

Abstract
This review the status of genetic laboratory testing in Prader-Willi syndrome (PWS) due to different genetic subtypes, most often a paternally derived 15q11-q13 deletion, with benefits and limitations related to prenatal screening. Medical literature was searched for prenatal screening and genetic laboratory testing methods in use or under development and discussed in relationship to PWS. Genetic testing includes six established laboratory diagnostic approaches for PWS with direct application to prenatal screening. Ultrasonographic, obstetric and cytogenetic reports were summarized in relationship to the cause of Prader-Willi syndrome and identification of specific genetic subtypes including maternal disomy 15. Advances in genetic technology were described for diagnosing PWS specifically DNA methylation and high-resolution chromosomal SNP microarrays as current tools for genetic screening and incorporating next generation DNA sequencing for noninvasive prenatal testing (NIPT) using cell-free fetal DNA. Positive experiences are reported with NIPT for detection of numerical chromosomal problems (aneuploidies) but not for structural problems (microdeletions). These reports will be discussed along with future directions for genetic screening of PWS. In summary, this review describes and discusses the status of established and ongoing genetic testing options for PWS applicable in prenatal screening including NIPT and future directions for early diagnosis in Prader-Willi syndrome.
INTRODUCTION AND BACKGROUND
Clinical Description of Prader-Willi Syndrome
Prader-Willi syndrome (PWS) is a complex neurodevelopmental disorder due to errors in genomic imprinting and first described in 1956 by Drs. Prader, Labhart and Willi in nine individuals with similar features.1 The major features in PWS include infantile hypotonia, a poor suck and feeding difficulties, hypogonadism/hypogenitalism, learning and behavior problems with morbid obesity beginning in early childhood. The genetic cause of this disorder was first reported in 1981 by Ledbetter and coworkers in their description of a subtle cytogenetic deletion of the proximal long arm of chromosome 15 in the 15q11-q13 region visible using high-resolution chromosome methods in four selected patients diagnosed clinically with PWS.2 Over the ensuing decade, alterations of the chromosome 15q11-q13 region were recognized using a variety of cytogenetic and molecular laboratory techniques in a range of PWS subjects presenting with abnormal chromosome findings including deletions, translocations, inversions, marker chromosomes and isochromosome formations.3–5
Using staining methods of the polymorphic short arm and centromeric regions of chromosome 15 to evaluate the parent of origin status of the deletion, Butler and Palmer in 1983 showed that the deletion was de novo in origin and preferentially inherited from the father.6 Using molecular genetic techniques and isolation of polymorphic DNA markers from the 15q11-q13 region, Nicholls, Butler and coworkers in 1989 analyzed two patients clinically diagnosed with PWS and without a recognized 15q11-q13 deletion by high-resolution chromosome analysis and found that both chromosome 15s came from the mother.7 No chromosome 15 was found from the father and no deletion was identified in either of the two chromosome 15s. The phenomenon of both chromosomes 15s from the mother was coined uniparental maternal disomy 15.
There are now three recognized genetic causes of PWS which ultimately lead to lack of expression of genes from the chromosome 15 contributed by the father. Approximately two thirds of those affected with PWS will show the paternally-contributed 15q11-q13 deletion; about 30% will have maternal uniparental disomy 15 and the remaining individuals will have a genetic or non-genetic anomaly in the imprinting center controlling the regulation or expression of genes on chromosome 15 termed an imprinting defect.8–16 Specifically, a microdeletion of the imprinting center can be inherited and passed to a PWS offspring if present on the father’s chromosome 15 leading to a 50% recurrence risk. If the defect is due to an epimutation and not a structural change affecting the methylation status in the imprinting center, then the risk is much lower.17–21 All three causes of PWS can be detected by genetic testing with the use of multiple techniques to detect the Prader-Willi critical region (PWCR). The DNA methylation test can detect 99% of individuals with PWS but will not identify the genetic subtype (deletion, maternal disomy or imprinting defect), information often required for genetic counseling and care management.15,16
Angelman syndrome (AS) was a second condition recognized during the 1980s with the same appearing chromosome 15q11-q13 deletion.22,23 The deletion found in AS was of maternal origin and in a similar percentage of cases (about 70%) as seen in PWS. Both PWS and AS became recognized as the first examples of errors in genomic imprinting in humans. Although the same chromosome 15q11-q13 region is altered in both disorders (i.e., paternal 15q11-q13 deletion in PWS and maternal deletion in AS), their clinical features are very distinct or different.
Common findings seen in PWS include severe infantile hypotonia, a poor suck with feeding problems leading to failure to thrive, underdeveloped sex organs (hypogonadism/hypogenitalism), growth and other hormone deficiencies leading to growth retardation, small hands/feet and endocrine disturbances. Additional findings include behavioral problems (OCD, self-injury, tantrums, stubbornness), mild intellectual disability and characteristic facial features (almond shaped eyes, narrow bifrontal diameter, short upturned nose, down turned corners of mouth with thin upper lip and decreased saliva). Later in early childhood, food seeking and hyperphagia leading to life-threatening obesity, if not controlled, become cardinal features of PWS.14–16,21,24–26 In comparison, AS is characterized with mild hypotonia, seizures, lack of speech, developmental delay and severe intellectual disability with characteristic facial features (broad nose, microcephaly, widely spaced teeth).22,23
PWS is now recognized as the most common known cause of life-threatening obesity in humans with an incidence of one in 10,000 to one in 30,000, as similarly seen in AS.3,27 The diagnosis of either genomic imprinting syndrome may not be readily appreciated at birth and genetic laboratory testing is often recommended to assist in the clinical suspicion of these two related but clinically distinct disorders by examination of chromosome anomalies with the use of various validated cytogenetic and molecular genetic techniques or assays. A major focus of this report will describe and discuss established and emerging genetic laboratory tests applicable to prenatal screening in PWS.
Genetics and Cytogenetics of Prader-Willi Syndrome
There are a number of genes in chromosome 15 causing features of PWS that are prevented normally from expression on the maternally inherited chromosome 15 and thus become potential gene candidates. The precise gene or genes causing manifestations of PWS are yet to be been determined but the most likely candidate causing several features seen in PWS is a specific small non-coding nucleolar-organizing RNA (snoRNA) called SNORD116.28–30
There are dozens of genes or transcripts located on chromosome 15q11-q13 with approximately 10 imprinted and paternally expressed genes (see Figure 1). These genes include SNRPN (small nuclear ribonucleoprotein polypeptide N)-SNURF (SNRPN upstream reading frame) and several copies of the C/D box snoRNAs located downstream to the imprinted SNRPN-SNURF gene complex including SNORD116. These snoRNAs are involved in RNA processing and encoded by an extended length of transcript from the SNRPN-SNURF gene complex located towards the middle of the 15q11-q13 region. The major snoRNAs in this region are: SNORD64, SNORD107, SNORD109A, SNORD115 and SNORD116.5,15,16,28–30
Figure 1.

Ideogram of chromosome 15 with representation of the 15q11-q13 region displaying genes with symbols in linear order from the centromeric (Cen) end of chromosome 15. Genes in the non-imprinted regions are shown in green while genes in the Prader-Willi syndrome region are shown in blue and genes in the Angelman syndrome region are shown in red. Chromosome 15q breakpoints (BP1, BP2, BP3, BP4, BP5) are shown.
Other paternally expressed imprinted genes located towards the centromeric end of the 15q11-q13 region are MKRN3, MAGEL2, NDN and C15orf2 (see Figure 1). Several of these genes are involved in neural development, brain function, infertility and circadian rhythm.12,14–16 Specifically, NDN or necdin is involved in outgrowth of central nervous system axons with expression in brain regions including the hypothalamus, thalamus and pons possibly influencing respiration function.14–16 Other recognized genes in the region include several gamma amino butyric acid (GABA) receptor subunits (GABRB3, GABRA5, GABRG3) which may show paternal biased expression.31 GABA is a major neurotransmitter with inhibitory function with alterations associated with behavior such as appetite control, obsessive-compulsions and memory.14 The UBE3A gene is imprinted and maternally expressed and located in this chromosome region but causing Angelman syndrome.22 The P gene involved with pigment production is located at the distal end of the 15q11-q13 region and when deleted leads to hypopigmentation in both PWS32 or Angelman syndrome.22 Mutations of this gene causes a recessive form of albinism.14,33 The HERC2 gene which is located at the common distal breakpoint (BP3) in the 15q11-q13 region encodes ubiquitin ligase.34,35 There are two recognized HERC2 pseudogenes located at the two common proximal 15q11-q13 breakpoints (BP1 and BP2) and are characterized as having low copy DNA repeats playing a role in chromosome 15 pairing during meiosis. When these chromosomes are mismatched then chromosomal anomalies may result such as deletions or duplications of the 15q11-q13 region.36–38 HERC2 gene polymorphisms have been linked to iris color in humans by impacting the function of the neighboring P gene.34
There are two typical 15q11-q13 deletions reported in PWS (and Angelman syndrome) referred to as Type I or Type II and differ by about 500kb in size.13,23,38–40 The Type I deletion is larger with loss of four genes (NIPA1, NIPA2, CGP5, CYFIP1) located at the centromeric end of the 15q11-q13 region. Those with this larger deletion generally have more severe problems such as obsessive-compulsions and self-injury (skin picking) with lower cognitive measures and poorer academic achievement than those with the smaller typical Type II deletion or those with maternal disomy 15.14,41 A small percentage (5%) of individuals with a 15q11-q13 deletion will have an atypical form, either smaller or larger than the typical Type I or Type II deletion.14,42 These individuals may have more unusual presentation of the PWS phenotype.
