

Abstract
The roles of βarrestins in regulating G protein coupling and receptor endocytosis following agonist stimulation of G protein-coupled receptors are well characterised. However, their ability to act on their own as direct modulators or activators of signalling remains poorly characterised. Here, βarrestin2 intrinsic signalling properties were assessed by forcing the recruitment of this accessory protein to vasopressin V1a or V2 receptors independently of agonist-promoted activation of the receptors. Such induction of a stable interaction with βarrestin2 initiated receptor endocytosis leading to intracellular accumulation of the βarrestin/receptor complexes. Interestingly, βarrestin2 association to a single receptor protomer was sufficient to elicit receptor dimer internalisation. In addition to recapitulating βarrestin2 classical actions on receptor trafficking, the receptor activity-independent recruitment of βarrestin2 activated the extracellular signal-regulated kinases. In the latter case, recruitment to the receptor itself was not required since kinase activation could be mediated by βarrestin2 translocation to the plasma membrane in the absence of any interacting receptor. These data demonstrate that βarrestin2 can act as a ‘bonafide' signalling molecule even in the absence of activated receptor.
Keywords: BRET, GPCR, MAPK, oligomerisation, trafficking
Introduction
Agonist-promoted desensitisation of G protein-coupled receptors (GPCRs) is a well-characterised process contributing to signal attenuation. In most cases, GPCRs are rapidly phosphorylated after agonist activation by G protein-coupled receptor kinases (GRKs), leading to the high-affinity binding of βarrestin to the ligand-occupied receptors, which promotes their uncoupling from their cognate heterotrimeric G proteins (Krupnick and Benovic, 1998; Pierce et al, 2002). The interaction of βarrestin with components of the endocytotic machinery next induces the sequestration of desensitised GPCRs from the plasma membrane into intracellular compartments through clathrin-coated pits. Two distinct classes of GPCRs could be distinguished on the basis of their interaction with βarrestin (Oakley et al, 1999, 2000). According to this classification, class A GPCRs are defined as receptors that release βarrestin rapidly following their targeting to clathrin-coated pits, while class B GPCRs are defined as receptors forming stable complexes with βarrestin that is redistributed with receptors to endosomes (this classification should not be confounded with the Frederickson family A, B and C that catalogues GPCRs according to their structural homologies). The stability of interaction with βarrestin has been proposed to influence the recycling efficiency of receptors. Indeed, while investigating the endocytosis/recycling pattern of numerous GPCRs, all class A GPCRs were found to rapidly recycle back to the cell surface following their internalisation, whereas all class B GPCRs were found to be trapped intracellularly with βarrestin and poorly recycled to the plasma membrane (Oakley et al, 1999, 2000; Anborgh et al, 2000). This hypothesis was, however, challenged by the observation that a mutant of the class B vasopressin V2 receptor (V2R), the V2RS363A, was efficiently recycled back to the cell surface despite its stable interaction with βarrestin (Innamorati et al, 2001). This led the authors to propose that the ability of an internalised GPCR to recycle to the plasma membrane is determined by specific molecular determinants intrinsic to the receptor rather than by the stability of its interaction with βarrestin. In addition, other proteins have been proposed to determine the fate of internalised GPCRs (Cao et al, 1999; Whistler et al, 2002). Thus, whether the interaction with βarrestin is sufficient by itself to determine the recycling efficiency of a GPCR remains an open question.
In addition to its role in signal termination and clathrin-mediated internalisation, βarrestin has been recently proposed to act as a scaffolding protein linking GPCRs to mitogen-activated protein kinase (MAPK) (DeFea et al, 2000b; McDonald et al, 2000; Luttrell et al, 2001; Hall and Lefkowitz, 2002; Perry and Lefkowitz, 2002; Tohgo et al, 2002, 2003). Such scaffolding properties of βarrestin are believed to play an important role in controlling the localisation and specific assembly of the MAPK signalling cascade. However, the role of βarrestin as a genuine signalling molecule that can activate the MAPK by itself remains incompletely characterised.
In the present study, we assessed the ability of βarrestin2 to determine, on its own, the signalling efficacy and trafficking patterns of GPCRs and investigated the ability of this scaffolding protein to activate the MAPK signalling cascade independently of ligand-mediated receptor activation.
Results and discussion
The receptor activity-independent recruitment of βarrestin2 induced by AP21967 treatment leads to receptor internalisation according to a ‘class B' GPCR endocytosis pattern
To determine whether βarrestin2 recruitment to a GPCR is sufficient by itself to mediate normal regulatory functions of βarrestin2, we took advantage of a cyclophilin-based approach that permits the translocation of βarrestin2 and its stable interaction with V1aR or V2R vasopressin receptors in the absence of receptor agonist-promoted activation. Such strategy is based on the capacity of a synthetic bivalent dimerising ligand, the ‘heterodimeriser' AP21967, to act as an adaptor to join the cyclophilin FKBP and FRB protein domains (Muthuswamy et al, 1999; Pollock et al, 2000). FKBP was thus fused at the C-terminus of both Myc-V2R and HA-V1aR, whereas FRB was attached at the N-terminus of βarrestin2 already tagged at its C-terminus by a YFP protein (Figure 1A). Fusion to FKBP did not significantly impair the binding or signalling properties of V2R and V1aR as assessed by their ability to promote AVP-stimulated cAMP (V2R) or inositol phosphate (V1aR) accumulation with EC50 values (Table I) similar to those previously reported with nontagged V2R and V1aR (Terrillon et al, 2003). It should also be emphasised that AP21967 did not lead to agonist-independent G protein activation since no adenylyl cyclase or phospholipase C response could be detected following AP21967 treatment of cells coexpressing FRB-βarrestin2-YFP with either V2R-FKBP or V1aR-FKBP, respectively (Table I).
Figure 1.
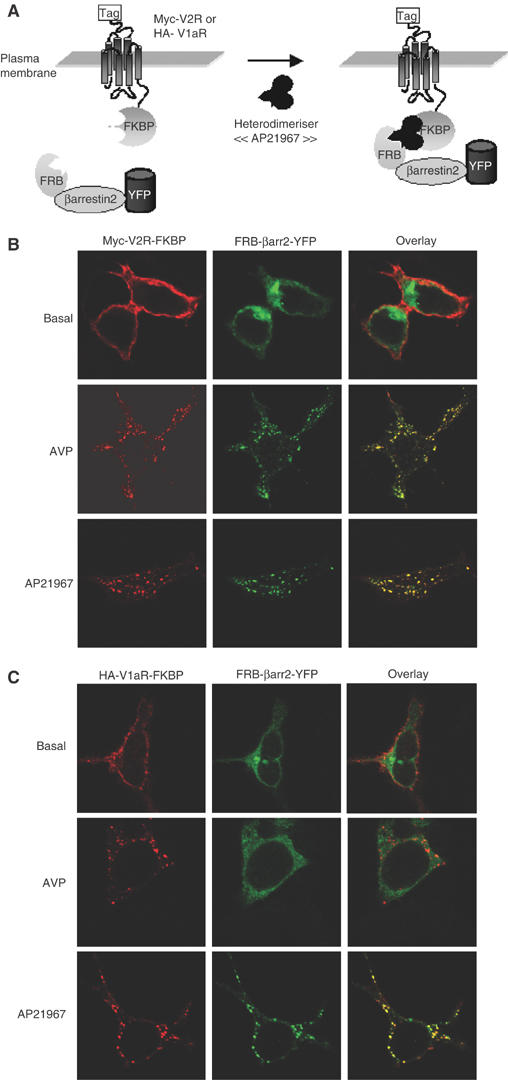
Cellular localisation of FRB-βarrestin2-YFP when coexpressed with either Myc-V2R-FKBP or HA-V1aR-FKBP following AVP or AP21967 treatment. Schematic representation of AP21967-induced translocation of FRB-βarrestin2-YFP to Myc-V2R-FKBP or HA-V1aR-FKBP (A). HEK 293T cells cotransfected with Myc-V2R-FKBP+FRB-βarrestin2-YFP (B) or HA-V1aR-FKBP+FRB-βarrestin2-YFP (C) were incubated with rabbit polyclonal antibody A14 or mouse monoclonal antibody 12CA5, for 1 h at 4°C. Next, cells were treated or not for 30 min at 37°C with the indicated ligand (100 nM AVP or 500 nM AP21967), fixed, permeabilised and labelled with goat anti-rabbit (V2R) or anti-mouse (V1aR) antibody coupled to Texas red. The samples were analysed by confocal laser-scanning microscopy.
