distal lung, resulting in chronic fibroproliferation and honeycomb cyst formation (Figure 11). Given the large size and extensive glycosylation of MUC5B/Muc5b protein, we postulate that its production presents a significant metabolic stress, which can interfere with differentiation of airway stem cells. This effect is enhanced in carriers of the gain-of-function MUC5B variant rs35705950 allele. Alternatively, excess MUC5B/Muc5b could affect KRT5/Krt5 cells, perhaps by modifying growth factor signaling pathways. While these disrupted development mechanisms may directly initiate fibroproliferative responses, it is notable that the murine bleomycin model can generate fibrosis without honeycombing (232). Although speculative, it is more likely that honeycomb cysts represent unsuccessful reparative responses, which then interfere with basic signaling events leading to the emergence of reprogrammed cells. These structures subsequently prevent establishment of normal alveolar homeostasis and may promote persistent foci of fibroproliferative activity.
FIGURE 11.
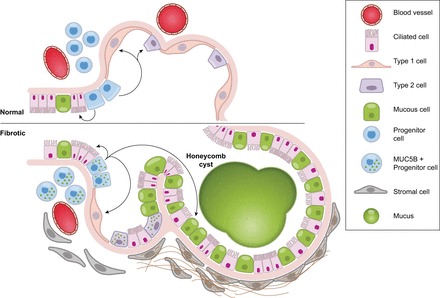
Model of stem cells repopulating bronchioles and alveoli under normal physiological conditions and when challenged with increased expression of MUC5B. We hypothesize that excessive production of MUC5B by stem cells that attempt to regenerate injured bronchiolar and alveolar epithelium disrupt normal developmental pathways and highjack the normal reparative mechanisms in the distal lung, resulting in chronic fibroproliferation and honeycomb cyst formation.
A second line of reasoning focuses on the possibility that IPF is a mucociliary disease caused by recurrent injury/inflammation/repair at the bronchoalveolar junction, which is initiated and exacerbated by overexpression of MUC5B and reduced mucociliary function, retention of particles, and enhanced lung injury (Figure 12). Based on the relationship between the MUC5B promoter variant rs35705950 and excess production of MUC5B specifically at the bronchoalveolar junction, we hypothesize that too much MUC5B impairs mucociliary function (14, 18), causes excessive retention of inhaled substances (air pollutants, particles and chemicals from cigarette smoke, microorganisms, etc.), and, over time, the foci of lung injury lead to scar tissue and persistent fibroproliferation that expands and displaces normal lung tissue. Excess concentrations of MUC5B may also physically impair ciliary function (18), further enhancing the retention of inhaled particles, dissolved chemicals, and microorganisms. Given the importance of environmental exposures, such as asbestos and silica, in the development of other forms of interstitial lung disease, it is logical to speculate that common inhaled particles, such as those associated with cigarette smoke or air pollution, might cause exaggerated interstitial injury in individuals who have defects in mucociliary clearance and mucosal host defense.
FIGURE 12.
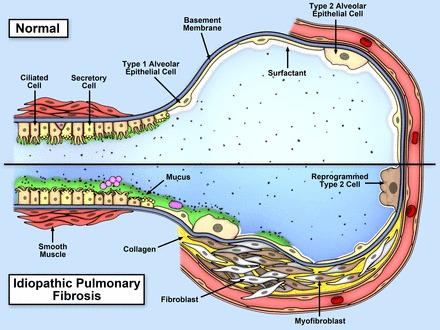
Model of recurrent inflammation/injury/repair at the bronchoalveolar junction that is initiated and exacerbated by overexpression of MUC5B, retention of inhaled particles, and enhanced lung injury. Top panel is the normal bronchoalveolar region, and bottom panel represents a bronchoalveolar region affected by IPF. We hypothesize that IPF is a mucociliary disease that is caused by recurrent injury/inflammation/repair at the bronchoalveolar junction that is initiated and exacerbated by overexpression of MUC5B leading to reduced ciliary function, retention of particles, and enhanced injury.
A third line of reasoning focuses on the relationship between MUC5B and motile cilia in the distal airway. We identified a cilium gene expression signature in the IPF lung contains both primary and motile cilia genes (Figure 10). A role of primary cilia in the pathogenesis of IPF would be consistent with the aberrant repair process following the injury to bronchiolo-alveolar regions of the lung and the concept that honeycomb cysts could be the result of aborted attempts to regenerate the terminal airways via activation of developmental pathways that signal in the primary cilium (55) and have been implicated extensively in IPF (181) (Figure 8). Alternatively, a role of cilium gene expression and function in multiciliated airway epithelial cells in the distal airways and bronchioles would be consistent with the idea that accumulation of mucus, caused by the MUC5B promoter variant, in the bronchoalveolar region of the lung impairs mucociliary function (Figure 8). Available mouse models are helping to clarify this somewhat, but more work is needed to define the pathobiology of MUC5B among carriers of the variant rs35705950 allele in humans.
Lastly, given the large size and heavily glycosylated characteristic of airway mucins (45), we have been particularly interested in the relationship between the MUC5B promoter variant rs35705950, enhanced transcription of MUC5B and posttranscriptional processing of MUC5B, endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), as a pathway to pulmonary fibrosis. In fact, ER stress is observed in a number of different diseases (62), including IPF (5, 26, 104–106, 117, 118, 125, 138, 201), and our preliminary findings indicate that excess production of Muc5b induces ER stress and activates UPR both at baseline and after profibrotic stimulation. The adaptive or homeostatic UPR attempts to restore normal protein folding by jointly coordinating three different responses: IRE-1/XBP1, the PERK/ATF4, and BiP/ATF6 (83). These three molecular pathways of UPR involve a specific membrane ligand and result in the activation of specific gene targets (57, 83). When proteostasis is abnormal, stress induced by the accumulation of unfolded proteins in the ER is a feature of secretory cells that undergo apoptosis. While ER stress has been observed in IPF (5, 26, 104–106, 117, 118, 125, 138, 201), in experimental models of lung fibrosis, activation of the UPR in alveolar epithelial cells can lead to EMT (202, 238) and fibroproliferative lung disease (117, 201). ER stress is also at play in the airway epithelium and is required for MUC5B production (127). Not surprisingly, ER stress and apoptosis interfere with the normal epithelial response to injury, wound healing, and tissue repair which are hallmarks of IPF (95), can be modeled in vitro (184), and may be particularly relevant to IPF that is associated with the gain-of-function MUC5B promoter variant.
