

Abstract
Nuclear accumulation of the serum response factor coactivator MAL/MKL1 is controlled by its interaction with G-actin, which results in its retention in the cytoplasm in cells with low Rho activity. We previously identified actin mutants whose expression promotes MAL nuclear accumulation via an unknown mechanism. Here, we show that actin interacts directly with MAL in vitro with high affinity. We identify a further activating mutation, G15S, which stabilises F-actin, as do the activating actins S14C and V159N. The three mutants share several biochemical properties, but can be distinguished by their ability to bind cofilin, ATP and MAL. MAL interaction with actin S14C is essentially undetectable, and that with actin V159N is weakened. In contrast, actin G15S interacts more strongly with MAL than the wild-type protein. Strikingly, the nuclear accumulation of MAL induced by overexpression of actin S14C is substantially dependent on Rho activity and actin treadmilling, while that induced by actin G15S expression is not. We propose a model in which actin G15S acts directly to promote MAL nuclear entry.
Keywords: actin, MAL, MKL1, Rho, SRF
Introduction
Actin has a long history of proposed connections with nuclear events as well as its classical role as a cytoskeletal component. Recent studies suggest potential roles for actin and its relatives in chromatin remodelling, transcription and RNA export (reviewed by Olave et al, 2002; Bettinger et al, 2004). Among these roles is that played by actin to control its own expression both transcriptionally and at the level of mRNA translation, localisation and stability (for references see Lyubimova et al, 1999; Sotiropoulos et al, 1999). Autoregulation of actin transcription occurs via a mechanism in which G-actin binds to and controls the activity of MAL/MKL1, a coactivator of the serum response factor (SRF) transcription factor (Miralles et al, 2003). SRF, a MADS-box protein, controls a large number of growth factor-inducible and muscle-specific genes through the mutually exclusive association of different SRF cofactors (Murai and Treisman, 2002; Miralles et al, 2003; Wang et al, 2004), and the expression of many of these genes is thus influenced by G-actin level. In fibroblasts, MAL and its relative MAL16/MKL2 are predominantly cytoplasmic in the absence of Rho signalling, but accumulate in the nucleus upon Rho activation (Cen et al, 2003; Miralles et al, 2003; Du et al, 2004; C Perez-Sanchez, unpublished data). MAL associates with G-actin in vivo, and depletion of the G-actin pool upon activation of Rho releases MAL for nuclear import (Sotiropoulos et al, 1999; Posern et al, 2002; Miralles et al, 2003).
Actin mutants have given useful insight into the mechanism by which Rho controls SRF activity. In support of the notion that G-actin is the regulator, the overexpression of nonpolymerising β-actin mutants such as actins R62D, G13R and actin-VP16 inhibits SRF activation and MAL nuclear accumulation (Posern et al, 2002). Consistent with this, MAL can be recovered in immunoprecipitates of these mutants and wild-type actin, and their overexpression prevents MAL nuclear accumulation (Miralles et al, 2003). It has remained unclear whether MAL interacts directly with G-actin. We also identified two actin mutants, actins S14C and V159N, which stabilise F-actin and whose expression strongly activates SRF (Posern et al, 2002). Curiously, however, although cells expressing these mutants accumulate MAL in the nucleus and exhibit an increase in the F-/G-actin ratio, their absolute level of G-actin is if anything slightly increased relative to that of untransfected cells (Posern et al, 2002; Miralles et al, 2003). We suggested two mechanisms to explain these observations: either the mutants were incapable of interacting with the SRF coactivator (i.e. MAL), allowing its release to the nucleus, or they acted independently of the actin treadmilling cycle to induce its activation (Posern et al, 2002).
In this paper, we study the connection between activating actin mutants and SRF activation in the light of our identification of MAL as an actin-regulated SRF coactivator. We show that wild-type actin binds the N-terminal RPEL domain of MAL directly in vitro, and identify a new activating actin mutant, actin G15S, in a two-hybrid screen with this domain. All three activating actin mutants, actins G15S, S14C and V159N, share the ability to stabilise F-actin. They all exhibit a decreased ability to bind the C-terminal half of gelsolin, and an enhanced affinity for profilin, suggesting that their structure mimics that of ATP-actin. However, they can also be distinguished biochemically by their ability to interact with nucleotide, cofilin and MAL. Although actin G15S binds MAL more strongly than wild-type actin, the S14C mutation greatly reduces the actin–MAL interaction. Strikingly, the ability of actin S14C to induce MAL nuclear accumulation is strongly dependent on the actin treadmilling cycle and basal Rho activity, while G15S- and V159N-induced MAL translocation is not. We propose that whereas actin S14C releases MAL from the inhibitory effect of wild-type G-actin by diluting the endogenous G-actin pool, actin G15S (and probably V159N) may act directly to induce MAL nuclear translocation.
Results
Actin interacts directly with the MAL RPEL motifs in vitro
We previously showed that MAL can be recovered in immunoprecipitates of cytoplasmic actin. The interaction was dependent on the integrity of the MAL N-terminal RPEL motifs (Miralles et al, 2003). It remained unclear, however, whether this complex involves a direct interaction between MAL and actin or requires additional proteins. To test whether the actin-MAL complex can form in vitro, we passed whole-cell extracts over affinity beads comprising MAL RPEL motifs 2 and 3 fused to GST. As controls we used mutant RPEL derivatives containing P → A or R → D mutations in each RPEL motif, which were previously shown to be defective for actin binding in vivo (Miralles et al, 2003). After extensive washing, bound proteins were eluted using the G-actin-binding drug swinholide A, which competes with MAL for actin binding (Miralles et al, 2003). A 42 kDa protein was prominent in the eluate from the wild-type but not mutant GST fusion protein; no other proteins in the 10–300 kDa Mr range bound specifically (Figure 1A). Similar results were obtained when proteins were eluted from the affinity beads by boiling in SDS–PAGE loading buffer (data not shown). An immunoblot experiment confirmed that the 42 kDa protein was indeed β-actin (Figure 1B). Purified β-actin also bound the wild-type but not the mutant MAL affinity beads, and this interaction was resistant to latrunculin B but sensitive to swinholide A (Figure 1C). These results complement previous experiments in which co-immunoprecipitation of MAL and actin was disrupted by cytochalasin D or swinholide A, which also promoted MAL nuclear accumulation, but not by latrunculin B, which did not (Miralles et al, 2003).
Figure 1.
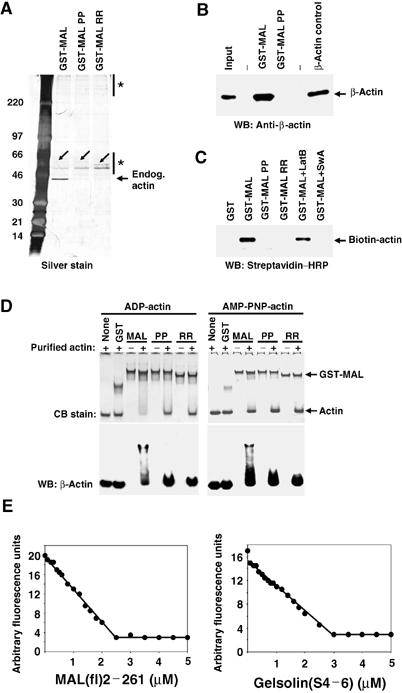
MAL binds directly to actin. (A) Actin does not associate with other cellular proteins on the RPEL motif. NIH3T3 cells were lysed by syringing in detergent-free buffer and the high-speed G-actin supernatant was affinity-precipitated using GST-MAL(met)(1–171) or its derivatives PP34/78AA (PP) or RR33/77DD (RR), which carry point mutations in each of the two RPEL motifs. Bound proteins were eluted with 500 nM swinholide A, separated by 6–16% gradient PAGE and detected by silver staining. The diagonal arrows indicate contaminating GST fusion proteins and asterisks mark nonspecifically retained polypeptides. (B) Precipitated proteins from a GST-MAL(met)(1–171) affinity precipitation experiment of the type shown in (A) were analysed by immunoblotting with β-actin antibody. (C) Affinity precipitation of purified biotinylated nonmuscle actin using GST-MAL(met)(1–171) or its derivatives. Where indicated, 1 μM latrunculin B or 100 nM swinholide A was included in the binding reaction. Bound actin was separated by 12% SDS–PAGE and detected by overlay with peroxidase-conjugated streptavidin. (D) Native 6% polyacrylamide gel electrophoresis assay of complex formation between GST-MAL(met)(1–171) derivatives and ADP- or AMP-PNP-loaded nonmuscle actin (left and right panels, respectively), with detection by Coomassie blue (CB) staining or anti-β-actin immunoblot. (E) Inhibition of skeletal muscle α-actin polymerisation by MAL(fl)2–261 (left), which contains all three RPEL motifs, and gelsolin(S4–6) (right). Data are mean of two independent experiments.