Table 1.
Functional properties of FKBP-fused receptors
| Receptor | Fold over basal | EC50 (nM) |
|---|---|---|
| V2R-FKBP | ||
| +AVP | 50.83±15.22 | 0.19±0.04 |
| +AP21967 | 0.96±0.02 | ND |
| V1aR-FKBP | ||
| +AVP | 3.03±0.21 | 0.36±0.07 |
| +AP21967 | 1.01±0.03 | ND |
| HEK 293T cells cotransfected with Myc-V2R-FKBP+FRB-βarrestin2-YFP or HA-V1aR-FKBP+FRB-βarrestin2-YFP were treated or not with increasing concentrations of AVP or 500 nM AP21967 before measuring the accumulation of cAMP (for V2R) or inositol phosphates (for V1aR). Data are expressed as the maximal response induced by AVP or AP21967 treatment (fold over basal) as well as the AVP concentration leading to 50% of the maximal response (EC50). All values correspond to the mean±s.e.m. calculated from at least three independent experiments. ND: not determined. | ||
To establish whether AP21967 can be used to induce the regulated translocation of βarrestin2 to V2R or V1aR and if such interaction between the proteins is sufficient to promote receptor endocytosis, confocal immunofluorescence microscopy was carried out on cells coexpressing FRB-βarrestin2-YFP+Myc-V2R-FKBP or FRB-βarrestin2-YFP+HA-V1aR-FKBP. Under basal conditions, Myc-V2R-FKBP and HA-V1aR-FKBP were observed at the plasma membrane, while FRB-βarrestin2-YFP was detected throughout the cytoplasm (Figure 1B and C). Upon exposure to saturating concentration of the agonist AVP for 30 min, both Myc-V2R-FKBP and HA-V1aR-FKBP were internalised and redistributed from the plasma membrane to endocytotic vesicles. But whereas FRB-βarrestin2-YFP extensively colocalised with Myc-V2R-FKBP in endosomes, it was excluded from those containing HA-V1aR-FKBP (Figure 1B and C). These results are consistent with the previously reported endocytotic properties defining V1aR and V2R as class A and class B GPCRs, respectively (Oakley et al, 1999; Innamorati et al, 2001). Taken with its cytoplasmic localisation under basal conditions, the ability of the FRB-βarrestin2-YFP construct to form a stable complex with Myc-V2R-FKBP but not HA-V1aR-FKBP strongly suggests that fusion to FRB did not significantly impair the biological properties of βarrestin2. Treatment with 500 nM AP21967 led to the internalisation of Myc-V2R-FKBP and HA-V1aR-FKBP (Figure 1B and C) but, in contrast to AVP stimulation, both receptors extensively colocalised with FRB-βarrestin2-YFP in endocytotic vesicles. This indicates that the addition of AP21967 is sufficient to induce βarrestin2 recruitment to V1aR or V2R and that βarrestin2 can engage the endocytotic machinery even in the absence of agonist-promoted activation of the receptors. This last observation is consistent with previous reports indicating that covalent fusion of βarrestin1 to the carboxyl terminal of neurokin NK1 receptor (Martini et al, 2002) or β1-adrenergic receptor (Shiina et al, 2000) promotes constitutive internalisation. More interestingly, V1aR, which behaves as a class A GPCR upon agonist activation, is converted into a class B GPCR by treatment with AP21967, reflecting the forced stable interaction between the receptor and βarrestin2.
Characteristics of V2R-FKBP and V1aR-FKBP internalisation when promoted by the receptor activity-independent recruitment of βarrestin2
The extent and kinetics of internalisation promoted by AP21967 were assessed by ELISA. As shown in Figure 2, AP21967 elicited a dose-dependent internalisation of the FKBP-tagged receptors with half-maximal internalisation achieved at similar concentration for both receptors. In contrast, AP21967 did not promote internalisation of Myc-V2R or HA-V1aR not fused to FKBP. When considering the rate of endocytosis, treatment with AVP (100 nM) led to a rapid internalisation of V2R-FKBP and V1aR-FKBP, the rate of V2R internalisation being somewhat slower than that of V1aR (internalisation half-time of 11.8±2.0 and 4.9±0.2 min, respectively) (Figure 3A and B). In contrast, Myc-V2R-FKBP and HA-V1aR-FKBP had similar rates of internalisation following treatment with AP21967 (500 nM) (internalisation half-time of 21.6±0.7 and 23.2±3.8 min, respectively). Although significantly slower than that observed following AVP stimulation, the rate of internalisation promoted by 500 nM AP21967 had reached its maximum since raising the concentration to 25 μM did not lead to further acceleration of Myc-V2R-FKBP endocytosis (data not shown). This slower kinetics of receptor internalisation promoted by the heterodimeriser is not the reflection of distinct endocytotic processes since no additivity between the extent of internalisation induced by AVP (100 nM) and AP21967 (25 μM) was observed (Figure 3C), indicating that the difference in kinetics between AVP- and AP21967-promoted endocytosis resulted from a lower efficacy of the heterodimeriser to initiate the endocytosis process (see below).
Figure 2.
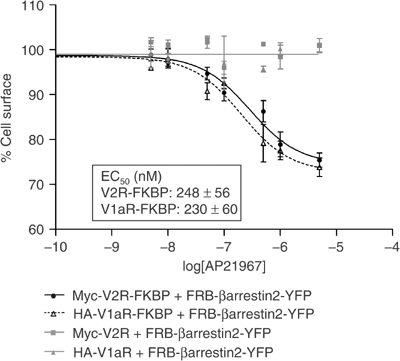
Quantitative assessment of Myc-V2R-FKBP and HA-V1aR-FKBP internalisation following AP21967-induced recruitment of FRB-βarrestin2-YFP. HEK 293T cells coexpressing FRB-βarrestin2-YFP along with either Myc-V2R-FKBP, HA-V1aR-FKBP, Myc-V2R or HA-V1aR were treated or not with increasing concentrations of AP21967 for 30 min at 37°C. The cell surface Myc epitope-tagged V2R or HA epitope-tagged V1aR was detected by ELISA before and after AP21967 treatment and expressed as % of control. All values correspond to the mean±s.e.m. calculated from three independent experiments.
Figure 3.