VI. PERSPECTIVE
The discovery that MUC5B plays a key role in the etiology of IPF, accounting for 30–35% of individuals who develop this is disease, is important for the following reasons. First, the assay for MUC5B promoter variant rs35705950 is an inexpensive genetic test that could be offered to high-risk asymptomatic individuals to identify an at-risk population or identify individuals in the early stages of disease. While high-risk populations, such as first-degree relatives of patients with either familial or sporadic IPF, are obvious targets for screening, it is possible that screening for IPF will be more widely deployed as therapeutic interventions become more effective in the early stages of disease and have fewer side effects. Preventing the destruction of lung tissue in an asymptomatic individual is a much more effective strategy than treating patients with IPF once extensive lung damage has occurred. Second, lung mucins may prove to be an effective therapeutic target for the treatment of IPF. We postulate that terminal bronchiolar and possibly alveolar epithelial inflammation/injury/repair as well as subsequent fibroproliferation induced by MUC5B can be overcome, in part, by decreasing the concentration of MUC5B in the distal airspace. Moreover, we think that our ability to restore MUC5B homeostasis, improve mucociliary clearance, and reduce the effects of excessive mucus accumulation may prove especially effective in limiting the progression of preclinical/mild stages of disease. However, given the spatial and temporal distribution of the microscopic lesions in IPF, reducing the concentration of MUC5B in the distal airspace may also prove effective in stabilizing established IPF. Third, since IPF subjects with the MUC5B promoter variant rs35705950 have improved survival compared with those without this variant (158), it is logical to control for the MUC5B genotype (and potentially other genotypes) when considering the impact of novel therapeutic agents. Lastly, the discovery that MUC5B plays a role in the etiology of IPF provides a novel direction in understanding the basic mechanisms involved in the pathogenesis of IPF. Although we have learned a great deal about many of the basic biological mechanisms involved in IPF, the disease is heterogeneous and the final common pathways of fibrogenesis are not well understood (145, 209). The MUC5B discovery not only identified a novel target to consider, this discovery also highlights the potential involvement of the distal airway, the bronchiolo-alveolar region, and mucociliary transport in the development of this complex and incurable disease.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01-HL097163, R21-HL120770, UH2-HL123442, P01-HL092870, and R25-ES025476 as well as Department of Veterans Affairs Merit Award 1I01BX001534. C. M. Evans was supported by NIH Grants R01-HL080396, R21-ES023384, and R01-HL130938 as well as American Heart Association Grant 14GRNT19990040.
DISCLOSURES
D. A. Schwartz has a patent on the use of the MUC5B promoter variant rs35705950 for the risk prediction, diagnosis, prognosis, and treatment of pulmonary disorders (US Patent No. 8,673,565); has a provisional patent on the early diagnosis and treatment of IPF; and has developed a company (Eleven P15) based on the early recognition and treatment of IPF.
ACKNOWLEDGMENTS
C. M. Evans, T. E. Fingerlin, and M. I. Schwarz contributed equally to this manuscript.
Address for reprint requests and other correspondence: D. A. Schwartz, Univ. of Colorado, 12631 East 17th Ave., B178, Aurora, CO 80045 (e-mail: david.schwartz@ucdenver.edu).
REFERENCES
- 1.Adelman AG, Chertkow G, Hayton RC. Familial fibrocystic pulmonary dysplasia: a detailed family study. Can Med Ass J 95: 603–610, 1966. [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis SE, Chakravarti A, Cohen JC, Hardy J. Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet 11: 380–384, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Araki T, Putman RK, Hatabu H, Gao W, Dupuis J, Latourelle JC, Nishino M, Zazueta OE, Kurugol S, Ross JC, Estepar RS, Schwartz DA, Rosas IO, Washko GR, O'Connor GT, Hunninghake GM. Development and progression of interstitial lung abnormalities in the Framingham Heart Study. Am J Respir Crit Care Med. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA 3rd, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356: 1317–1326, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Baek HA, Kim do S, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 46: 731–739, 2012. [DOI] [PubMed] [Google Scholar]
- 6.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating Centers. Am J Epidemiol 152: 307–315, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 155: 242–248, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, DeCamp MM, Mestan KK, Perlman H, Budinger GR, Misharin AV. Flow cytometry reveals similarities between lung macrophages in humans and mice. Am J Respir Cell Mol Biol 54: 147–149, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bitterman PB, Rennard SI, Keogh BA, Wewers MD, Adelberg S, Crystal RG. Familial idiopathic pulmonary fibrosis: evidence of lung inflammation in unaffected members. N Engl J Med 314: 1343–1347, 1986. [DOI] [PubMed] [Google Scholar]
- 11.Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of clara cells in normal human airway epithelium. Am J Respir Crit Care Med 159: 1585–1591, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Bonanni PP, Frymoyer JW, Jacox RF. A family study of idiopathic pulmonary fibrosis. A possible dysproteinemic and genetically determined disease. Am J Med 39: 411–421, 1965. [DOI] [PubMed] [Google Scholar]
- 13.Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B, Israel-Biet D, Cadranel J, Cottin V, Gazal S, Peljto AL, Varga J, Schwartz DA, Valeyre D, Grandchamp B. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the european caucasian population. PLoS One 8: e70621, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boucher RC. Idiopathic pulmonary fibrosis–a sticky business. N Engl J Med 364: 1560–1561, 2011. [DOI] [PubMed] [Google Scholar]
- 15.Brody A. Asbestos-induced lung disease. Environ Health Perspect 100: 21–30, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JM, Witman GB. Cilia and diseases. Bioscience 64: 1126–1137, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger-van Paassen N, van der Sluis M, Bouma J, Korteland-van Male AM, Lu P, Van Seuningen I, Boehm G, van Goudoever JB, Renes IB. Colitis development during the suckling-weaning transition in mucin Muc2-deficient mice. Am J Physiol Gastrointest Liver Physiol 301: G667–G678, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Button B, Cai LH, Ehre C, Kesimer M, Hill DB, Sheehan JK, Boucher RC, Rubinstein M. A periciliary brush promotes the lung health by separating the mucus layer from airway epithelia. Science 337: 937–941, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caramori G, Casolari P, Di Gregorio C, Saetta M, Baraldo S, Boschetto P, Ito K, Fabbri LM, Barnes PJ, Adcock IM, Cavallesco G, Chung KF, Papi A. MUC5AC expression is increased in bronchial submucosal glands of stable COPD patients. Histopathology 55: 321–331, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet 151C: 263–280, 2009. [DOI] [PubMed] [Google Scholar]