RESULTS AND DISCUSSION
Genetic Laboratory Testing for Prader-Willi Syndrome
Prader-Willi syndrome may be highly variable in its clinical presentation affecting multiple systems; hence, clinical consensus diagnostic criteria were established by a group of PWS experts in the past.43 Yet, the diagnosis may be difficult to establish based only on clinical grounds and the genetic basis is heterogeneous. The genetics of PWS is complicated and more often than not, additional testing may be required to confirm the diagnosis once suspected and to establish the genetic subtype. Since the advent of modern genetic laboratory techniques, the diagnosis when suspected clinically can be readily confirmed with genetic testing as the diagnosis may be difficult to establish based on clinical grounds.
Diagnostic genetic testing categories in Prader-Willi syndrome
High-resolution chromosome analysis and fluorescence in situ hybridization (FISH)
Chromosome testing has been a routine part of the clinical evaluation and laboratory testing of patients presenting for clinical genetic services including Prader-Willi syndrome for many years in order to rule out chromosomal anomalies such as translocations and deletions. The typical 15q11-q13 interstitial deletion seen in PWS or Angelman syndrome can be detected with high-resolution chromosome analysis at greater than 550 band level usually with G-banding methods established during the past 35 years. This cytogenetic testing alone is no longer considered sufficient for such interpretations. It generally has been replaced due to both false-negative and false-positive results but better than standard chromosome analysis at a lower band level as illustrated in a retrospective study of 26,041 reported amniocentesis samples over a 12 year period from 2001 to 2012.44 They found that 43 amniocentesis samples shared a suspicion of having a possible 15q11-q13 deletion by standard cytogenetic testing and had undergone more detailed molecular testing (FISH, DNA methylation analysis and/or MS-MLPA). Three fetuses (7%) were confirmed to have the 15q11-q13 deletion [PWS (N=1) and Angelman syndrome (N=2)]. However, cytogenetic studies can identify important translocations alone that predispose to 15q deletions and may identify other disorders that mimic both PWS and Angelman syndrome. In addition, many older patients may have had only a routine chromosome study done prior to high-resolution chromosome analysis or molecular genetic diagnostic techniques. High-resolution chromosome analysis of individual prometaphase or high-resolution chromosomes under a microscope by trained cytogeneticists with expertise in interpreting subtle chromosome anomalies such as deletions, duplications or translocations could diagnosis PWS or Angelman syndrome but not detect smaller deletions (e.g., < 5Mb in size), uniparental disomy or imprinting defects.
In the early 1990s fluorescence in situ hybridization (FISH) was established using chromosome 15 probes from the proximal long arm, centromeric regions and/or distal long arm to identify deletions of various sizes and became widely available.43,45,46 Probes of a different color for the centromeric region, for example, the chromosome 15 alpha- satellite area, would allow discrimination between those cases due to an interstitial deletion compared with the rare case involving a chromosome 15 translocation that leads to the cytogenetic deletion commonly seen in PWS. For those individuals with normal appearing chromosomes without evidence of a deletion with high-resolution chromosome analysis or with FISH probes, the advent of polymorphic chromosome 15 DNA markers became readily available in the mid 1990s after isolation of markers in the mid to late 1980s useful for genotyping.47 These DNA markers could trace the transmission of the chromosome 15 from each parent to the child important to identify maternal disomy 15 and imprinting defects in PWS. However, genotyping analysis requires DNA from both parents and the child for best accuracy.
To address concerns raised about the selection and ordering of genetic tests for diagnosis, the American College of Medical Genetics and the ASHG/ACMG Test and Technology Transfer Committee in 1996 was assigned the task to generate diagnostic testing approaches for both PWS and Angelman syndrome.43 When assessing the scientific validity of diagnostic testing for PWS and AS, high-resolution chromosome analysis was judged to be accurate in identifying chromosome 15 translocations that could predispose to 15q deletions in rare cases and to identify disorders that mimic both PWS and AS. However, newer molecular genetic techniques are more reliable, accurate and consistent in identifying the 15q11-q13 deletion, the major cause for both PWS and AS. The use of FISH was shown to be scientifically and clinically valid for the detection of deletions using commercially available probes (e.g., SNRPN) located within the common deletion boundaries in combination with any probe outside these boundaries such as the centromeric region. Advisably, a two color FISH probe approach was recommended. With advances in genetic technology and emerging high-resolution chromosomal microarrays with over 2 million separate DNA probes generated from the whole genome developed during the past few years, high-resolution chromosome analysis and/or FISH studies are ordered less frequently. High-resolution chromosomal microarray studies are now more available and utilize single nucleotide polymorphism (SNP) probes but are more expensive than high-resolution chromosome analysis. Microarrays will establish a diagnosis more quickly and with more detail by identifying the specific PWS genetic subtypes (15q11-q13 deletions-typical and atypical); maternal disomy 15 subclasses (heterodisomy, segmental isodisomy, total isodisomy) or imprinting defects (see Figure 2). They prove to be more accurate than using stepwise approaches with other chromosome testing options.48
Figure 2.


High-resolution chromosomal SNP microarray results for Prader-Willi syndrome (PWS) using copy number variant (CNV) and single nucleotide polymorphism (SNP) probes to identify the typical 15q11-q13 deletions in PWS classified as Type I involving breakpoints BP1 and BP3 or smaller Type II involving breakpoints BP2 and BP3 (A); High-resolution chromosomal SNP microarray results showing loss of heterozygosity status for chromosome 15 and maternal disomy 15 subclasses (segmental isodisomy 15, isodisomy 15, and heterodisomy 15) in PWS (B).
High-resolution chromosomal SNP microarray analysis
Chromosomal microarrays have been available for genetic testing for the past 10 years and have significantly changed or evolved over time. The earlier microarrays were based on only a few thousand oligonucleotide DNA probes placed on microscope slides. Based on today’s technology, the early microarrays were primitive with probes unevenly and widely spaced but yet useful in identifying known microdeletion syndromes such as PWS or Angelman syndrome. Nonetheless, these early microarrays could determine the copy number status (deletion or duplication) at a resolution manyfold greater than with high-resolution chromosome analysis detecting chromosomal anomalies from 3 to 5 Mb in size via examination of chromosomes with light microscopy by a trained cytogeneticist. The current microarrays contain over 2 million probes, a combination of both copy number and SNPs, distributed throughout the genome. Ulitlzing the SNP probe patterns, these microarrays can also identify regions of homozygosity (ROH) or loss of heterozygosity (LOH) and when greater than 8 Mb in size becomes an efficient tool to diagnose uniparental disomy (e.g., maternal disomy 15 in PWS) as well as deletions.48,49 In addition, the level of absence of heterozygosity noted throughout the genome can be used to calculate the inbreeding coefficient or consanguinity status in an individual to determine if common ancestry or inbreeding is present.50–52 The LOH pattern seen in chromosome 15 in PWS can be used to not only identify maternal disomy 15 but also the specific disomic subclass (i.e., segmental isodisomy, total isodisomy or heterodisomy) as those individuals with PWS with segmental or total isodisomy 15 could be at increased risk of developing a second genetic condition such as an autosomal recessive disorder if the mother carried a recessive gene allele in the LOH region and the PWS child would have two identical copies5 (see Figure 2).
High-resolution chromosomal SNP microarray analysis decreases the need of PCR amplification and genotyping polymorphic chromosome 15 DNA markers, particularly in those individuals with features of PWS and a non-deletion chromosome 15 status by conventional testing. However, chromosomal microarrays will not identify translocations or inversions. Genotyping informative chromosome 15 DNA markers may still be required if the high-resolution chromosomal microarray results are normal and therefore not helpful in determining whether the child demonstrates a normal (biparental) inheritance pattern for the chromosome pair 15 due to not having PWS or from an imprinting defect causing PWS. Confirmation of paternity should also be considered by the use of DNA markers from other chromosomes to further support the presence of two maternal chromosome 15s.
DNA methylation analysis of chromosome 15
The smallest region of overlap of the chromosome 15 deletion among individuals with Prader-Willi syndrome is estimated at 4.3 kb in size and includes the location of the imprinting center which lies at the 5′ end of the bicistronic SNURF-SNRPN complex loci.18,40 CpG islands are located in the promoter, exon 1 and intron 1 of this gene and involved with gene regulation and are found in clusters throughout the genome. When the CpG islands are not methylated in their respective genes then the lack of methylation renders the gene active or expressed. The methylation pattern therefore becomes useful to study regarding the gene activity status in the diagnosis of PWS.13,15,16,53–56 Southern hybridization, and later, PCR methods have been used to determine whether specific chromosome 15 probes (e.g., SNRPN) show differential methylation representing gene activity to detect the parent of origin in both PWS and Angelman syndrome. This technology was first used in 1992 to genetically confirm the diagnosis of PWS.8,53–55 Parental DNA samples are also not required as this test can demonstrate a PWS pattern (maternal chromosome 15 signal only) or an Angelman syndrome pattern (paternal chromosome 15 signal only) in the DNA sample from the patient. DNA methylation analysis of chromosome 15 using only methylation specific markers for both PWS and AS is considered very accurate (99% in PWS; 85% in AS) and becomes a gold standard for diagnostic studies in these two rare genomic imprinting disorders.54 However, DNA methylation testing will not allow for determination of the specific genetic subtype (deletion, uniparental disomy 15, imprinting defects, translocations) seen in either condition which is required for accurate genetic counseling for family members.