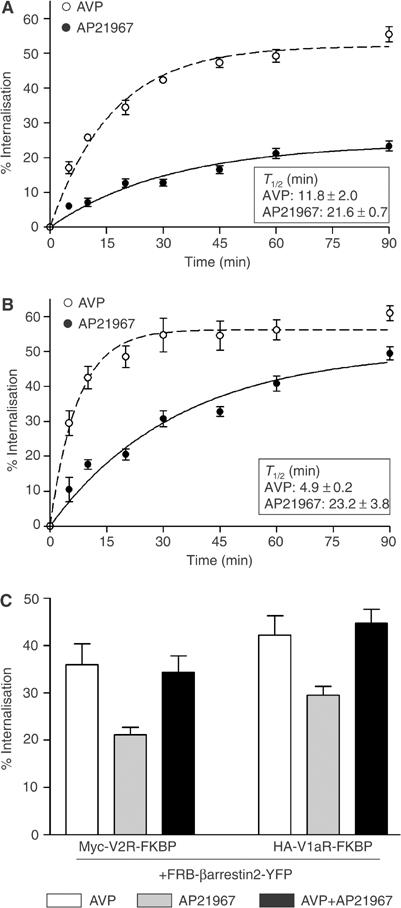
Kinetics of Myc-V2R-FKBP and HA-V1aR-FKBP endocytosis. HEK 293T cells coexpressing Myc-V2R-FKBP+FRB-βarrestin2-YFP (A) or HA-V1aR-FKBP+FRB-βarrestin2-YFP (B) were treated or not with 100 nM AVP or 500 nM AP21967 at 37°C for the indicated periods of time. In (C), the cells were treated for 30 min at 37°C with 100 nM AVP and 25 μM AP21967 added separately or in combination. The cell surface Myc epitope-tagged V2R and HA epitope-tagged V1aR were detected by ELISA. The extent of internalisation was determined by measuring the cell surface receptor before and after ligand treatment and expressed as the loss of cell surface expression (% of control). All values correspond to the mean±s.e.m. calculated from three independent experiments.
The interaction of βarrestin2 with either V1aR or V2R is sufficient to promote the internalisation of the V1a/V2R heterodimer independently of ligand-mediated receptor activation
Several studies have suggested that GPCR homo- and heterodimerisation could play important roles in regulating receptor functions (Angers et al, 2002; George et al, 2002). We have recently reported that V1aR and V2R can be internalised as a stable heterodimer with trafficking properties determined by the identity of the activated protomer and its ability to stably interact with βarrestin2 (Terrillon et al, 2004). Although strong evidence supports the idea that this cointernalisation of both receptors is dictated by their heterodimerisation, the possibility that it could result from signalling crosstalk between one activated receptor and the cointernalised GPCR could not be entirely excluded. To demonstrate directly that the cotrafficking of V1aR and V2R truly results from their heterodimerisation, cells were transfected with FRB-βarrestin2-YFP in the presence of the two receptors, with only one being fused to FKBP. Following AP21967 treatment of cells coexpressing HA-V1aR with Myc-V2R-FKBP and FRB-βarrestin2-YFP, both receptors were internalised and extensively colocalised in βarrestin2-positive endosomes (Figure 4A). In contrast, when expressed alone with FRB-βarrestin2-YFP, HA-V1aR remained at the cell surface (see Figure 2). Reciprocally, following coexpression with HA-V1aR-FKBP+FRB-βarrestin2-YFP, AP21967 treatment promoted Myc-V2R endocytosis in intracellular vesicles where the three proteins colocalised (Supplementary Figure 1). Quantitative assessment of the AP21967-promoted cointernalisation confirmed that βarrestin2 recruitment to one protomer is sufficient to induce the internalisation of the V1aR/V2R heterodimer in the absence of agonist-induced activation of the receptors (Figure 4B). The selectivity of this cointernalisation was confirmed by the fact that AP21967-promoted internalisation of HA-V1aR-FKBP was not accompanied by the endocytosis of a coexpressed Myc opioid receptor (Figure 4B). The fact that βarrestin2 interaction with either V2R or V1aR is sufficient to promote the specific internalisation of both receptors in the absence of their agonist-promoted activation clearly demonstrates that the two receptors are not internalised as separate entities but rather as a stable heterodimeric complex. It also confirms our previous observation that βarrestin2 recruitment to a single protomer is sufficient to support dimer internalisation (Terrillon et al, 2004).
Figure 4.

Internalisation of the V2R/V1aR heterodimer following the translocation of FRB-βarrestin2-YFP to a single FKBP-fused receptor. HEK 293T cells transfected with FRB-βarrestin2-YFP in the presence of Myc-V2R-FKBP+HA-V1aR (A) were incubated with rabbit polyclonal anti-Myc antibody A14 and mouse monoclonal anti-HA antibody 12CA5 for 1 h at 4°C. After treatment with 500 nM AP21967 for 30 min at 37°C, cells were fixed, permeabilised and labelled with Texas red-conjugated goat anti-rabbit and Alexa 633-conjugated goat anti-mouse antibodies to visualise V2R and V1aR, respectively. In (B), the internalisation extent of coexpressed HA epitope-tagged V1aR and Myc epitope-tagged receptor (V2R or δOR) was determined by measuring the cell surface receptor before and after AP21967 treatment by ELISA and expressed as the loss of cell surface expression (% of control). All values correspond to the mean±s.e.m. calculated from at least three independent experiments.
βarrestin2 can form stable oligomeric complexes with GPCR dimers
To determine whether the AP21967-promoted colocalisation of βarrestin2 with V1aR and V2R truly reflects the formation of a stable oligomer between βarrestin2 and the V1aR/V2R heterodimer, bioluminescence resonance energy transfer (BRET) studies allowing the detection of a trimeric complex between receptor dimers and βarrestin2 were performed in living cells. For this purpose, V1aR or V2R fused at its C-terminus to Rluc (V1aR-Rluc or V2R-Rluc) (Terrillon et al, 2003) was coexpressed with either HA-V1aR-FKBP+FRB-βarrestin2-YFP or Myc-V2R-FKBP+FRB-βarrestin2-YFP, respectively. The occurrence of stable intermolecular interactions between βarrestin2 and both V1aR and V2R homo- and heterodimers was assessed by determining the transfer of energy between Rluc fused to the protomer lacking the FKBP moiety and YFP fused to FRB-βarrestin2 (Figure 5A). To promote βarrestin2 recruitment, attached cells were first treated with 100 nM AVP or 500 nM AP21967 for 90 min at 37°C. The occurrence of BRET was then assessed following extensive washing and resuspension of the cells. The inclusion of washing steps before energy transfer measurement is expected to allow the discrimination between class B GPCRs that form a very stable interaction with βarrestin2 and thus would lead to strong BRET signals and class A GPCRs that transiently interact with βarrestin2 thus producing a weaker signal. In agreement with this assumption, when the class B V2R-Rluc was coexpressed with FRB-βarrestin2-YFP, activation with AVP promoted its stable association with FRB-βarrestin2-YFP leading, to robust BRET signals (Figure 5B). For the class A V1aR-Rluc, the significantly weaker AVP-promoted BRET signals reflected the transient nature of its interaction with FRB-βarrestin2-YFP (Figure 5C). It should be emphasised in the particular case of the V1aR-V2R heterodimer that, whereas the nonselective activation of the two protomers would result in a transient interaction of βarrestin2 with the class A V1aR, it would lead to a stable interaction of βarrestin2 with the class B V2R. It follows that the interaction with FRB-βarrestin2-YFP will not be symmetrical for the two heterodimer configurations (i.e. V2R-FKBP+V1aR-Rluc and V1aR-FKBP+V2R-Rluc). Given that the extent of BRET is dependent on the distance and the relative orientation between the energy donor and acceptor, one would expect the extent of BRET to be different for the two heterodimer orientations. The strong BRET observed for the V1aR-FKBP+V2R-Rluc combination most likely results from the direct stable interaction of FRB-βarrestin2-YFP with V2R-Rluc and a weaker BRET cross-signal between V2R-Rluc and FRB-βarrestin2-YFP that transiently interacts with V1aR-FKBP. The weaker BRET observed for the reverse combination (V2R-FKBP+V1aR-Rluc) would result from the crossinteraction between V1aR-Rluc and FRB-βarrestin2-YFP stably associated with V2R-FKBP and the transient direct interaction between V1aR-Rluc and FRB-βarrestin2-YFP.