Some actin-binding proteins strongly discriminate between different nucleotide-bound forms of actin. We therefore tested the nucleotide-binding requirements of actin–MAL interaction using a native gel electrophoresis assay. Wild-type or mutant GST-MAL fusion proteins were mixed with ADP- or AMP-PNP-bound actin, separated on a native gel and actin detected by immunoblotting. Actin–MAL complex formation was detected regardless of the bound nucleotide (Figure 1D). These results show that additional accessory proteins are not required for binding of actin to the MAL RPEL motifs.
Finally, we determined the apparent affinity of the intact RPEL domain, by exploiting its ability to inhibit actin polymerisation (Hertzog et al, 2002). We used skeletal muscle α-actin since it is readily purified and recovers MAL as efficiently as β-actin in co-immunoprecipitation experiments (see Supplementary Figure S1). As a positive control, we evaluated gelsolin segments 4–6 (gelsolin(S4–6)). Increasing amounts of the MAL RPEL domain or gelsolin(S4–6) led to complete inhibition of F-actin assembly at comparable concentrations (Figure 1E). The data are consistent with an apparent Kd of 24 nM for MAL–actin, and an apparent Kd of 76 nM for gelsolin(S4–6), in good agreement with published data (Way et al, 1989). Profilin overexpression induces MAL nuclear accumulation and SRF activation (Sotiropoulos et al, 1999; Miralles et al, 2003), and consistent with this, MAL is not recovered in profilin immunoprecipitates (Supplementary Figure S1). Thus, high-affinity MAL–actin interaction allows it to compete effectively with profilin for free G-actin.
Identification of actin G15S, a novel MAL-activating protein
To identify cDNAs encoding proteins that interact with the MAL N-terminal region in an unbiased way, we set up a yeast two-hybrid assay. A GAL4 fusion protein containing MAL residues 1–631 was used to screen an NIH3T3 cDNA library, and all cDNAs recovered were counterscreened against a mutant MAL containing RPEL motif point mutations. From 600 000 clones screened, we recovered a mouse γ-actin cDNA containing the mutation G15S and two other cDNAs whose characterisation will be described in full elsewhere. Colony growth and liquid β-galactosidase assays showed that binding of γ-actin G15S to MAL was dependent on the RPEL motifs: it was severely reduced by R → D or P → A mutations at single RPEL motifs, and undetectable upon mutation of both motifs (Figure 2A). Intrigued by this observation, and our previous finding that mutations at the β-actin nucleotide-binding pocket can substantially affect its activity in the SRF activation assay (Posern et al, 2002), we tested whether the G15S mutation affects the ability of actin to repress SRF activity. Expression of γ-actin G15S strongly potentiated activity of the SRF reporter gene 3D.A-Luc in serum-starved cells; in contrast, expression of wild-type γ-actin, as with wild-type β-actin, suppressed activation of the reporter following serum stimulation (Figure 2B).
Figure 2.
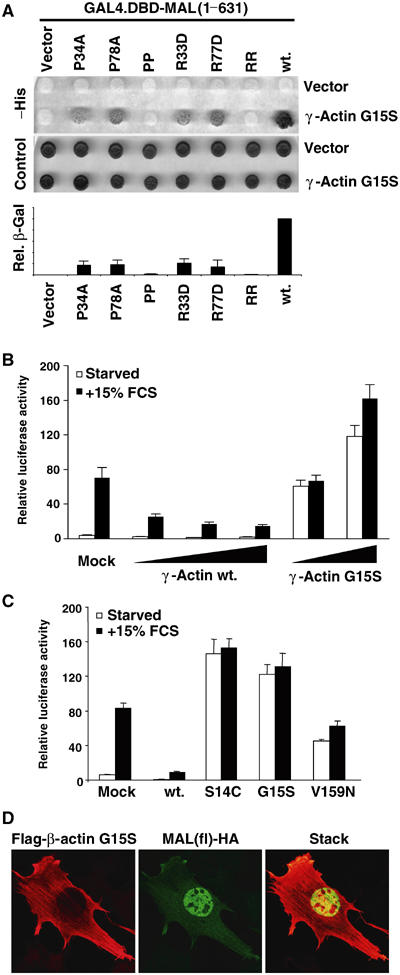
Identification of actin G15S, an activator of MAL and SRF. (A) Yeast two-hybrid interactions between γ-actin G15S and MAL(met)(1–631) or mutant derivatives containing point changes in either or both RPEL motifs. Upper panels: trans-illuminated images of colony growth on selective (−His) or nonselective (control) medium. Lower histogram: interaction quantification by liquid-culture Gal4-lacZ reporter gene assay (WT γ-actin=100; error bars: s.e.m.; n=3) (B) γ-Actin G15S expression activates SRF. Cells were transfected with SRF reporter 3D.A-Luc (40 ng), together with wild-type γ-actin (100, 250 or 500 ng) or γ-actin G15S (100 or 250 ng), maintained in 0.3% FCS for 40 h and then serum-stimulated where indicated. Data are means of three independent experiments; error bars: s.e.m. (C) Reporter activation by β-actin G15S and other β-actin mutants. Cells were transfected with reporter and the indicated β-actin mutants and treated as in (B). (D) β-Actin G15S expression induces MAL nuclear accumulation. Cells expressing MAL(fl)-HA (50 ng) and β-actin G15S (500 ng) were processed for immunofluorescence 24 h after transfection. Confocal sections of 0.3 μm thickness show actin G15S (anti-Flag; red) and nuclear accumulation of MAL(fl) (anti-HA; green). The merged picture shows the stack of both actin and MAL sections.
We next introduced the G15S mutation into β-actin. Expression of β-actin G15S also substantially activated expression of the SRF reporter in serum-starved cells, to levels comparable to those achieved by expression of the activating mutants actins S14C and V159N (Figure 2C; Posern et al, 2002). Consistent with its ability to activate SRF, β-actin G15S expression induced nuclear accumulation of the SRF coactivator MAL in NIH3T3 cells, but did not itself accumulate in the nucleus (Figure 2D). To allow comparison with our other ‘activating' actin mutants, all further studies were performed with β-actin G15S.