To distinguish between deletion or uniparental disomy status in PWS other studies such as FISH, genotyping, methylation specific-multiplex ligation-probe amplification (MS-MLPA) and/or high-resolution chromosomal SNP microarrays will be required. High-resolution microarrays will not only identify the chromosome 15 deletion or maternal disomy 15 in PWS but will yield information regarding the size of the deletion (typical vs atypical) and the disomic subclass (e.g., segmental isodisomy 15 due maternal nondisjuncton and recombination during meiosis I; total isodisomy 15 due to nondisjunction in meiosis II or maternal heterodisomy 15 without recombination events in meiosis I). High-resolution microarrays with SNP probes are available commercially in laboratories nationwide.
Methylation specific-multiplex ligation-dependent probe amplification (MS-MLPA)
Methylation specific-multiplex ligation-dependent probe amplification (MS-MLPA) for genetic testing of PWS was first reported in 200657 and later validated by our research group.58,59 MS-MLPA is an advanced molecular genetic semi-quantitative technique for determining the copy number of genes involved in the causation of PWS and also for the chromosome 15 DNA methylation status.57–59 It incorporates a single multiplex ligation-dependent PCR-based reaction using up to 60 markers, with unique DNA sequences in a commercially available kit (MRC-Holland, Amsterdam, Netherlands) to detect genes of interest in the chromosome 15q11-q13 region or reference genes elsewhere for determination of copy number and chromosome 15 methylation status. The adjacent probes are then ligated into one single probe structure before amplification. A PCR reaction format is used to generate fragment sizes varying from 150 – 500 bp incorporating a common fluorescently labeled PCR primer pair. Each fragment is separated by capillary electrophoresis that represents a specific gene or reference sequence. The peak height of each generated fragment of the targeted DNA sequence then is compared to peak heights identified in reference DNA signals. A deletion or duplication is determined from the relative decrease (deletion) or increase (duplication) in peak height in the non-reference DNA signal and figures generated and analyzed accordingly to represent the copy number status (1=deletion, 2=normal, 3=duplication) of genes under study (see Figure 3).
Figure 3.
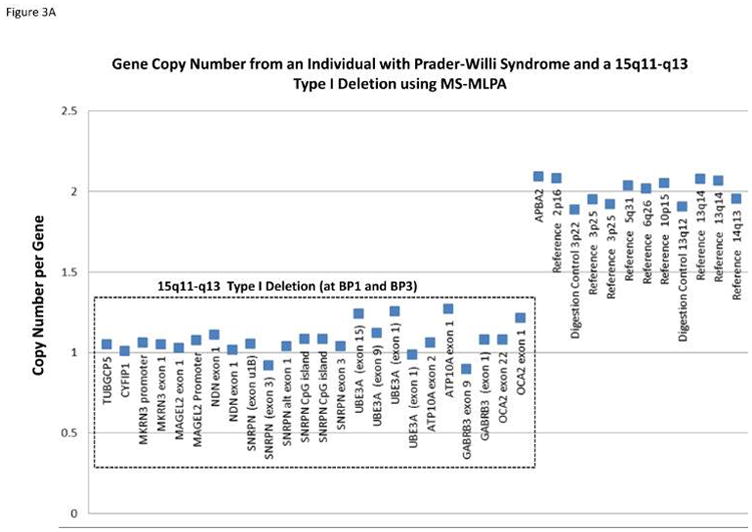
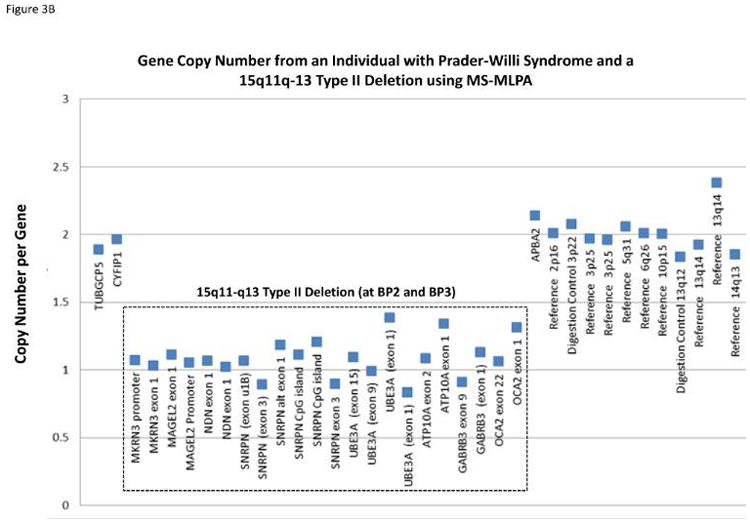
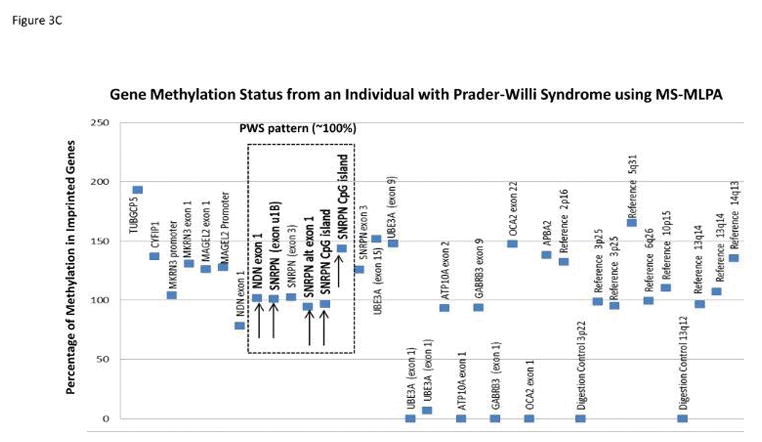
Gene copy number data compiled from the methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) kit assay for individuals with Prader-Willi syndrome (PWS) having either the 15q11-q13 Type I deletion showing a gene copy number of 1 for probes within 15q11-q13 region located between breakpoints BP1 and BP3 (A); the smaller 15q11-q13 Type II deletion is shown with a gene copy number of 1 for probes within the 15q11-q13 region located between breakpoints BP2 and BP3 (B); MS-MLPA gene methylation pattern from an individual with PWS showing an approximate 100 percent methylation status (abnormal PWS pattern) indicating gene inactivity for five 15q11-q13 probes selected for methylation analysis (C)
A basic principle of the methylation specific (MS) component of the MS-MLPA method is to determine the methylation status of active (unmethylated) or inactive (methylated) genes. Specific MS – MLPA probes identify DNA containing restriction enzyme sites at the gene level and with the use of methylation sensitive enzymes can determine the status by the ratio of the peak height for each target probe following PCR amplification before and after digestion. Therefore, the methylation statuses for imprinted genes in the chromosome 15q11-q13 region play a role in the causation of PWS. It can be measured and used for diagnosis [i.e., 100% methylation pattern for DNA sequences representing imprinted maternally inactivated genes in PWS (see Figure 3) due to the missing active paternal gene allele and a 50% methylation pattern in normal control DNA with one normally active allele (0% methylated) from the father and one normally inactive allele (100% methylated) from the mother]. Another advantage of the MS – MLPA method is the ability to test methylation status at multiple loci on chromosome 15 at once reducing the risk for false positive or false negative results. This robust MS-MLPA method and kits are now widely used for diagnosis of imprinting disorders such as PWS, Angelman syndrome and Albright hereditary osteodystrophy.
In those cases where the suspicion of PWS is weak or the ordering physician is not familiar with the clinical presentation or selection of genetic testing options available to order in identifying the specific genetic subtypes, the DNA methylation test may be the best approach with a 99% correct diagnostic rate. DNA methylation is now widely available individually or as a component of MS-MLPA. Historically, the first step at many clinical sites until the last ten years was to undertake chromosome testing with FISH probe analysis for identifying the deletion seen in PWS.5,43,45 If the methylation test was positive, then additional genetic testing was recommended to determine the specific PWS genetic subtype.
Clinically, PWS may be misdiagnosed, some patients may have Angelman syndrome instead, as both conditions will show hypotonia and developmental delay as infants and share the same chromosome 15 deletion in the majority of cases but of different parental origin. Starting with the DNA methylation test will avoid the diagnostic dilemma in this particular clinical scenario during infancy as the methylation patterns are distinctively different in PWS and Angelman syndrome. Subsequently, the risk for additional children to be similarly affected is less than 1% if the affected child has a 15q11-q13 deletion or uniparental disomy 15, up to 50% if an imprinting defect is present and up to 25% if a parental chromosome translocation is found leading to PWS.5,15,16,24 Prenatal testing and diagnosis are possible for pregnancies at increased risk if these underlying genetic mechanisms are known to exist in the at-risk pregnancy.
There are six established cytogenetic and molecular approaches for undertaking genetic laboratory testing in PWS (or AS). These include high-resolution chromosome analysis (at ≥550 band level); commercially available FISH probes for chromosome 15; genotyping polymorphic chromosome 15 markers; DNA methylation analysis of SNRPN gene on chromosome 15; methylation specific- multiplex ligation-dependent probe amplification (MS-MLPA) and high-resolution chromosomal SNP microarrays.5 Historically, high-resolution chromosome analysis has been used but currently replaced by more advanced technology with much higher resolution and accuracy (i.e., high-resolution chromosomal SNP microarrays)48 and/or MS-MLPA. However, routine chromosome analysis is performed to rule out chromosome translocations or rearrangements that would be missed using high-resolution microarrays and significantly impact genetic counseling of family members. These six established genetic laboratory methods have been applied successfully for both postnatal and prenatal diagnosis of PWS (see Figure 4).