Figure 5.
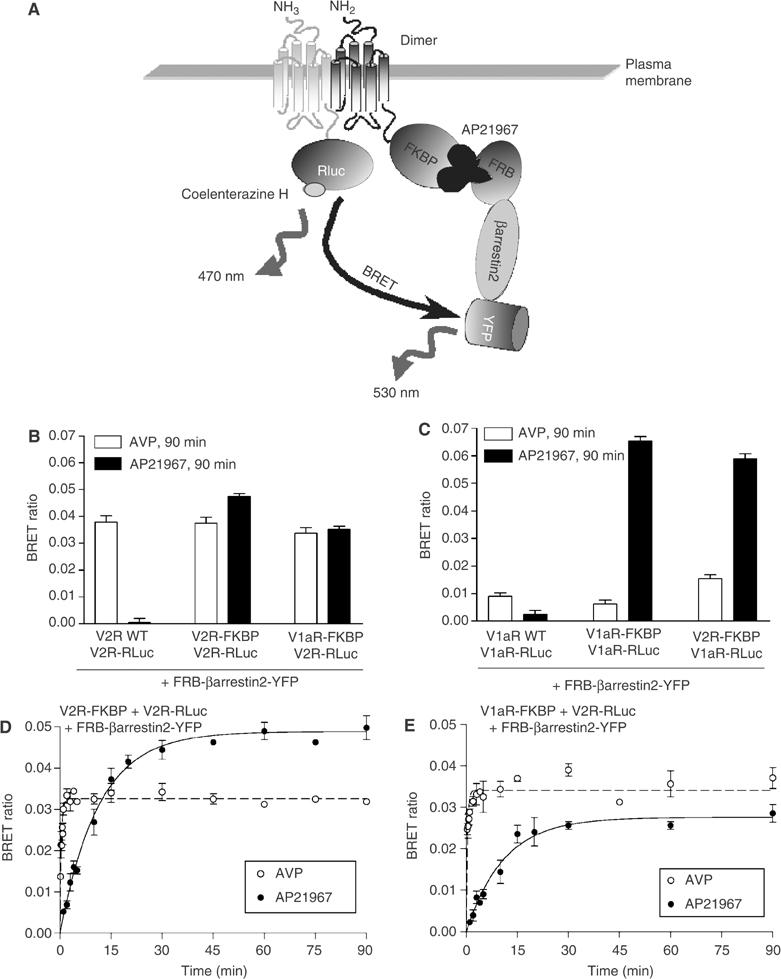
Monitoring FRB-βarrestin2-YFP stable association with V1aR and V2R homo- and heterodimers by BRET. Schematic representation of the experimental paradigm used to monitor the AP21967-promoted recruitment of βarrestin2 to V1aR and V2R homo- and heterodimers using BRET in cells coexpressing FRB-βarrestin2-YFP and a combination of receptors harbouring the FKBP moiety (V1aR-FKBP or V2R-FKBP) and the Renilla luciferase (V1aR-Rluc or V2R-Rluc) (A). HEK 293T cells transfected with FRB-βarrestin2-YFP+FKBP-fused receptor in the presence of V2R-Rluc (B, D, E) or V1aR-Rluc (C) were treated or not with 100 nM AVP or 500 nM AP21967 at 37°C for the indicated periods of time. Following extensive washing, cells were detached and BRET measured. All values correspond to the mean±s.e.m. calculated from at least three independent experiments.
When considering treatment of cells with AP21967, the receptor activity-independent translocation of FRB-βarrestin2-YFP to one protomer fused to FKBP within the V1aR and V2R homo- and heterodimer led to the detection of robust BRET signals between FRB-βarrestin2-YFP and the second protomer fused to Rluc (Figure 5B and C). In contrast, no BRET was detected when V2R-Rluc or V1aR-Rluc was coexpressed with either V2R or V1aR not fused to FKBP in the presence of FRB-βarrestin2-YFP (Figure 5B and C). These data clearly show that the interaction of βarrestin2 with only one protomer within a GPCR dimer is not only sufficient to support the internalisation of the whole dimeric assembly, but that it also leads to the engagement of βarrestin2 in the formation of stable oligomeric complexes involving at least two GPCR molecules.
As indicated above, the kinetics of AP21967-promoted endocytosis was somewhat slower than that induced by AVP (see Figure 3A and B). This could result in part from a slower rate of βarrestin2 translocation to the receptors following AP21967 treatment as compared to that induced by AVP. To test this hypothesis, the kinetics of βarrestin2 interaction with V1aR and V2R homo- and heterodimer was assessed by BRET following cell treatment with AVP or AP21967. As shown in Figure 5D, when the V2R homodimer was considered (V2R-FKBP+V2R-Rluc+FRB-βarrestin2-YFP), AVP stimulation promoted the formation of a stable oligomeric complex with a half-time of 0.25±0.06 min as compared to 8.1±1.0 min upon AP21967 treatment (Table II). A similar difference in βarrestin2 translocation was observed for the V2R/V1aR heterodimer (V1aR-FKBP+V2R-Rluc+FRB-βarrestin2-YFP) since half-times of 0.25±0.02 and 7.8±0.6 min were obtained with AVP and AP21967 treatment, respectively (Figure 5E and Table II). Interestingly, AP21967 treatment induced βarrestin2 recruitment to V1aR-Rluc engaged in the assembly of either heterodimers with V2R-FKBP or homodimers with V1aR-FKBP with similar slow kinetics (half-time of 8.6±0.6 and 8.6±1.5 min, respectively) (Table II). Taken together, these results indicate that the rate of βarrestin2 recruitment to the receptor homo- and heterodimers was considerably reduced when promoted by AP21967 as compared to AVP, and may explain, in part, the slower endocytotic process observed following AP21967 treatment. In addition, one cannot exclude the possibility that the βarrestin2–receptor complex may not reach its optimal internalisation-prone conformation following the artificial AP21967-induced recruitment of βarrestin2. Indeed, in the case of agonist-promoted activation, the conformational change of βarrestin resulting from receptor phosphorylation by GRKs is believed to contribute to the fast recruitment of βarrestin to the receptor (Hirsch et al, 1999; Vishnivetskiy et al, 1999), and to allow the high-affinity binding of the endocytotic machinery components to the C-terminal domain of βarrestin2 (Goodman et al, 1997; Krupnick et al, 1997; Laporte et al, 2000, 2002; Kim and Benovic, 2002) and possibly to the activated receptor itself (Fan et al, 2001; Diviani et al, 2003).
Table 2.
