

Abstract
Phospholipase C-ε (PLC-ε) is a unique PLC isoform that can be regulated by multiple signaling inputs from both Ras family GTPases and heterotrimeric G proteins and has primary sites of expression in the heart and lung. Whereas the role of PLC-ε in cardiac function and pathology has been documented, its relevance in acute lung injury (ALI) is unclear. We used PLC-ε−/− mice to address the role of PLC-ε in regulating lung vascular inflammation and injury in an aerosolized bacterial LPS inhalation mouse model of ALI. PLC-ε−/− mice showed a marked decrease in LPS-induced proinflammatory mediators (ICAM-1, VCAM-1, TNF-α, IL-1β, IL-6, macrophage inflammatory protein 2, keratinocyte-derived cytokine, monocyte chemoattractant protein 1, and granulocyte-macrophage colony-stimulating factor), lung neutrophil infiltration and microvascular leakage, and loss of VE-cadherin compared with PLC-ε+/+ mice. These data identify PLC-ε as a critical determinant of proinflammatory and leaky phenotype of the lung. To test the possibility that PLC-ε activity in endothelial cells (EC) could contribute to ALI, we determined its role in EC inflammation and barrier disruption. RNAi knockdown of PLC-ε inhibited NF-κB activity in response to diverse proinflammatory stimuli, thrombin, LPS, TNF-α, and the nonreceptor agonist phorbol 13-myristate 12-acetate (phorbol esters) in EC. Depletion of PLC-ε also inhibited thrombin-induced expression of NF-κB target gene, VCAM-1. Importantly, PLC-ε knockdown also protected against thrombin-induced EC barrier disruption by inhibiting the loss of VE-cadherin at adherens junctions and formation of actin stress fibers. These data identify PLC-ε as a novel regulator of EC inflammation and permeability and show a hitherto unknown role of PLC-ε in the pathogenesis of ALI.
Keywords: endothelial cells, adhesion molecules, transcription factors, signal transduction, lung inflammation
the acquisition of proinflammatory phenotype and disruption of vascular endothelial barrier leading to inflammatory cell infiltration and protein-rich edema formation are prominent pathogenic features of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) (3, 31, 50). The major mechanisms of inflammatory cell extravasation and increased vascular permeability in the inflamed lung and other tissues include activation of the transcription factor NF-κB, a master regulator of inflammation, and disassembly of VE-cadherin homodimers, the main constituent of inter-EC adherens junctions (AJs), respectively (7, 10, 38). Activation of NF-κB is initiated by IKK-β-mediated phosphorylation of Ser32 and Ser36 of Iκ-Bα, an inhibitory protein that associates with NF-κB to retain it as an inactive complex in the cytoplasm (22, 38). Phosphorylation of Iκ-Bα marks it for ubiquitination and proteasome-mediated degradation, causing the release of NF-κB. The released NF-κB [predominantly RelA/p65 homodimer in endothelial cells (EC) (35)] undergoes rapid nuclear translocation and binding to the promoter of proinflammatory genes. Phosphorylation of RelA/p65 at Ser536 serves to confer transcriptional competency to NF-κB bound to the promoter (22, 38). Activated NF-κB renders the otherwise “antiadhesive” lung microvasculature into a “proadhesive” one via activation of proinflammatory genes such as adhesion molecules (ICAM-1, VCAM-1, E-selectin), cytokines (TNF-α, IL-1β, IL-6), and chemokines [IL-8, monocyte chemoattractant protein (MCP-1)] (13, 33, 34, 39, 53, 54).
Disassembly of AJs is primarily regulated by loss of VE-cadherin from AJs, which is further aided by the contractile forces generated by actin-myosin interaction (actin stress fiber formation) (10, 11, 31). Thus VE-cadherin disassembly and actin-myosin interaction are important determinants of AJ disruption and increased endothelial permeability caused by proinflammatory mediators (10, 11, 30, 31). The coordinated and concerted actions of these events (induction of proinflammatory genes via NF-κB activation and disruption of endothelial AJs via VE-cadherin disassembly) serve to facilitate adhesion and transendothelial migration of inflammatory cells, particularly neutrophils (polymorphonuclear leukocytes, PMN) (29, 39, 43), and to increase endothelial permeability (6, 11, 32, 44) associated with ALI/ARDS (3, 5, 14, 18, 20, 45, 50).
Phosphoinositide-specific phospholipase C-ε (PLC-ε) is the most recently described member of the PLC family that hydrolyzes phosphatidylinositol 4, 5-bisphosphate (PIP2) to generate the second messengers inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) (8, 26). The IP3 and DAG in turn lead to generation of Ca2+ and activation of protein kinase C (PKC), respectively (8, 26). PLC-ε contains Ras association (RA) domains that confer regulation by Ras family members and a guanine nucleotide exchange factor (GEF) domain that regulates activity of the small GTPase, Rap (8, 26). PLC-ε is directly regulated by small GTPases including Rho, Rac, Ras, and Rap downstream of G protein-coupled receptors (GPCRs), including thrombin and lysophosphatidic acid receptors, as well as receptor tyrosine kinases (RTK) such as insulin growth factor receptor (8, 12, 26, 41). PLC-ε is implicated to play a role in the development and function of heart as well as in skin inflammation and tumor promotion (24, 28, 49). In cardiac myocytes, PLC-ε hydrolyzes phosphatidylinositol 4-phosphate as a substrate instead of PIP2 at the Golgi apparatus to generate local DAG (55). Other work indicates that PLC-β isoforms respond to acute receptor signals, whereas PLC-ε is responsible for longer-duration signaling (25). These data indicate that PLC-ε may play an important role in chronic receptor signaling and DAG generation at intracellular compartments. Studies using PLC-ε−/− mice have identified novel PLC-ε-dependent pathways for adrenergic regulation of cardiac contraction and GPCR- and RTK-dependent regulation of cardiac hypertrophy (49, 55). Lung is another primary site of PLC-ε expression; however, its role in the lung, particularly in the context of ALI, has not been studied. In this study, we provide evidence that PLC-ε is an important component of lung vascular inflammation and injury and that this action of PLC-ε relies, at least in part, on its ability to promote EC inflammation and barrier disruption.