Figure 4.

Genetic laboratory testing diagram showing an approach with subsequent genetic methods for detecting the specific Prader-Willi syndrome (PWS) genetic subtype beginning with DNA methylation testing in an individual suspected to have PWS.
Prenatal Diagnosis Experience in Prader-Willi Syndrome
Ultrasonographic and obstetric reports
Prenatal diagnosis in PWS is important particularly for genetic counseling in those families having an imprinting defect identified in the father with a 50% recurrence risk or if a familial translocation involving chromosome 15 is present. In these scenarios, there is usually a prior identification of the disorder causing an abnormality (e.g., chromosomal anomaly) and hence preimplantation genetic diagnosis, noninvasive prenatal diagnosis or testing of fetal tissue may be warranted. However, the vast majority (over 95%) of cases with PWS are sporadic and without specific fetal malformations. Nonetheless small case series have retrospectively analyzed the ultrasonographic findings in cases diagnosed post-natally as having PWS.60–64 Antenatal diagnosis of PWS has also been made prospectively in families without risk factors for PWS based uniquely on ultrasound features identified in the third trimester.61
The most common ultrasonographic feature noted in the published reports is the association of fetal growth restriction (typically a fetal abdominal circumference below the 5th centile) and polyhydramnios, which in some cases can be severe enough to require amnioreduction.62,64 Both features are typically diagnosed in the third trimester. The polyhydramnios may be related to lack of fetal deglutition, as described in one case.64 Maternal perception of decreased fetal movements in the early part of the third trimester is also a commonly reported, though less specific, observation and it may relate to the hypotonia noted in the affected fetuses and frequently in the neonates. Fetal malformations are typically absent at prenatal ultrasonography of PWS; however, the 2 cases prospectively diagnosed with the syndrome via invasive testing were noted to have subtle malpositions of the extremities, with extended feet and flexed toes, in addition to fetal abdominal circumferences below the 5th centile and polyhydramnios.61 Therefore, when presented with the unusual association of a fetal abdominal circumference below the 10th centile and polyhydramnios, attention should be paid to unusual position of the extremities as supporting evidence which may prompt genetic testing to evaluate for PWS. It is unclear at present what proportion of PWS cases exhibit the above sonographic features.
In an attempt to provide an answer to such a question, Gross et al.65 interviewed the mothers of 106 individuals with PWS and reviewed the obstetrical records for those less than 10 years of age (N=47). They compared obstetric records with those of the siblings and the general population. The authors found a significant difference (p<0.001) in rates of decreased fetal movements (88%), small for gestational age (SGA) (65%), asymmetrical intrauterine growth with increased head/abdomen circumference ratio (43%), and polyhydramnios (34%) in PWS compared to the controls. No major congenital anomalies were found in the PWS pregnancies. A combination of 2, 3 or 4 of the above factors was present in 27%, 29% and 24% of the pregnancies, respectively. Fourteen of the 15 PWS pregnancies with umbilical artery Doppler studies were within normal limits. The combination of asymmetric growth patterns and polyhydramnios was identified in 34% of the PWS pregnancies. The investigators concluded that prenatal genetic screening for PWS with the use of DNA methylation would be indicated whenever combination of polyhydramnios, SGA or asymmetric intrauterine growth without morphological abnormalities is noted in the fetus by ultrasound. Normal findings at umbilical artery Doppler and abnormal positioning of the extremities may further support the diagnosis. Prenatal diagnosis of PWS may be desirable even in the third trimester of pregnancy as affected infants may require admission to the neonatal intensive care unit due to severe hypotonia and respiratory distress associated with oxygen dependence.
Cytogenetic reports
Cytogenetic testing for chromosomal problems for prenatal diagnosis has been used for several decades specifically chorionic villi sampling (CVS) performed in the first trimester at about 10 weeks gestation and later with amniocentesis. Most notably and regarding PWS, a report by Cassidy et al.66 in 1992 demonstrated new information about the causation of maternal disomy 15 and PWS. They summarized the results of chromosome testing from CVS and later by amniocentesis in a single pregnancy with the fetus showing trisomy 15 with CVS only and the infant was born with classical features of PWS. Furthermore, L’Hermine et al.67 reported the PWS fetal phenotype and maternal disomy 15 mosaicism found in CVS performed at 13 weeks gestation. Amniocentesis results showed cells with a normal karyotype. Molecular analysis with DNA methylation showed the typical methylation pattern seen in PWS and maternal disomy 15. Fetal ultrasound showed slightly enlarged lateral ventricles and hypoplastic male external genitalia but without intrauterine growth retardation. The report points out that a karyotypically normal fetus with ambiguous genitalia and cerebral anomalies would warrant a detailed cytogenetic and molecular genetic study because of the risk of PWS.
Trisomy 15 is a common chromosomal abnormality causing spontaneous abortions in early pregnancy. However, the pregnancy reported by Cassidy et al.66 did not spontaneously abort and the female infant was delivered with features of PWS including hypotonia, a poor suck and feeding difficulties. Genetic testing identified maternal disomy 15 and PWS. This case was explained by a trisomic rescue event in early pregnancy of a fetal cell with trisomy 15 that lost a chromosome 15 inherited from the father. The fetus had trisomy 15 due to a chromosome 15 nondisjunction event occurring in meiosis from the mother leading to two maternal chromosome 15s in the egg. When this egg was fertilized by a normal sperm with a single chromosome 15 then trisomy 15 resulted. In early pregnancy, if a trisomic 15 rescue event occurs then a and miscarriage may not occur. DNA methylation testing can be performed for prenatal diagnosis of PWS when requested but will not determine the PWS genetic subtype.
As maternal disomy 15 occurs in about 25% of cases with PWS and mostly due to errors (nondisjunction) in maternal meosis I correlated with advanced maternal age,68–70 an early attempt to study maternal disomy 15 and trisomy 15 mosaicism in pregnancies was undertaken by Christian et al.71 They used molecular genetic tools to analyze seven cases of trisomy 15 mosaicism identified in amniotic fluid (N=3) or CVS (N=4). In all cases the mother was older than 35 years of age. The genetic testing results showed that two of the seven cases of trisomy 15 mosaicism resulted in maternal disomy 15 consistent with the theoretical expectation of one-third indicating a high risk of uniparental disomy in such pregnancies. Hence, uniparental disomy should be considered in all cases of trisomy 15 mosaicism by cytogenetic testing when encountered in CVS or amniocentesis and more detailed genetic testing offered.
Hahnemann and Vejersley72 further summarized cytogenetic data in 1997 from the European collaborative research on mosaicism in cytotrophoblast, villus mesenchyme and one or more fetal tissues in 192 gestations with noted mosaicism or non-mosaic fetoplacental discrepancy involving a single autosomal trisomy in CVS. They identified predictors of confined placental mosaicism, generalized mosaicism and/or uniparental disomy and distribution of mosaic and non-mosaic aneuploid cell lines in the different fetal and extrafetal tissue sources. Trisomies 15 and 16 were most often found in both cytotrophoblast and villus mesenchyme and not in fetal cells compared with other trisomies. Their report supported that mosaicism discrepancy for both trisomies 15 and 16 result from trisomic rescue events thereby increasing the likelihood for uniparental disomy (i.e., maternal disomy 15 in PWS). Heidemann et al.73 also reported a rare fetus with PWS resulting from a balanced familial translocation involving chromosomes 2 and 15 leading to maternal disomy 15. The mother was found to be a carrier of the cytogenetically identical balanced translocation involving chromosomes 2 and 15 at the 15q11.2 band. The maternal disomy 15 in the fetus in this report was due to a 3:1 chromosome segregation during meiosis with loss of the normal paternal chromosome 15; therefore, genetic information for chromosome 15 in the fetus was maternal in origin only. They summarized related findings from 14 cases with PWS involving non-Robertsonian translocations in the literature.
Noninvasive prenatal testing (NIPT)
The development of sequenced-based noninvasive prenatal testing (NIPT) to date was initially triggered by the identification of fetal DNA in maternal plasma and serum allowing for recovery of this DNA source for testing.74,75 Cell-free DNA has been used as a source of DNA for (NIPT) to screen pregnant women during the past few years. It was first applied to screen for aneuploidies (i.e., trisomy 21) in 201176 and has advanced. It continues to evolve with a high detection rate for identification of aneuploidies with over 99% detection for trisomy 21 with improved sensitivity and specificity for other trisomies (i.e., 13, 18) and sex chromosome aneuploidy.77 A number of laboratories are validating different technical procedures when using cell-free DNA for screening fetal chromosomal aneuploidy but results cannot be reported in all women screened due to indeterminate or no call test result and genetic counseling should be offered to discuss other testing options. Hence, cell-free fetus DNA screening does not replace the precision of other more established prenatal diagnostic testing at this time and is limited in the type of chromosomal anomalies detected. In addition, cell-free DNA testing does not assess risk of fetal anomalies such as neural tube defects and hence should not be considered as a test in isolation from other approved methods. There are limitations of cell-free DNA screening performance and limited data in cost effectiveness and range of chromosomal anomalies identified. It continues to be an emerging field for investigation but not in the first line of screening for most women in the general obstetric population – conventional genetic screening remain most appropriate choice for prenatal diagnosis, specifically for PWS and most other genetic disorders. Yet, it has the potential to revolutionize prenatal screening for genetic disorders.
There are two NIPT approaches that are currently available including massive or targeted parallel DNA sequencing based on quantitative methods for counting or interpreting datasets and SNP-based studies that depend on identification of fetal and maternal gene allele distributions and differences. Both approaches can detect Down syndrome or other trisomic conditions or sex chromosomal aneuploidies.78–83 NIPT was initially focused on identifying aneuploidies for chromosomes 13, 18, 21, X and Y which account for proximally 30% of all livebirths with chromosomal anomalies.84–86 The use of SNP-based approach can also detect triploidy.87
A recent review of 6,697 women was reported with screening during the first trimester over the past ten years with cell-free fetal DNA was reported.88 Fetal aneuploidies are now detected with high precision facilitating a reduction in the number of invasive diagnostic procedures performed. 84,89–91 The investigators assumed a possible scenario implementing first trimester screening based on identification of high risk prenatal factors such as specific abnormal ultrasound and maternal serum findings and concluded that this new technology may result in a 6-fold reduction in the number of invasive procedures.
Commercially available approaches have now been developed to use both massive or targeted parallel DNA sequencing or SNPs to identify maternal allele distribution and patterns.83,85 With direct application for aneuploidy detection prenatally, there is growing interest to identify other chromosomal anomalies such as microdeletions or duplications which occur in 1.7% of all pregnancies without fetal abnormalities.92 However, identification of structural chromosomal problems is more challenging and requires higher depth of next generation sequencing reads than currently used for aneuploidy detection.79 The performance of NIPT was reviewed using low-coverage (0.1X) whole-genome sequencing of maternal plasma DNA at a single center with 1982 consecutive cases. NIPT was positive for common trisomies in 29 cases including 23 with trisomy 21, four with trisomy 18 and two with trisomy 13 and were all confirmed with prenatal karyotyping. Eleven showed sex chromosomal abnormalities (SCA) and nine had other aneuploidies or deletions/duplications. Overall, 86% of the NIPT suspected SCA were of fetal origin and 67% of the other abnormalities were caused by confined placental mosaicism. Follow up studies found that three chromosomal abnormalities were not detected by NIPT including a case of triploidy, unbalanced translocation and balanced translocation. There were no known false negatives indicating 100% sensitivity.93
DNA sequencing results are also complicated by identification of variants of unknown clinical significance but gaining ground for widespread use in genomic screening. However, no standards have been firmly established to ensure maximum benefit to participants when screening for genetic conditions and to minimize risk of harm in relationship to false positives with true clinical sensitivity and specificity data.94 A very high threshold should be incorporated when calling for pathogenic gene variants and care must be taken to understand the potential impact and negative consequences in genetic screening; however, these concerns are not as critical in the interpretation of prenatal screening or testing results in PWS as PWS is due to errors is genomic imprinting generally from a paternally derived chromosome 15q11-q13 deletion and not due to single gene nucleotide basepair changes or allelic variants as seen in most single gene disorders. Hence, progress is being made utilizing advances in genetic technology and incorporating bioinformatics with computer accessible programs and algorithms in the diagnosis of structural chromosomal anomalies such as microdeletion syndromes. For example, Wapner et al.83 reported their experience using a SNP-based non-invasive prenatal testing with five microdeletion syndromes accounting for a population incidence of about one in 1000. They studied 469 plasma samples (from 352 pregnant women with DiGeorge, 5p or 1p36 related deletions in fetuses and 356 unaffected pregnancies; 111 artificial plasma mixtures of DiGeorge, 5p, PWS or Angelman related deletions) and collected DNA amplified with the use of massive multiplexed PCR then sequenced and analyzed with the Next generation Aneuploidy Test Using SNPs algorithm for the presence or absence of deletions seen in chromosomes 22q11.2 (N=46 samples), 1p36 (N=1), distal 5p (or cri-du-chat) (N=24) and Prader-Willi/Angelman region [PWS (N=15) and Angelman syndrome (N=21)]. The detection rates were 100% for PWS, Angelman syndrome, 1p36 deletion and cri-du-chat and 97.8% for 22q11.2 deletion. No false positives were detected using this technology for PWS (0/428); Angelman syndrome (0/442) or 1p36 deletion (0/422) and less than 1% for the other deletion syndromes studied. They concluded that SNP-based noninvasive prenatal microdeletion screening appears accurate and could be considered in the general population particularly for screening purposes of clinically relevant and well-characterized microdeletions and duplications that occur in greater than 1% of pregnancies regardless of maternal age. They used a PlasmART method as a means to produce a mixture of artificial pregnancy plasma mixtures with a range of fetal DNA concentrations but has not been fully validated as a testing model for rare genetic disorders. However, Zhao et al.91 reported their experience with detection of fetal subchromosomal abnormalities by DNA sequencing procedures using small quantities of circulating cell-free DNA from the fetus found in maternal plasma and based their conclusions on 17 cases of PWS. They made comparisons with other microdeletion syndromes such as 22q11.2 and 1p36 then concluded that challenging factors limit detection for structural chromosomal problems. They reported that limits in detection of any given microdeletion or microduplication syndrome found in the fetus may include fetal DNA fraction compared with the mother source as plasma cell-free DNA consists of both maternal and fetal fragments and fetal DNA accounted for 3.4% of total DNA in maternal plasma in early gestation and 6.2% in late gestation.95 Fetal DNA fragments are mainly from placental apoptosis.96 The size of the chromosome anomaly under investigation and DNA (exon) sequencing coverage of the targeted region with biological and technical variability inherent to the methodology are also factors along with instrumentation used. More recently, Valderramas et al.97 reported their experience with cell-free DNA screening in clinical practice for aneuploidies and microdeletions from 2013–2015 in 121 patients with abnormal cell-free DNA results confirmed by prenatal or postnatal karyotype or microarray. They found 105 with trisomy 21, 18 or 13 and 92 (88%) were positive and 13 (12%) were non-reportable. The positive predictive value of cell-free DNA results was 73.5% for all trisomies. Twenty-six patients had positive (N=9) or non-reportable (N=17) microdeletion results. Seven of nine screens positive for microdeletions underwent confirmatory testing and all were false positives with a predictive value for microdeletion testing of 0%. They concluded that diagnostic testing is needed to confirm abnormal cell-free DNA results for both aneuploidy and microdeletions.
Gross et al.98 recently reported their experience in 21,948 samples with SNP-based NIPT and detection of fetal 22q11.2 deletion in clinical practice. Ninety-five cases were reported at high risk for having a fetal 22q11.2 deletion using this technology and diagnostic testing was available for 61 (64%) cases. Eleven (18%) of the cases were confirmed as true positives while 50 (82%) cases were identified as false positives resulting in a positive predictive value (PPV) of 18%. Ultrasound abnormalities were present in 82% of true-positive and 18% of false-positive cases. They concluded that their clinical experience with SNP-based NIPT for the 22q11.2 deletion syndrome indicated that this screening method requires counseling and other management resources for these high-risk pregnancies. Tan et al.99 also reported their experience over a two year period in 565 pregnant women with NIPT with twin pregnancies after assisted reproductive technology (ART) and screening for trisomy 21, 18 and 13 by sequencing cell-free DNA in maternal plasma. Positive NIPT results were confirmed by karyotyping. The NIPT failure rate was 0.9% (5/565 cases). Four cases of trisomy 21 were identified by NIPT and confirmed by karyotyping which resulted in a 100% (95% CI, 39.8% – 100%) positive productive value. Of 556 cases with reported NIPT negative results, 506 cases (91%) were confirmed by follow-up studies. No false negative results were reported. They concluded that their experience using NIPT with a high positive predictive value and low false positive rate could imply its use as a good alternative approach to conventional prenatal screening during early first trimester pregnancy in twin pregnancies utilizing ART.
The emerging field of noninvasive prenatal screening using cell-free DNA for identification of chromosomal problems particularly aneuploidy in the fetus has gained popularity in both the public and medical provider communities for prenatal care. Until recently, noninvasive prenatal screening for aneuploidy was dependent on maternal serum measures and/or ultrasound studies with false-positive rates of 5% and detection rates of 50–95% depending on the specific screening method.100
Advances in genomic technology, instrumentation and bioinformatics have enabled these methodologies to be used in the clinical setting and disclosure of results to the patient with the aim of providing pertinent information that can help optimize pregnancy outcomes.101 To assist the healthcare providers and patients with information updates, the American College of Medical Genetics and Genomics (ACMG) has published policy statements related to prenatal screening.102–104 Since the previous position statement published in 2013 on non-invasive prenatal screening, ACMG has now released an update for 2016.100 The readers are referred to this recent publication designed primarily as an educational resource for clinicians to help provide quality medical services related to noninvasive prenatal screening for fetal aneuploidy.
CONCLUSION
Prenatal genetic testing for PWS has been recognized for the past 25 years either in the form of chromosome analysis to examine for the chromosome 15q11-q13 deletion or other chromosomal related anomalies such as trisomy 15 or chromosome 15 translocations. Early results in prenatal diagnostic testing for PWS were based on concerns raised or obstetrical observations during pregnancy using CVS or amniocentesis and high-resolution chromosome analysis and/or FISH. In addition, serendipity has played a role in identifying abnormal chromosome 15 findings leading to PWS using prenatal genetic testing as reported by Cassidy et al.66 in a fetus with trisomy 15 found in the CVS sample. Trisomy 15 was then followed by a trisomic 15 rescue event in early pregnancy leading to PWS. The infant was diagnosed postnatally with hyponia, a poor suck and feeding problems and the presence of maternal disomy 15. The risk of having a second child affected with PWS is less than 1% for the typical 15q11-q13 deletion or maternal disomy 15, but a higher risk (e.g., 50%) can occur for those with an imprinting defect, indicating the importance of prenatal diagnosis in families with imprinting defects or a familial chromosome 15 abnormality causing PWS.
Since discovery of circulating cell-free fetal DNA in maternal plasma and use of genetic based noninvasive prenatal tests (NIPT), chromosomal aneuploidies (i.e., trisomies 13, 18, 21, X and Y) have been detected with high sensitivity and specificity. There is growing interest with new reports to expand NIPT to include microdeletion syndromes.90,91 However, chromosomal microdeletions or duplications are harder to detect and the application of this technology will need to be more widely studied before adopted105 and applicable for prenatal diagnosis of PWS or other structural chromosomal abnormalities. At this stage, the methodology is considered novel at the level of proof of concept but emerging. Currently, it is not economically feasible for large-scale clinical application due to limitations and technical factors.
Discussions are underway to incorporate microdeletion syndromes such as PWS, Angelman syndrome and other genetic disorders into expanded newborn screening (NBS) protocols utilizing next generation (NexGen) sequencing of multiple PCR-based fragments from chromosomal regions for determining copy number status. In addition, methylation specific-quantitative melt analysis (MS – QMA) with DNA collected from blood on NBS filter paper could be used for diagnosing certain genetic conditions as indicated in early studies with fragile X syndrome.106 Other disorders in the future may include PWS and Angelman syndrome but more research is needed. Digital PCR and quantitative microsphere hybridization (QMH) utilizing nanoparticle technology to identify copy number status are under investigation for both prenatal and postnatal diagnostic purposes and further supported by a report from Newkirk et al.107 in identifying submicroscopic deletions of chromosome 15q11-q13 region in PWS.
Prenatal genetic testing has provided timely and accurate results over the past decades using invasive procedures such as CVS and amniocentesis with established cytogenetic and molecular techniques for both numerical and structural chromosomal problems. These prenatal results often provide reassurance for the family regarding advanced maternal age or concerns about the fetus such as decreased movement or polyhydraminios most often seen in PWS. Ultimately, the use of noninvasive prenatal tests are gaining popularity with improved accuracy and detection rates for numerical chromosomal problems. Progress is needed and studies are underway in the area of cell-free DNA and NIPT for identification of structural chromosomal anomalies by incorporating advanced genetic technology, computer programs and databases with better methods and instrumentation (e.g., high-resolution SNP microarrays and next generation sequencing). Families seeking a diagnosis or have concerns about PWS should seek out an experienced clinician preferably a certified medical geneticist with the assistance of a trained genetic counselor to discuss the testing options, benefits of prenatal diagnosis in terms of parental preparation and medical approaches available in the prenatal clinical setting and health care needs and management of the infant after delivery. As several features in PWS relate to growth hormone deficiency, common in PWS, early diagnosis and growth hormone therapy is needed. Similarly, early diagnosis may also be beneficial in addressing behavioral aspects seen in PWS if anticipated and allow the parents to implement behavioral support with better awareness and education as quickly as possible. Specifically, eating disorders and increasingly recognized mood disorders can be confirmed that often accompany individuals with PWS. Ultimately, a goal of prenatal screening would be early diagnosis of PWS thereby impacting medical care after delivery and quality of life for the patient and their family.
What’s already known about this topic?
Genetic laboratory testing approaches for the diagnosis of Prader-Willi syndrome with different genetic subtypes do exist and other genetic testing options under development for application in prenatal screening.
Early diagnosis of Prader-Willi syndrome leads to early treatment with improved quality of life, decreased comorbidities and reduced medical costs.
What does this study add?
A current review and summary of the literature relating to genetic testing in Prader-Willi syndrome with description of benefits and limitations for prenatal screening.
Discussion of genetic testing approaches applied to prenatal screening with existing and emerging technologies leading to early diagnosis of Prader-Willi syndrome.
Ultrasonographic features which may prompt suspicion of Prader-Willi syndrome.
No statement is required regarding the ethical background for this study or any institutional or national ethical committee approval.
Acknowledgments
The National Institute of Child Health and Human Development (NICHD) grant HD02528 is acknowledged.
Funding Source: National Institute of Child Health and Human Development (NICHD) grant HD02528
Footnotes
There are no conflicts of interest to report for the author.
References
- 1.Prader A, Labhart A, Willi H. Ein sydnrom von adipositas, kleinwuchs, kryptochismus und oligophrenie nach myatonieartigem zustand im neugeborenenalter. Schweizerische Medizinishce Wochenschrift. 1956;6(3):1260–1. [Google Scholar]
- 2.Ledbetter DH, Riccardi VM, Airhart SD, et al. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304(6):325–9. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- 3.Butler MG. Prader-Willi syndrome: Current understanding of cause and diagnosis. Am J Med Genet. 1990;35(3):319–32. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler MG, Thompson T. Prader-Willi syndrome: Clinical and genetic findings. Endocrinol. 2000;10:3S–16S. doi: 10.1097/00019616-200010041-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler MG. Single gene and syndromic causes of obesity: Illustrative examples. Prog Mol Biol Transl Sci. 2016;140:1–45. doi: 10.1016/bs.pmbts.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler MG, Palmer CG. Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet. 1983;1(8336):1285–6. doi: 10.1016/s0140-6736(83)92745-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholls RD, Knoll JH, Butler MG, et al. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. 1989;342(6247):281–5. doi: 10.1038/342281a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Driscoll DJ, Waters MF, Williams CA, et al. A DNA methylation imprint, determined by the sex of the parent, distingushes the Angelman and Prader-Willi syndromes. Genomics. 1992;13(4):917–24. doi: 10.1016/0888-7543(92)90001-9. [DOI] [PubMed] [Google Scholar]
- 9.Nicholls RD. Genomic imprinting and candidate genes in Prader-Willi and Angelman syndromes. Curr Opin Genet Dev. 1993;3(3):445–56. doi: 10.1016/0959-437x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 10.Reis A, Dittrich B, Greger V, et al. Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader-Willi syndromes. Am J Hum Genet. 1994;54(5):741–7. [PMC free article] [PubMed] [Google Scholar]
- 11.Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: Potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Preprod. 1997;3(4):321–32. doi: 10.1093/molehr/3.4.321. [DOI] [PubMed] [Google Scholar]
- 12.Hanel ML, Wevrick R. The role of genomic imprinting in human developmental disorders: lessons from Prader-Willi syndrome. Clin Genet. 2001;59(3):156–64. doi: 10.1034/j.1399-0004.2001.590303.x. [DOI] [PubMed] [Google Scholar]
- 13.Bittel DC, Butler MG. Prader-Willi syndrome: Clinical genetics, cytogenetics and molecular biology. Expert Rev Mol Med. 2005;7:1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler MG, Lee PDK, Whitman BY. Management of Prader-Willi Syndrome. New York: Springer-Verlag; 2006. [Google Scholar]
- 15.Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 16.Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249–63. doi: 10.1007/s40618-015-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buiting K, Saitoh S, Gross S, et al. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9(4):395–400. doi: 10.1038/ng0495-395. [DOI] [PubMed] [Google Scholar]
- 18.Saitoh S, Buiting K, Rogan PK, et al. Minimal definition of the imprinting center and fixation of chromosome 15q11-q13 epigenotype by imprinting mutations. Proc Natl Acad Sci USA. 1996;93(15):7811–5. doi: 10.1073/pnas.93.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buiting K, Dittrich B, Gross S, et al. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: Implications for imprinting switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet. 1998;63(1):170–80. doi: 10.1086/301935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buiting K, Gross S, Lich C, et al. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am J Hum Genet. 2003;72(3):571–7. doi: 10.1086/367926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler MG, Cassidy SB. Genetic basis, genetic testing and genetic counseling for Prader-Willi syndrome. In: Hoybye C, editor. Prader-Willi Syndrome. Nova Science Publishers, Inc; 2013. [Google Scholar]
- 22.Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12(7):385–95. doi: 10.1097/GIM.0b013e3181def138. [DOI] [PubMed] [Google Scholar]
- 23.Sachdeva R, Donkers SJ, Kim SY. Angelman syndrome: A review highlighting musculoskeletal and anatomical aberrations. Clin Anat. 2016;29(5):561–7. doi: 10.1002/ca.22659. [DOI] [PubMed] [Google Scholar]
- 24.Butler MG. Prader-Willi syndrome: Obesity due to genomic imprinting. Curr Genomics. 2011;12:204–15. doi: 10.2174/138920211795677877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics. 1993;91(2):398–402. [PMC free article] [PubMed] [Google Scholar]
- 26.Clarke DJ, Boer H, Whittington J, et al. Prader-Willi syndrome, compulsive and ritualistic behaviours: the first population-based survey. Br J Psychiatry. 2002;180:358–62. doi: 10.1192/bjp.180.4.358. [DOI] [PubMed] [Google Scholar]
- 27.Butler J, Whittington JE, Holland AJ, et al. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Dev Med Child Neurol. 2002;44(4):248–55. doi: 10.1017/s001216220100202x. [DOI] [PubMed] [Google Scholar]
- 28.Sahoo T, del Gaudio D, German JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40(6):719–21. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Smith AJ, Purmann C, Walters RG, et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet. 2009;18(17):3257–65. doi: 10.1093/hmg/ddp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lassi G, Priano L, Maggi S, et al. Deletion of the Snord116/SNORD116 alters sleep in mice and patients with Prader-Willi syndrome. Sleep. 2016;39(3):637–44. doi: 10.5665/sleep.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bittel DC, Kibiryeva N, Talebizadeh Z, et al. Microarray analysis of gene/transcript expression in Prader-Willi syndrome: Deletion versus UPD. J Med Genet. 2003;40(9):568–74. doi: 10.1136/jmg.40.8.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler MG. Hypopigmentation: A common feature of Prader-Labhart-Willi syndrome. Am J Hum Genet. 1989;45(1):140–6. [PMC free article] [PubMed] [Google Scholar]
- 33.Spritz RA, Bailin T, Nicholls RD, et al. Hypopigmentation in the Prader-Willi syndrome correlates with P gene deletion but not with haplotype of the hemizygous P allele. Am J Med Genet. 1997;71(1):57–62. doi: 10.1002/(sici)1096-8628(19970711)71:1<57::aid-ajmg11>3.0.co;2-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kayser M, Liu F, Janssens AC, et al. Three genome-wide association studies and a linkage analysis identify HERC2 as a human iris color gene. Am J Hum Genet. 2008;82(2):411–23. doi: 10.1016/j.ajhg.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; 65(2):370–86. [Google Scholar]
- 35.Visser M, Kayser M, Palstra RJ. HERC2 rs12913832 modulates human pigmentation by attenuating chromatinloop formation between a long-range enhancer and the OCA2 promoter. Genome Res. 2012;22(3):446–55. doi: 10.1101/gr.128652.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amos-Landgraf JM, Ji Y, Gottlieb W, et al. Chromosome breakage in the Prader-Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet. 1999;65(2):370–86. doi: 10.1086/302510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pujana MA, Nadal M, Guitart M, et al. Human chromosome 15q11-q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet. 2002;10(1):26–35. doi: 10.1038/sj.ejhg.5200760. [DOI] [PubMed] [Google Scholar]
- 38.Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73(4):898–925. doi: 10.1086/378816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christian SL, Fantes JA, Mewborn SK, et al. Large genomic duplicons map to sites of instability in the Prader-Willi/Angleman syndrome chromosome region (15q11-q13) Hum Mol Genet. 1999;8(6):1025–37. doi: 10.1093/hmg/8.6.1025. [DOI] [PubMed] [Google Scholar]
- 40.Nicholls RD, Knepper JL. Genome organization, function, and imprinting in Prader-Willi and Angelman syndromes. Annu Rev Genomics Hum Genet. 2001;2:153–75. doi: 10.1146/annurev.genom.2.1.153. [DOI] [PubMed] [Google Scholar]
- 41.Butler MG, Bittel DC, Kibiryeva N, et al. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics. 2004;113:565–73. doi: 10.1542/peds.113.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SJ, Miller JL, Kuipers PJ, et al. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20(3):283–90. doi: 10.1038/ejhg.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cassidy SB, Beaudet AL, Knoll JHM, et al. Diagnostic testing for Prader-Willi and Angelman syndromes: Report of ASHG/ACMG test and technology transfer committee. Am J Hum Genet. 1996;58:1085–8. [PMC free article] [PubMed] [Google Scholar]
- 44.Chang CW, Hsu HK, Kao CC, et al. Prenatal diagnosis of Prader-Willi syndrome and Angelman syndrome for fetuses with suspicious deletion of chromosomal region 15q11-q13. Int J Gynecol Obstet. 2014;125(1):18–21. doi: 10.1016/j.ijgo.2013.09.028. [DOI] [PubMed] [Google Scholar]
- 45.Kuwano A, Mutirangura A, Dittrich B, et al. Molecular dissection of the Prader-Willi/Angelman syndrome region (15q11-13) by YAC cloning and FISH analysis. Hum Mol Genet. 1992;1(6):417–25. doi: 10.1093/hmg/1.6.417. [DOI] [PubMed] [Google Scholar]
- 46.Delach JA, Rosengren SS, Kaplan L, et al. Comparison of high resolution chromosome banding and fluorescence in situ hybridization (FISH) for the laboratory evaluation of Prader-Willi syndrome and Angelman syndrome. Am J Med Genet. 1994;52(1):85–91. doi: 10.1002/ajmg.1320520117. [DOI] [PubMed] [Google Scholar]
- 47.Donlon TA, Lalande M, Wyman A, et al. Isolation of molecular probes associated with the chromosome 15 instability in the Prader-Willi syndrome. Proc Natl Acad Sci USA. 1986;83(12):4408–12. doi: 10.1073/pnas.83.12.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Zhang R, Wei J, et al. Rapid diagnosis of imprinting disorders involving copy number variation and uniparental disomy using genome-wide SNP microarrays. Cytogenet Genome Res. 2015;146(1):9–18. doi: 10.1159/000435847. [DOI] [PubMed] [Google Scholar]
- 49.Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with SNP genotyping microarray. Am J Med Genet. 2011;155(4):757–68. doi: 10.1002/ajmg.a.33939. [DOI] [PubMed] [Google Scholar]
- 50.Bittles AH, Black ML. The impact of consanguinity of neonatal and infant health. Early Hum Dev. 2010;86(11):737–41. doi: 10.1016/j.earlhumdev.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Kearney HM, Kearney JB, Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med. 2011;31(4):595–613. doi: 10.1016/j.cll.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Butler MG, Usrey K, Roberts JL, et al. Clinical presentation and microarray analysis of Peruvian children with atypical development and/or aberrant behavior. Genet Res Int. 2014;2014:408516. doi: 10.1155/2014/408516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dittrich B, Robinson WP, Knoblauch H, et al. Molecular diagnosis of the Prader-Willi and Angelman syndromes by detection of parent-of-origin specific DNA methylation in 15q11-13. Hum Genet. 1992;90(3):313–5. doi: 10.1007/BF00220089. [DOI] [PubMed] [Google Scholar]
- 54.Kubota T, Sutcliffe JS, Aradhya S, et al. Validation studies of SNRPN methylation as a diagnostic test for Prader-Willi syndrome. Am J Med Genet. 1996;66(1):77–80. doi: 10.1002/(SICI)1096-8628(19961202)66:1<77::AID-AJMG18>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 55.Muralidhar B, Butler MG. Methylation PCR analysis of Prader-Willi syndrome, Angelman syndrome, and control subjects. Am J Med Genet. 1998;80(3):263–5. [PMC free article] [PubMed] [Google Scholar]
- 56.Ohta T, Gray TA, Rogan PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet. 1999;64(2):379–413. doi: 10.1086/302233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Procter M, Chou LS, Tang W, et al. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006;52(7):1276–83. doi: 10.1373/clinchem.2006.067603. [DOI] [PubMed] [Google Scholar]
- 58.Bittel DC, Kibiryeva N, Butler MG. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet Test. 2007;(4):467–75. doi: 10.1089/gte.2007.0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henkhaus RS, Kim SJ, Kimonis VE, Gold JA, et al. Methylation-specific multiplex ligation-dependent probe amplification and identification of deletion genetic subtypes in Prader-Willi syndrome. Genet Test Mol Biomarkers. 2012;16(3):178–86. doi: 10.1089/gtmb.2011.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geysenbergh B, De Catte L, Vogels A. Can fetal ultrasound result in prenatal diagnosis of Prader-Willi syndrome? Genet Couns. 2011;22(2):207–16. [PubMed] [Google Scholar]
- 61.Bigi N, Faure JM, Coubes C, et al. Prader-Willi syndrome: Is there a recognizable fetal phenotype? Prenat Diagn. 2008;28(9):796–9. doi: 10.1002/pd.1973. [DOI] [PubMed] [Google Scholar]
- 62.Fong BF, De Vries J. Obstetric aspects of the Prader-Willi syndrome. Ultrasound Obstet Gynecol. 2003;21(4):389–92. doi: 10.1002/uog.90. [DOI] [PubMed] [Google Scholar]
- 63.Hiroi H, Kozuma S, Hayashi N, et al. A fetus with Prader-Willi syndrome showing normal diurnal rhythm and abnormal ultradian rhythm on heart rate monitoring. Fetal Diagn Ther. 2000;15(5):304–7. doi: 10.1159/000021026. [DOI] [PubMed] [Google Scholar]
- 64.Denizot S, Boscher C, Le Vaillant C, et al. Distal arthrogryposis and neonatal hypotonia: an unusual presentation of Prader-Willi syndrome (PWS) J Perinatol. 2004;24(11):733–4. doi: 10.1038/sj.jp.7211185. [DOI] [PubMed] [Google Scholar]
- 65.Gross N, Rabinowitz R, Gross-Tsur V, et al. Prader-Willi syndrome can be diagnosed prenatally. Am J Med Genet A. 2015;167A(A):80–5. doi: 10.1002/ajmg.a.36812. [DOI] [PubMed] [Google Scholar]
- 66.Cassidy SB, Lai LW, Erickson RP, et al. Trisomy 15 with loss of the paternal 15 as a cause of Prader-Willi syndrome due to maternal disomy. Hum Mol Genet. 1992;51(4):701–8. [PMC free article] [PubMed] [Google Scholar]
- 67.L’Herminé AC, Aboura A, Brisset S, et al. Fetal phenotype of Prader-Willi syndrome due to maternal disomy for chromosome 15. Prenat Diagn. 2003;23(11):938–43. doi: 10.1002/pd.732. [DOI] [PubMed] [Google Scholar]
- 68.Robinson WP, Langlois S, Schuffenhauer S, et al. Cytogenetic and age-dependent risk factors associated with uniparental disomy 15. Prenat Diagn. 1996;16(9):837–44. doi: 10.1002/(SICI)1097-0223(199609)16:9<837::AID-PD956>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 69.Los FJ, van Opstal D, van den Berg C, et al. Uniparental disomy with and without confined placental mosaicism: A model for trisomic zygote rescue. Prenat Diagn. 1998;18(7):659–68. [PubMed] [Google Scholar]
- 70.Kalousek DK, Vekemans M. Confined placental mosaicism and genomic imprinting. Baillieres Best Pract Res Clin Obstet Gynaecol. 2000;14(4):723–30. doi: 10.1053/beog.2000.0107. [DOI] [PubMed] [Google Scholar]
- 71.Christian SL, Smith AC, Macha M, et al. Prenatal diagnosis of uniparental disomy 15 following trisomy 15 mosaicism. Prenat Diagn. 1996;16(4):323–32. doi: 10.1002/(SICI)1097-0223(199604)16:4<323::AID-PD856>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 72.Hahnemann JM, Vejerslev LO. European collaborative research on mosaicism in CVS (EUCROMIC)—fetal and extrafetal cell lineages in 192 gestations with CVS mosaicism involving single autosomal trisomy. Am J Med Genet. 1997;70(2):179–87. doi: 10.1002/(sici)1096-8628(19970516)70:2<179::aid-ajmg15>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 73.Heidemann S, Plendl H, Vater I, et al. Maternal uniparental disomy 15 in a fetus resulting from a balanced familial translocation t(215)(p11;q11. 2) Prenat Diagn. 2010;30(2):183–5. doi: 10.1002/pd.2436. [DOI] [PubMed] [Google Scholar]
- 74.Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350(9076):485–7. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 75.Bianchi DW. Circulating fetal DNA: Its origin and diagnostic potential-a review. Placenta. 2004;25(Suppl A):S93–S101. doi: 10.1016/j.placenta.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 76.Yaron Y, Jani J, Schmid M, Oepkes D. Current status of testing for microdeletion syndromes and rare autosomal trisomies using cell-free DNA technology. Obstet Gynecol. 2015;126(5):1095–9. doi: 10.1097/AOG.0000000000001091. [DOI] [PubMed] [Google Scholar]
- 77.Committee Opinion No. 640: Cell-Free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126(3):e31–7. doi: 10.1097/AOG.0000000000001051. [DOI] [PubMed] [Google Scholar]
- 78.Kitzman JO, Snyder MW, Ventura M, et al. Noninvasive whole-genome sequencing of a human fetus. Sci Transl Med. 2012;4:137ra76. doi: 10.1126/scitranslmed.3004323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srinivasan A, Bianchi DW, Huang H, et al. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet. 2013;92:167–76. doi: 10.1016/j.ajhg.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levy B, Norwitz E. Non-invasive prenatal aneuploidy testing: Technologies and clinical implication. MLO Med Lab Obs. 2013;45:8, 10, 12. [PubMed] [Google Scholar]
- 81.Nicolaides KH, Sungelaki A, Gil M, et al. Validation of targeted sequencing of single-nucleotide polymorphisms for non-invasive prenatal detection of aneuploidy of chromosomes 13, 18, 21, X, and Y. Prenat Diagn. 2013;33:575–9. doi: 10.1002/pd.4103. [DOI] [PubMed] [Google Scholar]
- 82.Pergament E, Cuckle H, Zimmermann B, et al. Single-nucleotide polymorphism-based non-invasive prenatal testing in a high- and low-risk cohort. Obstet Gynecol. 2014;124:210–8. doi: 10.1097/AOG.0000000000000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: Detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015;212(3):332, e1–9. doi: 10.1016/j.ajog.2014.11.041. [DOI] [PubMed] [Google Scholar]
- 84.Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study. Genet Med. 2011;13:913–20. doi: 10.1097/GIM.0b013e3182368a0e. [DOI] [PubMed] [Google Scholar]
- 85.Bianchi DW, Platt LD, Goldberg JD, et al. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119:890–901. doi: 10.1097/AOG.0b013e31824fb482. [DOI] [PubMed] [Google Scholar]
- 86.Sparks AB, Struble CA, Wang ET, et al. Noninvasive prenatal detection and selective analysis of cell-free DNA obtained from maternal blood: Evaluation for trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;206:319, e1–9. doi: 10.1016/j.ajog.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 87.Nicolaides K, Syngelaki A, Gil M, et al. Prenatal detection of fetal triploidy from cell-free DNA testing in maternal blood. Fetal Diagn Ther. 2014;35:212–7. doi: 10.1159/000355655. [DOI] [PubMed] [Google Scholar]
- 88.Rosignoli L, Tonni G. Should cell-free fetal DNA be included in first trimester screening (FTS) for common trisomy? A possible scenario on 6697 women screened over 10 years. J Eval Clin Pract. 2016 doi: 10.1111/jep.12557. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 89.Porreco RP, Garite TJ, Maurel K, et al. Noninvasive prenatal screening for fetal trisomies 21, 18, 13 and the common sex chromosome aneuploidies from maternal blood using massively parallel genomic sequencing of DNA. Am J Obstet Gynecol. 2014;211(4):365e1–12. doi: 10.1016/j.ajog.2014.03.042. [DOI] [PubMed] [Google Scholar]
- 90.Gil MM, Quezada MS, Revello R, et al. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: Updated meta-analysis. Ultrasound Obstet Gynecol. 2015;45(3):249–66. doi: 10.1002/uog.14791. [DOI] [PubMed] [Google Scholar]
- 91.Zhao C, Tynan J, Ehrich M, et al. Detection of fetal subchromosomal abnormalities by sequencing circulating cell-free DNA from maternal plasma. Clin Chem. 2015;61(4):608–16. doi: 10.1373/clinchem.2014.233312. [DOI] [PubMed] [Google Scholar]
- 92.Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–84. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lau TK, Cheung SW, Lo PS, et al. Re: Non-invasive prenatal testing for fetal chromosomal abnormalities by low-coverage whole-genome sequencing of maternal plasma DNA: review of 1982 consecutive cases in a single center. Ultrasound Obstet Gynecol. 2014;43:254–264. doi: 10.1002/uog.13277. [DOI] [PubMed] [Google Scholar]
- 94.Adams MC, Evans JP, Henderson GE, et al. The promise and peril of genomic screening in the general population. Genet Med. 2016;18(6):593–9. doi: 10.1038/gim.2015.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chan KC, Zhang J, Hui AB, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem. 2004;50(1):88–92. doi: 10.1373/clinchem.2003.024893. [DOI] [PubMed] [Google Scholar]
- 96.Li Y, Zimmermann B, Rusterholz C, et al. Size separation of circulatory DNA in maternal plasma permits ready detection of fetal DNA polymorphisms. Clin Chem. 2004;50(6):1002–11. doi: 10.1373/clinchem.2003.029835. [DOI] [PubMed] [Google Scholar]
- 97.Valderramos SG, Rao RR, Scibetta EW, et al. Cell-free DNA screening in clinical practice: Abnormal autosomal aneuploidy and microdeletion results. Am J Obstet Gynecol. 2016 doi: 10.1016/j.ajog.2016.06.039. pii:S0002-9378(16)30384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gross SJ, Stosic M, McDonald-McGinn DM, et al. Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11. 2 deletion syndrome. Ultrasound Obstet Gynecol. 2016;47(2):177–83. doi: 10.1002/uog.15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tan Y, Gao Y, Lin G, et al. Noninvasive prenatal testing (NIPT) in twin pregnancies with treatment of assisted reproductive techniques (ART) in a single center. Prenat Diagn. 2016;36(7):672–9. doi: 10.1002/pd.4837. [DOI] [PubMed] [Google Scholar]
- 100.Gregg AR, Skotko BG, Benkendorf JL, et al. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016 doi: 10.1038/gim.2016.97. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 101.Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Socienty of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125:653–662. doi: 10.1097/AOG.0000000000000666. [DOI] [PubMed] [Google Scholar]
- 102.Driscoll DA, Gross SJ Professional Practice and Guidelines Committee. First trimester diagnosis and screening for fetal aneuploidy. Genet Med. 2008;10:73–75. doi: 10.1097/GIM.0b013e31815efde8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grody WW, Thompson BH, Gregg AR, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013;15:482–483. doi: 10.1038/gim.2013.47. [DOI] [PubMed] [Google Scholar]
- 104.Gregg AR, Gross SJ, Best RG, et al. ACMG statement on noninvasive prenatal screening for fetal aneuploidy. Genet Med. 2013;15:395–398. doi: 10.1038/gim.2013.29. [DOI] [PubMed] [Google Scholar]
- 105.Vora NL, O’Brien BM. Noninvasive prenatal testing for microdeletion syndromes and expanded trisomies: Proceed with caution. Obstet Gynecol. 2014;123(5):1097–9. doi: 10.1097/AOG.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 106.Inaba Y, Schwartz CE, Bui QM, et al. Early detection of fragile X syndrome: applications of a novel approach for improved quantitive methylation analysis in venous blood and newborn blood spots. Clin Chem. 2014;60(7):963–73. doi: 10.1373/clinchem.2013.217331. [DOI] [PubMed] [Google Scholar]
- 107.Newkirk HL, Bittel DC, Butler MG. Analysis of the Prader-Willi syndrome chromosome region using quantitative microsphere hybridization (QMH) array. Am J Med Genet. 2008;146A(18):2346–54. doi: 10.1002/ajmg.a.32459. [DOI] [PMC free article] [PubMed] [Google Scholar]