Half-time of FRB-βarrestin2-YFP recruitment to V1aR and V2R homo- and heterodimers upon AVP or AP21967 treatment
|
T1/2 (min) |
|||
|---|---|---|---|
| Dimers | AVP | AP21967 | |
| V1aR-FKBP | V1aR-RLuc | ND | 8.6±1.5 |
| V1aR-FKBP | V2R-Rluc | 0.25±0.02 | 7.8±0.6 |
| V2R-FKBP | V1aR-RLuc | ND | 8.6±0.6 |
| V2R-FKBP | V2R-Rluc | 0.25±0.06 | 8.1±1.0 |
| ND: not determined. | |||
Stable interaction between receptor and βarrestin2 determines the recycling pattern
The observation that the AP21967 treatment promotes a stable interaction between V1aR and βarrestin2 while AVP stimulation only induces the formation of a transient complex provided an excellent tool to assess directly the hypothesis that the stability of the interaction between the receptor and βarrestin determines the receptor recycling properties. Thus, the recycling patterns of V2R and V1aR following AVP- and AP21967-induced endocytosis were assessed by measuring the cell surface reappearance of Myc-V2R-FKBP and HA-V1aR-FKBP by ELISA. As expected, after AVP-promoted internalisation, the class B Myc-V2R-FKBP was unable to efficiently recycle to the cell surface even 120 min after agonist removal (Figure 6A). Indeed, following AVP removal (time 0), some of the activated receptors remaining at the cell surface undergo internalisation leading to a decrease in the receptor number at the cell surface that cannot be compensated by the inefficient recycling during the first 40 min. The slow recycling could overcome this late endocytotic process only 2 h after AVP removal barely allowing to recover the cell surface expression level that was reached at time 0, thus leading to a net recycling of 0% 120 min after AVP removal. In contrast, the efficient recycling of the class A HA-V1aR-FKBP rapidly predominated over the late internalisation following AVP removal, leading to an increase in the number of receptors at the cell surface as soon as 40 min and to a net recycling of 14±9 and 59±18% after 40 and 120 min, respectively (Figure 6B). When the internalisation was promoted by AP21967 treatment, both Myc-V2R-FKBP and HA-V1aR-FKBP were trapped intracellularly (Figure 6) and no recycling could be observed. This observation confirmed that the AP21967-promoted stable interaction with βarrestin2 is sufficient to impair the recycling properties of a class A GPCR and clearly demonstrates that the recycling properties of a given GPCR are directly determined by the stability of its association with βarrestin2. Consistent with this notion, when AVP and AP21967 were added together, the AP21967-promoted stabilisation of interaction between FRB-βarrestin2-YFP and the AVP-stimulated receptors completely inhibited the recycling that was observed following AVP stimulation alone. These results nonambiguously confirm the hypothesis formulated by Oakley et al (1999, 2000) but may appear to contradict the observation that a mutant form of V2R (V2RS363A), which was proposed to bind stably to βarrestin, recycles efficiently to the plasma membrane (Innamorati et al, 2001). In the latter study, the stability of the interaction between V2RS363A and βarrestin2 was inferred from their colocalisation in endosomes following AVP stimulation. However, the V2RS363A recycling observed could result from a partial reduction of its affinity for βarrestin2 that could allow late dissociation in the endosomes and receptor recycling.
Figure 6.
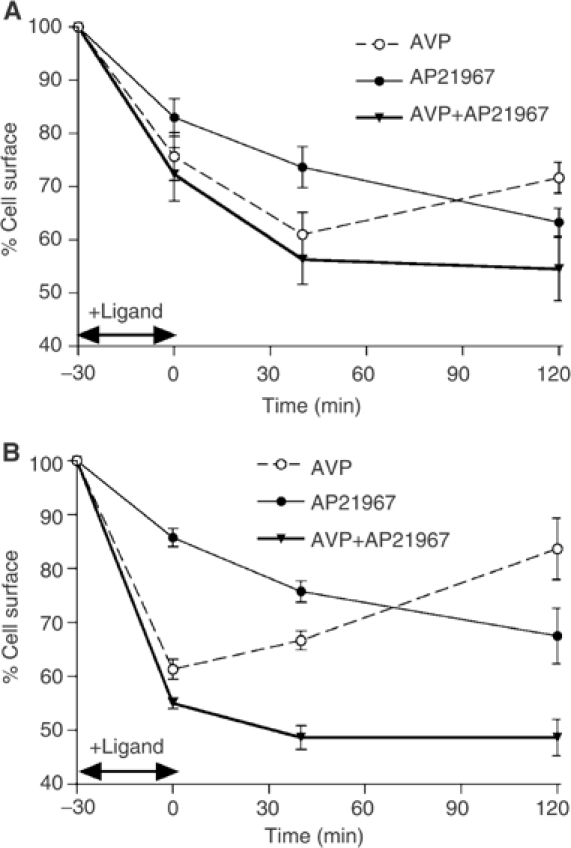
Recycling efficiency of Myc-V2R-FKBP and HA-V1aR-FKBP following AVP- or AP21967-promoted internalisation. HEK 293T cells cotransfected with Myc-V2R-FKBP+FRB-βarrestin2-YFP (A) or HA-V1aR-FKBP+FRB-βarrestin2-YFP (B) were treated for 30 min at 37°C with 100 nM AVP or 500 nM AP21967 added separately or in combination, to promote the internalisation of the receptors. The ligand remaining after treatment was removed by cold PBS and acidic washes. Fresh media were then added and cells reincubated at 37°C for 40 or 120 min to allow the recycling of the receptors. The differentially epitope-tagged receptors present at the cell surface were assessed by ELISA at different periods of time following the addition and the removal of the ligands and expressed as % of the basal level. All values correspond to the mean±s.e.m. calculated from at least three independent experiments.
The receptor activity-independent recruitment of βarrestin2 leads to the activation of extracellular signal-regulated kinases 1 and 2
Besides their roles in GPCR trafficking, mounting evidence suggests that βarrestins may also contribute to GPCR signalling by acting as scaffolding proteins that bring MAPK cascade components in close vicinity of the agonist-occupied receptors (DeFea et al, 2000b; McDonald et al, 2000; Luttrell et al, 2001; Hall and Lefkowitz, 2002; Perry and Lefkowitz, 2002; Tohgo et al, 2002, 2003). In several instances, this MAPK activation has been proposed to be independent of any G protein engagement (Azzi et al, 2003; Wei et al, 2003). However, whether βarrestin2 translocation is sufficient, on its own, to activate extracellular signal-regulated kinases (ERKs) in the absence of any ligand-promoted activation of the receptor itself has never been investigated. To address this question, we studied the effects of AVP- and AP21967-promoted βarrestin2 recruitment on ERK1/2 phosphorylation. As shown in Figure 7A, when Myc-V2R-FKBP and HA-V1aR-FKBP were separately coexpressed with FRB-βarrestin2-YFP, AVP stimulation led to a time-dependent increase in ERK1/2 activity that reached its maximum after 2 min. The AP21967-promoted translocation of FRB-βarrestin2-YFP to either FKBP-fused vasopressin receptors also led to robust ERK1/2 activation (Figure 7B), indicating that the recruitment of βarrestin2 to the receptor in the absence of its activation by an agonist is sufficient to activate the MAPK pathway. As was the case for the AP21967-induced βarrestin2 recruitment and endocytosis (see Figures 4 and 5), the kinetics of ERK1/2 activation by the heterodimeriser was significantly slower than that induced by AVP, reaching its maximum at 20–30 min. Taken with the fact that AP21967 treatment did not promote adenylyl cyclase or phospholipase C response (see Table I), these results clearly support the existence of a G protein-independent signalling pathway that only requires the translocation of the scaffolding protein βarrestin2. This conclusion is consistent with recent findings that receptor ligands that cannot activate classical G protein-mediated signalling are nevertheless able to activate the MAPK pathway in a βarrestin-dependent manner (Azzi et al, 2003; Wei et al, 2003).
Figure 7.