Actin G15S stabilises F-actin
Upon transient overexpression in fibroblasts, actins S14C and V159N exhibit properties suggesting that they stabilise F-actin (Posern et al, 2002), consistent with structural studies of yeast actin V159N (Belmont and Drubin, 1998; Belmont et al, 1999a). We therefore measured intracellular levels of F-actin and G-actin in cells expressing the different actins. Cells were stained with phalloidin, which specifically binds F-actin, or DNase I, which specifically binds G-actin, and staining was quantified in transfected cells relative to untransfected cells in the same population using the FACS (Geneste et al, 2002; Posern et al, 2002). Cells expressing wild-type actin exhibit an increase of approximately 40% in both F- and G-actin levels. In this assay, overexpression of actin G15S altered the balance between F- and G-actin in favour of F-actin, as observed previously for actins S14C and V159N (Figure 3A). To corroborate this result, we used detergent extraction and centrifugation to produce F-actin- and G-actin-enriched supernatant and pellet fractions from cells expressing Flag-tagged actin. In this assay, serum stimulation led to increased recovery of wild-type actin in the F-actin fraction (Figure 3B). In contrast, depolymerisation of the cytoskeleton by the actin-binding drugs swinholide A or latrunculin B, or coexpression of C3 transferase or the G-actin-binding protein profilin, caused Flag-actin to accumulate in the G-actin fraction (Figure 3B). Actin G15S was recovered in increased amounts in the F-actin fraction compared to wild-type actin, as were actins S14C and V159N (Figure 3C; Posern et al, 2002). Moreover, coexpression of actin G15S and wild-type actin led to an increased recovery of wild-type actin in the F-actin fraction (Figure 3D). These results show that like actins S14C and V159N, actin G15S can copolymerise with wild-type actin to generate F-actin of increased stability.
Figure 3.
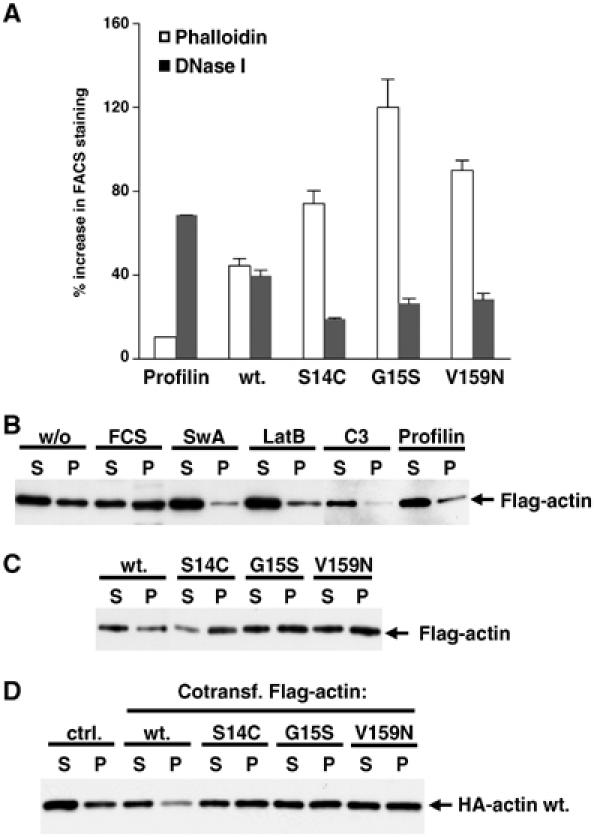
Actin G15S stabilises filament formation. (A) Transfected cells expressing the indicated Flag-tagged actins (1 μg) were stained for the Flag epitope and either TRITC-phalloidin (for F-actin) or FITC-DNase I (for G-actin). The FACS was used to quantify mean levels of F- or G-actin in the transfected population relative to those of the untransfected population (error bars: s.e.m.; n=3). (B) Actin fractionation lysates were prepared from cells expressing wild-type Flag-actin (1 μg) with C3 transferase (50 ng) or profilin (500 ng) coexpression, or treatment with latrunculin B (0.3 μM, 1 h), swinholide A (100 nM, 1 h) or FCS (15%, 10 min) as indicated. Flag-actin in each supernatant (S) and pellet (P) fraction was detected by immunoblotting using anti-Flag antibodies. (C) Actin fractionation lysates were prepared from cells expressing the indicated Flag-tagged actin mutants (1 μg) and analysed by anti-Flag immunoblotting as in (B). (D) Activating actin mutants copolymerise with and stabilise wild-type F-actin. Actin fractionation lysates were prepared from cells expressing the indicated Flag-tagged actins (1 μg) together with HA-tagged wild-type actin (500 ng), and analysed by anti-HA immunoblot.
Activating actins share altered gelsolin- and profilin-binding properties
To gain insight into the changed conformation of the activating actins, we studied their interaction with different actin-binding proteins, initially focusing on proteins that discriminate between different nucleotide-bound states of the molecule. The C-terminal half of the F-actin severing protein gelsolin, comprising segments 4–6, specifically binds wild-type ADP-bound G-actin (Laham et al, 1993). We used the GST-gelsolin(S4–6) fusion protein in pulldown assays with high-speed supernatant extracts of transiently transfected NIH3T3 cells expressing Flag-tagged wild-type or mutant actins. All the activating mutants were recovered inefficiently in comparison to wild-type actin and the nonpolymerisable mutant R62D (Figure 4A, upper); in a control experiment, purified wild-type ADP-actin, but not AMP-PNP-actin, effectively bound GST-gelsolin(S4–6) (Figure 4A, lower). The decreased affinity of the activating actins for GST-gelsolin(S4–6) suggests that their structure does not readily adopt a conformation characteristic of ADP-actin.
Figure 4.
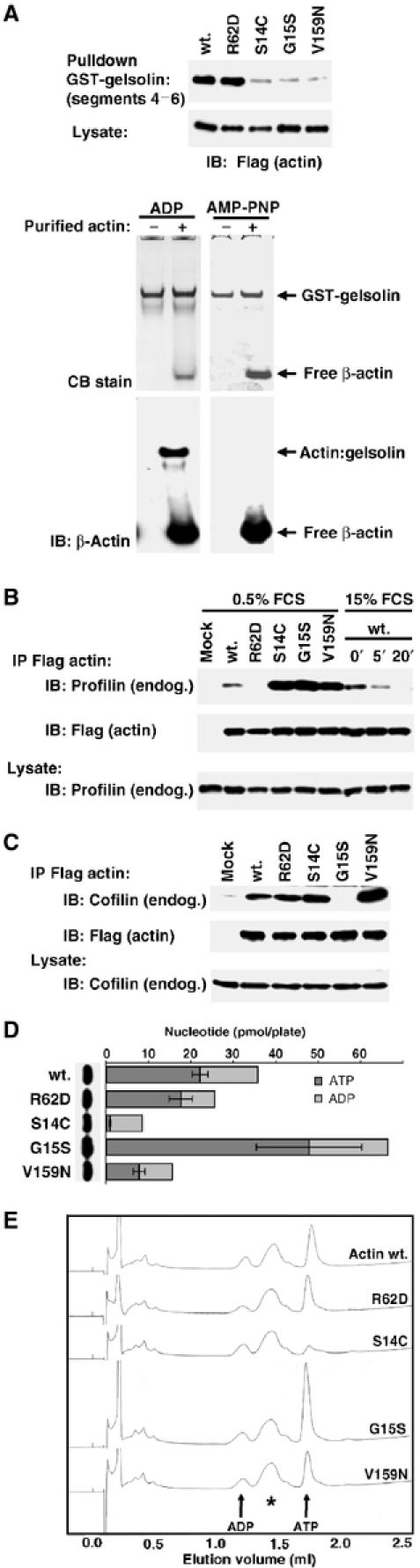
Activating actin mutants have both shared and distinct properties. (A) Activating actins exhibit reduced affinity for the C-terminal half of gelsolin. Upper panel: G-actin supernatants from cells expressing the indicated actins (2 μg) were affinity-precipitated using GST-gelsolin(S4–6), and bound proteins were detected by immunoblotting with anti-Flag antibodies. Lower panels: native gel electrophoresis assay of complex formation between GST-gelsolin(S4–6) and ADP- or AMP-PNP-loaded nonmuscle actin, with detection by Coomassie blue or anti-β-actin immunoblot. (B) Interaction of actins with endogenous profilin. Upper panels: Extracts as in (A) were immunoprecipitated with anti-Flag and analysed for profilin (anti-profilin; upper) and actin (anti-Flag; lower). Cells were in 0.5% serum except where stimulated with 15% serum for the indicated times (min). Lower panel: Control immunoblot for profilin in the lysate. (C) Interaction of actins with endogenous cofilin. Extracts were prepared and analysed as in (B). (D) Nucleotide binding by actin mutants. Actin immunoprecipitates were prepared as in (B), followed by nucleotide determination using the luciferase-based ATP assay. Immunoblot: recovered actin in each sample. Data are mean±s.e.m. (n=4). (E) Nucleotide binding by actin mutants. Actins were prepared as in (D) and nucleotide was identified by Mono-Q anion exchange chromatography. UV absorbance at 254 nm from a representative experiment is shown, with elution volumes for ADP and ATP indicated. The asterisk indicates a nonspecifically recovered component also present in precipitates from mock-transfected cells.
We next used the co-immunoprecipitation assay to examine the ability of the mutants to associate with profilin, which favours nucleotide exchange on actin and preferentially binds ATP-actin (see Perelroizen et al, 1995; Pollard and Borisy, 2003). Extracts were prepared from cells expressing the Flag-tagged actins, immunoprecipitated using anti-Flag antibodies and recovery of cellular profilin was monitored by immunoblotting. Profilin was recovered in immunoprecipitates of wild-type actin, but not the nonpolymerisable mutant R62D (Figure 4B). In this assay, profilin was more effectively recovered by the activating actin mutants than by the wild-type protein (Figure 4B). Serum stimulation led to a rapid decrease in the recovery of profilin associated with actin (Figure 4B, right).
Taken together, these results suggest that the activating actins cannot enter a conformation readily adopted by ADP-bound wild-type G-actin, and an increased tendency to enter a conformation preferred by ATP-bound wild-type G-actin. In contrast, the nonpolymerising mutant R62D, which retains the ability to inhibit MAL nuclear accumulation, appears to adopt a conformation more characteristic of ADP-bound G-actin.
The activating mutants interact differentially with cofilin and nucleotide
Cofilin binding discriminates in favour of ADP-bound actin (Carlier et al, 1997). We therefore also evaluated the ability of the mutant actins to associate with cellular cofilin using the co-immunoprecipitation assay. In contrast to their similar interactions with gelsolin(S4–6) and profilin, however, the activating actins exhibited very different behaviour in this assay: cofilin was efficiently recovered in all the actin immunoprecipitates save that of the activating mutant G15S (Figure 4C).
Given that the behaviour of the activating mutants in the gelsolin(S4–6)- and profilin-binding assays suggested that they might adopt an ‘ATP-like' conformation more readily than wild-type actin, we next sought to confirm the identity of the nucleotide associated with the different mutants. Each of the actins was recovered from transfected cells by anti-Flag immunoprecipitation (IP) and bound ATP was quantified using a luciferase enzymatic assay (Chen et al, 1995; Chen and Rubenstein, 1995; Schuler et al, 1999). In this assay, less ATP was recovered with the nonpolymerisable R62D and the activating V159N actin mutants than with wild-type actin, while more ATP was recovered with the activating mutant G15S; in contrast, activating actin S14C was recovered virtually free of ATP (Figure 4D). The relatively high amounts of ADP recovered in these assays may reflect hydrolysis during actin isolation. Essentially identical results were obtained when nucleotide association was evaluated by HPLC assay (Figure 4E; Rosenblatt et al, 1995).
Taken together with the results in the preceding section, these data show that although all three activating mutants share a number of properties, they nevertheless can be distinguished biochemically according to their ability to bind nucleotide (S14C defective) and cofilin (G15S defective).
Activating actins also exhibit distinct MAL-binding properties
We next examined the interaction between the activating actins and MAL. Actins were immunoprecipitated from extracts of cells transfected with Flag-tagged actins and recovery of endogenous MAL was assessed by immunoblotting. In this assay, the different activating actins again behaved differently. MAL binding to actin S14C was essentially undetectable; in contrast, actin G15S bound MAL more effectively than wild-type actin (Figure 5A). To increase the sensitivity of the assay, we repeated it, this time overexpressing MAL as well as the actins. Again, MAL binding to actin S14C was undetectable, while actin G15S bound more efficiently than wild-type actin, and V159N less effectively (Figure 5B). Neither wild-type nor mutant actins associated with the MAL mutant PP34/78AA, which carries P → A changes in its RPEL motifs (Figure 5B; Miralles et al, 2003). MAL nuclear accumulation in response to serum stimulation is dependent on two basic sequence elements (Miralles et al, 2003). MALΔB1ΔB2, which lacks these basic sequences, remained cytoplasmic upon expression of actins S14C, G15S and V159N, indicating that the basic sequences are also required for MAL nuclear accumulation induced by the activating actins (Figure 5C). A control experiment confirmed that MALΔB1ΔB2 nevertheless remains competent to bind wild-type actin as well as the R62D and G15S mutants (Figure 5C).
Figure 5.
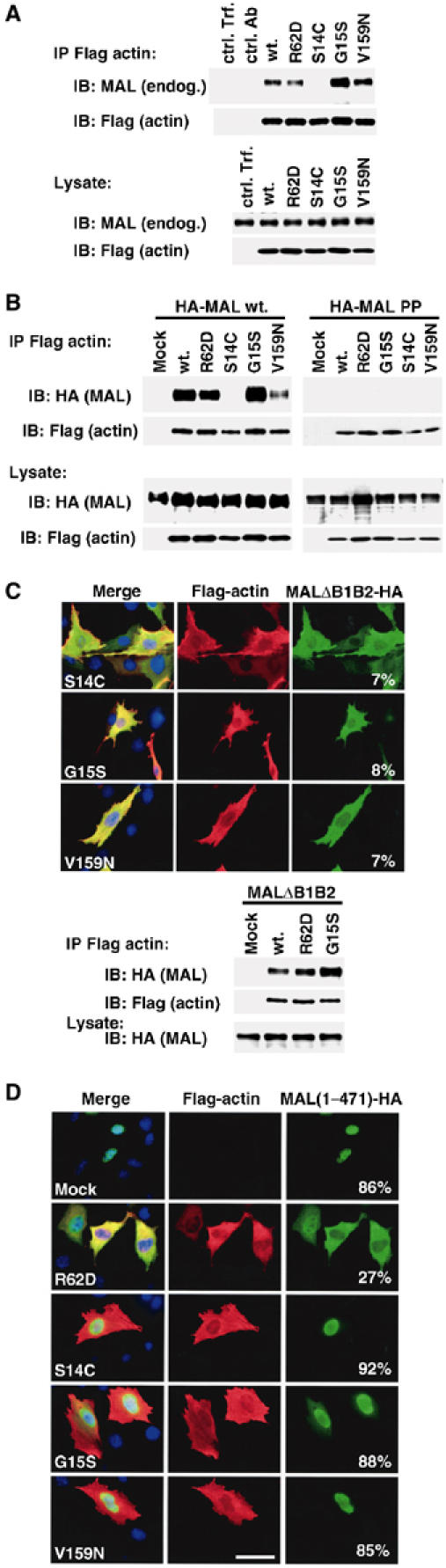
Interaction of MAL with wild-type and mutant actins. (A) Interaction of actins with endogenous MAL from NIH3T3 cells. Upper panels: extracts from cells expressing the indicated Flag-tagged actin mutants (1 μg) were immunoprecipitated with anti-Flag or control antibodies, and analysed for MAL (anti-MAL; upper) and actin (anti-Flag; lower). Lower panels: control immunoblots for MAL (anti-MAL; upper) and actin expression (anti-Flag) in the lysate. ctrl. Trf.: no actin; ctrl. Ab: wild-type actin, anti-HA IP. (B) Interaction of actins with overexpressed HA-tagged MAL(met) and HA-MAL(met) PP34/78AA (containing mutated RPEL motifs) (1 μg). Cells were transfected and processed as in (A). Data in lanes 1–3 are from Miralles et al (2003). (C) Nuclear accumulation induced by the activating actins requires MAL basic box regions. Upper panels: cells in 0.5% FCS expressing MALΔB1B2-HA (0.1 μg), which lacks the basic regions, and activating actins (1 μg) were fixed and stained for MAL (anti-HA; green), actins (anti-Flag; red) and DNA (Hoechst 33258; blue). Numbers show proportion of cells with predominantly nuclear MAL. Lower panels: control co-immunoprecipitation experiments performed as in (B). (D) Overexpression of nonpolymerisable actin R62D, but not the F-actin-stabilising mutants, promotes relocalisation of MAL(met)1–471 to the cytoplasm. Cells expressing HA-tagged MAL(met)1–471 with vector (mock) or Flag-tagged actins were stained and scored as in (C).