- 21.Cha SI, Ryerson CJ, Lee JS, Kukreja J, Barry SS, Jones KD, Elicker BM, Kim DS, Papa FR, Collard HR, Wolters PJ. Cleaved cytokeratin-18 is a mechanistically informative biomarker in idiopathic pulmonary fibrosis. Respir Res 13: 105, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Korfhagen TR, Karp CL, Impey S, Xu Y, Randell SH, Kitzmiller J, Maeda Y, Haitchi HM, Sridharan A, Senft AP, Whitsett JA. Foxa3 induces goblet cell metaplasia and inhibits innate antiviral immunity. Am J Respir Crit Care Med 189: 301–313, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen G, Korfhagen TR, Xu Y, Kitzmiller J, Wert SE, Maeda Y, Gregorieff A, Clevers H, Whitsett JA. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest 119: 2914–2924, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Lim JH, Jono H, Gu XX, Kim YS, Basbaum CB, Murphy TF, Li JD. Nontypeable Haemophilus influenzae lipoprotein P6 induces MUC5AC mucin transcription via TLR2-TAK1-dependent p38 MAPK-AP1 and IKKbeta-IkappaBalpha-NF-kappaB signaling pathways. Biochem Biophys Res Commun 324: 1087–1094, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Zhao YH, Wu R. Differential regulation of airway mucin gene expression and mucin secretion by extracellular nucleotide triphosphates. Am J Respir Cell Mol Biol 25: 409–417, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res 162: 156–173, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162: 1495–1502, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung JH, Chawla A, Peljto AL, Cool CD, Groshong SD, Talbert JL, McKean DF, Brown KK, Fingerlin TE, Schwarz MI, Schwartz DA, Lynch DA. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest 147: 450–459, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cogan JD, Kropski JA, Zhao M, Mitchell DB, Rives L, Markin C, Garnett ET, Montgomery KH, Mason WR, McKean DF, Powers J, Murphy E, Olson LM, Choi L, Cheng DS, Blue EM, Young LR, Lancaster LH, Steele MP, Brown KK, Schwarz MI, Fingerlin TE, Schwartz DA, Lawson WE, Loyd JE, Zhao Z, Phillips JA 3rd, Blackwell TS, University of Washington Center for Mendelian Genetics. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med 191: 646–655, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collard HR, Anstrom KJ, Schwarz MI, Zisman DA. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest 131: 897–899, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 168: 538–542, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Muller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ, Idiopathic Pulmonary Fibrosis Clinical Research Network. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176: 636–643, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med 174: 654–658, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corfield AP. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim Biophys Acta 1850: 236–252, 2015. [DOI] [PubMed] [Google Scholar]
- 36.Coultas DB, Zumwalt RE, Black WC, Sobonya R. The epidemiology of interstitial lung diseases. Am J Respir Crit Care Med 150: 967–972, 1994. [DOI] [PubMed] [Google Scholar]
- 37.Davies JR, Svitacheva N, Lannefors L, Kornfalt R, Carlstedt I. Identification of MUC5B, MUC5AC and small amounts of MUC2 mucins in cystic fibrosis airway secretions. Biochem J 344: 321–330, 1999. [PMC free article] [PubMed] [Google Scholar]
- 38.Deane JA, Ricardo SD. Polycystic kidney disease and the renal cilium. Nephrology 12: 559–564, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, King TE Jr, Lancaster L, Noble PW, Sahn SA, Thomeer M, Valeyre D, Wells AU. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 184: 1382–1389, 2011. [DOI] [PubMed] [Google Scholar]
- 40.Duan F, Song C, Dai L, Cui S, Zhang X, Zhao X. The effect of MUC1 rs4072037 functional polymorphism on cancer susceptibility: evidence from published studies. PLoS One 9: e95651, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elliot TL, Lynch DA, Newell JD Jr, Cool C, Tuder R, Markopoulou K, Veve R, Brown KK. High-resolution computed tomography features of nonspecific interstitial pneumonia and usual interstitial pneumonia. J Comput Assist Tomogr 29: 339–345, 2005. [DOI] [PubMed] [Google Scholar]
- 42.Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P, Type 1 Diabetes Genetics Consortium. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57: 1084–1092, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Erwteman TM, Braat MC, van Aken WG. Interstitial pulmonary fibrosis: a new side effect of practolol. Br Med J 2: 297–298, 1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans CM, Williams OW, Tuvim MJ, Nigam R, Mixides GP, Blackburn MR, DeMayo FJ, Burns AR, Smith C, Reynolds SD, Stripp BR, Dickey BF. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol 31: 382–394, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fahy JV, Dickey BF. Airway mucus function and dysfunction. N Engl J Med 363: 2233–2247, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fell CD, Martinez FJ, Liu LX, Murray S, Han MK, Kazerooni EA, Gross BH, Myers J, Travis WD, Colby TV, Toews GB, Flaherty KR. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 181: 832–837, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, Collard HR, Wolters PJ, Bradford WZ, Kossen K, Seiwert SD, du Bois RM, Garcia CK, Devine MS, Gudmundsson G, Isaksson HJ, Kaminski N, Zhang Y, Gibson KF, Lancaster LH, Cogan JD, Mason WR, Maher TM, Molyneaux PL, Wells AU, Moffatt MF, Selman M, Pardo A, Kim DS, Crapo JD, Make BJ, Regan EA, Walek DS, Daniel JJ, Kamatani Y, Zelenika D, Smith K, McKean D, Pedersen BS, Talbert J, Kidd RN, Markin CR, Beckman KB, Lathrop M, Schwarz MI, Schwartz DA. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 45: 613–620, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease-associated interstitial lung disease: a call for clarification. Chest 138: 251–256, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, Travis WD, Flint A, Toews GB, Lynch JP 3rd, Martinez FJ. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 168: 543–548, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Flaherty KR, Thwaite EL, Kazerooni EA, Gross BH, Toews GB, Colby TV, Travis WD, Mumford JA, Murray S, Flint A, Lynch JP 3rd, Martinez FJ. Radiological versus histological diagnosis in UIP and NSIP: survival implications. Thorax 58: 143–148, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol 8: 880–893, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Fujishima S, Shiomi T, Yamashita S, Yogo Y, Nakano Y, Inoue T, Nakamura M, Tasaka S, Hasegawa N, Aikawa N, Ishizaka A, Okada Y. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med 134: 1136–1142, 2010. [DOI] [PubMed] [Google Scholar]
- 53.Furuhashi K, Suda T, Nakamura Y, Inui N, Hashimoto D, Miwa S, Hayakawa H, Kusagaya H, Nakano Y, Nakamura H, Chida K. Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir Med 104: 1204–1210, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature 526: 68–74, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greene KE, King TE Jr, Kuroki Y, Bucher-Bartelson B, Hunninghake GW, Newman LS, Nagae H, Mason RJ. Serum surfactant proteins-A and -D as biomarkers in idiopathic pulmonary fibrosis. Eur Respir J 19: 439–446, 2002. [DOI] [PubMed] [Google Scholar]
- 57.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Helgason A, Palsson S, Thorleifsson G, Grant SF, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Schafer H, Faruque M, Doumatey A, Zhou J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Sigurdsson G, Hebebrand J, Pedersen O, Thorsteinsdottir U, Gulcher JR, Kong A, Rotimi C, Stefansson K. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 39: 218–225, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Henderson WR Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, Kahn M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA 107: 14309–14314, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, Feingold E, Juan-Guardela BM, Richards TJ, Lussier Y, Huang Y, Vij R, Lindell KO, Xue J, Gibson KF, Shapiro SD, Garcia JG, Kaminski N. Peripheral Blood Mononuclear Cell Gene Expression Profiles May Predict Poor Outcome in Idiopathic Pulmonary Fibrosis. Sci Transl Med 5: 205ra136, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature Rev Drug Discovery 12: 703–719, 2013. [DOI] [PubMed] [Google Scholar]
- 63.Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, Forest MG. A biophysical basis for mucus solids concentration as a candidate biomarker for airways disease. PLoS One 9: e87681, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirakawa H, Pierce RA, Bingol-Karakoc G, Karaaslan C, Weng M, Shi GP, Saad A, Weber E, Mariani TJ, Starcher B, Shapiro SD, Cataltepe S. Cathepsin S deficiency confers protection from neonatal hyperoxia-induced lung injury. Am J Respir Crit Care Med 176: 778–785, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax 57: 338–342, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Horimasu Y, Ohshimo S, Bonella F, Tanaka S, Ishikawa N, Hattori N, Kohno N, Guzman J, Costabel U. MUC5B promoter polymorphism in Japanese patients with idiopathic pulmonary fibrosis. Respirology 20: 439–444, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Hubbard R, Cooper M, Antoniak M, Venn A, Khan S, Johnston I, Lewis S, Britton J. Risk of cryptogenic fibrosing alveolitis in metal workers. Lancet 355: 466–467, 2000. [DOI] [PubMed] [Google Scholar]
- 68.Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet 347: 284–289, 1996. [DOI] [PubMed] [Google Scholar]
- 69.Hubbard R, Venn A, Smith C, Cooper M, Johnston I, Britton J. Exposure to commonly prescribed drugs and the etiology of cryptogenic fibrosing alveolitis: a case-control study. Am J Respir Crit Care Med 157: 743–747, 1998. [DOI] [PubMed] [Google Scholar]
- 70.Hughes EW. Familial interstitial pulmonary fibrosis. Thorax 19: 515–525, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huh JW, Kim DS, Oh YM, Shim TS, Lim CM, Lee SD, Koh Y, Kim WS, Kim WD, Kim KR. Is metalloproteinase-7 specific for idiopathic pulmonary fibrosis? Chest 133: 1101–1106, 2008. [DOI] [PubMed] [Google Scholar]
- 72.Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, Nishino M, Araki T, Zazueta OE, Kurugol S, Ross JC, San Jose Estepar R, Murphy E, Steele MP, Loyd JE, Schwarz MI, Fingerlin TE, Rosas IO, Washko GR, O'Connor GT, Schwartz DA. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med 368: 2192–2200, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hunninghake GW, Lynch DA, Galvin JR, Gross BH, Muller N, Schwartz DA, King TE Jr, Lynch JP 3rd, Hegele R, Waldron J, Colby TV, Hogg JC. Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest 124: 1215–1223, 2003. [DOI] [PubMed] [Google Scholar]
- 74.Hunninghake GW, Zimmerman MB, Schwartz DA, King TE Jr, Lynch J, Hegele R, Waldron J, Colby T, Muller N, Lynch D, Galvin J, Gross B, Hogg J, Toews G, Helmers R, Cooper JA Jr, Baughman R, Strange C, Millard M. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 164: 193–196, 2001. [DOI] [PubMed] [Google Scholar]
- 75.Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Ann Am Thoracic Soc 11: 1176–1185, 2014. [DOI] [PubMed] [Google Scholar]
- 76.Idiopathic Pulmonary Fibrosis Clinical Research Network, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 363: 620–628, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Innes AL, Carrington SD, Thornton DJ, Kirkham S, Rousseau K, Dougherty RH, Raymond WW, Caughey GH, Muller SJ, Fahy JV. Ex vivo sputum analysis reveals impairment of protease-dependent mucus degradation by plasma proteins in acute asthma. Am J Respir Crit Care Med 180: 203–210, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Innes AL, McGrath KW, Dougherty RH, McCulloch CE, Woodruff PG, Seibold MA, Okamoto KS, Ingmundson KJ, Solon MC, Carrington SD, Fahy JV. The H antigen at epithelial surfaces is associated with susceptibility to asthma exacerbation. Am J Respir Crit Care Med 183: 189–194, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Innes AL, Woodruff PG, Ferrando RE, Donnelly S, Dolganov GM, Lazarus SC, Fahy JV. Epithelial mucin stores are increased in the large airways of smokers with airflow obstruction. Chest 130: 1102–1108, 2006. [DOI] [PubMed] [Google Scholar]
- 80.Ishii H, Mukae H, Kadota J, Kaida H, Nagata T, Abe K, Matsukura S, Kohno S. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax 58: 52–57, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iwai K, Mori T, Yamada N, Yamaguchi M, Hosoda Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am J Respir Crit Care Med 150: 670–675, 1994. [DOI] [PubMed] [Google Scholar]
- 82.Jain R, Pan J, Driscoll JA, Wisner JW, Huang T, Gunsten SP, You Y, Brody SL. Temporal relationship between primary and motile ciliogenesis in airway epithelial cells. Am J Respir Cell Mol Biol 43: 731–739, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol 15: 910–919, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Javaheri S, Lederer DH, Pella JA, Mark GJ, Levine BW. Idiopathic pulmonary fibrosis in monozygotic twins. The importance of genetic predisposition. Chest 78: 591–594, 1980. [DOI] [PubMed] [Google Scholar]
- 85.Jonsdottir HR, Arason AJ, Palsson R, Franzdottir SR, Gudbjartsson T, Isaksson HJ, Gudmundsson G, Gudjonsson T, Magnusson MK. Basal cells of the human airways acquire mesenchymal traits in idiopathic pulmonary fibrosis and in culture. Lab Invest 95: 1418–1428, 2015. [DOI] [PubMed] [Google Scholar]
- 86.Jung SH, Chung WC, Lee KM, Paik CN, Jung JH, Lee MK, Lee YK, Chung IS. Risk factors in malignant transformation of gastric epithelial neoplasia categorized by the revised Vienna classification: endoscopic, pathological, and immunophenotypic features. Gastric Cancer 13: 123–130, 2010. [DOI] [PubMed] [Google Scholar]
- 87.Kadota J, Mizunoe S, Mito K, Mukae H, Yoshioka S, Kawakami K, Koguchi Y, Fukushima K, Kon S, Kohno S, Saito A, Uede T, Nasu M. High plasma concentrations of osteopontin in patients with interstitial pneumonia. Respir Med 99: 111–117, 2005. [DOI] [PubMed] [Google Scholar]
- 88.Kamio K, Matsushita I, Hijikata M, Kobashi Y, Tanaka G, Nakata K, Ishida T, Tokunaga K, Taguchi Y, Homma S, Nakata K, Azuma A, Kudoh S, Keicho N. Promoter analysis and aberrant expression of the MUC5B gene in diffuse panbronchiolitis. Am J Respir Crit Care Med 171: 949–957, 2005. [DOI] [PubMed] [Google Scholar]
- 89.Karimi-Shah BA, Chowdhury BA. Forced vital capacity in idiopathic pulmonary fibrosis–FDA review of pirfenidone and nintedanib. N Engl J Med 372: 1189–1191, 2015. [DOI] [PubMed] [Google Scholar]
- 90.Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol 11: 463–483, 1996. [PubMed] [Google Scholar]
- 91.Kim V, Kelemen SE, Abuel-Haija M, Gaughan JP, Sharafkaneh A, Evans CM, Dickey BF, Solomides CC, Rogers TJ, Criner GJ. Small airway mucous metaplasia and inflammation in chronic obstructive pulmonary disease. COPD 5: 329–338, 2008. [DOI] [PubMed] [Google Scholar]
- 92.King T, Costabel U, Cordier J, DoPico G, du Bois R, Lynch D, Myers J, Panos R, Raghu G, Schwartz D, Smith C, Corn J. International consensus statement idiopathic pulmonary fibrosis: diagnosis and treatment. Am J Respir Crit Care Med 161: 646–664, 2000. [DOI] [PubMed] [Google Scholar]
- 93.King T, Costabel U, Cordier J, DoPico G, DuBois R, Lynch D, Lynch J, Myers J, Panos R, Raghu G, Schwartz D, Smith C. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med 161: 646–664, 2000. [DOI] [PubMed] [Google Scholar]
- 94.King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370: 2083–2092, 2014. [DOI] [PubMed] [Google Scholar]
- 95.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961, 2011. [DOI] [PubMed] [Google Scholar]