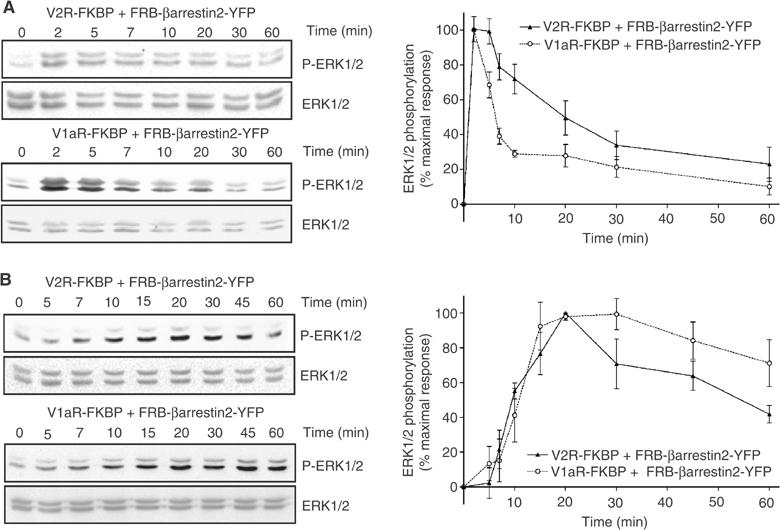
Time course of AVP- and AP21967-induced phosphorylation of ERK1/2. HEK 293T cells cotransfected with Myc-V2R-FKBP+FRB-βarrestin2-YFP or HA-V1aR-FKBP+FRB-βarrestin2-YFP were treated with 100 nM AVP (A) or 500 nM AP21967 (B) at 37°C for the indicated periods of time. ERK1/2 activation was determined by immunoblotting with a phospho-ERK1/2-specific antibody (P-ERK1/2). Expression levels of ERK1/2 were controlled using an antibody directed against the total kinase population (ERK1/2). The levels of phosphorylated ERK1/2 are expressed as the percentage of the maximal increase of phosphorylation obtained during the time course. The graphs represent the mean±s.e.m. calculated from at least six independent experiments.
Plasma membrane translocation of cytosolic βarrestin2 is sufficient on its own to promote signalling pathway activation leading to ERK1/2 phosphorylation
The observation that βarrestin2 translocation to the receptors is sufficient to lead to ERK1/2 phosphorylation begs the question whether βarrestin2 interaction with receptors is required to promote ERK1/2 activation or if βarrestin2 translocation to the plasma membrane is sufficient. To answer this question, FRB-βarrestin2-YFP was coexpressed with a chimeric fusion protein (MyrFKBP2) containing an amino-terminal myristoylation signal (Myr) and two copies of FKBP (FKBP2). Due to the presence of the myristoylation signal, FKBP2 would be targeted to the cytoplasmic face of membranes and AP21967 treatment should promote the specific recruitment of βarrestin2 to the cell surface in the absence of interaction with any receptor. As expected, when FRB-βarrestin2-YFP was coexpressed with MyrFKBP2, AP21967 treatment led to the translocation of FRB-βarrestin2-YFP to the plasma membrane (Figure 8A). This conditional plasma membrane translocation of FRB-βarrestin2-YFP in response to AP21967 led to robust time-dependent ERK1/2 activation that reached its maximum at 15 min (Figure 8B). The selectivity of this effect was demonstrated by the absence of ERK1/2 activation following AP21967 treatment of cells expressing either MyrFKBP2 or FRB-βarrestin2-YFP separately (data not shown). These results clearly demonstrate that βarrestin2 translocation to the plasma membrane is sufficient on its own to promote the activation of signalling pathway leading to ERK1/2 phosphorylation. Moreover, given that, following its artificial AP21967-induced dimerisation with MyrFKBP2, FRB-βarrestin2-YFP remains localised at the plasma membrane and is not internalised into endosomal vesicles (see Figure 8A), these results discredit the hypothesis that endocytosis of βarrestin2–GPCR complex is a prerequisite for ERK1/2 activation. Recruitment to the plasma membrane has also been demonstrated to constitute a key step for the activation of other intracellular signalling molecules such as Akt (Burgering and Coffer, 1995; Kohn et al, 1996; Andjelkovic et al, 1997). Two general mechanisms could be considered to explain how the simple recruitment of βarrestin2 at the plasma membrane can lead to the MAPK activation. First, since a fraction of βarrestin was suggested to exist as a preformed complex with the nonreceptor tyrosine kinase c-Src (Luttrell et al, 1999; Barlic et al, 2000; DeFea et al, 2000a; Miller et al, 2000), one could propose that βarrestin translocation brings c-Src to the plasma membrane where it could induce the Ras-dependent activation of the ERK pathway (Luttrell et al, 1996). Second, as a scaffolding unit for some of the ERK kinase components (DeFea et al, 2000b; Luttrell et al, 2001; Tohgo et al, 2002, 2003), βarrestin could bring a preassembled complex to the plasma membrane where the ERK signalling cascade could be completed by a partner(s) that is confined to this compartment by its binding to cytoskeletal elements such as filamin (Marti et al, 1997). In any case, it would appear that in the context of βarrestin-dependent MAPK activation, GPCR may primarily serve as a means to bring βarrestin to the plasma membrane and may not be required as an integral part of the signalling module.
Figure 8.
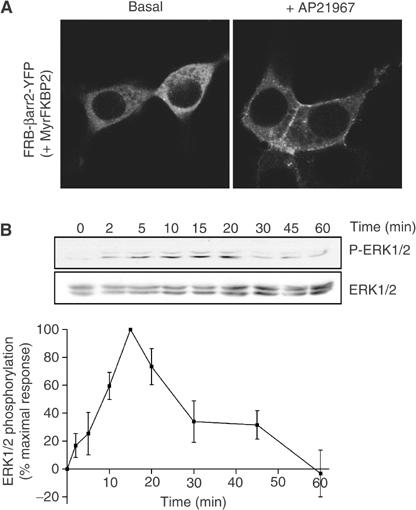
Conditional plasma membrane translocation of FRB-βarrestin2-YFP to myristoylated FKBP2 and ERK1/2 activation. HEK 293T cells cotransfected with MyrFKBP2+FRB-βarrestin2-YFP were treated with 500 nM AP21967 at 37°C for the indicated periods of time, so as to induce the plasma membrane translocation of FRB-βarrestin2-YFP. Both the cellular localisation of FRB-βarrestin2-YFP (A) and the ERK1/2 activation (B) were then determined by confocal microscopy and immunoblotting with a phospho-ERK1/2-specific antibody (P-ERK1/2), respectively. Expression levels of ERK1/2 were controlled using an antibody directed against the total kinase population (ERK1/2). The levels of phosphorylated ERK1/2 are expressed as the percentage of the maximal increase of phosphorylation obtained during the time course. The graphs represent the mean±s.e.m. calculated from six independent experiments.
Although βarrestin recruitment to the plasma membrane appears to be sufficient to scaffold the required signalling elements leading to ERK1/2 activation, the association with specific receptors could determine the efficiency of βarrestin2-mediated activation of ERK1/2 and regulates its spatiotemporal pattern. For example, class B GPCRs have been proposed to activate a βarrestin-bound pool of ERK1/2 more efficiently than class A GPCRs, thus leading to a cytosolic retention of active ERK1/2 and a diminished transcriptional response (Tohgo et al, 2003). Such a cytosolic retention of βarrestin-dependent activated ERK1/2 also leads to a more sustained signalling, whereas nuclear translocation of activated ERK1/2 results in a transient response (Ahn et al, 2004a). When considering the present study, the ERK1/2 activity is persistent following AP21967-mediated βarrestin2 recruitment to V1aR or V2R (see Figure 7B) but more transient following the receptor-independent localisation of βarrestin2 to the plasma membrane using MyrFKBP2 (see Figure 8B). One could thus assume that the AP21967-mediated receptor–βarrestin2 interaction would lead to a cytosolic retention of the complex containing activated ERK1/2, whereas, in the absence of receptor interaction, βarrestin2 may not achieve its optimal conformation for persistent scaffolding of phospho-ERK1/2, resulting in their rapid translocation to the nucleus and subsequent dephosphorylation. Alternatively, it is possible that the activated ERK1/2 remained associated with βarrestin2 translocated to MyrFKBP2 at the plasma membrane but that the conformation reached by βarrestin2 would not allow it to shield the phosphorylated ERKs from cytosolic phosphatases. Additional studies will be required to determine the precise subcellular localisation of activated ERK1/2 following βarrestin2 recruitment to the receptor and translocation to the plasma membrane.