We previously showed that MAL(1–471), a MAL mutant lacking its C-terminal sequences, which include the dimerisation interface, accumulates in the nucleus under conditions of basal Rho signalling (Miralles et al, 2003). Overexpression of nonpolymerisable actin R62D, which binds MAL, is sufficient to relocalise MAL(1–471) to the cytoplasm under these conditions (Figure 5D, top rows). In contrast, none of the activating actins, even actins G15S and V159N, which can interact with MAL, relocalised MAL(1–471) to the cytoplasm (Figure 5D, lower rows). Binding of these actins to MAL is thus incapable of retaining it in the cytoplasm.
MAL activation by actins exhibits differential dependence on actin treadmilling
We previously proposed that activating actin mutants might potentiate SRF activation either because of their inability to bind a putative coactivator (MAL) or because their binding activates it (Posern et al, 2002). The different MAL-binding properties of the mutants described above are consistent with the notion that the mutants use different mechanisms to induce MAL nuclear accumulation. To investigate this issue, we tested the dependence of SRF reporter gene activation on basal Rho activity, using C3 transferase coexpression to inactivate endogenous Rho (Hill et al, 1995). As previously observed, expression of C3 transferase completely abolished serum-induced activation of the SRF reporter gene, but did not affect its activation by coexpression of an activated mDia mutant (Hill et al, 1995; Copeland and Treisman, 2002). Expression of C3 transferase substantially inhibited reporter activation induced by actin S14C expression but had no significant effect upon activation by actin G15S (Figure 6A). In these experiments, the effect of C3 transferase upon activation by actin V159N was of borderline statistical significance, however.
Figure 6.
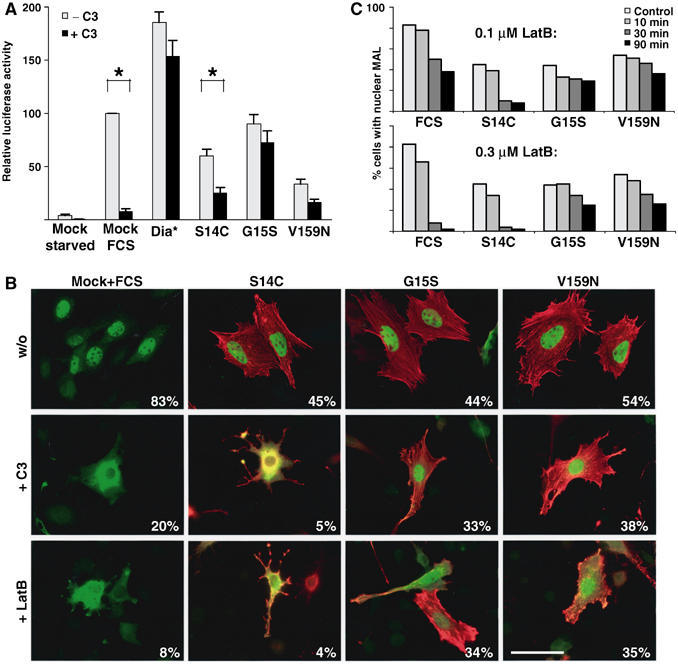
Activating actins exhibit different requirements for actin treadmilling in SRF activation. (A) SRF reporter activation by activating actins is differentially sensitive to Rho inactivation. Cells transfected with SRF reporter expressed activating actins (500 ng), C3 transferase (20 ng) and activated mDia (50 ng) as indicated. Data are the mean of four independent experiments (error bars: s.e.m.; asterisks: statistical significance at P<0.01, unpaired Student's t-test). (B) Immunofluorescence analysis of MAL nuclear accumulation induced by serum stimulation or coexpression with activating actin mutants. Cells expressed MAL(met) (50 ng) and activating actins (500 ng), with C3 transferase coexpression (20 ng) or latrunculin B treatment as indicated, and were maintained in 0.5% FCS unless stimulated with 15% FCS for 30 min as indicated. Cells were fixed and stained for MAL (anti-HA; green) and actins (anti-Flag; red). Latrunculin treatment (0.3 μM) was for 30 min prior to staining. Around 100 cells each were analysed and scored for predominantly nuclear MAL staining. (C) Kinetics of MAL cytoplasmic reaccumulation. Cells were transfected as in (B) and treated with either 0.1 or 0.3 μM latrunculin B for the times indicated. Data are the mean percentage of predominantly nuclear MAL staining in two independent experiments.
We next tested whether MAL nuclear translocation induced by the activating actins required functional Rho and actin treadmilling. Coexpression of C3 transferase substantially inhibited MAL nuclear accumulation in response to serum stimulation (Figure 6B; Miralles et al, 2003). The activating actins induced MAL nuclear accumulation less effectively than serum, with about half of the cell population exhibiting predominantly nuclear fluorescence. Inactivation of Rho by expression of C3 transferase, however, prevented nuclear accumulation of MAL in cells expressing actin S14C, but had little effect on MAL in cells expressing actins V159N and G15S (Figure 6B). To test whether Rho dependence reflected a requirement for actin treadmilling, we used latrunculin B treatment to inhibit actin treadmilling following activation. Although MAL remains in the nucleus for several hours following serum stimulation, treatment with latrunculin B after only 1 h induces its rapid cytoplasmic reaccumulation (Figure 6B and C). In cells expressing actin S14C, nuclear MAL was rapidly redistributed to the cytoplasm following addition of latrunculin B (Figure 6B and C). In contrast, in cells expressing actins G15S and V159N, addition of latrunculin B had little effect on the proportion of cells with nuclear MAL (Figure 6B and C). Expression of C3 transferase or latrunculin B treatment had a more pronounced effect on F-actin staining in cells expressing actin S14C than the other activating actins (Figure 6B); this may reflect both differential effects of the mutants on F-actin stability and the fact that chronic SRF activation itself appears to increase cellular F-actin level (Schratt et al, 2002).
Taken together with the biochemical data, these results show that while the three activating actins all stabilise F-actin, they must use distinct mechanisms to induce MAL nuclear accumulation. The behaviour of actin S14C is consistent with a model in which MAL nuclear accumulation results from replacement of the cytoplasmic G-actin pool by a mutant form incompetent to bind MAL, while that of G15S is consistent with a model in which binding of the actin itself directly promotes nuclear MAL accumulation independent of operation of the actin treadmilling cycle (Figure 7; see Discussion).
Figure 7.
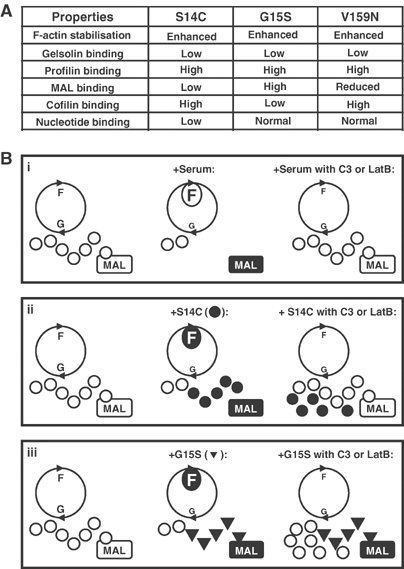
(A) Summary of mutant actin properties. Biochemical characteristics of the indicated point mutants are compared to those of the wild-type protein. (B) Models for activation of MAL by mutant actins. (i) Normal cells in low serum exhibit a basal actin treadmilling (circle; G: G-actin; F: F-actin). MAL is retained in the cytoplasm (open box) by interaction with G-actin (white circles), which somehow inhibits its nuclear accumulation. Upon serum stimulation, the G-actin pool is depleted and MAL released for nuclear import (black box); blockade of treadmilling by latrunculin B or Rho inactivation prevents this, inhibiting MAL release (open box). (ii) Actin S14C (black circles) stabilises F-actin; its overexpression increases F-actin but does not deplete the G-actin pool. However, actin S14C itself does not effectively bind MAL, which is therefore released for nuclear accumulation (black box). Upon blockade of treadmilling, wild-type G-actin reaccumulates, and MAL is again inhibited (open box). (iii) Actin G15S (black triangles) stabilises F-actin; its overexpression increases F-actin but does not deplete the G-actin pool. Binding of actin G15S to MAL directly promotes MAL nuclear accumulation (black box). MAL therefore remains active upon inhibition of actin treadmilling.
Discussion
We have investigated the functional and physical interaction of wild-type and mutant actins with the SRF coactivator MAL. MAL is a member of two protein families that contain the so-called RPEL motif (interpro IPR004018; pfam PF02755). Integrity of the RPEL motif is required for association with β-actin in vivo, and its disruption results in MAL nuclear accumulation (Miralles et al, 2003; S Ellis, unpublished data). We find that actin binds directly to MAL through its RPEL motifs in vitro, and that this interaction is compatible with binding either to ATP (AMP-PNP) or ADP. The affinity of the actin–MAL interaction (apparent Kd 24 nM) is comparable to gelsolin. This, and the fact that MAL and profilin bind to actin mutually exclusively, is consistent with a model in which MAL competes effectively with profilin for free G-actin. The nonpolymerising actin mutant actin R62D, which nevertheless retains the ability to regulate MAL activity (Posern et al, 2002; Miralles et al, 2003), has similar MAL-binding properties to the wild-type protein and our studies indicate that it may adopt a structure characteristic of ADP-bound wild-type G-actin.
We have identified a third actin mutant whose overexpression activates rather than inhibits MAL nuclear accumulation and SRF activation. This mutant, actin G15S, shares a number of properties with two other ‘activating' actins, actins V159N and S14C, including the ability to stabilise F-actin (Posern et al, 2002). The three mutants nevertheless exhibit distinct behaviour in several biochemical assays, including binding to nucleotide, cofilin and MAL (Figure 7A). Our functional studies suggest that these activating mutants use different mechanisms to promote MAL nuclear accumulation, and that interaction of actin G15S with MAL may directly promote MAL nuclear accumulation.
MAL is at least in part regulated by a G-actin titration mechanism in which binding of MAL to G-actin somehow inhibits its nuclear accumulation, perhaps by occluding its nuclear import signals (Figure 7B, i; Miralles et al, 2003). How then might expression of F-actin-stabilising actin mutants lead to MAL nuclear accumulation and SRF activation? We previously proposed two nonmutually exclusive mechanisms by which expression of such mutants might lead to activation of SRF regulatory cofactors (MAL at that time remained uncharacterised; Posern et al, 2002). One class of mutants might exhibit reduced affinity for MAL but stabilise F-actin. Although expression of such mutants might leave G-actin level substantially unaffected, it would lead to dilution of the wild-type G-actin pool with mutant actin incompetent to interact with MAL, thereby releasing MAL for nuclear import (Figure 7B, ii). Activation by these mutants would require operation of the actin treadmilling cycle to equilibrate with the cellular actin pool, so their ability to drive MAL accumulation and SRF activation would be expected to be heavily dependent on operation of the actin treadmilling cycle. An alternative and more novel type of actin mutation might alter the way in which actin interacts with MAL, such that even when these mutants bind MAL, the complex is somehow actively targeted for nuclear import (Figure 7B, iii). (Note that mutations of this type need not necessarily result in F-actin stabilisation.) Such mutants might mimic the function of a subpopulation of wild-type actin, which is normally required for MAL nuclear accumulation. Their ability to promote MAL nuclear accumulation directly would obviate any requirement for operation of the actin treadmilling cycle in SRF activation.
The activating actin mutants described here apparently fall into both of the above classes. Actin S14C exhibits substantially reduced interaction with MAL, and actin S14C-induced MAL nuclear accumulation and SRF activation are indeed substantially dependent on actin treadmilling, consistent with the titration model. In contrast, actin G15S retains high-affinity interaction with MAL, and its ability to promote MAL nuclear accumulation and SRF activation is largely independent of actin treadmilling, consistent with the ‘direct activation' model (Figure 7B, iii). Actin V159N is also likely to be of this type, even though it exhibits weakened interaction with MAL, since it promotes MAL nuclear accumulation substantially independently of actin treadmilling. We note that although the behaviour of actin S14C is consistent with the titration model, we cannot exclude the possibility that a direct-acting subpopulation of endogenous wild-type actin also remains required for MAL nuclear accumulation upon overexpression of this mutant.
Further experiments are necessary to substantiate the direct activation hypothesis. As yet we have not detected altered interactions between MAL and actin G15S in limited proteolysis experiments (G Posern, unpublished). Moreover, actin G15S does not accumulate in the nucleus, even in the presence of overexpressed MAL, suggesting that although this actin–MAL complex appears targeted for nuclear import, its actin component must either be stripped off during import or immediately re-exported from the nucleus upon MAL entry. Biophysical approaches will allow the dynamics of the interaction between actin and MAL to be studied in living cells.
Our data suggest that mammalian β-actin G15S generates F-actin of increased stability, as previously proposed for yeast actin G15S on the basis that diploid cells expressing both wild-type and G15S actin exhibit increased resistance to latrunculin A (Belmont et al, 1999b). The mechanism by which such stabilisation might arise remains obscure, however. Previous studies have shown that the yeast actin V159N generates F-actin that retains structural characteristics of the ADP+Pi F-actin structure even after departure of the hydrolysed phosphate (Belmont et al, 1999a). Residues S14 and G15 make intimate contacts with the ATP γ- and β-phosphates, and form part of a loop highly conserved throughout the actin superfamily (Kabsch and Holmes, 1995). ATP hydrolysis may allow S14 to switch to its preferred rotamer, allowing establishment of new hydrogen bonds with R183 in subdomain 4, and thereby transmit conformational changes to distant parts of the molecule (Otterbein et al, 2001; Graceffa and Dominguez, 2003). We previously suggested that replacement of serine 14 by cysteine might inhibit such changes (Posern et al, 2002). The G15S mutation might also stabilise an ATP-bound actin conformation perhaps through the introduction of new hydrogen bonds between G15S and either R183 or D157 (N McDonald, personal communication), or by constraining the mobility of the conserved loop α-carbon chain. Structural studies might resolve these questions.
The three activating actins share the ability to stabilise F-actin and activate SRF; biochemically, they possess both shared and distinct properties (Figure 7A). Studies of the corresponding yeast mutants also suggest that these mutations are not functionally identical, since the G15S and S14C mutations are recessive lethal, while the V159N mutation remains viable (Chen and Rubenstein, 1995; Belmont and Drubin, 1998; Belmont et al, 1999b). Under G-actin conditions, all the mutants share a reduced ability to interact with gelsolin segments S4–S6, and an increased affinity for profilin. These observations suggest that the mutants less readily adopt conformations exclusively available to wild-type ADP-actin, and perhaps more readily enter a conformation accessible to wild-type ATP-actin. Nevertheless, under G-actin conditions, the three mutants exhibit differential affinity for MAL and for cofilin. Unlike actins G15S and V159N, actin S14C exhibits greatly reduced affinity for ATP, consistent with previous studies of the yeast actins S14C and S14A (Chen et al, 1995; Chen and Rubenstein, 1995; Schuler et al, 1999).
What is the relevance of the activating mutations to the cellular physiology of MAL activation? An exciting possibility is that the actin G15S (and possibly V159N) mutation mimics a directly activating conformation that is adopted by the wild-type protein during normal MAL regulation, either alone or through its association with other proteins. For example, the actin G15S mutation might in some ways be considered equivalent to activating mutations in regulatory GTPases, which lock the proteins in the activated state by inhibiting GTP hydrolysis (Bourne et al, 1991). Actin-related proteins (Arps) are involved in protein complexes controlling numerous cytoplasmic and nuclear processes. Moreover, ATP hydrolysis by Arps has been associated with changes in functional state, such as destabilisation of the branching interaction of the arp2/3 complex with F-actin following ATP hydrolysis on arp2 (Le Clainche et al, 2003). It might be interesting to test the effect of the G15S mutation on the activity of actin-related proteins.
Materials and methods
Plasmids and proteins
Reporters, and expression plasmids for actins, Rho effectors, C3, profilin and cofilin were described previously (Sotiropoulos et al, 1999; Copeland and Treisman, 2002; Miralles et al, 2003). MAL(met) residues 1–631 in pGBT9 were used to screen an NIH3T3 cDNA library in pACT2 (Clontech no. ML4009AH) in Saccharomyces cerevisiae strain Y190 (Clontech), which allows selection on medium with 15–30 mM 3-aminotriazole and quantification of interactions by liquid culture β-galactosidase assay. Point mutant derivatives of the MAL N-terminal RPEL motifs are as specified in the figure legends. GST-MAL(met)(1–171) and its RPEL mutant derivatives were expressed using pET41. MAL(fl)2–261 (including all three RPEL repeats) and gelsolin(S4–6) (Way et al, 1989) were cleaved from appropriate GST fusions and purified by standard techniques.
Transfections and reporter assays
Transient transfections of NIH3T3 cells were carried out using Lipofectamine (Invitrogen) and reporter assays performed as described (Posern et al, 2002). Amounts of expression plasmid used are indicated in the figure legends. For co-immunoprecipitation assays, cells were transfected in 10 cm plates with 1 μg of each MAL and actin plasmid; cells were starved for only 24 h prior to lysis in RIPA buffer. For immunofluorescence assays, cells were seeded on coverslips in six-well plates and starved for 24 h following transfection with 50 ng MAL and 0.5 μg actin constructs.
Actin polymerisation assay
Rabbit skeletal α-actin monomers (2.5 μM, 20% pyrene-labelled) were incubated with test protein for 30 min in G buffer (2 mM Tris–HCl pH 8, 0.3 mM MgCl2, 0.2 mM ATP, 0.2 mM EGTA, 0.2 mM DTT). Critical concentration was estimated as 0.27 μM. For gelsolin measurements, G buffer contained 0.2 mM CaCl2 instead of MgCl2 and EGTA. Actin polymerisation was initiated by addition of 1/20 volume of initiation buffer at room temperature (2 M NaCl, 60 mM MgCl2, 10 mM ATP); end-point pyrene fluorescence was measured 2 h later (Hertzog et al, 2002).
Immunoprecipitations and affinity precipitations
For co-immunoprecipitation assays, extracts of half a 10 cm plate were precipitated for 2 h at 4°C in IP buffer (20 mM Tris pH 8.0, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1% Triton X-100, protease inhibitors) with M2 anti-Flag or anti-HA agarose (Sigma), followed by extensive washing. G-actin supernatant fractions for GST pulldowns, profilin-actin, cofilin-actin and nucleotide determinations were prepared by syringing cells from one 10 cm plate in 500 μl EDTA-free 10 mM Tris pH 8, 50 mM NaCl, 0.2 mM CaCl2 and 0.2 mM DTT containing protease inhibitors (Roche) followed by centrifugation at 436 000 g. Precipitations (200 μl) used 20 μl GST-gelsolin(S4–6) beads, or antibodies in the same buffer at 4°C with three washes. Purified biotinylated nonmuscle actin (1 μg, Cytoskeleton Inc.) was affinity-precipitated using 5 μg GST-MAL precoupled to glutathione-agarose beads in 500 μl 10 mM Tris pH 8, 100 mM NaCl, 0.2 mM CaCl2, 0.2 mM DTT, 5% glycerol and 0.5% Triton X-100 supplemented with 1.6 mg/ml dialysed Escherichia coli protein lysate (Posern et al, 1998). Binding was for 2 h at 4°C, followed by four washes. Following PAGE and transfer to PVDF membrane, biotin-actin was detected by streptavidin–peroxidase conjugate (Sigma). GST-MAL-binding proteins were separated by 6–16% gradient SDS–PAGE and detected by silver staining. Immunoblotting was by standard techniques using anti-Flag peroxidase conjugate (M2, Sigma), anti-HA peroxidase conjugate (3F10, Roche), anti-β-actin (AC15, Sigma), anti-MAL (Miralles et al, 2003), anti-cofilin (Cytoskeleton Inc.) and anti-profilin (Immunoglobe, Wuerzburg, Germany).
Native gels
To generate ADP-actin, nonmuscle actin (100 μg and 99% pure, Cytoskeleton Inc.) in G buffer (5 mM Tris pH 8.0, 0.2 mM CaCl2, 0.2 mM DTT) was treated with hexokinase-agarose (Sigma), 5 mM glucose and 0.5 mM ADP for 2 h at 4°C (Ojala et al, 2002). AMP-PNP-actin was made by incubation of 100 μg actin with 1.2 mM AMP-PNP for 2 h in G buffer. Following centrifugation at 200 000 g for 5 min, the supernatant G-actin fraction was mixed with equimolar amounts of GST fusions in 5 mM Tris pH 8, 0.2 mM CaCl2, 0.2 mM DTT and 0.2 mM ADP or AMP-PNP for 30 min on ice. Samples were loaded with 10% glycerol on prerun 6% native gels in 25 mM Tris, 194 mM glycine, 0.1 mM CaCl2, 0.5 mM DTT, pH 8.0, containing either 0.2 mM ADP or 0.02 mM AMP-PNP. Protein detection was by Coomassie blue staining or immunoblotting.
Nucleotide determination
For nucleotide analysis by luciferase assay (Chen et al, 1995; Chen and Rubenstein, 1995; Schuler et al, 1999), Flag-actin immunoprecipitate from a 10 cm plate was hydrolysed in 6% perchloric acid (100 μl) and neutralised with 5 M K2CO3 (14 μl). The luciferase enzymatic ATP assay (ApoGlow, Cambrex Biosciences) was performed according to the manufacturer's instructions, with ADP determination by conversion to ATP prior to measurement and calibration with nucleotide standards. For nucleotide determination by HPLC (Rosenblatt et al, 1995), precipitates were boiled in 130 μl 10 mM Tris pH 7.4 and 8 M urea for 5 min, filtered through 10 kDa microspin columns (Amicon Inc.) and resolved on a Mono-Q 1.6 column (0.1 ml bed) eluted with 100–500 mM NH4HCO3 in HPLC-grade water at 6°C (flow rate 0.1 ml/min) on a Pharmacia SMART apparatus. Quantification of the chromatographs was hampered by low sensitivity of ADP detection, but gave essentially the same results as the luciferase-based assay.
FACS analysis, actin fractionation and immunofluorescence
FACS quantification of F-actin or G-actin content, actin fractionation lysates, and immunofluorescence were as described (Geneste et al, 2002; Posern et al, 2002). Micrographs were taken either by a conventional Zeiss Axioplan II microscope or a Zeiss LSM 510 confocal laser scanning microscope using a Plan-Neofluar × 63 objective. For quantification, 80–120 cells were assessed under each condition for predominantly nuclear MAL staining.
Supplementary Material
Supplementary Figure S1
Acknowledgments
We thank members of the laboratory, Michael Way and Caroline Hill for helpful discussions and/or comments on the manuscript. We thank Derek Davies for FACS services and Graham Clark for assistance with F-actin quantification and ATP determinations. GP, FM and SG were supported by fellowships from EMBO, EU and Boehringer Ingelheim Fonds, respectively. This work was funded by Cancer Research UK.
References
- Belmont LD, Drubin DG (1998) The yeast V159N actin mutant reveals roles for actin dynamics in vivo. J Cell Biol 142: 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont LD, Orlova A, Drubin DG, Egelman EH (1999a) A change in actin conformation associated with filament instability after Pi release. Proc Natl Acad Sci USA 96: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont LD, Patterson GM, Drubin DG (1999b) New actin mutants allow further characterization of the nucleotide binding cleft and drug binding sites. J Cell Sci 112: 1325–1336 [DOI] [PubMed] [Google Scholar]
- Bettinger BT, Gilbert DM, Amberg DC (2004) Actin up in the nucleus. Nat Rev Mol Cell Biol 5: 410–415 [DOI] [PubMed] [Google Scholar]
- Bourne HR, Sanders DA, McCormick F (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117–127 [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol 136: 1307–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cen B, Selvaraj A, Burgess RC, Hitzler JK, Ma Z, Morris SW, Prywes R (2003) Megakaryoblastic leukemia 1, a potent transcriptional coactivator for serum response factor (SRF), is required for serum induction of SRF target genes. Mol Cell Biol 23: 6597–6608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Peng J, Pedram M, Swenson CA, Rubenstein PA (1995) The effect of the S14A mutation on the conformation and thermostability of Saccharomyces cerevisiae G-actin and its interaction with adenine nucleotides. J Biol Chem 270: 11415–11423 [DOI] [PubMed] [Google Scholar]
- Chen X, Rubenstein PA (1995) A mutation in an ATP-binding loop of Saccharomyces cerevisiae actin (S14A) causes a temperature-sensitive phenotype in vivo and in vitro. J Biol Chem 270: 11406–11414 [DOI] [PubMed] [Google Scholar]
- Copeland J, Treisman R (2002) The Diaphanous-Related Formin mDia1 controls Serum Response Factor (SRF) activity through its effects on actin polymerisation. Mol Biol Cell 13: 4088–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du KL, Chen M, Li J, Lepore JJ, Mericko P, Parmacek MS (2004) Megakaryoblastic leukemia factor-1 (MKL1) transduces cytoskeletal signals and induces smooth muscle cell differentiation from undifferentiated embryonic stem cells. J Biol Chem 17: 17. [DOI] [PubMed] [Google Scholar]
- Geneste O, Copeland JW, Treisman R (2002) LIM kinase and Diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol 157: 831–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graceffa P, Dominguez R (2003) Crystal structure of monomeric actin in the ATP state. Structural basis of nucleotide-dependent actin dynamics. J Biol Chem 278: 34172–34180, Epub 32003 Jun 34117 [DOI] [PubMed] [Google Scholar]
- Hertzog M, Yarmola EG, Didry D, Bubb MR, Carlier MF (2002) Control of actin dynamics by proteins made of beta-thymosin repeats: the actobindin family. J Biol Chem 277: 14786–14792 [DOI] [PubMed] [Google Scholar]
- Hill CS, Wynne JK, Treisman RH (1995) The Rho family GTPases RhoA, Rac1 and CDC42hs regulate transcriptional activation by SRF. Cell 81: 1159–1170 [DOI] [PubMed] [Google Scholar]
- Kabsch W, Holmes KC (1995) The actin fold. FASEB J 9: 167–174 [DOI] [PubMed] [Google Scholar]
- Laham LE, Lamb JA, Allen PG, Janmey PA (1993) Selective binding of gelsolin to actin monomers containing ADP. J Biol Chem 268: 14202–14207 [PubMed] [Google Scholar]
- Le Clainche C, Pantaloni D, Carlier MF (2003) ATP hydrolysis on actin-related protein 2/3 complex causes debranching of dendritic actin arrays. Proc Natl Acad Sci USA 100: 6337–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubimova A, Bershadsky AD, Ben-Ze'ev A (1999) Autoregulation of actin synthesis requires the 3′-UTR of actin mRNA and protects cells from actin overproduction. J Cell Biochem 76: 1–12 [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou A-I, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113: 329–342 [DOI] [PubMed] [Google Scholar]
- Murai K, Treisman R (2002) Interaction of Serum Response Factor (SRF) with the Elk-1 B-Box inhibits RhoA–actin signalling to SRF and potentiates transcriptional activation by Elk-1. Mol Cell Biol 22: 7083–7092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala PJ, Paavilainen VO, Vartiainen MK, Tuma R, Weeds AG, Lappalainen P (2002) The two ADF-H domains of twinfilin play functionally distinct roles in interactions with actin monomers. Mol Biol Cell 13: 3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olave IA, Reck-Peterson SL, Crabtree GR (2002) Nuclear actin and actin-related proteins in chromatin remodeling. Annu Rev Biochem 71: 755–781 [DOI] [PubMed] [Google Scholar]
- Otterbein LR, Graceffa P, Dominguez R (2001) The crystal structure of uncomplexed actin in the ADP state. Science 293: 708–711 [DOI] [PubMed] [Google Scholar]
- Perelroizen I, Carlier MF, Pantaloni D (1995) Binding of divalent cation and nucleotide to G-actin in the presence of profilin. J Biol Chem 270: 1501–1508 [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112: 453–465 [DOI] [PubMed] [Google Scholar]
- Posern G, Sotiropoulos A, Treisman R (2002) Mutant actins reveal a role for unpolymerised actin in control of transcription by Serum Response Factor. Mol Biol Cell 13: 4167–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G, Zheng J, Knudsen BS, Kardinal C, Muller KB, Voss J, Shishido T, Cowburn D, Cheng G, Wang B, Kruh GD, Burrell SK, Jacobson CA, Lenz DM, Zamborelli TJ, Adermann K, Hanafusa H, Feller SM (1998) Development of highly selective SH3 binding peptides for Crk and CRKL which disrupt Crk-complexes with DOCK180, SoS and C3G. Oncogene 16: 1903–1912 [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Peluso P, Mitchison TJ (1995) The bulk of unpolymerized actin in Xenopus egg extracts is ATP-bound. Mol Biol Cell 6: 227–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A (2002) Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol 156: 737–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler H, Korenbaum E, Schutt CE, Lindberg U, Karlsson R (1999) Mutational analysis of Ser14 and Asp157 in the nucleotide-binding site of beta-actin. Eur J Biochem 265: 210–220 [DOI] [PubMed] [Google Scholar]
- Sotiropoulos A, Gineitis D, Copeland J, Treisman R (1999) Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 98: 159–169 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428: 185–189 [DOI] [PubMed] [Google Scholar]
- Way M, Gooch J, Pope B, Weeds AG (1989) Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J Cell Biol 109: 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1