- 96.King TE Jr, Schwarz MI, Brown K, Tooze JA, Colby TV, Waldron JA Jr, Flint A, Thurlbeck W, Cherniack RM. Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 164: 1025–1032, 2001. [DOI] [PubMed] [Google Scholar]
- 97.King TE Jr, Tooze JA, Schwarz MI, Brown KR, Cherniack RM. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 164: 1171–1181, 2001. [DOI] [PubMed] [Google Scholar]
- 98.Kirkham S, Kolsum U, Rousseau K, Singh D, Vestbo J, Thornton DJ. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 178: 1033–1039, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kirkham S, Sheehan JK, Knight D, Richardson PS, Thornton DJ. Heterogeneity of airways mucus: variations in the amounts and glycoforms of the major oligomeric mucins MUC5AC and MUC5B. Biochem J 361: 537–546, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kiwamoto T, Katoh T, Evans CM, Janssen WJ, Brummet ME, Hudson SA, Zhu Z, Tiemeyer M, Bochner BS. Endogenous airway mucins carry glycans that bind Siglec-F and induce eosinophil apoptosis. J Allergy Clin Immunol 135: 1329–1340 e1321–1329, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 188: 913–922, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Konigshoff M, Balsara N, Pfaff EM, Kramer M, Chrobak I, Seeger W, Eickelberg O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS One 3: e2142, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Konigshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, Rose F, Fink L, Seeger W, Schaefer L, Gunther A, Eickelberg O. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest 119: 772–787, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 178: 838–846, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korfei M, Schmitt S, Ruppert C, Henneke I, Markart P, Loeh B, Mahavadi P, Wygrecka M, Klepetko W, Fink L, Bonniaud P, Preissner KT, Lochnit G, Schaefer L, Seeger W, Guenther A. Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res 10: 2185–2205, 2011. [DOI] [PubMed] [Google Scholar]
- 106.Korfei M, von der Beck D, Henneke I, Markart P, Ruppert C, Mahavadi P, Ghanim B, Klepetko W, Fink L, Meiners S, Kramer OH, Seeger W, Vancheri C, Guenther A. Comparative proteome analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF), non-specific interstitial pneumonia (NSIP) and organ donors. J Proteomics 85: 109–128, 2013. [DOI] [PubMed] [Google Scholar]
- 107.Korfhagen TR, Kitzmiller J, Chen G, Sridharan A, Haitchi HM, Hegde RS, Divanovic S, Karp CL, Whitsett JA. SAM-pointed domain ETS factor mediates epithelial cell-intrinsic innate immune signaling during airway mucous metaplasia. Proc Natl Acad Sci USA 109: 16630–16635, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Korthagen NM, van Moorsel CH, Barlo NP, Ruven HJ, Kruit A, Heron M, van den Bosch JM, Grutters JC. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med 105: 106–113, 2011. [DOI] [PubMed] [Google Scholar]
- 109.Koyama S, Sato E, Haniuda M, Numanami H, Nagai S, Izumi T. Decreased level of vascular endothelial growth factor in bronchoalveolar lavage fluid of normal smokers and patients with pulmonary fibrosis. Am J Respir Crit Care Med 166: 382–385, 2002. [DOI] [PubMed] [Google Scholar]
- 110.Kropski JA, Lawson WE, Young LR, Blackwell TS. Genetic studies provide clues on the pathogenesis of idiopathic pulmonary fibrosis. Dis Model Mech 6: 9–17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET, Degryse AL, Mitchell DB, Polosukhin VV, Rickman OB, Choi L, Cheng DS, McConaha ME, Jones BR, Gleaves LA, McMahon FB, Worrell JA, Solus JF, Ware LB, Lee JW, Massion PP, Zaynagetdinov R, White ES, Kurtis JD, Johnson JE, Groshong SD, Lancaster LH, Young LR, Steele MP, Phillips Iii JA, Cogan JD, Loyd JE, Lawson WE, Blackwell TS. Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease. Am J Respir Crit Care Med 191: 417–426, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang de Y, Lim B, Chow VT, Crum CP, Xian W, McKeon F. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147: 525–538, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nature Med 8: 885–889, 2002. [DOI] [PubMed] [Google Scholar]
- 114.Kuperman DA, Huang X, Nguyenvu L, Holscher C, Brombacher F, Erle DJ. IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J Immunol 175: 3746–3752, 2005. [DOI] [PubMed] [Google Scholar]
- 115.Kuyper LM, Pare PD, Hogg JC, Lambert RK, Ionescu D, Woods R, Bai TR. Characterization of airway plugging in fatal asthma. Am J Med 115: 6–11, 2003. [DOI] [PubMed] [Google Scholar]
- 116.Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, Veeraraghavan S, Hansell DM, Wells AU. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 168: 531–537, 2003. [DOI] [PubMed] [Google Scholar]
- 117.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 108: 10562–10567, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol 294: L1119–L1126, 2008. [DOI] [PubMed] [Google Scholar]
- 119.Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB, Markin C, Renzoni E, Lympany P, Thomas AQ, Roldan J, Scott TA, Blackwell TS, Phillips JA 3rd, Loyd JE, du Bois RM. Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 59: 977–980, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee HL, Ryu JH, Wittmer MH, Hartman TE, Lymp JF, Tazelaar HD, Limper AH. Familial idiopathic pulmonary fibrosis: clinical features and outcome. Chest 127: 2034–2041, 2005. [DOI] [PubMed] [Google Scholar]
- 121.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183: 431–440, 2011. [DOI] [PubMed] [Google Scholar]
- 122.Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr, Collard HR. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Internal Med 156: 684–691, 2012. [DOI] [PubMed] [Google Scholar]
- 123.Li J, Lin LH, Wang J, Peng X, Dai HR, Xiao H, Li F, Wang YP, Yang ZJ, Li L. Interleukin-4 and interleukin-13 pathway genetics affect disease susceptibility, serum immunoglobulin E levels, and gene expression in asthma. Ann Allergy Asthma Immunol 113: 173–179, 2014. [DOI] [PubMed] [Google Scholar]
- 124.Liu JY, Morris GF, Lei WH, Hart CE, Lasky JA, Brody AR. Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am J Respir Cell Mol Biol 17: 129–140, 1997. [DOI] [PubMed] [Google Scholar]
- 125.Mahavadi P, Henneke I, Ruppert C, Knudsen L, Venkatesan S, Liebisch G, Chambers RC, Ochs M, Schmitz G, Vancheri C, Seeger W, Korfei M, Guenther A. Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone-induced lung fibrosis. Toxicol Sci 142: 285–297, 2014. [DOI] [PubMed] [Google Scholar]
- 126.Marshall R, Puddicombe A, Cookson W, Laurent G. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax 55: 143–146, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, O'Neal WK, Ribeiro CM. The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol 6: 639–654, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mathai SK, Schwartz DA, Warg LA. Genetic susceptibility and pulmonary fibrosis. Curr Opin Pulm Med 20: 429–435, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mathieson JR, Mayo JR, Staples CA, Muller NL. Chronic diffuse infiltrative lung disease: comparison of diagnostic accuracy of CT and chest radiography. Radiology 171: 111–116, 1989. [DOI] [PubMed] [Google Scholar]
- 130.Mayo JR. CT evaluation of diffuse infiltrative lung disease: dose considerations and optimal technique. J Thorac Imaging 24: 252–259, 2009. [DOI] [PubMed] [Google Scholar]
- 131.McKusick VA, Fisher AM. Congenial cystic disease of the lung with progressive pulmonary fibrosis and carcinomatosis. Ann Internal Med 48: 774–790, 1958. [DOI] [PubMed] [Google Scholar]
- 132.Mermigkis C, Chapman J, Golish J, Mermigkis D, Budur K, Kopanakis A, Polychronopoulos V, Burgess R, Foldvary-Schaefer N. Sleep-related breathing disorders in patients with idiopathic pulmonary fibrosis. Lung 185: 173–178, 2007. [DOI] [PubMed] [Google Scholar]
- 133.Messina MS, O'Riordan TG, Smaldone GC. Changes in mucociliary clearance during acute exacerbations of asthma. Am Rev Respir Dis 143: 993–997, 1991. [DOI] [PubMed] [Google Scholar]
- 134.Meyer KC, Cardoni A, Xiang ZZ. Vascular endothelial growth factor in bronchoalveolar lavage from normal subjects and patients with diffuse parenchymal lung disease. J Lab Clin Med 135: 332–338, 2000. [DOI] [PubMed] [Google Scholar]
- 135.Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49: 503–510, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miyake Y, Sasaki S, Yokoyama T, Chida K, Azuma A, Suda T, Kudoh S, Sakamoto N, Okamoto K, Kobashi G, Washio M, Inaba Y, Tanaka H. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Ann Occup Hyg 49: 259–265, 2005. [DOI] [PubMed] [Google Scholar]
- 137.Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz DA, Wells AU, Cookson WO, Maher TM, Moffatt MF. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 190: 906–913, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mulugeta S, Nureki S, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 309: L507–L525, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Musk AW, Pollard JA. Pindolol and pulmonary fibrosis. Br Med J 2: 581–582, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Musk AW, Zilko PJ, Manners P, Kay PH, Kamboh MI. Genetic studies in familial fibrosing alveolitis. Possible linkage with immunoglobulin allotypes (Gm). Chest 89: 206–210, 1986. [DOI] [PubMed] [Google Scholar]
- 141.Nakano Y, Yang IV, Walts AD, Watson AM, Helling BA, Fletcher AA, Lara AR, Schwarz MI, Evans CM, Schwartz DA. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 193: 464–466, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Navaratnam V, Fleming KM, West J, Smith CJ, Jenkins RG, Fogarty A, Hubbard RB. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 66: 462–467, 2011. [DOI] [PubMed] [Google Scholar]
- 143.Neurohr C, Huppmann P, Thum D, Leuschner W, von Wulffen W, Meis T, Leuchte H, Baumgartner R, Zimmermann G, Hatz R, Czerner S, Frey L, Ueberfuhr P, Bittmann I, Behr J, Munich Lung Transplant Group. Potential functional and survival benefit of double over single lung transplantation for selected patients with idiopathic pulmonary fibrosis. Transpl Int 23: 887–896, 2010. [DOI] [PubMed] [Google Scholar]
- 144.Ng W, Loh AX, Teixeira AS, Pereira SP, Swallow DM. Genetic regulation of MUC1 alternative splicing in human tissues. Br J Cancer 99: 978–985, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122: 2756–2762, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi PG, Scuirba J, Richards T, Juan-Guardela B, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae DL, Kaminski N, Garcia JG. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 1: 309–317, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.O'Dwyer DN, Armstrong ME, Trujillo G, Cooke G, Keane MP, Fallon PG, Simpson AJ, Millar AB, McGrath EE, Whyte MK, Hirani N, Hogaboam CM, Donnelly SC. The Toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 188: 1442–1450, 2013. [DOI] [PubMed] [Google Scholar]
- 148.O'Riordan TG, Zwang J, Smaldone GC. Mucociliary clearance in adult asthma. Am Rev Respir Dis 146: 598–603, 1992. [DOI] [PubMed] [Google Scholar]
- 149.Obonyo M, Sabet M, Cole SP, Ebmeyer J, Uematsu S, Akira S, Guiney DG. Deficiencies of myeloid differentiation factor 88, Toll-like receptor 2 (TLR2), or TLR4 produce specific defects in macrophage cytokine secretion induced by Helicobacter pylori. Infect Immun 75: 2408–2414, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, Hiwada K, Kohno N. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med 165: 378–381, 2002. [DOI] [PubMed] [Google Scholar]
- 151.Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med 176: 277–284, 2007. [DOI] [PubMed] [Google Scholar]
- 152.Ordonez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y, Novikov A, Dolganov G, Fahy JV. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med 163: 517–523, 2001. [DOI] [PubMed] [Google Scholar]
- 153.Pardo A, Gibson K, Cisneros J, Richards TJ, Yang Y, Becerril C, Yousem S, Herrera I, Ruiz V, Selman M, Kaminski N. Up-regulation and profibrotic role of osteopontin in human idiopathic pulmonary fibrosis. PLoS Med 2: e251, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Park JS, Yeom JS, Seo JH, Lim JY, Park CH, Woo HO, Youn HS, Jun JS, Park JH, Ko GH, Baik SC, Lee WK, Cho MJ, Rhee KH. Immunohistochemical rxpressions of MUC2, MUC5AC, and MUC6 in normal, Helicobacter pylori infected and metaplastic gastric mucosa of children and adolescents. Helicobacter 20: 260–268, 2015. [DOI] [PubMed] [Google Scholar]
- 155.Park KS, Korfhagen TR, Bruno MD, Kitzmiller JA, Wan H, Wert SE, Khurana Hershey GK, Chen G, Whitsett JA. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest 117: 978–988, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Peljto AL, Selman M, Kim DS, Murphy E, Tucker L, Pardo A, Lee JS, Ji W, Schwarz MI, Yang IV, Schwartz DA, Fingerlin TE. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest 147: 460–464, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Peljto AL, Steele MP, Fingerlin TE, Hinchcliff ME, Murphy E, Podlusky S, Carns M, Schwarz M, Varga J, Schwartz DA. The pulmonary fibrosis-associated MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest 142: 1584–1588, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd JE, Gibson KF, Seibold MA, Brown KK, Talbert JL, Markin C, Kossen K, Seiwert SD, Murphy E, Noth I, Schwarz MI, Kaminski N, Schwartz DA. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 309: 2232–2239, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, Whitsett JA. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 66: 651–657, 2011. [DOI] [PubMed] [Google Scholar]
- 160.Ponnusamy MP, Seshacharyulu P, Vaz A, Dey P, Batra SK. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J Ovarian Res 4: 7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Prasse A, Pechkovsky DV, Toews GB, Schafer M, Eggeling S, Ludwig C, Germann M, Kollert F, Zissel G, Muller-Quernheim J. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheumatism 56: 1685–1693, 2007. [DOI] [PubMed] [Google Scholar]
- 162.Putman RK, Hatabu H, Araki T, Gudmundsson G, Gao W, Nishino M, Okajima Y, Dupuis J, Latourelle JC, Cho MH, El-Chemaly S, Coxson HO, Celli BR, Fernandez IE, Zazueta OE, Ross JC, Harmouche R, Estepar RS, Diaz AA, Sigurdsson S, Gudmundsson EF, Eiriksdottir G, Aspelund T, Budoff MJ, Kinney GL, Hokanson JE, Williams MC, Murchison JT, MacNee W, Hoffmann U, O'Donnell CJ, Launer LJ, Harrris TB, Gudnason V, Silverman EK, O'Connor GT, Washko GR, Rosas IO, Hunninghake GM. Association between interstitial lung abnormalities and all-cause mortality. JAMA 315: 672–681, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Putman RK, Rosas IO, Hunninghake GM. Genetics and early detection in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 189: 770–778, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Raghu G. Idiopathic pulmonary fibrosis: guidelines for diagnosis and clinical management have advanced from consensus-based in 2000 to evidence-based in 2011. Eur Respir J 37: 743–746, 2011. [DOI] [PubMed] [Google Scholar]
- 165.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ, Fibrosis AEJACoIP. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, Alfredsson L, Padyukov L, Klareskog L, Worthington J, Siminovitch KA, Bae SC, Plenge RM, Gregersen PK, de Bakker PI. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet 44: 291–296, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Reis CA, David L, Correa P, Carneiro F, de Bolos C, Garcia E, Mandel U, Clausen H, Sobrinho-Simoes M. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Res 59: 1003–1007, 1999. [PubMed] [Google Scholar]
- 169.Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, Li K, Choi J, Vuga LJ, Lindell KO, Klesen M, Zhang Y, Gibson KF. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 185: 67–76, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Richeldi L, Cottin V, Flaherty KR, Kolb M, Inoue Y, Raghu G, Taniguchi H, Hansell DM, Nicholson AG, Le Maulf F, Stowasser S, Collard HR. Design of the INPULSIS trials: two phase 3 trials of nintedanib in patients with idiopathic pulmonary fibrosis. Respir Med 108: 1023–1030, 2014. [DOI] [PubMed] [Google Scholar]
- 171.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 108: E1475–1483, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rock JR, Onaitis MW, Rawlins EL, Lu Y, Clark CP, Xue Y, Randell SH, Hogan BL. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA 106: 12771–12775, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rom W, Travis W, Brody AR. Cellular and molecular basis of the asbestos-related diseases. Am Rev Respir Dis 143: 408–422, 1991. [DOI] [PubMed] [Google Scholar]
- 174.Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, Lindell KO, Cisneros J, Macdonald SD, Pardo A, Sciurba F, Dauber J, Selman M, Gochuico BR, Kaminski N. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 5: e93, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Roy MG, Rahmani M, Hernandez JR, Alexander SN, Ehre C, Ho SB, Evans CM. Mucin production during prenatal and postnatal murine lung development. Am J Respir Cell Mol Biol 44: 755–760, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schwarz M. Autoimmunity: a pathway to usual interstitial pneumonia? Chest 148: 1367–1369, 2015. [DOI] [PubMed] [Google Scholar]
- 177.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, Reynolds SD. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One 8: e58658, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 364: 1503–1512, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Internal Med 134: 136–151, 2001. [DOI] [PubMed] [Google Scholar]
- 180.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 3: 364–372, 2006. [DOI] [PubMed] [Google Scholar]
- 181.Selman M, Pardo A, Kaminski N. Idiopathic pulmonary fibrosis: aberrant recapitulation of developmental programs? PLoS Med 5: e62, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sheehan J, Richardson P, Fung D, Howard M, Thornton D. Analysis of respiratory mucus glycoproteins in asthma: a detailed study from a patient who died in status asthmaticus. Am J Respir Cell Mol Biol 13: 748–756, 1995. [DOI] [PubMed] [Google Scholar]
- 183.Silva CI, Muller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, Chung MP, Churg A. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 246: 288–297, 2008. [DOI] [PubMed] [Google Scholar]
- 184.Soderholm J, Heald R. Scratch n' screen for inhibitors of cell migration. Chem Biol 12: 263–265, 2005. [DOI] [PubMed] [Google Scholar]
- 185.Solliday NH, Williams JA, Gaensler EA, Coutu RE, Carrington C. Familial chronic interstitial pneumonia. Am Rev Respir Dis 108: 193–204, 1973. [DOI] [PubMed] [Google Scholar]
- 186.Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 37: 356–363, 2011. [DOI] [PubMed] [Google Scholar]
- 187.Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci USA 101: 10143–10148, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA 3rd, Sporn TA, McAdams HP, Schwarz MI, Schwartz DA. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med 172: 1146–1152, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stewart JP, Egan JJ, Ross AJ, Kelly BG, Lok SS, Hasleton PS, Woodcock AA. The detection of Epstein-Barr virus DNA in lung tissue from patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 159: 1336–1341, 1999. [DOI] [PubMed] [Google Scholar]
- 191.Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, Abraham DJ, Hansell DM, Nicholson AG, Maher TM, Wells AU, Lindahl GE, Renzoni EA. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax 68: 436–441, 2013. [DOI] [PubMed] [Google Scholar]
- 192.Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, Weissler J, Fitzgerald J, Kershaw C, Klesney-Tait J, Mageto Y, Shay JW, Ji W, Bilguvar K, Mane S, Lifton RP, Garcia CK. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet 47: 512–517, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Swaye P, van Ordstrand HS, McCormack LJ, Wolpaw SE. Familial Hamman-Rich syndrome. Chest 55: 7–12, 1969. [DOI] [PubMed] [Google Scholar]
- 194.Sylvester PA, Myerscough N, Warren BF, Carlstedt I, Corfield AP, Durdey P, Thomas MG. Differential expression of the chromosome 11 mucin genes in colorectal cancer. J Pathol 195: 327–335, 2001. [DOI] [PubMed] [Google Scholar]
- 195.Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, Shiratori M, Kuroki Y, Abe S. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 162: 1109–1114, 2000. [DOI] [PubMed] [Google Scholar]
- 196.Takeyama K, Dabbagh K, Lee HM, Agusti C, Lausier JA, Ueki IF, Grattan KM, Nadel JA. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA 96: 3081–3086, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tamura Y, Higashi M, Kitamoto S, Yokoyama S, Osako M, Horinouchi M, Shimizu T, Tabata M, Batra SK, Goto M, Yonezawa S. MUC4 and MUC1 expression in adenocarcinoma of the stomach correlates with vessel invasion and lymph node metastasis: an immunohistochemical study of early gastric cancer. PLoS One 7: e49251, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tanaka N, Newell JD, Brown KK, Cool CD, Lynch DA. Collagen vascular disease-related lung disease: high-resolution computed tomography findings based on the pathologic classification. J Comput Assist Tomogr 28: 351–360, 2004. [DOI] [PubMed] [Google Scholar]
- 199.Tang S, Tan S, Yao L, Li F, Li L, Guo X, Liu Y, Hao X, Li Y, Ding X, Zhang Z, Tong L, Huang J. Risk factors for poor treatment outcomes in patients with MDR-TB and XDR-TB in China: retrospective multi-center investigation. PLoS One 8: e82943, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tang YW, Johnson JE, Browning PJ, Cruz-Gervis RA, Davis A, Graham BS, Brigham KL, Oates JA Jr, Loyd JE, Stecenko AA. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J Clin Microbiol 41: 2633–2640, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 302: L721–L729, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem 290: 3277, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Taskar V, Coultas D. Exposures and idiopathic lung disease. Semin Respir Crit Care Med 29: 670–679, 2008. [DOI] [PubMed] [Google Scholar]
- 204.Ten Have-Opbroek AA, Otto-Verberne CJ, Dubbeldam JA, Dykman JH. The proximal border of the human respiratory unit, as shown by scanning and transmission electron microscopy and light microscopical cytochemistry. Anat Rec 229: 339–354, 1991. [DOI] [PubMed] [Google Scholar]
- 205.Thomas AQ, Lane K, Phillips J, Prince M 3rd, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 165: 1322–1328, 2002. [DOI] [PubMed] [Google Scholar]
- 206.Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol 70: 459–486, 2008. [DOI] [PubMed] [Google Scholar]
- 207.Travis W, King T, Bateman E, Lynch D, Capron F, Center D, Colby T, Cordier J, DuBois R, Galvin J, Grenier P, Hansell D, Hunninghake G, Kitaichi M, Muller N, Myers J, Nagai S, Nicholson A, Raghu G, Wallaert B. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 165: 277–304, 2002. [DOI] [PubMed] [Google Scholar]
- 208.Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D. An official american thoracic society/european respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188: 733–748, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D, ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188: 733–748, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Travis WD, Hunninghake G, King TE Jr, Lynch DA, Colby TV, Galvin JR, Brown KK, Chung MP, Cordier JF, du Bois RM, Flaherty KR, Franks TJ, Hansell DM, Hartman TE, Kazerooni EA, Kim DS, Kitaichi M, Koyama T, Martinez FJ, Nagai S, Midthun DE, Muller NL, Nicholson AG, Raghu G, Selman M, Wells A. Idiopathic nonspecific interstitial pneumonia: report of an American Thoracic Society project. Am J Respir Crit Care Med 177: 1338–1347, 2008. [DOI] [PubMed] [Google Scholar]
- 211.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA 104: 7552–7557, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tsukahara M, Kajii T. Interstitial pulmonary fibrosis in two sisters. Possible autosomal recessive inheritance. Jinrui Idengaku Zasshi 28: 263–267, 1983. [DOI] [PubMed] [Google Scholar]
- 213.Van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, van Es HW, van den Bosch JM, Grutters JC. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med 182: 1419–1425, 2010. [DOI] [PubMed] [Google Scholar]
- 214.Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, Matthay MA, Rock JR, Chapman HA. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517: 621–625, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Vij R, Noth I. Peripheral blood biomarkers in idiopathic pulmonary fibrosis. Transl Res 159: 218–227, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Vincent A, Perrais M, Desseyn JL, Aubert JP, Pigny P, Van Seuningen I. Epigenetic regulation (DNA methylation, histone modifications) of the 11p15 mucin genes (MUC2, MUC5AC, MUC5B, MUC6) in epithelial cancer cells. Oncogene 26: 6566–6576, 2007. [DOI] [PubMed] [Google Scholar]
- 217.Vourlekis JS, Schwarz MI, Cool CD, Tuder RM, King TE, Brown KK. Nonspecific interstitial pneumonitis as the sole histologic expression of hypersensitivity pneumonitis. Am J Med 112: 490–493, 2002. [DOI] [PubMed] [Google Scholar]
- 218.Vuga LJ, Ben-Yehudah A, Kovkarova-Naumovski E, Oriss T, Gibson KF, Feghali-Bostwick C, Kaminski N. WNT5A is a regulator of fibroblast proliferation and resistance to apoptosis. Am J Respir Cell Mol Biol 41: 583–589, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Vuorinen K, Myllarniemi M, Lammi L, Piirila P, Rytila P, Salmenkivi K, Kinnula VL. Elevated matrilysin levels in bronchoalveolar lavage fluid do not distinguish idiopathic pulmonary fibrosis from other interstitial lung diseases. APMIS 115: 969–975, 2007. [DOI] [PubMed] [Google Scholar]
- 220.Wakatsuki K, Yamada Y, Narikiyo M, Ueno M, Takayama T, Tamaki H, Miki K, Matsumoto S, Enomoto K, Yokotani T, Nakajima Y. Clinicopathological and prognostic significance of mucin phenotype in gastric cancer. J Surg Oncol 98: 124–129, 2008. [DOI] [PubMed] [Google Scholar]
- 221.Walsh MD, Clendenning M, Williamson E, Pearson SA, Walters RJ, Nagler B, Packenas D, Win AK, Hopper JL, Jenkins MA, Haydon AM, Rosty C, English DR, Giles GG, McGuckin MA, Young JP, Buchanan DD. Expression of MUC2, MUC5AC, MUC5B, and MUC6 mucins in colorectal cancers and their association with the CpG island methylator phenotype. Mod Pathol 26: 1642–1656, 2013. [DOI] [PubMed] [Google Scholar]
- 222.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, Whitsett JA. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131: 953–964, 2004. [DOI] [PubMed] [Google Scholar]
- 223.Wang C, Zhuang Y, Guo W, Cao L, Zhang H, Xu L, Fan Y, Zhang D, Wang Y. Mucin 5B promoter polymorphism is associated with susceptibility to interstitial lung diseases in Chinese males. PLoS One 9: e104919, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, DiMaio JM, Kinch LN, Grishin NV, Garcia CK. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 84: 52–59, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Washko GR, Hunninghake GM, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, Ross JC, Estepar RS, Lynch DA, Brehm JM, Andriole KP, Diaz AA, Khorasani R, D'Aco K, Sciurba FC, Silverman EK, Hatabu H, Rosas IO, CO Investigators. Lung volumes and emphysema in smokers with interstitial lung abnormalities. N Engl J Med 364: 897–906, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wei R, Li C, Zhang M, Jones-Hall YL, Myers JL, Noth I, Liu W. Association between MUC5B and TERT polymorphisms and different interstitial lung disease phenotypes. Transl Res 163: 494–502, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wells AU, Desai SR, Rubens MB, Goh NS, Cramer D, Nicholson AG, Colby TV, du Bois RM, Hansell DM. Idiopathic pulmonary fibrosis: a composite physiologic index derived from disease extent observed by computed tomography. Am J Respir Crit Care Med 167: 962–969, 2003. [DOI] [PubMed] [Google Scholar]
- 228.Williams KA, Terry KL, Tworoger SS, Vitonis AF, Titus LJ, Cramer DW. Polymorphisms of MUC16 (CA125) and MUC1 (CA15.3) in relation to ovarian cancer risk and survival. PLoS One 9: e88334, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 166: 1321–1332, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Woodruff PG, Modrek B, Choy DF, Jia G, Abbas AR, Ellwanger A, Koth LL, Arron JR, Fahy JV. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180: 388–395, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Yang IV. The polymeric mucin Muc5ac is required for allergic airway hyperreactivity. Am J Trop Med Hyg 6: 6281, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, Rosen R, Neidermyer AJ, McKean DF, Groshong SD, Cool C, Cosgrove GP, Lynch DA, Brown KK, Schwarz MI, Fingerlin TE, Schwartz DA. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax 68: 1114–1121, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yonemaru M, Kasuga I, Kusumoto H, Kunisawa A, Kiyokawa H, Kuwabara S, Ichinose Y, Toyama K. Elevation of antibodies to cytomegalovirus and other herpes viruses in pulmonary fibrosis. Eur Respir J 10: 2040–2045, 1997. [DOI] [PubMed] [Google Scholar]
- 234.Young HW, Williams OW, Chandra D, Bellinghausen LK, Perez G, Suarez A, Tuvim MJ, Roy MG, Alexander SN, Moghaddam SJ, Adachi R, Blackburn MR, Dickey BF, Evans CM. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5' elements. Am J Respir Cell Mol Biol 37: 273–290, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zappala CJ, Latsi PI, Nicholson AG, Colby TV, Cramer D, Renzoni EA, Hansell DM, du Bois RM, Wells AU. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J 35: 830–836, 2010. [DOI] [PubMed] [Google Scholar]
- 236.Zhang Y, Noth I, Garcia GN, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med 364: 1576–1577, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhen G, Park SW, Nguyenvu LT, Rodriguez MW, Barbeau R, Paquet AC, Erle DJ. IL-13 and epidermal growth factor receptor have critical but distinct roles in epithelial cell mucin production. Am J Respir Cell Mol Biol 36: 244–253, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, Kim KJ, duBois RM, Crandall ED, Beers MF, Borok Z. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol 45: 498–509, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zuo F, Kaminski N, Eugui E, Allard J, Yakhini Z, Ben-Dor A, Lollini L, Morris D, Kim Y, DeLustro B, Sheppard D, Pardo A, Selman M, Heller RA. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci USA 99: 6292–6297, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]