Whether our observation can be generalised to βarrestin1 remains to be investigated. Given that βarrestin1 has also been proposed to act as a scaffolding protein for c-Src (Luttrell et al, 1999; Barlic et al, 2000; DeFea et al, 2000a; Miller et al, 2000) and components of the ERK pathway (DeFea et al, 2000b; Tohgo et al, 2002), one could predict that βarrestin1 translocation to the plasma membrane could also be sufficient to induce the ERK1/2 activation. However, the recent finding that, at physiological levels, βarrestin1 but not βarrestin2 acts as a negative regulator of the angiotensin AT1A receptor-mediated ERK1/2 phosphorylation (Ahn et al, 2004b) raises the possibility of distinct regulatory influences for the two molecules.
Our results clearly show that the interaction between receptors and βarrestin2 is sufficient on its own to mediate the normal regulatory function of this scaffolding adaptor protein, independently of the classical conformational changes associated with agonist-promoted receptor activation. This represents a proof of principle that allosteric regulation of the βarrestin–GPCR interaction constitutes a mean to control specific aspects of GPCR signalling independently of receptor ligand binding. The observation that translocation of βarrestin2 to the plasma membrane in the absence of direct interaction with the receptor is sufficient to activate MAPK further demonstrates that βarrestins are genuine signalling molecules that have the potential to act independently of GPCR. Whether βarrestin/receptor interaction or βarrestin translocation to the plasma membrane could be amenable to allosteric pharmacological manipulation in native environment where fusion proteins cannot be used remains an open question that will deserve further investigation.
Materials and methods
Heterodimeriser-induced βarrestin2 recruitment to receptor/plasma membrane
Heterodimerisation of FRB-βarrestin2 and receptor-FKBP constructs was induced by cell treatment with the heterodimeriser AP21967, a chemically modified derivative of rapamycin (see Figure 1A). Such chemical inducer of dimerisation is a cell-permeant organic molecule with two separate motifs, binding with high affinity FKBP and FRB respectively (http://www.ariad.com/regulationkits). βarrestin2 translocation to plasma membrane in a receptor-independent manner was induced by the AP21967-promoted heterodimerisation of βarrestin2 with a chimeric fusion protein containing an amino-terminal myristoylation signal followed by two copies of FKBP (MyrFKBP2).
Immunofluorescence microscopy
At 48 h after transfection, HEK 293T cells were incubated with rabbit polyclonal antibody A14 and/or mouse monoclonal antibody 12CA5 for 1 h at 4°C. Following washes at 4°C, cells were treated for 30 min at 37°C in the presence or absence of the appropriate ligand (100 nM AVP or 500 nM AP21967). Cells were then washed, fixed and permeabilised before the incubation with a secondary goat anti-rabbit antibody coupled to Texas red (for the Myc-V2R-FKBP) or goat anti-mouse antibody coupled to Texas red (for the HA-V1aR-FKBP) for 30 min at room temperature (RT). For experiments where both FKBP-fused and nonfused receptors were coexpressed, a secondary goat anti-rabbit antibody coupled to Texas red (for the Myc-V2R) and a goat anti-mouse antibody coupled to Alexa 633 (for the HA-V1aR) were used in combination. The samples were analysed by confocal laser-scanning microscopy utilising a Leica TCS SP1 confocal microscope, and colocalisation was performed by overlay of the images using the Leica Confocal Software LCS (Heidelberg, Germany). Excitation and emission filters for the different labelled dyes were as follows: YFP (green): λex=488 nm, λem=540/25 nm; Texas red (red): λex=568 nm, λem=610/30 nm; Alexa 633 (blue): λex=633 nm, λem=705/45 nm.
ELISA
At 48 h post-transfection, cells were treated or not with the appropriate ligand (AVP or AP21967) for 30 min at 37°C. After two washes, cells were fixed and incubated in blocking solution (PBS/0.2% BSA) for 15 min at RT. Cells were kept at RT for all subsequent steps. Cells were then incubated with anti-Myc (9E10) or anti-HA (12CA5) antibodies for 30 min. After three PBS/0.2% BSA washes, cells were incubated with anti-mouse/horseradish peroxidase (HRP) conjugate (Amersham Pharmacia Biotech, Little Chalfont, UK). After extensive washing, the HRP substrate o-phenylenediamine dihydrochloride (Sigma, St Louis, MO) was added and optical density (OD) was measured at 492 nm. For each experiment, mock conditions corresponding to cells transfected with empty vector were included. The percentage of internalisation is defined as 100 × ((ODBasal−ODMock)−(ODStimulated−ODMock))/(ODBasal−ODMock), where ODStimulated and ODBasal correspond to the OD obtained with ligand-treated and nontreated cells, respectively. For kinetic analysis of receptor recycling, the ligand treatment was followed by two washes with PBS, two washes with acidic buffer (150 mM NaCl/5 mM acetic acid) and again three washes with PBS at 4°C to remove all bound ligand. Cells were then transferred back to 37°C in DMEM for different times of recycling (40 or 120 min). The percentage of receptors at the cell surface is defined as 100 × (ODStimulated−ODMock)/(ODBasal−ODMock). Triplicates were performed for each condition within an experiment.
BRET assays
At 48 h post-transfection, cells were treated or not with either 100 nM AVP or 500 nM AP21967 at 37°C for the indicated period of times before being washed with PBS, detached in PBS/glucose (1 g/l) and distributed in a 96-well microplate. Coelenterazine H was added to a final concentration of 5 μM and readings were collected using a multidetector plate reader FUSION™ (Packard Instrument Company, Meriden, CT), allowing the sequential integration of the signals detected in the 440–500 and 510–590 nm windows. The BRET values were determined by calculating the ratio of the fluorescence signal emitted by FRB-βarrestin2-YFP (emission at 510–590) over the luminescence signal emitted by the Rluc-fused V1aR or V2R (emission at 440–500). These values were corrected by subtracting the background signal detected when the V1aR-Rluc or V2R-Rluc construct was expressed alone.
Detection of phosphorylated ERK1/2 (p42/p44 ERK)
HEK 293T cells were serum-starved for 24 h and treated for the indicated times with 100 nM AVP or 500 nM AP21967 at 37°C. The reaction was stopped in sample buffer (60 mM Tris–HCl pH 7.4/2% SDS/15% glycerol/50 mM dithiothreitol) and the samples were resolved by SDS/PAGE. The mouse monoclonal anti-P-ERK1/2 (E4) 1/2000 was used to detect the phosphorylated ERK1/2, and the immunoreactivity was revealed using HRP-coupled anti-mouse antibody 1/5000. The blots were then stripped and reblotted with the rabbit polyclonal anti-ERK1/2 (K23) 1/20 000 and the HRP-coupled anti-rabbit antibody 1/10 000, to control for the total amount of kinases loaded. Data from separate experiments were analysed using Quantity One (Bio-Rad) software and ERK1/2 phosphorylation was normalised according to the loading of proteins by expressing the data as a ratio of P-ERK1/2 to total ERK1/2.
For all other materials and methods, see Supplementary data.