MATERIALS AND METHODS
Reagents.
Human thrombin was purchased from Enzyme Research Laboratories (South Bend, IN). LPS of Escherichia coli (E. coli) origin, diethylaminoethyl (DEAE)-dextran, and an anti-PLC-ε antibody were from Sigma-Aldrich Chemical (St. Louis, MO). Antibodies to VCAM-1, ICAM-1, RelA/p65, Iκ-Bα, and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to VE-cadherin were obtained from Abcam (Cambridge, MA) and BD Biosciences (San Jose, CA). Antibodies to phospho-(Ser32 and Ser36)-Iκ-Bα and phospho-(Ser536)-RelA/p65 were obtained from Cell Signaling Technology (Beverly, MA). Plasmid maxi kit was from Qiagen (Valencia, CA). All other materials were from Fisher Scientific (Pittsburg, PA) or VWR Scientific Products (Gaithersburg, MD).
Mouse model of ALI.
PLC-ε−/− (knockout, KO) mice were generated as described previously (49). Age-matched C57BL/6 PLC-ε+/+ (wild-type, WT) mice were used as controls (Jackson Laboratory, Bar Harbor, ME). WT and KO mice were exposed to aerosol of saline alone or saline containing E. coli LPS (0.5 mg/ml) for 30 min as described (40, 52). After 18 h, bronchoalveolar lavage (BAL) fluids and lungs from these mice were collected and analyzed for the various markers of lung inflammation and injury as described (5, 14, 16). All mice care and treatment procedures were approved by the University of Rochester Committee on Animal Resources and performed in adherence to the National Institute of Health guidelines.
EC.
Human pulmonary artery ECs (HPAEC) were obtained from Lonza (Walkersville, MD) and cultured as described (4) in gelatin-coated flasks using endothelial basal medium 2 (EBM2) with bullet kit additives (BioWhittaker, Walkersville, MD). Experiments were performed in HPAEC < passage 6.
Measurement of lung inflammation and injury.
Lung homogenates were prepared in radioimmune precipitation (RIPA) buffer [50.00 mM Tris·HCl, pH 7.4, 150.00 mM NaCl, 0.25 mM EDTA, pH 8.0, 1.00% deoxycholic acid, 1.00% Triton X-100, 5.00 mM NaF, 1.00 mM sodium orthovanadate supplemented with protease inhibitor cocktail (Sigma-Aldrich)] as described (5, 14). The levels of proinflammatory mediators [TNF-α, MCP-1, keratinocyte-derived cytokine (KC), macrophage inflammatory protein 2 (MIP2), and granulocyte-macrophage colony-stimulating factor (GM-CSF)] in mouse lung homogenates were measured by bead-based multiplex immune assay (Millipore, Billerica, MA) using a Luminex FlexMAP3D instrument (Luminex, Austin, TX) according to manufacturers' recommendations. The levels of IL-1β, IL-6, MCP-1, and KC in BAL fluids were determined using ELISA kits from R&D Systems (Minneapolis, MN) as described (5, 14). The levels of ICAM-1 and VCAM-1 in lung homogenates were determined by ELISA as described (42). The recruitment of PMN in the lung was determined by monitoring the differential cell counts in BAL fluids and myeloperoxidase activity in the lung tissues as described (5, 14, 52). Total protein levels in BAL fluids were determined using bicinchoninic acid (BCA) kit (Pierce, Rockford, IL).
RNAi knockdown.
Predesigned short interfering RNA (siRNA) specific for human PLC-ε (si-PLC-ε) and a nontargeting siRNA control (si-Con) were obtained from Dharmacon (Lafayette, CO). HPAEC were transfected with si-PLC-ε or si-Con using DharmaFect1 siRNA transfection reagent (Dharmacon) essentially as described (4). Briefly, 50–100 nM siRNA and DharmaFect1 were mixed together and then added to cells that were 50–60% confluent. At 24–36 h after transfection, cells were challenged with thrombin or other agonists and then lysed after the indicated time periods for various analyses.
Immunoblot analysis.
Immunoblotting was performed as described previously (15). In brief, cells after appropriate treatments were lysed in RIPA buffer or phosphorylation lysis buffer [50.0 mM HEPES, 150.0 mM NaCl, 200.0 μM sodium orthovanadate, 10.0 mM sodium pyrophosphate, 100.0 mM sodium fluoride, 1.0 mM EDTA, 1.5 mM magnesium chloride, 10.0% glycerol, 0.5–1.0% Triton X-100, 1.0 mM phenylmethylsulfonyl fluoride (PMSF), and protease inhibitor cocktail]. Equal amounts of protein from the cell lysates as determined by the BCA assay (Pierce) were subjected to SDS-PAGE and then transferred onto nitrocellulose membranes for Western blotting as described (15). Representative blots presented in results come from the same membrane that may have more samples in various groups.
Nuclear extract preparation and assessment of NF-κB DNA binding.
Nuclear extracts were prepared and analyzed for NF-κB DNA binding activity as described (1). After appropriate treatments, cells were washed twice with ice-cold PBS and resuspended in 400 μl of buffer A (10.0 mM HEPES, pH 7.9, 10.0 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1.0 mM DTT, and 0.5 mM PMSF). After 15 min, NP-40 was added to a final concentration of 0.6%, and the samples were centrifuged to collect the supernatants containing the cytoplasmic proteins. The pelleted nuclei were resuspended in 50 μl of buffer B (20.0 mM HEPES, pH 7.9, 0.4 M NaCl, 1.0 mM EDTA, 1.0 mM EGTA, 1.0 mM DTT, and 1.0 mM PMSF). After 0.5 h at 4°C, lysates were centrifuged, and supernatants containing the nuclear proteins were collected. The nuclear DNA-binding activity of RelA/p65 was determined using an ELISA-based DNA binding assay kit (Cayman Chemical, Ann Arbor, MI) as described (5, 27).
Reporter gene constructs and luciferase assay.
The reporter plasmid pNF-κB-LUC (Stratagene, La Jolla, CA) containing five copies of consensus NF-κB sequences linked to a minimal E1B promoter-luciferase gene was used to determine the transcriptional activity of NF-κB. The pTKRLUC plasmid (Promega, Madison, WI) containing Renilla luciferase gene driven by the constitutively active thymidine kinase promoter was used to normalize the transfection efficiencies. Reporter gene transfections and luciferase assays were performed essentially as described (36). Briefly, DEAE-dextran (50 μg/ml) in serum-free EBM2 was mixed with 5 μg pNF-κB-LUC and 0.125 μg pTKRLUC. The resulting mixture was applied onto cells that were 60–70% confluent. After 1 h, cells were exposed to 10% DMSO in serum-free EBM2 for 4 min and then washed twice with PBS and allowed to grow in EBM2-10% FBS. The confluent monolayers were treated with appropriate agonists and then lysed in passive reporter lysis buffer (Promega). The cell extracts were assayed for firefly and Renilla luciferase activities using dual luciferase reporter assay system (Promega), and the data were expressed as a ratio of firefly to Renilla luciferase activity. In experiments evaluating the effect of PLC-ε knockdown on NF-κB transcriptional activity, cells were first transfected with PLC-ε siRNA and 24 h later cells were again transfected with pNF-κB-LUC and pTKRLUC using DEAE-dextran as described (4).
Immunofluorescence.
Confluent HPAEC monolayers grown on coverslips were subjected to immunofluorescence staining as described (15). To localize F-actin filaments, cells were incubated with Alexa Fluor 488-phalloidin. VE-cadherin antibody (BD Biosciences) was used to visualize AJ. DNA was stained using Hoechst dye to visualize nuclei. Images were obtained using a Nikon fluorescence microscope (Nikon Instech, Tokyo, Japan).
Measurement of endothelial permeability by transendothelial electrical resistance.
The endothelial barrier integrity was measured by monitoring transendothelial electrical resistance (TER) across confluent monolayers using the highly sensitive electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as described (32). Briefly, cells were grown to confluence on gelatin-coated gold microelectrodes in complete culture medium (EBM2 containing 10% FBS and bullet kit additives). After 24 h, culture medium was replaced with EBM2 containing 1% FBS, and 2 h later thrombin was added and the TER measured over a period of 4 h. Resistance was normalized to the initial voltage and expressed as a fraction of the normalized resistance value.
Statistical analysis.
Data presented are means ± SE and were analyzed by using standard one-way ANOVA. The significance between the groups was determined using Tukey's test (Prism 5.0; GraphPad Software, San Diego, CA). A P value <0.05 between two groups was considered statistically significant.
RESULTS
Role of PLC-ε in lung vascular inflammation and injury.
We used PLC-ε−/− (KO) mice to address the role of PLC-ε in regulating lung inflammatory injury in an aerosolized bacterial LPS inhalation mouse model of ALI (40, 52). LPS inhalation induced the levels of proinflammatory mediators (ICAM-1, VCAM-1, TNF-α, MCP-1, KC, MIP-2, and GM-CSF) in the lungs of PLC-ε+/+ (WT) mice, but these responses were significantly inhibited in KO mice (Fig. 1A, a–g). Similarly, the levels of proinflammatory mediators (IL-1β, IL-6, MCP-1, and KC) in the BAL fluids of KO mice were also attenuated compared with WT mice after LPS challenge (Fig. 1B, a–d). Consistent with this, KO mice showed a marked decrease in LPS-induced lung PMN recruitment and microvascular leakage compared with WT mice (Fig. 2, A–C). Because the levels of VE-cadherin, a major regulator of endothelial barrier integrity (10, 47), are decreased in inflamed lungs (18, 20), we determined whether deficiency of PLC-ε influences VE-cadherin levels in mice challenged with LPS. Results showed that KO mice were protected against LPS-induced decrease in VE-cadherin levels (Fig. 3). These data identify PLC-ε as a critical determinant of proinflammatory and leaky phenotype of the lung.
Fig. 1.
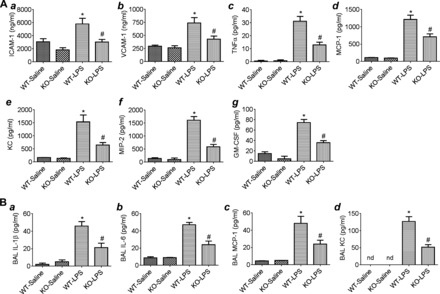
Effect of phospholipase C-ε (PLC-ε) deficiency on LPS-induced proinflammatory mediators in the lung. Age-matched C57BL/6L wild-type (WT) and PLC-ε−/− (knockout, KO) mice were aerosolized with saline alone or saline containing Escherichia coli LPS as described in materials and methods. At 18 h after LPS inhalation, lungs and bronchoalveolar lavage (BAL) fluids from these mice were collected, and levels of proinflammatory mediators were measured. A: lung homogenates were analyzed for ICAM-1 and VCAM-1 (a and b) levels by ELISA and TNF-α, monocyte chemoattractant protein 1 (MCP-1), keratinocyte-derived cytokine (KC), macrophage inflammatory protein 2 (MIP2), and granulocyte-macrophage colony-stimulating factor (GM-CSF) levels (c–g) by multiplex immune assay system. B: BAL fluids were analyzed for IL-1β, IL-6, MCP-1, and KC levels (a–d) by ELISA. Data are means ± SE (n = 3–5 for each condition). *P <0.05 vs. saline WT; #P vs. LPS WT. nd, not detected.
Fig. 2.
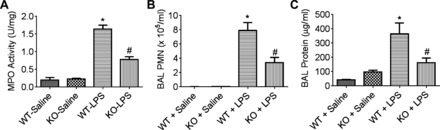
Effect of PLC-ε deficiency on LPS-induced lung polymorphonuclear leukocyte (PMN) infiltration and injury. Lung homogenates and BAL fluids were collected from WT and KO mice exposed to LPS as in Fig. 1. Lung homogenates were analyzed for tissue myeloperoxidase (MPO) activity (A), and BAL fluids were analyzed for PMN counts (B) and total protein (C). Data are means ± SE (n = 3–5 for each condition). *P <0.05 vs. saline WT; #P vs. LPS WT.
Fig. 3.
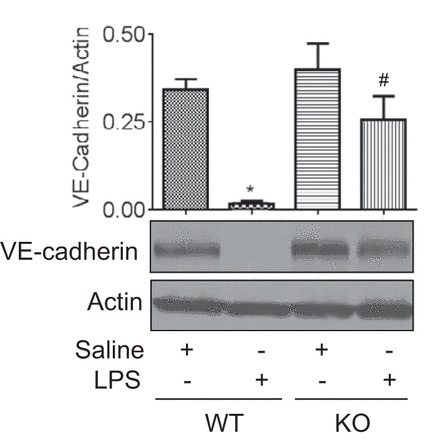
Effect of PLC-ε deficiency on LPS-induced decrease in VE-cadherin levels. Lung homogenates from WT and KO mice exposed to LPS as in Fig. 1 were analyzed for VE-cadherin levels by immunoblotting. Actin levels were used to monitor loading. The bar graph represents the effect of PLC-ε deficiency on LPS-induced decrease in VE-cadherin levels normalized to actin levels. Data are means ± SE (n = 3 for each condition). *P <0.05 vs. saline WT; #P vs. LPS WT.
PLC-ε regulates EC inflammation via NF-κB activation.
To test the possibility that PLC-ε activity in EC could contribute to the pathogenesis of ALI, we determined its role in mediating EC inflammation. Cells were transfected with siRNA targeting PLC-ε (si-PLC-ε) or control si-RNA (si-Con), and VCAM-1 level was measured as a marker of EC inflammation. Transfection of EC with si-PLC-ε resulted in significant depletion of PLC-ε (Fig. 4A) and inhibition of VCAM-1 expression in response to thrombin (Fig. 4B), an important edemagenic and proinflammatory agonist whose concentration is elevated in plasma and lavage fluids of patients with ALI/ARDS (17, 23) and known to induce PLC-ε activity (9). Because VCAM-1 is primarily regulated by NF-κB (38), we determined the possibility that PLC-ε mediates EC inflammation by activating NF-κB. To this end, cells were transfected with si-PLC-ε or si-Con in combination with pNF-κB-Luc, and luciferase activity was measured as an indicator of NF-κB activity. We observed that depletion of PLC-ε prevented thrombin-induced NF-κB activity (Fig. 4C), consistent with its effect on VCAM-1 expression (Fig. 4B). We also examined whether PLC-ε depletion affects NF-κB activity following stimulation of EC with other proinflammatory stimuli. We found that PLC-ε depletion was effective in preventing NF-κB activity in response to stimuli as diverse as LPS, TNF-α, and the nonreceptor agonist phorbol 13-myristate 12-acetate (phorbol esters) (Fig. 4, D–F). These data indicate that PLC-ε is an important regulator of NF-κB activity in the endothelium.
Fig. 4.
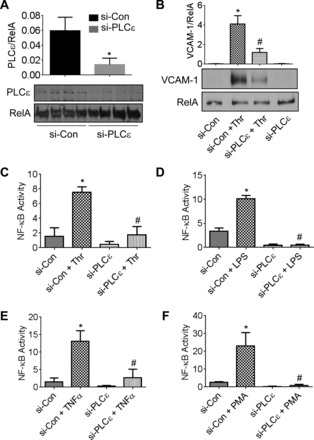
PLC-ε regulates NF-κB signaling in endothelial cells (EC). A: human pulmonary artery EC (HPAEC) were transfected with control siRNA (si-Con) or PLC-ε-specific siRNA (si-PLC-ε). After 36 h, total cell lysates were immunoblotted with an antibody to PLC-ε. RelA/p65 levels were used to monitor loading. The bar graph represents the effect of siRNA on PLC-ε level normalized to RelA/p65 level. Data are means ± SE (n = 4 for each condition). *P < 0.05 vs. si-Con control. B: HPAEC were transfected with si-Con or si-PLC-ε. After 36 h, cells were challenged with thrombin (5 U/ml) for 6 h, and the cell lysates were immunoblotted with an antibody to VCAM-1. RelA/p65 levels were used to monitor loading. The bar graph represents the effect of PLC-ε knockdown on thrombin-induced VCAM-1 expression normalized to actin level. Data are means ± SE (n = 3 for each condition). *P < 0.05 vs. si-Con untreated controls; #P < 0.05 vs. si-Con thrombin-treated controls. C–F: HPAEC were transfected with si-PLC-ε or si-Con using DharmaFect1. After 24 h, cells were again transfected with NF-κBLUC construct using diethylaminoethyl-dextran as described in materials and methods. Eighteen hours later, cells were challenged with thrombin (5 U/ml) (C), LPS (0.1 mg/ml) (D), TNF-α (100 U/ml) (E), or phorbol 13-myristate 12-acetate (PMA) (50 nm) (F) for 6 h. Cell extracts were assayed for firefly and Renilla luciferase activities as a measure of NF-κB activity. Data are means ± SE (n = 3–6 for each condition). *P < 0.05 vs. si-Con untreated controls; #P < 0.05 vs. si-Con thrombin-treated controls.
We analyzed NF-κB activation pathway to determine the mechanism by which PLC-ε regulates NF-κB activity. We first evaluated the effect of PLC-ε depletion on Iκ-Bα degradation, a prerequisite for the release of NF-κB (predominantly RelA/p65 homodimer in EC) (15, 35) for its translocation to the nucleus, where its binds to the promoters of target genes to activate their transcription (7, 22, 38). We found that thrombin induced Iκ-Bα degradation in cells transfected with si-Con, but this response was markedly reduced in cells transfected with si-PLC-ε (Fig. 5A). Consistent with this, depletion of PLC-ε was also effective in inhibiting thrombin-induced nuclear DNA binding of RelA/p65 (Fig. 5B). We next assessed the role of PLC-ε in mediating phosphorylation of Ser536 within the transactivation domain 1 of RelA/p65, a critical event conferring transcriptional competency to the bound NF-κB (22, 38). Results showed that thrombin-induced Ser536 phosphorylation of RelA/p65 was attenuated in cells depleted of PLC-ε (Fig. 5C). Considered together, these data show that PLC-ε controls NF-κB activation and thereby VCAM-1 expression by a dual mechanism involving Iκ-Bα degradation-dependent nuclear DNA binding and phosphorylation-dependent increase in transcriptional capacity of the bound RelA/p65.
Fig. 5.
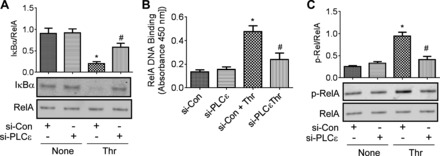
PLC-ε regulates NF-κB activity by promoting degradation of Iκ-Bα, nuclear DNA biding, and phosphorylation of RelA/p65. HPAEC were transfected with si-Con or si-PLC-ε. After 36 h, cells were treated with thrombin (5 U/ml) for 1.5 h. A: total cell lysates were prepared and immunoblotted with an anti-Iκ-Bα antibody. RelA/p65 levels were used to monitor loading. B: nuclear extracts were prepared and analyzed for RelA/p65 nuclear DNA binding using Cayman's NF-κB transcription factor assay kit as described in materials and methods. C: total cell lysates were immunoblotted with an anti-phospho-RelA/p65 (Ser536) antibody. RelA/p65 levels were used to monitor loading. Data are means ± SE (n = 6–8 for each condition). *P < 0.05 vs. si-Con untreated controls; #P < 0.05 vs. si-Con thrombin-treated controls.
PLC-ε mediates EC barrier disruption by promoting VE-cadherin disassembly and actin stress fiber formation.
We also determined whether PLC-ε is required for EC barrier disruption. Real-time measurement of TER showed that thrombin induced the characteristic time-dependent change in TER (32). The maximal decrease occurred around 0.5 h after thrombin challenge, which gradually recovered to baseline TER by 4 h (Fig. 6A). Depletion of PLC-ε had no effect on baseline TER but protected against thrombin-induced decrease in TER (Fig. 6A). These data identify PLC-ε as a novel regulator of EC barrier function.
Fig. 6.
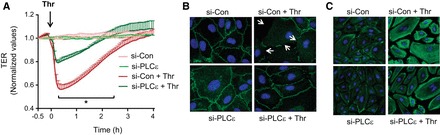
PLC-ε regulates endothelial barrier disruption by decreasing cell surface VE-cadherin and increasing actin stress fiber formation. HPAEC were transfected with si-Con or si-PLC-ε. A: after 24 h, cells were reseeded on gold electrode plates and cultured for an additional 48 h. The confluent monolayers were then treated with thrombin, and endothelial barrier disruption was determined by measuring transendothelial electrical resistance (TER). Data are means ± SE (n = 3–5 for each condition). *Difference between si-Con thrombin-treated vs. si-PLC-ε thrombin-treated controls (P < 0.05). B: after 36 h, cells were challenged with thrombin for 5 min, and immunofluorescence was performed using VE-cadherin antibody to visualize adherens junctions. Arrows indicate the sites of disruption of VE-cadherin staining. C: after 36 h, cells were challenged with thrombin for 15 min and labeled with Alexa 488-labeled phalloidin to visualize the actin stress fibers by fluorescence microscopy. Results are representative of 2–3 experiments.
We next determined whether the barrier-disruptive action of PLC-ε derives from its ability to mediate the loss of VE-cadherin at AJs and formation of actin stress fibers. Visualization of VE-cadherin junctions showed that thrombin induced a decrease in immunostaining of VE-cadherin at AJs in HPAEC transfected with si-Con, and this response was prevented in HPAEC transfected with si-PLC-ε (Fig. 6B). Similarly, thrombin-induced actin stress fiber formation was reduced in PLC-ε-depleted EC (Fig. 6C). Together, these data identify a novel role of PLC-ε in regulating EC barrier disruption via its ability to induce VE-cadherin disassembly and actin cytoskeleton rearrangement.
DISCUSSION
In this study, we have identified a novel role of PLC-ε in mediating EC inflammation and barrier disruption and in causing lung inflammatory injury in mice. Our in vivo experiments using PLC-ε KO mice establish PLC-ε as a key mediator of lung vascular inflammation and injury. Our in vitro data using cultured EC reveal that PLC-ε is an important regulator of EC inflammation and barrier disruption. We show that PLC-ε mediates EC inflammation by promoting NF-κB signaling via Iκ-Bα degradation-dependent DNA binding and phosphorylation-dependent increase in transcriptional activity of the bound RelA/p65. We also demonstrate that PLC-ε promotes endothelial barrier disruption by inducing actin stress fiber formation and loss of VE-cadherin at AJs. Thus the pathogenic role of PLC-ε in ALI derives, at least in part, from its ability to induce EC inflammation and permeability.
The vascular endothelium represents a critical interface that serves to regulate vascular homeostasis by virtue of regulating leukocyte trafficking, controlling vessel wall permeability, and maintaining blood fluidity. The extravasation of PMN and loss of endothelial barrier function are key early events in the pathogenesis of ALI and other inflammatory diseases (3, 31, 50, 51). Our results indicate that PLC-ε serves to promote lung vascular inflammation and leakage associated with ALI. Genetic ablation of PLC-ε rendered the mice resistant to lung vascular leak caused by LPS. Moreover, PLC-ε−/− mice showed a significant protection against the loss of VE-cadherin in the lungs of mice exposed to LPS. Similarly, we also observed that PLC-ε deficiency was associated with reduced lung PMN sequestration and migration in mice exposed to LPS. Consistent with this, the levels of adhesion molecules, cytokines, and chemokines, which act in concert to aid the adhesion of PMN to the endothelium and subsequently extravasation of adherent PMN into the surrounding tissues, were reduced in the lungs of PLC-ε−/− mice compared with PLC-ε+/+ mice challenged with LPS. Further support for the proinflammatory role of PLC-ε comes from previous reports showing a role of PLC-ε in skin and intestinal inflammation and tumorigenesis (24, 28).
Studies have shown that activation of NF-κB in EC plays an important role in ALI and other inflammatory diseases by its ability to control the expression of proinflammatory genes (53). Conditional blockade of NF-κB signaling in the endothelium is sufficient to reduce multiple-organ (including lung) inflammation, prevent vascular leak, and improve survival in murine models of sepsis (53). Similarly, EC-specific inhibition of NF-κB protects mice from atherosclerosis (19). Hence, we addressed the possibility that PLC-ε contributes, at least in part, to lung inflammatory injury via activation of NF-κB in the endothelium. Indeed, our data show that PLC-ε is an important mediator of NF-κB activation in EC. Furthermore, PLC-ε-mediated activation of NF-κB was noted for several major physiological proinflammatory mediators, such as thrombin, LPS, and TNF-α, an effect that may allow for enhanced NF-κB signaling in the endothelium. Analysis of NF-κB signaling pathway revealed that PLC-ε regulates thrombin-induced NF-κB activation by a dual mechanism whereby it mediates Iκ-Bα degradation-dependent nuclear uptake and DNA binding of RelA/p65 and also phosphorylation of RelA/p65 to increase its transcriptional capacity. Consistent with its role in NF-κB activation, PLC-ε knockdown was associated with a significant decrease in VCAM-1 levels following thrombin challenge of EC. Together, these data identify PLC-ε as an important mediator of EC inflammation by its ability to control NF-κB signaling.
Our data also support a previously unrecognized role of PLC-ε in regulating endothelial barrier function. EC depleted of PLC-ε showed a decrease in EC barrier disruption caused by thrombin. We found that PLC-ε mediates EC barrier disruption by inducing loss of VE-cadherin from AJs and contractile forces generated by actin-myosin interaction (actin stress fiber formation). Together, these data underscore the importance of PLC-ε in causing EC inflammation and permeability by its ability to induce NF-κB signaling and disassembly of AJs.
There are several possible mechanisms by which PLC-ε may control EC inflammation and permeability. First, the facts that PLC-ε, unlike other PLC isoforms, is an important effector of Rho and Ras superfamily of GTPases (8, 9, 26, 41) and that these GTPases are implicated in EC inflammation and barrier disruption (1, 27, 31) raise the possibility that PLC-ε may serve to link small GTPases to NF-κB activation and EC barrier disruption. One possible link may involve generation of second messengers by PLC-ε such as DAG and subsequent PKC activation (25, 41), which has been shown to be important in mediating EC inflammation and permeability (31, 37, 46). Relative to PLC-β, which transduces physiological responses primarily via rapid IP3-mediated calcium mobilization (2, 12, 48), PLC-ε appears to be involved in longer-term second messenger generation and may be primarily involved in chronic DAG production (9, 12, 25, 55). These observations raise the interesting possibility that PLC-β is engaged to initiate, whereas PLC-ε serves to prolong, EC dysfunction caused by GPCRs such as proteinase-activated receptor 1. Moreover, because PKC can also mediate activation of small GTPases (31, 32), the other possibility that PLC-ε may act upstream of these GTPases to induce NF-κB activation and EC barrier disruption cannot be excluded. Thus PLC-ε may contribute to NF-κB activation and EC barrier disruption by multiple pathways; however, elucidation of the precise mechanism underlying PLC-ε-mediated EC inflammation and permeability requires additional comprehensive studies.
In summary, our integrated in vitro and in vivo studies establish PLC-ε as a critical regulator of EC dysfunction (inflammation and permeability) and lung inflammatory injury. Several lines of evidence are consistent with an important role of endothelial PLC-ε in lung vascular inflammation and injury. These include 1) in vitro evidence showing the importance of PLC-ε in EC inflammation and barrier disruption, 2) in vivo results showing protection against LPS-induced decrease in VE-cadherin levels, which is exclusively expressed in the endothelium, and 3) reduction in LPS-induced expression of adhesion molecules in PLC-ε−/− lungs, particularly that of VCAM-1, whose expression is largely restricted to the endothelium. However, it should be emphasized that the present study does not exclude the involvement of PLC-ε derived from other cells, particularly epithelial and inflammatory cells, in this model of lung inflammation. Although our preliminary experiments show that depletion of PLC-ε in A549 lung epithelial cells fails to inhibit TNF-α-induced NF-κB activity (data not shown), a role of PLC-ε in causing inflammation in some epithelial cells, particularly keratinocytes, has been reported (21). Moreover, PLC-ε also reduces the levels of cytokines/chemokines (TNF-α, MCP-1, KC, MIP-2, and GM-CSF) that are also produced by epithelial and inflammatory cells, in addition to EC. However, PLC-ε is highly expressed in lung and poorly expressed in tissues rich in immune cells such as spleen or thymus (26). Nevertheless, addressing the precise contribution of PLC-ε derived from endothelial vs. epithelial or inflammatory cells in ALI requires additional comprehensive studies using mice with cell-specific deletion of PLC-ε.
GRANTS
This work was supported in part by National Heart, Lung, and Blood Institute grants HL116632 and HL096907. This study was also supported in part by National Institute of Environmental Health Sciences Center (EHSC) grant ES-01247 and an SDG Grant from American Heart Association (K. Bijli).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.M.B., F.F., A.V.S., and A.R. conception and design of research; K.M.B., F.F., S.A.S., A.L., V.G., and W.B.A. performed experiments; K.M.B., F.F., S.A.S., A.L., and V.G. analyzed data; K.M.B., F.F., S.A.S., A.L., A.V.S., and A.R. interpreted results of experiments; K.M.B. and A.R. prepared figures; K.M.B., F.F., S.A.S., A.L., W.B.A., A.V.S., and A.R. approved final version of manuscript; F.F., A.V.S., and A.R. edited and revised manuscript; A.R. drafted manuscript.
ACKNOWLEGMENTS
Present address for K. Bijli: Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Medicine, Atlanta Veterans Affairs and Emory University Medical Centers, Atlanta, GA 30033.
REFERENCES
- 1.Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol 173: 6965–6972, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 361: 315–325, 1993. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol 75: 593–615, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Bijli KM, Fazal F, Rahman A. Regulation of Rela/p65 and endothelial cell inflammation by proline-rich tyrosine kinase 2. Am J Respir Cell Mol Biol 47: 660–668, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bijli KM, Kanter BG, Minhajuddin M, Leonard A, Xu L, Fazal F, Rahman A. Regulation of endothelial cell inflammation and lung polymorphonuclear lymphocyte infiltration by transglutaminase 2. Shock 42: 562–569, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birukova AA, Meng F, Tian Y, Meliton A, Sarich N, Quilliam LA, Birukov KG. Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta 1852: 778–791, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280–288, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Bunney TD, Katan M. Phospholipase C epsilon: linking second messengers and small GTPases. Trends Cell Biol 16: 640–648, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Citro S, Malik S, Oestreich EA, Radeff-Huang J, Kelley GG, Smrcka AV, Brown JH. Phospholipase Cepsilon is a nexus for Rho and Rap-mediated G protein-coupled receptor-induced astrocyte proliferation. Proc Natl Acad Sci USA 104: 15543–15548, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dejana E, Vestweber D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci 116: 119–144, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 91: 1487–1500, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Dusaban SS, Kunkel MT, Smrcka AV, Brown JH. Thrombin promotes sustained signaling and inflammatory gene expression through the CDC25 and Ras-associating domains of phospholipase Cϵ. J Biol Chem 290: 26776–26783, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fazal F, Bijli KM, Minhajuddin M, Rein T, Finkelstein JN, Rahman A. Essential role of cofilin-1 in regulating thrombin-induced RelA/p65 nuclear translocation and intercellular adhesion molecule 1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 21047–21056, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fazal F, Bijli KM, Murrill M, Leonard A, Minhajuddin M, Anwar KN, Finkelstein JN, Watterson DM, Rahman A. Critical role of non-muscle myosin light chain kinase in thrombin-induced endothelial cell inflammation and lung PMN infiltration. PLoS One 8: e59965, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fazal F, Minhajuddin M, Bijli KM, McGrath JL, Rahman A. Evidence for actin cytoskeleton-dependent and -independent pathways for RelA/p65 nuclear translocation in endothelial cells. J Biol Chem 282: 3940–3950, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Fu P, Usatyuk PV, Lele A, Harijith A, Gregorio CC, Garcia JG, Salgia R, Natarajan V. c-Abl mediated tyrosine phosphorylation of paxillin regulates LPS-induced endothelial dysfunction and lung injury. Am J Physiol Lung Cell Mol Physiol 308: L1025–L1038, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gando S, Nanzaki S, Morimoto Y, Kobayashi S, Kemmotsu O. Systemic activation of tissue-factor dependent coagulation pathway in evolving acute respiratory distress syndrome in patients with trauma and sepsis. J Trauma 47: 719–723, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Gao R, Ma Z, Hu Y, Chen J, Shetty S, Fu J. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am J Physiol Lung Cell Mol Physiol 308: L847–L853, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab 8: 372–383, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Gong H, Rehman J, Tang H, Wary K, Mittal M, Chaturvedi P, Zhao YY, Chatturvedi P, Zhao Y, Komarova YA, Komorova YA, Vogel SM, Malik AB. HIF2α signaling inhibits adherens junctional disruption in acute lung injury. J Clin Invest 125: 652–664, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harada Y, Edamatsu H, Kataoka T. PLC-ε cooperates with the NF-κB pathway to augment TNFα-stimulated CCL2/MCP1 expression in human keratinocyte. Biochem Biophys Res Commun 414: 106–111, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev 18: 2195–2224, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Idell S, Gonzalez K, Bradford H, MacArthur CK, Fein AM, Maunder RJ, Garcia JG, Griffith DE, Weiland J, Martin TR. Procoagulant activity in bronchoalveolar lavage in the adult respiratory distress syndrome. Contribution of tissue factor associated with factor VII. Am Rev Respir Dis 136: 1466–1474, 1987. [DOI] [PubMed] [Google Scholar]
- 24.Ikuta S, Edamatsu H, Li M, Hu L, Kataoka T. Crucial role of phospholipase C epsilon in skin inflammation induced by tumor-promoting phorbol ester. Cancer Res 68: 64–72, 2008. [DOI] [PubMed] [Google Scholar]
- 25.Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. G-protein-coupled receptor agonists activate endogenous phospholipase Cepsilon and phospholipase Cbeta3 in a temporally distinct manner. J Biol Chem 281: 2639–2648, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley GG, Reks SE, Ondrako JM, Smrcka AV. Phospholipase C(epsilon): A novel Ras effector. EMBO J 20: 743–754, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonard A, Marando C, Rahman A, Fazal F. Thrombin selectively engages LIM kinase 1 and slingshot-1L phosphatase to regulate NF-κB activation and endothelial cell inflammation. Am J Physiol Lung Cell Mol Physiol 305: L651–L664, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M, Edamatsu H, Kitazawa R, Kitazawa S, Kataoka T. Phospholipase Cepsilon promotes intestinal tumorigenesis of Apc(Min/+) mice through augmentation of inflammation and angiogenesis. Carcinogenesis 30: 1424–1432, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Liu SF, Malik AB. NF-κB activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 290: L622–L645, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol 77: 1763–1772, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 86: 279–367, 2006. [DOI] [PubMed] [Google Scholar]
- 32.Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 276: 22614–22620, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Minami T, Aird WC. Endothelial cell gene regulation. Trends Cardiovasc Med 15: 174–184, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Minhajuddin M, Bijli KM, Fazal F, Sassano A, Nakayama KI, Hay N, Platanias LC, Rahman A. Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells. J Biol Chem 284: 4052–4061, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman A, Anwar KN, True AL, Malik AB. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol 162: 5466–5476, 1999. [PubMed] [Google Scholar]
- 36.Rahman A, Anwar KN, Uddin S, Xu N, Ye RD, Platanias LC, Malik AB. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Mol Cell Biol 21: 5554–5565, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rahman A, Bando M, Kefer J, Anwar KN, Malik AB. Protein kinase C-activated oxidant generation in endothelial cells signals intercellular adhesion molecule-1 gene transcription. Mol Pharmacol 55: 575–583, 1999. [PubMed] [Google Scholar]
- 38.Rahman A, Fazal F. Blocking NF-κB: An inflammatory issue. Proc Am Thorac Soc 8: 497–503, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahman A, Fazal F. Hug tightly and say goodbye: role of endothelial ICAM-1 in leukocyte transmigration. Antioxid Redox Signal 11: 823–839, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reutershan J, Basit A, Galkina EV, Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 289: L807–L815, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Smrcka AV, Brown JH, Holz GG. Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal 24: 1333–1343, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Speyer CL, Rancilio NJ, McClintock SD, Crawford JD, Gao H, Sarma JV, Ward PA. Regulatory effects of estrogen on acute lung inflammation in mice. Am J Physiol Cell Physiol 288: C881–C890, 2005. [DOI] [PubMed] [Google Scholar]
- 43.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76: 301–314, 1994. [DOI] [PubMed] [Google Scholar]
- 44.Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB. Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13: 693–708, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, Cheppudira BP, Mishra RK, Debroy A, Qian Z, Bachmaier K, Zhao YY, Christman JW, Vogel SM, Ma A, Malik AB. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol 15: 239–247, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandenbroucke St Amant E, Tauseef M, Vogel SM, Gao XP, Mehta D, Komarova YA, Malik AB. PKCα activation of p120-catenin serine 879 phospho-switch disassembles VE-cadherin junctions and disrupts vascular integrity. Circ Res 111: 739–749, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vestweber D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol 28: 223–232, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, Harden TK. Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330: 974–980, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Phospholipase C epsilon modulates beta-adrenergic receptor-dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 97: 1305–1313, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 51.Weis SM. Vascular permeability in cardiovascular disease and cancer. Curr Opin Hematol 15: 243–249, 2008. [DOI] [PubMed] [Google Scholar]
- 52.Yao H, Yang SR, Edirisinghe I, Rajendrasozhan S, Caito S, Adenuga D, O'Reilly MA, Rahman I. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol 39: 7–18, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 205: 1303–1315, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.You J, Peng W, Lin X, Huang QL, Lin JY. PLC/CAMK IV-NF-kappaB involved in the receptor for advanced glycation end products mediated signaling pathway in human endothelial cells. Mol Cell Endocrinol 320: 111–117, 2010. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Phospholipase C epsilon scaffolds to muscle-specific A kinase anchoring protein (mAKAPbeta) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem 286: 23012–23021, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]