Supplementary Material
SUPPLEMENTARY MATERIAL
Acknowledgments
We are grateful to Dr Victor Rivera from Pharmaceuticals Ariad for the generous gift of the expression vectors pC4EN-F1 (FKBP), pC4-RHE vectors (FRB) and pC4M-F2 (MyrFKBP2) and for kindly providing the heterodimeriser AP21967 (http://www.ariad.com/regulationkits). We thank Stéphanie Pontier and Dr Ali Salahpour for helpful discussion, as well as Dr Riad Qanbar and Dr Monique Lagacé for critical reading of the manuscript. This work was supported by a grant from the Canadian Institute for Health Research and the Kidney Foundation of Canada (MB). MB is the holder of the Hans Selye Chair in Molecular and Cell Biology and holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ (2004a) Differential kinetic and spatial patterns of beta-arrestin2 and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem 279: 35518–35525 [DOI] [PubMed] [Google Scholar]
- Ahn S, Wei H, Garrison TR, Lefkowitz RJ (2004b) Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J Biol Chem 279: 7807–7811 [DOI] [PubMed] [Google Scholar]
- Anborgh PH, Seachrist JL, Dale LB, Ferguson SS (2000) Receptor/beta-arrestin complex formation and the differential trafficking and resensitization of beta2-adrenergic and angiotensin II type 1A receptors. Mol Endocrinol 14: 2040–2053 [DOI] [PubMed] [Google Scholar]
- Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA (1997) Role of translocation in the activation and function of protein kinase B. J Biol Chem 272: 31515–31524 [DOI] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Bouvier M (2002) Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol 42: 409–435 [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, Pineyro G (2003) {Beta}-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA 100: 11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlic J, Andrews JD, Kelvin AA, Bosinger SE, DeVries ME, Xu L, Dobransky T, Feldman RD, Ferguson SS, Kelvin DJ (2000) Regulation of tyrosine kinase activation and granule release through beta-arrestin by CXCRI. Nat Immunol 1: 227–233 [DOI] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376: 599–602 [DOI] [PubMed] [Google Scholar]
- Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M (1999) A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature 401: 286–290 [DOI] [PubMed] [Google Scholar]
- DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Dery O, Bunnett NW (2000a) The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc Natl Acad Sci USA 97: 11086–11091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW (2000b) Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148: 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D, Lattion AL, Abuin L, Staub O, Cotecchia S (2003) The adaptor complex 2 directly interacts with the alpha 1b-adrenergic receptor and plays a role in receptor endocytosis. J Biol Chem 278: 19331–19340 [DOI] [PubMed] [Google Scholar]
- Fan GH, Yang W, Wang XJ, Qian Q, Richmond A (2001) Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalisation. Biochemistry 40: 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SR, O'Dowd BF, Lee SP (2002) G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov 1: 808–820 [DOI] [PubMed] [Google Scholar]
- Goodman OB Jr, Krupnick JG, Gurevich VV, Benovic JL, Keen JH (1997) Arrestin/clathrin interaction. Localization of the arrestin binding locus to the clathrin terminal domain. J Biol Chem 272: 15017–15022 [DOI] [PubMed] [Google Scholar]
- Hall RA, Lefkowitz RJ (2002) Regulation of G protein-coupled receptor signalling by scaffold proteins. Circ Res 91: 672–680 [DOI] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB (1999) The 2.8 Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell 97: 257–269 [DOI] [PubMed] [Google Scholar]
- Innamorati G, Le Gouill C, Balamotis M, Birnbaumer M (2001) The long and the short cycle. Alternative intracellular routes for trafficking of G-protein-coupled receptors. J Biol Chem 276: 13096–13103 [DOI] [PubMed] [Google Scholar]
- Kim YM, Benovic JL (2002) Differential roles of arrestin-2 interaction with clathrin and adaptor protein 2 in G protein-coupled receptor trafficking. J Biol Chem 277: 30760–30768 [DOI] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA (1996) Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem 271: 31372–31378 [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Benovic JL (1998) The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38: 289–319 [DOI] [PubMed] [Google Scholar]
- Krupnick JG, Goodman OB Jr, Keen JH, Benovic JL (1997) Arrestin/clathrin interaction. Localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J Biol Chem 272: 15011–15016 [DOI] [PubMed] [Google Scholar]
- Laporte SA, Miller WE, Kim KM, Caron MG (2002) Beta-arrestin/AP-2 interaction in G protein-coupled receptor internalisation: identification of a beta-arrestin binging site in beta 2-adaptin. J Biol Chem 277: 9247–9254 [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG (2000) The interaction of beta-arrestin with the AP-2 adaptor is required for the clustering of beta 2-adrenergic receptor into clathrin-coated pits. J Biol Chem 275: 23120–23126 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ (1999) Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283: 655–661 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ (1996) Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem 271: 19443–19450 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA 98: 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti A, Luo Z, Cunningham C, Ohta Y, Hartwig J, Stossel TP, Kyriakis JM, Avruch J (1997) Actin-binding protein-280 binds the stress-activated protein kinase (SAPK) activator SEK-1 and is required for tumor necrosis factor-alpha activation of SAPK in melanoma cells. J Biol Chem 272: 2620–2628 [DOI] [PubMed] [Google Scholar]
- Martini L, Hastrup H, Holst B, Fraile-Ramos A, Marsh M, Schwartz TW (2002) NK1 receptor fused to beta-arrestin displays a single-component, high-affinity molecular phenotype. Mol Pharmacol 62: 30–37 [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ (2000) Beta-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290: 1574–1577 [DOI] [PubMed] [Google Scholar]
- Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ (2000) Beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem 275: 11312–11319 [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Gilman M, Brugge JS (1999) Controlled dimerization of ErbB receptors provides evidence for differential signalling by homo- and heterodimers. Mol Cell Biol 19: 6845–6857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG (1999) Association of beta-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem 274: 32248–32257 [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS (2000) Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275: 17201–17210 [DOI] [PubMed] [Google Scholar]
- Perry SJ, Lefkowitz RJ (2002) Arresting developments in heptahelical receptor signalling and regulation. Trends Cell Biol 12: 130–138 [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639–650 [DOI] [PubMed] [Google Scholar]
- Pollock R, Issner R, Zoller K, Natesan S, Rivera VM, Clackson T (2000) Delivery of a stringent dimerizer-regulated gene expression system in a single retroviral vector. Proc Natl Acad Sci USA 97: 13221–13226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiina T, Kawasaki A, Nagao T, Kurose H (2000) Interaction with beta-arrestin determines the difference in internalisation behaviour between beta1- and beta2-adrenergic receptors. J Biol Chem 275: 29082–29090 [DOI] [PubMed] [Google Scholar]
- Terrillon S, Barberis C, Bouvier M (2004) Heterodimerization of V1a and V2 vasopressin receptors determines the interaction with beta-arrestin and their trafficking patterns. Proc Natl Acad Sci USA 101: 1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrillon S, Durroux T, Mouillac B, Breit A, Ayoub MA, Taulan M, Jockers R, Barberis C, Bouvier M (2003) Oxytocin and vasopressin V1a and V2 receptors form constitutive homo- and heterodimers during biosynthesis. Mol Endocrinol 17: 677–691 [DOI] [PubMed] [Google Scholar]
- Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, Oakley RH, Caron MG, Lefkowitz RJ, Luttrell LM (2003) The stability of the G protein-coupled receptor–beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem 278: 6258–6267 [DOI] [PubMed] [Google Scholar]
- Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM (2002) Beta-arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem 277: 9429–9436 [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV (1999) How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem 274: 11451–11454 [DOI] [PubMed] [Google Scholar]
- Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ (2003) Independent {beta}-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA 100: 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda P, Murray SR, von Zastrow M (2002) Modulation of postendocytic sorting of G protein-coupled receptors. Science 297: 615–620 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL