

Abstract
Phospholipase Cγ1 (PLCγ1) has been reported to be expressed predominantly in T cells and to play an important role in T-cell receptor signaling. Here we show that PLCγ1 is expressed throughout B-cell development, with high expression in B-cell progenitors, and is involved in pre-B-cell receptor (pre-BCR) signaling. Reduced expression of PLCγ1, in the absence of PLCγ2 (PLCγ1+/−PLCγ2−/−), impedes early B-cell development at the pro-B- to pre-B-cell transition and impairs immunoglobulin heavy chain allelic exclusion, hallmarks of defective pre-BCR signaling. In contrast, early B-cell development is largely normal, whereas late B-cell maturation is impaired in the absence of PLCγ2 alone (PLCγ2−/−) and overexpression of PLCγ1 in PLCγ2−/− mice fails to restore BCR-mediated B-cell proliferation and maturation. These studies reveal an essential role of PLCγ1, distinct from that of PLCγ2, in B-cell development.
Keywords: allelic exclusion, B-cell development, phospholipase Cγ1, pre-B-cell receptor
Introduction
B-cell development and maturation depend on transduction of signals by the pre-B-cell receptor (pre-BCR) and BCR. Pro-B cells begin the process of immunoglobulin (Ig) heavy (H) gene rearrangement, and successful rearrangement of Ig H chain variable (V), diversity (D), and joining (J) gene segments leads to the formation of the pre-BCR, which contains the newly generated H chain in complex with the VpreB/λ5 surrogate light (L) chain. Signals from the pre-BCR instruct pre-B cells to expand and undergo rearrangement of Ig L chain V and J gene segments. A successfully rearranged L chain in combination with the previously rearranged H chain generates a surface IgM form of the BCR, which marks the cells as immature B cells (Healy and Goodnow, 1998; Hardy and Hayakawa, 2001). Immature B cells emerge from the bone marrow into the spleen. In the spleen, signals transduced by the BCR direct immature B cells to mature through transitional B cells of type 1 (T1) and type 2 (T2) stages, and thereafter to long-lived follicular (FO) B cells (Martin and Kearney, 2001). Disruption of the pre-BCR or BCR arrests B-cell development at the pro-B to pre-B or at the immature to mature B-cell transitions, respectively (Kitamura et al, 1991; Lam et al, 1997).
The pre-BCR and BCR complexes both contain Igα and Igβ signal transduction subunits and have signal transduction pathway components in common (Hombach et al, 1990). Thus, pre-BCR/BCR signaling relies on sequential activation of members of three distinct families of cytoplasmic protein tyrosine kinases, including Lyn, Syk, and Btk, on recruitment and tyrosine phosphorylation of the adapter protein, B-cell linker protein (BLNK), and on recruitment and activation of the lipid kinase, phosphatidylinositol 3-kinase (PI3K) (Kurosaki, 1999; Reth et al, 2000; Niiro and Clark, 2002). An important consequence of these events is activation of phospholipase Cγ (PLCγ), which hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), both of which are required second messengers for cellular responses (Rhee and Bae, 1997).
PLCγ has two isoforms, PLCγ1 and PLCγ2, which display 50% identity at the amino-acid level. PLCγ1 is ubiquitously expressed, while PLCγ2 is predominantly expressed in hematopoietic cells (Rhee and Bae, 1997). Studies of PLCγ2-deficient mice revealed profoundly impaired late B-cell development and disrupted B-cell function, demonstrating that PLCγ2 plays an essential role in B-cell development and function (Hashimoto et al, 2000; Wang et al, 2000). PLCγ1-deficient mice die at midgestation during embryogenesis (Ji et al, 1997), which precludes their analysis to determine the role, if any, of PLCγ1 in B-cell development and function in vivo. Nevertheless, although PLCγ1 is predominantly expressed in T cells and plays an important role in T-cell receptor (TCR) signaling (Park et al, 1991; Secrist et al, 1991; Irvin et al, 2000), studies of cell lines suggest that PLCγ1 may also be involved in BCR signaling (Coggeshall et al, 1992; Roifman and Wang, 1992). We report here studies of PLCγ2-deficient mice that are heterozygous for PLCγ1 deficiency. Our results demonstrate that PLCγ1 plays an important and as yet unappreciated role in pre-BCR-mediated early B-cell development.
Results
Expression patterns and activation of PLCγ1 and PLCγ2 following pre-BCR and BCR stimulation in primary B cells
Previous studies with cell lines have shown that PLCγ2 is the predominant PLCγ isoform in B cells (Coggeshall et al., 1992; Takata et al, 1995), leading to the notion that PLCγ2 is sufficient for BCR signaling (Kurosaki et al, 2000; Kurosaki, 2002). This perception was substantiated by studies of PLCγ2-deficient mice, which demonstrated that PLCγ2 deficiency results in defective B-cell development and function (Hashimoto et al, 2000; Wang et al, 2000). However, only late B-cell development is affected in PLCγ2-deficient mice, suggesting the existence of a redundant pathway to that of PLCγ2 in pre-BCR and even in BCR signaling. PLCγ1 is the only other PLCγ family member and its role in B-cell development and function is not known. To assess the role of PLCγ1 in B-cell development, we first examined the level of PLCγ1 expression in primary B cells at different stages of development. Pro-B/pre-B (B220+IgM−), immature (B220+IgM+), and mature (B220hiIgM+) B cells were purified from mouse bone marrow, whereas transitional T1 (CD23−CD21loIgMhi), T2 (CD23+CD21hiIgMhi), and mature (CD23+CD21intIgMlo) B cells were purified from mouse spleen by FACS sorting. Both PLCγ1 and PLCγ2 were expressed in each of these subsets of B cells (Figure 1A). Interestingly, PLCγ1 was more highly expressed in pro/pre-B cells than in immature and mature B cells, whereas PLCγ2 was highly expressed in all subsets of B cells (Figure 1A). These data demonstrate that, in addition to PLCγ2, PLCγ1 is expressed at all stages of B-cell development with higher expression in early B-cell progenitors.
Figure 1.
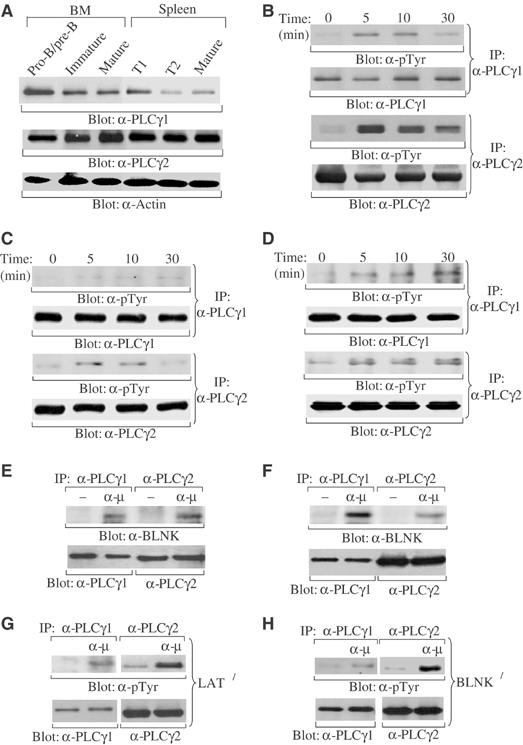
Expression of PLCγ1 and PLCγ2 during B-cell development and their activation by pre-BCR or BCR. (A) PLCγ1 and PLCγ2 protein expression in primary B cells at different developmental stages. Pro-B/pre-B (B220+IgM−), immature (B220+IgM+), and mature (B220hiIgM+) B cells were sorted from bone marrow cells, whereas T1 (CD23−CD21−IgM+), T2 (CD23+CD21hiIgMhi), and FO mature (CD23+CD21intIgMlo) B cells were sorted from splenocytes. Total cell lysates (4 μg) were subjected to direct Western blot with anti-PLCγ1 (α-PLCγ1), anti-PLCγ2 (α-PLCγ2), or anti-actin (α-actin) antibodies. (B) Activation of both PLCγ1 and PLCγ2 by BCR engagement in immature/mature B cells. Purified splenic B cells, which consist of immature/mature B cells, were stimulated with anti-μ antibodies for the indicated time. Cell lysates were immunoprecipitated (IP) with antibodies to PLCγ1 or PLCγ2. Precipitated proteins were immunoblotted with anti-phosphorylated tyrosine (α-pTyr), anti-PLCγ1 (α-PLCγ1), or anti-PLCγ2 (α-PLCγ2) antibodies. (C) Activation of both PLCγ1 and PLCγ2 by pre-BCR engagement in late pro-B cells. Bone marrow-derived pro-B cells were stimulated with anti-μ antibodies for the indicated time. Cell lysates were subjected to Western blot as described in (B). (D) Activation of both PLCγ1 and PLCγ2 by pre-BCR engagement in a pre-B-cell line. 70Z/3 cells were stimulated with anti-μ antibodies for the indicated time. Cell lysates were subjected to Western blot as described in (B). (E) Association of PLCγ1 and PLCγ2 with BLNK upon pre-BCR engagement. Bone marrow-derived pro-B cells were stimulated with anti-μ antibodies for 5 min. Cell lysates were immunoprecipitated with antibodies specific for PLCγ1 or PLCγ2. Precipitated proteins were immunoblotted with anti-BLNK (α-BLNK), anti-PLCγ1, or anti-PLCγ2 antibodies as indicated. (F) Association of PLCγ1 and PLCγ2 with BLNK upon BCR engagement. Purified splenic B cells were stimulated with anti-μ antibodies for 5 min. Cell lysates were subjected to Western blot as described in (E). (G) Activation of both PLCγ1 and PLCγ2 by BCR engagement in LAT-deficient B cells. Purified splenic B cells from LAT−/− mice were stimulated with anti-μ antibodies for 5 min. Cell lysates were subjected to Western blot as described in (B). (H) Activation of both PLCγ1 and PLCγ2 by pre-BCR engagement in BLNK-deficient pro-B cells. Bone marrow-derived pro-B cells from BLNK−/− mice were stimulated with anti-μ antibodies for 5 min. Cell lysates were subjected to Western blot as described in (B).
Expression of PLCγ1 in immature and mature B cells raises the possibility that both PLCγ1 and PLCγ2 are involved in BCR signaling. Therefore, we examined activation of PLCγ1 and PLCγ2 by BCR engagement in primary B cells. Primary immature/mature B cells were isolated from mouse spleen and stimulated with antibodies to μ proteins, a component of the BCR complex. Although the level of PLCγ1 expression is relatively low in immature/mature B cells, BCR engagement activated both PLCγ2 and PLCγ1 as measured by protein tyrosine phosphorylation (Figure 1B). The kinetics of PLCγ1 and PLCγ2 activation following BCR ligation were comparable (Figure 1B). These data demonstrate that, in addition to PLCγ2, PLCγ1 can be activated by BCR in primary immature/mature B cells.
High expression of PLCγ1 and PLCγ2 in pro/pre-B cells suggests that both PLCγs might also be involved in pre-BCR signaling. To test this possibility, we examined activation of PLCγ1 and PLCγ2 by pre-BCR engagement in B-cell progenitors. Mouse bone marrow cells were cultured in the presence of IL-7 for 5 days to derive pro-B cells, which were shown by FACS analysis to be late pro-B cells (B220+CD43+), as previously reported (Ray et al, 1998; Flemming et al, 2003) (data not shown). Engagement of the pre-BCR was accomplished using antibodies to μ proteins, a component of pre-BCR complex, which engage the pre-BCR and initiate signaling from this receptor (Flemming et al, 2003). Both PLCγ1 and PLCγ2 were activated, albeit to a lesser extent than was observed in immature/mature B cells upon BCR ligation, following pre-BCR engagement and the kinetics of PLCγ1 and PLCγ2 activation were comparable (Figure 1C). In addition, activation of PLCγ1 and PLCγ2 by pre-BCR engagement was confirmed in a pre-B-cell line, 70Z/3 (Paige et al, 1981), in which antibodies to μ proteins activated not only PLCγ2 but also PLCγ1 with comparable kinetics (Figure 1D). These data demonstrate that engagement of the pre-BCR activates both PLCγ1 and PLCγ2 in primary pre-B cells and in a pre-B-cell line, although the level of their activation is low. However, later studies show that the low-level activation of PLCγ1 and PLCγ2 is sufficient for pre-BCR functions (see below).
It is known that the adaptor protein BLNK is critical for activation of PLCγ2 upon BCR engagement whereas LAT, together with SLP-76, plays an essential role in activation of PLCγ1 upon TCR ligation. Recent studies have demonstrated that LAT and SLP-76 are expressed along with BLNK in pre-B cells and participate in pre-BCR signaling (Su and Jumaa, 2003). Thus, we examined whether activation of PLCγ1 in B cells is via BLNK or LAT/SLP-76. Engagement of the pre-BCR in wild-type pre-B cells induced association of PLCγ1 and PLCγ2 with BLNK (Figure 1E), whereas ligation of BCR induced association of PLCγ1 and PLCγ2 with BLNK in immature/mature B cells (Figure 1F). Moreover, BCR engagement activated PLCγ1 and PLCγ2 in LAT-deficient B cells (Figure 1G). Thus, engagement of either the pre-BCR or BCR could activate PLCγ1 as well as PLCγ2 via BLNK. We failed to detect association of LAT or SLP-76 with PLCγ1/PLCγ2 upon pre-BCR or BCR engagement (data not shown), probably due to low levels of expression of LAT and SLP-76 in B cells (Su and Jumaa, 2003). Nonetheless, activation of PLCγ1 and PLCγ2 upon pre-BCR engagement was observed in BLNK-deficient pre-B cells (Figure 1H). In addition, previous studies have shown that pre-BCR or BCR engagement was capable of inducing Ca2+ flux in BLNK-deficient B cells, although to a lesser extent relative to wild-type B cells (Jumaa et al, 1999; Pappu et al, 1999; Su and Jumaa, 2003). Importantly, this residual Ca2+ flux was abolished in BLNK and LAT double-deficient pre-B cells, demonstrating that LAT is able to compensate for BLNK deficiency to activate PLCγs upon pre-BCR engagement (Su and Jumaa, 2003). Taken together, both BLNK and LAT/SLP-76 adapter systems are involved in recruitment and activation of PLCγ1 and PLCγ2 upon pre-BCR or BCR engagement.
Early B-cell development is severely impaired in PLCγ1+/−PLCγ2−/− mice
Studies of PLCγ2-deficient mice have revealed that PLCγ2 is essential for BCR-mediated B-cell maturation and function (Hashimoto et al, 2000; Wang et al, 2000). However, PLCγ1-deficient mice die at day 9 of embryonic gestation, which prevents in vivo analysis of the role of PLCγ1 in B-cell development (Ji et al, 1997).
To assess the role of PLCγ1 in B-cell development, we generated PLCγ1+/−PLCγ2−/− mice by crossing PLCγ1+/− with PLCγ2+/− mice. The level of expression of either PLCγ isoform was gene dosage dependent in that PLCγ1 protein level was reduced in splenocytes of PLCγ1+/− relative to PLCγ1+/+ mice (Figure 2A), as previously observed (Ji et al, 1997), and PLCγ2 protein was reduced in splenocytes of PLCγ2+/− relative to PLCγ2+/+ mice (Figure 2A). The reduction of PLCγ protein levels was quantified by densitometry, which showed that each PLCγ isoform was reduced by approximately 50% in heterozygous (+/−) compared to wild-type (+/+) splenocytes (Figure 2B).
Figure 2.
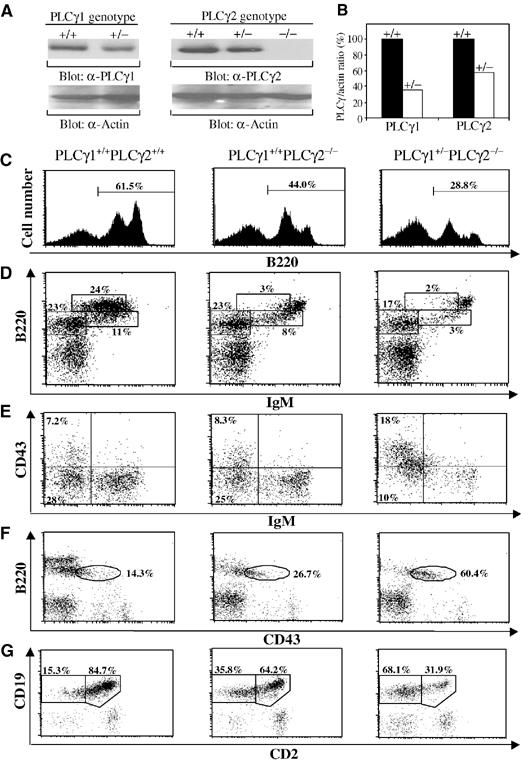
Gene dosage-dependent expression of PLCγ protein and impaired early B-cell development in PLCγ1+/−PLCγ2−/− mice. (A) Reduced expression levels of PLCγ1 and PLCγ2 protein in PLCγ1+/− and PLCγ2+/− splenocytes, respectively. PLCγ protein levels were determined by direct Western blot of cell lysates (20 μg) from splenocytes with wild-type (+/+), heterozygous PLCγ-deficient (+/−), or homozygous PLCγ-deficient (−/−) genotype using anti-PLCγ1 (α-PLCγ1), anti-PLCγ2 (α-PLCγ2), or anti-actin (α-actin) antibodies. (B) Quantitation of levels of PLCγ1 and PLCγ2 protein. The band densities of PLCγ and actin in (A) were quantified by densitometry. The ratios of the band densities of PLCγs versus actin were determined in wild-type (+/+) and heterozygous (+/−) splenocytes. PLCγ:actin ratios in +/+ splenocytes were assigned a value of 100% and the corresponding ratios in +/− splenocytes were calculated accordingly. (C–G) Bone marrow cells from mice of the indicated genotypes were stained with a combination of antibodies to B220, IgM, and CD43, or to CD19 and CD2. (C) Reduction of total B cells in bone marrow of PLCγ1+/−PLCγ2−/− mice. Histograms show the percentage of B220+ cells within the lymphoid cell gate. (D) FACS analysis with B220 and IgM staining. Percentages indicate cells in the gated lymphoid populations. (E) FACS analysis with CD43 and IgM staining of B220+ gated cells. Percentages indicate cells in the gated lymphoid populations. (F) FACS analysis with B220 and CD43 staining. Percentages indicate cells in B220+ gated cells. (G) FACS analysis with CD19 and CD2 staining. Percentages indicate cells in CD19+ gated cells. The FACS data shown are representative of eight mice per genotype.
To determine the effects of reduced expression of PLCγ1 in the absence of PLCγ2 on BCR signaling, we compared BCR-induced Ca2+ flux in wild-type, PLCγ2−/−, and PLCγ1+/−PLCγ2−/− B cells. Reduction in PLCγ1 expression levels by disruption of one allele further diminished BCR-induced Ca2+ mobilization in PLCγ2-deficient B cells (Supplementary Results and Supplementary Figure 1).
To determine whether PLCγ1 is involved in B-cell development, we tested whether reduced levels of PLCγ1 in the absence of PLCγ2 affected B-cell development. First, we examined B-cell development in bone marrow derived from wild-type, PLCγ2−/−, and PLCγ1+/−PLCγ2−/− mice. Although the total number of bone marrow cells was comparable among these mice, the population of B220+ cells in bone marrow was decreased in PLCγ2−/− relative to wild-type mice and was further markedly decreased in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice (Table I and Figure 2C). Specifically, the mature B-cell population (B220hiIgM+) was dramatically decreased, whereas immature B-cell population (B220+IgM+) was normal in PLCγ2−/− relative to wild-type mice as in previous studies (Table I and Figure 2D) (Wang et al, 2000). In contrast, the immature B-cell population (B220+IgM+) was dramatically decreased in the bone marrow of PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− or wild-type mice (Table I and Figure 2D). In addition, the population of mature (B220hiIgM+) B cells was further decreased in the bone marrow of PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice, which already had a dramatic decrease in mature B cells (Table I and Figure 2D).
Table 1.
B-cell population in the bone marrow and spleens derived from wild-type, PLCγ2−/−, and PLCγ1+/−PLCγ2−/− mice (in millions)
| Bone marrow | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B-lineage cells (B220+) |
Pro-B cells (B220+CD43+) |
Pre-B cells (B220+CD43−IgM−) |
Immature (B220+IgM+) |
Mature (B220hiIgM+) |
||||||
| Number | Percent | Number | Percent | Number | Percent | Number | Percent | Number | Percent | |
 | ||||||||||
| Spleen | ||||||||||
| B-lineage cells (B220+) |
MZ (CD23−CD21hiIgMhi) |
T1 (CD23−CD21loIgMhi) |
T2 (CD23+CD21hiIgMhi) |
FO (CD23+CD21intIgMlo) |
||||||
| |
Number |
Percent |
Number |
Percent |
Number |
Percent |
Number |
Percent |
Number |
Percent |
| The phenotype of lymphocytes was determined by flow cytometry. The numbers of each subset of B cells and their percentages in gated lymphoid cells were determined for each mouse, and the mean value and standard deviation were calculated. For bone marrow n = 8 and for spleen n=7. *P<0.01, **P<0.05 (Student's t-test). | ||||||||||
A decrease in the number and percentage of immature B cells in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice suggested impaired development prior to the immature B-cell stage. To pinpoint the stage at which B-cell development is impaired in PLCγ1+/−PLCγ2−/− mice, expression of cell surface markers that distinguish pro-B (B220+CD19+CD43+CD2−) from pre-B (B220+CD19+CD43−CD2+) cells was examined. The pre-B-cell population (B220+CD43−IgM−) was slightly decreased in PLCγ2−/− but markedly decreased in PLCγ1+/−PLCγ2−/− relative to wild-type mice (Table I and Figure 2E). Although the number of pro-B cells (B220+CD43+IgM−) was comparable among mice with all three genotypes (Table I), the proportion of these cells among bone marrow lymphocytes was markedly increased in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− or wild-type mice (Table I and Figure 2E). Among B220+ cells, the increase in the proportion of pro-B cells (B220+CD43+) in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− or wild-type mice was even more obvious (Figure 2F). Consistent with the increased proportion of pro-B cells and decreased pre-B-cell population in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− or wild-type mice, the ratio of early B-cell progenitors (CD19+CD2−) versus B cells at a later developmental stage (CD19+CD2+) was dramatically increased in PLCγ1+/−PLCγ2−/− relative to wild-type or even PLCγ2−/− mice (Figure 2G). However, in PLCγ1+/− mice, which have reduced levels of PLCγ1 but normal levels of PLCγ2, no abnormal B-cell development was detected (data not shown). Taken together, in the absence of PLCγ2, reduced expression of PLCγ1 results in impaired early B-cell development at the pro-B-cell stage immediately prior to the pre-BCR checkpoint. This developmental block is not observed in mice in missing PLCγ2 alone.
Defective IL-7 receptor signaling could also account for impaired B-cell development at the pro-B-cell stage in PLCγ1+/−PLCγ2−/− mice (Peschon et al, 1994). Therefore, we examined IL-7 receptor signaling in PLCγ1+/−PLCγ2−/− mice. Defective IL-7 receptor signaling is characterized by failure to utilize the normally frequently used VHJ558 family of VH genes, which lie furthest away from DH and JH genes at the 5′ end of the VH cluster, in rearranged Ig H chain genes (Corcoran et al, 1998). PCR analysis, performed using primers specific for the VHJ558 family of VH genes, identified rearranged genes containing VHJ558 gene segments in bone marrow cells derived from wild-type, PLCγ2−/−, and PLCγ1+/−PLCγ2−/− mice (Figure 3A). In contrast, VHJ558-containing rearranged Ig H chain genes were barely detectable in JAK3-deficient bone marrow cells, which have defective IL-7 receptor signaling, or in embryonic stem cells, which have no Ig gene rearrangement (Figure 3A). Thus, IL-7 receptor signaling in PLCγ1+/−PLCγ2−/− mice appears to be normal, suggesting that the arrest of B-cell development in these mice at the pro-B-cell stage is not due to defective IL-7 receptor signaling.
Figure 3.
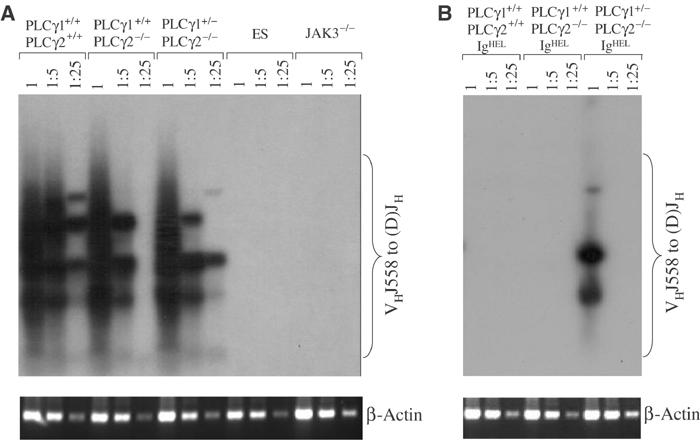
Normal Ig H chain rearrangement but impaired allelic exclusion in PLCγ1+/−PLCγ2−/− mice. (A) Normal Ig H chain rearrangement in PLCγ1+/−PLCγ2−/− mice. Serial dilutions of genomic DNA extracted from bone marrow cells derived from mice of the indicated genotypes were subjected to two rounds of PCR using primers designed to amplify rearranged VHJ558DJH genes. PCR products were subjected to Southern blot with a fragment spanning JH3 and JH4 as a probe. PCR products from the β-actin gene served as controls for the quantity of the genomic DNA. (B) Impaired Ig H chain allelic exclusion in PLCγ1+/−PLCγ2−/− mice. Serial dilutions of genomic DNA extracted from bone marrow cells derived from IgHEL BCR transgenic mice of the indicated genotypes were subjected to PCR and Southern blot analysis as described in (A). PCR products from the β-actin gene served as controls for the quantity of the genomic DNA. The figure shown is representative of two independent experiments.
Pre-BCR signals guide the transition from pro-B to pre-B cells (Shinkai et al, 1992). Our observation that the pre-B-cell population was reduced in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice suggested an impaired transition from pro-B to pre-B cells and, therefore, defective signaling from pre-BCR. The fact that we could detect endogenous Ig H chain gene rearrangements in wild-type, PLCγ2−/−, and PLCγ1+/−PLCγ2−/− mice made it possible for us to examine directly the effect of PLCγ2 deficiency, with or without PLCγ1 reduction, on pre-BCR signals by evaluating the ability of these mice to undergo Ig H chain allelic exclusion. For these experiments, we made use of mice that are transgenic for IgHEL, which is the hen egg lysozyme (HEL)-specific BCR. Expression of the IgHEL BCR initiates in pro-B cells and should efficiently shut off rearrangement of endogenous Ig H chain genes in wild-type mice, indicative of allelic exclusion. PCR analysis using VHJ558-specific primers showed that no VHJ558-containing Ig H gene rearrangements were detectable in bone marrow cells derived from IgHEL transgenic wild-type and PLCγ2−/− mice (Figure 3B), indicating that signals from the pre-BCR were sufficient, even in the absence of PLCγ2, to turn off rearrangement of endogenous Ig H chain genes. In contrast, VHJ558-containing Ig H chain genes were detectable in bone marrow cells derived from IgHEL transgenic PLCγ1+/−PLCγ2−/− mice (Figure 3B), although to a lesser extent than was observed in bone marrow cells derived from nontransgenic mice (Figure 3A). In addition, upsurge of IgM+ but non-HEL binding B cells, an indication of endogenous Ig gene rearrangement, was observed in PLCγ1+/−PLCγ2−/− IgHEL transgenic mice (Supplementary Results and Supplementary Figure 2). Taken together, these data show that endogenous Ig H chain gene rearrangement continues in PLCγ1+/−PLCγ2−/− mice, despite the presence of a productively rearranged transgenic BCR. Thus, signaling from the pre-BCR is impaired in pro-B cells that are both missing PLCγ2 and have reduced levels of PLCγ1, leading to a leakage of allelic exclusion.
B-cell maturation is more severely impaired in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice
Impaired early B-cell development could affect late B-cell maturation. Therefore, we examined the effect of PLCγ1 reduction in the absence of PLCγ2 on B-cell maturation in the spleen. As previously observed, PLCγ2 deficiency alone resulted in a decrease in the total number of B220+ B cells in the spleen (Table I and Figure 4A) (Wang et al, 2000). However, the population of B220+ B cells was further dramatically decreased in spleens derived from PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice (Table I and Figure 4A). To further analyze the impairment of B-cell maturation in PLCγ1+/−PLCγ2−/− mice, the expression of cell surface markers that distinguish different stages of B-cell maturation was examined. Splenic B cells can be separated into T1, T2, FO, and marginal zone (MZ) subpopulations on the basis of staining with anti-IgM, anti-CD21, and anti-CD23 (Oliver et al, 1999; Martin and Kearney, 2000). CD23+ cells include CD21hiIgMhi T2 and CD21intIgMlo FO B cells. CD23− B cells include CD21loIgMhi T1 and CD21hiIgMhi MZ B cells. The population of FO mature B cells (CD23+CD21intIgMlo) was dramatically decreased, whereas the population of T2 B cells (CD23+CD21hiIgMhi) was increased, in PLCγ2−/− relative to wild-type mice (Table I and Figure 4B). In contrast, FO mature B cells were further decreased and T2 B cells were also markedly decreased in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice (Table I and Figure 4B). In addition, T1 B cells (CD23−CD21loIgMhi) were relatively normal in PLCγ2−/− mice, but noticeably decreased in PLCγ1+/−PLCγ2−/− relative to wild-type mice (Table I and Figure 4C). However, MZ B cells (CD23−CD21hiIgMhi) were comparable among mice of all genotypes (Figure 4C). Based on expression of IgD and IgM, splenocytes can also be separated into IgMhiIgD− (T1), IgMhiIgD+ (T2), and IgMloIgD+ (FO) B cells (Loder et al, 1999). FACS analysis of splenic lymphocytes revealed that PLCγ2 deficiency alone led to a marked decrease in the population of FO mature B cells, a slight increase in the population of T2 B cells, but relatively normal population of T1 B cells in the spleen (Figure 4D). Whereas the population of T1 B cells was comparable, T2 and FO B-cell populations were further decreased in the spleens of PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice (Figure 4D). In addition, more severely impaired B-cell maturation was also observed in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice based on expression of AA4, CD23, and IgM (Supplementary Results and Supplementary Figure 3). Although the three staining methods used identified different percentages of T1 and T2 B cells, the results, when taken together, reveal that transitional B cells were noticeably decreased in PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice. Thus, in the absence of PLCγ2, reduction of PLCγ1 further impedes B-cell maturation.
Figure 4.
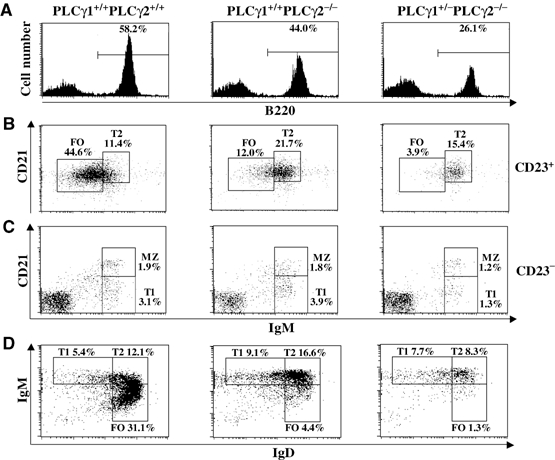
More severely impeded B-cell maturation in PLCγ1+/−PLCγ2−/− than in PLCγ2−/− mice. Splenocytes from mice of the indicated genotypes were stained with a combination of antibodies to IgM, CD21, and CD23, or to IgM, IgD, and B220. (A) Further reduction of total B cells in the spleens of PLCγ1+/−PLCγ2−/− relative to PLCγ2−/− mice. Histograms show the percentage of B220+ cells within the lymphoid cell gate. (B) FACS analysis with CD21 and IgM staining of CD23+ gated cells. (C) FACS analysis with CD21 and IgM staining of CD23− gated cells. (D) FACS analysis with IgM and IgD staining of B220+ gated cells. The numbers indicate the percentage of gated cells within the lymphoid populations for (B–D). The data shown are representative of eight mice per genotype.
Moreover, we demonstrated that severely impaired early B-cell development and late maturation in PLCγ1+/−PLCγ2−/− mice were B-cell autonomous (Supplementary Results and Supplementary Figure 4).
PLCγ1 and PLCγ2 play distinct roles in B-cell development
Although we have demonstrated that, in addition to PLCγ2, PLCγ1 plays an important role in B-cell development, it is not known whether the role of each PLCγ isoform is redundant or unique. Given that early B-cell development is relatively normal in PLCγ2−/− (Figure 2 and Table I) and PLCγ1+/− (data not shown) mice, we conclude that normal levels of PLCγ1 in the absence of PLCγ2 as well as normal levels of PLCγ2 in the presence of reduced PLCγ1 are able to support pre-BCR-mediated early B-cell development. These results suggest that PLCγ1 and PLCγ2 play redundant roles in pre-BCR signaling. If this is also the case for BCR signaling, overexpression of PLCγ1 should be able to overcome the normally low level of PLCγ1 expression in immature/mature B cells and compensate for PLCγ2 deficiency in BCR-mediated B-cell maturation. To test this hypothesis, we assessed the ability of PLCγ1 to restore BCR function in PLCγ2-deficient B cells. To achieve high-level gene transfer into murine primary B cells, we employed a previously developed retrovirus-mediated gene transfer with bone marrow reconstitution strategy (Wen et al, 2001). First, PLCγ2-deficient bone marrow cells were infected in vitro with a retrovirus encoding PLCγ1 or PLCγ2, an internal ribosome entry site (IRES), and green fluorescent protein (GFP). In addition, PLCγ2-deficient or wild-type bone marrow cells transduced with a retrovirus encoding GFP alone served as negative or positive controls. Next, retrovirally transduced bone marrow cells were transplanted into sublethally irradiated JAK3-deficient mice. Subsequently, the retrovirally transduced bone marrow repopulated the lymphocytes, including B cells, in recipient JAK3-deficient mice. Virus-transduced cells were identified by virtue of the GFP gene. For example, GFP-positive B cells in JAK3-deficient recipients of PLCγ1-IRES-GFP retrovirus-transduced PLCγ2-deficient bone marrow represented PLCγ1-overexpressing PLCγ2-deficient B cells. Cells transduced with PLCγ1-IRES-GFP or PLCγ2-IRES-GFP virus, sorted by FACS on the basis of expression of GFP and B220, exhibited higher levels of PLCγ1 or PLCγ2 expression, respectively, relative to wild-type cells transduced with GFP virus (Figure 5A). To determine whether high expression of PLCγ1 or PLCγ2 has an effect on BCR signaling, we examined BCR-induced Ca2+ flux in these GFP-positive, virally transduced PLCγ2-deficient B cells. Interestingly, enforced expression of high levels of PLCγ1 in PLCγ2-deficient B cells restored BCR-induced Ca2+ flux to the same level as was observed in wild-type B cells transduced with GFP alone, although to a lesser extent than was observed upon reintroduction of PLCγ2 (Figure 5B). However, enforced expression of GFP alone did not restore BCR-mediated Ca2+ flux in PLCγ2-deficient B cells (Figure 5B). Thus, overexpression of PLCγ1 can compensate for the absence of PLCγ2 in BCR-mediated Ca2+ flux.
Figure 5.
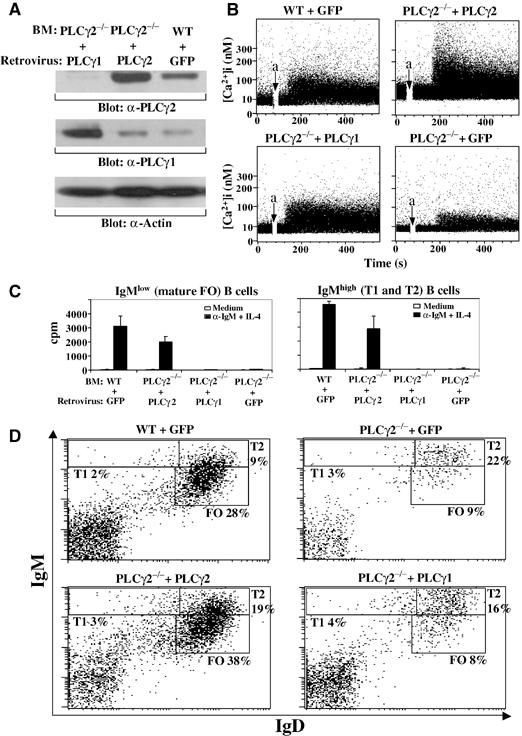
Restoration of BCR-induced Ca2+ flux but not proliferation or development of PLCγ2-deficient B cells by enforced expression of PLCγ1. JAK3-deficient mice were reconstituted with wild-type bone marrow transduced with IRES-GFP retroviruses (WT+GFP) or with PLCγ2-deficient bone marrow transduced with IRES-GFP retroviruses (PLCγ2−/−+GFP), PLCγ2-IRES-GFP retroviruses (PLCγ2−/−+PLCγ2), or PLCγ1-IRES-GFP retroviruses (PLCγ2−/−+PLCγ1). The recipient mice were analyzed 2 months after the reconstitution. (A) Enforced expression of PLCγ1 or PLCγ2 in PLCγ2-deficient B cells. Total cell lysates (4 μg) of B cells derived from the recipient mice were subjected to direct Western blot with anti-PLCγ1 (α-PLCγ1), anti-PLCγ2 (α-PLCγ2), or anti-actin (α-actin) antibodies. (B) Restoration of BCR-induced Ca2+ flux in PLCγ2-deficient B cells by enforced expression of PLCγ1 or PLCγ2. Splenocytes derived from the recipient mice were incubated with indo-1AM and PE-conjugated anti-B220 antibody. Then, cells were washed and stimulated with anti-μ antibodies. Induction of Ca2+ mobilization was determined in GFP/B220 double-positive cells by flow cytometry. Anti-μ antibodies were added at the time indicated by arrow a. (C) Failure of enforced expression of PLCγ1 to restore BCR-mediated proliferation of PLCγ2-deficient transitional and mature B cells. Transitional (GFP+/IgMhi) and mature (GFP+/IgMlo) B cells were sorted from the splenocytes of recipient mice and stimulated with anti-IgM antibodies plus IL-4. Proliferation was determined by incorporation of [3H]thymidine. (D) Failure of enforced expression of PLCγ1 to restore development of PLCγ2-deficient B cells. Splenocytes derived from the recipient mice were stained with antibodies to IgM and IgD. GFP-positive lymphocytes were gated and the percentages of cells in gated GFP+ cells are indicated. The figure shown is representative of three independent analyses.
We next examined whether restoration of BCR-induced Ca2+ flux in PLCγ2-deficient B cells by overexpression of PLCγ1 is able to support BCR-mediated proliferation of two B-cell populations in the spleen, including IgMhi B cells, which contain transitional B cells (T1 and T2), and IgMlo cells, which contain mature FO B cells. Splenocytes from the recipient mice were sorted into transitional (GFP+/IgMhi) and mature (GFP+/IgMlo) B cells. Whereas reintroduction of PLCγ2 restored proliferation of both populations of B cells, enforced expression of PLCγ1 surprisingly failed to restore the ability of PLCγ2-deficient transitional and mature B cells to proliferate (Figure 5C). In addition, we demonstrated that enforced expression of PLCγ1 could only partially compensate for the absence of PLCγ2 in BAFF-mediated B-cell survival (Supplementary Results and Supplementary Figure 5).
Lastly, we sought to determine whether BCR-mediated signaling via PLCγ1 could compensate for the absence of PLCγ2 in B-cell development. We examined B-cell development in JAK3-deficient recipients that were transplanted with wild-type bone marrow transduced with IRES-GFP retrovirus or with PLCγ2-deficient bone marrow transduced with either PLCγ2-IRES-GFP, PLCγ1-IRES-GFP, or IRES-GFP retrovirus. The mature FO B-cell (IgMloIgD+) population was as low in JAK3-deficient mice transplanted with PLCγ1-IRES-GFP retrovirus-transduced PLCγ2-deficient bone marrow as it was in control JAK3-deficient recipient mice that had received IRES-GFP retrovirus-transduced PLCγ2-deficient bone marrow (Figure 5D). In contrast, the mature FO B-cell population developed normally in JAK3-deficient recipient mice transplanted with either PLCγ2-IRES-GFP retrovirus-transduced PLCγ2-deficient bone marrow or IRES-GFP retrovirus-transduced wild-type bone marrow (Figure 5D). Therefore, enforced expression of PLCγ1 failed to replace PLCγ2 in providing signals required for B-cell development. Taken together, these results suggest that BCR-mediated Ca2+ flux is an insufficient stimulus for B-cell proliferation and development and that PLCγ1 cannot replace PLCγ2 in BCR-mediated B-cell proliferation and development.
Discussion
It has been thought that PLCγ1 plays an important role in TCR signaling, whereas PLCγ2 plays an important role in BCR signaling (Kurosaki et al, 2000; Kurosaki, 2002). This concept is based on the observation that PLCγ1 is predominantly expressed in T cells and activated upon TCR ligation (Park et al, 1991; Secrist et al, 1991; Irvin et al, 2000) whereas PLCγ2 is predominantly expressed in B cells and activated upon BCR ligation (Coggeshall et al, 1992; Hempel et al, 1992). This concept is further reinforced by the finding that disruption of the PLCγ2 gene in the DT40 chicken B-cell line impairs BCR signal transduction (Takata et al, 1995), whereas disruption of PLCγ1 in the Jurkat T-cell line severely impairs TCR-induced Ca2+ flux and NFAT activation (Irvin et al, 2000). In addition, B-cell, but not T-cell, development and function are severely impaired in PLCγ2-deficient mice (Hashimoto et al, 2000; Wang et al, 2000). Nevertheless, the observations that PLCγ1 is expressed at moderate levels in B-cell lines (Coggeshall et al, 1992; Roifman and Wang, 1992) and that PLCγ2 deficiency results in only an incomplete block in B-cell development at the immature to mature B-cell transition point (Hashimoto et al, 2000; Wang et al, 2000) raises the possibility that PLCγ1 makes an important contribution to signal transduction by the pre-BCR and/or BCR. Unfortunately, evaluation of the effect of PLCγ1 deficiency on T-cell versus B-cell development and function has not been possible because of the early embryonic lethality of PLCγ1-deficient mice (Ji et al, 1997).
The present studies demonstrate that PLCγ1 is highly expressed in early B-cell progenitors and weakly expressed in more mature B cells, which represents a difference between immortalized B-cell lines (Coggeshall et al, 1992; Roifman and Wang, 1992) and primary B cells, and that engagement of the pre-BCR or BCR activates not only PLCγ2 but also PLCγ1. In studies of PLCγ2-deficient mice that are also heterozygous for PLCγ1 deficiency, we can discern a role for PLCγ1 in pre-BCR signaling when PLCγ2 is missing. Thus, in the absence of PLCγ2, reduction of PLCγ1 expression by disruption of one PLCγ1 allele results in partial escape from allelic exclusion and severely impaired B-cell development at an earlier stage than is observed in the absence of PLCγ2 alone.
Previous studies have shown that LAT together with SLP-76 plays a central role in TCR-induced PLCγ1 activation (Jackman et al, 1995; Zhang et al, 1998) whereas BLNK specifically plays a critical role in pre-BCR/BCR-mediated PLCγ2 activation (Fu et al, 1998; Wienands et al, 1998). Recent studies demonstrate that LAT and SLP-76 are also expressed in pre-B cells and participate in pre-BCR-mediated signaling (Su and Jumaa, 2003). It is tempting to speculate that ligation of pre-BCR/BCR activates PLCγ1 via LAT/SLP-76 and PLCγ2 via BLNK. However, our findings demonstrate that both BLNK and LAT/SLP-76 adapter systems participate in activation of PLCγ1 and PLCγ2 in B cells, although their participations are not equal. It seems that activation of PLCγ1 and PLCγ2 depends more on BLNK than LAT/SLP-76. BLNK deficiency severely affects pre-BCR/BCR-induced Ca2+ flux (Jumaa et al, 1999; Pappu et al, 1999) whereas LAT deficiency has no detectable defects in pre-BCR/BCR signaling (Zhang et al, 1999). However, the contribution of LAT to pre-BCR-mediated PLCγs activation is revealed by the finding that the residual pre-BCR-mediated Ca2+ flux in BLNK-deficient B cells is completely abolished by BLNK and LAT double deficiency (Su and Jumaa, 2003).
Another striking phenotype of the PLCγ1+/−PLCγ2−/− mice is their partial failure, when made transgenic for a productively rearranged Ig H chain gene, to exclude rearrangement of endogenous Ig H chain genes. Failure to undergo Ig H chain allelic exclusion has previously been described in μMT mice (Kitamura and Rajewsky, 1992) and more recently in mice deficient in both Syk and ZAP-70 kinases (Schweighoffer et al, 2003); however, our study is the first to identify signaling molecules, that is, PLCγ1 and PLCγ2, downstream of tyrosine kinases that are required for Ig H chain allelic exclusion. Interestingly, the finding that Ig H chain allelic exclusion fails totally in Syk−/−ZAP-70−/− mice, is only partially inhibited in Syk−/− mice, and is completely unaffected in ZAP-70−/− mice (Schweighoffer et al, 2003) is consistent with the notion that the signaling threshold required for the pre-BCR to drive the cessation of Ig H chain gene rearrangement is low. In this context, our observation that allelic exclusion proceeds normally in PLCγ2−/− mice and is partially inhibited in PLCγ1+/−PLCγ2−/− mice suggests that, in the absence of PLCγ2, the weak signal transmitted via the pre-BCR by PLCγ1 is sufficient to terminate rearrangement of endogenous Ig H chain genes but only when PLCγ1 is expressed at normal levels.
An important remaining question raised by the results of our studies is whether the contributions made by PLCγ1 and PLCγ2 to pre-BCR/BCR signal transduction are quantitatively different but qualitatively identical or, instead, are qualitatively different. Studies have shown that the magnitude and duration of Ca2+ flux and PKC activation, both of which are dependent on the phospholipase activity of PLCγ, differentially affect activation of subsets of transcription regulators that ultimately determine the survival, proliferation, and differentiation of B cells (Healy and Goodnow, 1998). Thus, the extent of BCR ligation may be integrated at the level of the degree of both PLCγ1 and PLCγ2 activation, thereby converting the quantity of BCR ligation into the total amount of PLCγ phospholipase activity, hence into the magnitude and duration of Ca2+ flux and PKC activation, and ultimately into the nature of the B-cell response. However, our observation that overexpression of PLCγ1 in PLCγ2-deficient B cells restores BCR-induced Ca2+ flux but is unable to support B-cell proliferation or maturation and only slightly restores BAFF-mediated B-cell survival raises the possibility that PLCγ1 and PLCγ2 make qualitatively different contributions to pre-BCR/BCR signal transduction. In this context, PLCγ1 may couple the pre-BCR/BCR signaling pathway to different downstream signaling molecules than does PLCγ2. This possibility presumes, of course, that PLCγ1 and PLCγ2 have signaling functions apart from their activity as phospholipases. Evidence that such is the case has been provided by studies showing that the mitogenic activity of PLCγ1 in fibroblasts is independent of its phospholipase activity (Smith et al, 1994, 1996), in that a catalytically inactive mutant of PLCγ1 elicited a full mitogenic response (Smith et al, 1994; Huang et al, 1995). Studies of structural units other than the catalytic domain that can support signaling by PLCγ1 have thus far focused on its SH3 domain. The SH3 domain of PLCγ1 has been shown to promote growth of PC12 cells (Bae et al, 1998) and NIH 3T3 (Smith et al, 1994; Huang et al, 1995). Importantly, a recent study has demonstrated that the SH3 domain of PLCγ1 displays physiological guanine nucleotide exchange factor (GEF) activity for the nuclear GTPase, PIKE, which accounts for the mitogenic properties of PLCγ1 (Ye et al, 2002). These observations are consistent with the notion that the GEF activities of PLCγ1 and/or PLCγ2 SH3 domains, either in addition to or instead of their activity as phospholipases, may be required for pre-BCR/BCR-mediated B-cell proliferation and development. The extent to which the PLCγ2 SH3 domain also exhibits GEF activity and, if so, whether PLCγ1 and PLCγ2 SH3 domains activate different GTPases are currently unknown. Furthermore, it is not known whether differential GEF activities of PLCγ1 versus PLCγ2 explains their differential ability to support B-cell proliferation and development. These represent important remaining questions that continue to be focal points of investigation.
Materials and methods
Mice
PLCγ1+/−PLCγ2−/− mice were generated by appropriate backcrosses of PLCγ2+/− mice (Wang et al, 2000) with PLCγ1+/− mice (Ji et al, 1997). IgHEL transgenic mice with different PLCγ1+/− and PLCγ2−/− backgrounds were generated by appropriate backcrosses of PLCγ1+/−PLCγ2+/− mice with IgHEL transgenic mice (C57BL/6 MD4) (Goodnow et al, 1988). LAT−/− mice were as described (Zhang et al, 1999). BLNK−/− mice were purchased from Jackson Laboratories (Bar Harbor, ME).
VDJ rearrangement
Bone marrow cells were treated with Gey's solution to remove red blood cells. A total of 107 cells were lysed in 100 mM Tris–HCl (pH 8.5), 200 mM NaCl, 5 mM EDTA, 0.2% SDS and 200 μg/ml proteinase K at 55°C for 12 h. Genomic DNA was precipitated with 0.7 volume isopropanol, washed once with 70% ethanol, air dried, and resuspended in 500 μl TE. The genomic DNA was quantified by semiquantitative PCR amplification of the β-actin gene with the following primers: 5′ primer, ACTCCTATGTGGGTGACGAG; 3′ primer, CAGGTCCAGACGCAGGATGGC. For VHJ558 to (D)JH rearrangement, two rounds of PCR amplification were used as previously described (Corcoran et al, 1998). First-round PCR reactions were set up using genomic DNA. For the second-round reaction, 1 μl of the first-round PCR product was used. Primers employed in the PCR were the same as previously described (ten Boekel et al, 1995; Corcoran et al, 1998). Specific, 5′ primers corresponding to VHJ558 segments were ACCATGGGATGGAGCTGKATCWTBC (first round) and GTGARGCCTGGGRCTTCAGTGAAG (second round). 3′ primers from the intron downstream of JH4 were AGGCTCTGAGATCCCTAGACAG (first round) and GGGTCTAGACTCTCAGCCGGCTCCCTCAGGG (second round). (K is (G/T); W is (A/T); B is (C/G/T); R is (A/G).) PCR products were separated on 2% agarose gels, transferred to Nytran plus membranes (Schleicher & Schuell), and probed with a 2 kb BamHI–EcoRI fragment spanning JH3 and JH4.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material
Acknowledgments
This work is supported in part by NIH grants RO1 AI52327 (RW) and R01 HL073284 (DW), and by American Cancer Society grant RSG CCG-106204 (DW). We thank Dr Graham Carpenter for PLCγ1+/− mice. We thank Jack A Gorski for critical review of this manuscript.
References
- Bae SS, Lee YH, Chang JS, Galadari SH, Kim YS, Ryu SH, Suh PG (1998) Src homology domains of phospholipase C γ1 inhibit nerve growth factor-induced differentiation of PC12 cells. J Neurochem 71: 178–185 [DOI] [PubMed] [Google Scholar]
- Coggeshall KM, McHugh JC, Altman A (1992) Predominant expression and activation-induced tyrosine phosphorylation of phospholipase C-γ 2 in B lymphocytes. Proc Natl Acad Sci USA 89: 5660–5664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran AE, Riddell A, Krooshoop D, Venkitaraman AR (1998) Impaired immunoglobulin gene rearrangement in mice lacking the IL-7 receptor. Nature 391: 904–907 [DOI] [PubMed] [Google Scholar]
- Flemming A, Brummer T, Reth M, Jumaa H (2003) The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat Immunol 4: 38–43 [DOI] [PubMed] [Google Scholar]
- Fu C, Turck CW, Kurosaki T, Chan AC (1998) BLNK: a central linker protein in B cell activation. Immunity 9: 93–103 [DOI] [PubMed] [Google Scholar]
- Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K (1988) Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334: 676–682 [DOI] [PubMed] [Google Scholar]
- Hardy RR, Hayakawa K (2001) B cell development pathways. Annu Rev Immunol 19: 595–621 [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Takeda K, Inaba M, Sekimata M, Kaisho T, Ikehara S, Homma Y, Akira S, Kurosaki T (2000) Cutting edge: essential role of phospholipase C-γ 2 in B cell development and function. J Immunol 165: 1738–1742 [DOI] [PubMed] [Google Scholar]
- Healy JI, Goodnow CC (1998) Positive versus negative signaling by lymphocyte antigen receptors. Annu Rev Immunol 16: 645–670 [DOI] [PubMed] [Google Scholar]
- Hempel WM, Schatzman RC, DeFranco AL (1992) Tyrosine phosphorylation of phospholipase C-γ 2 upon cross-linking of membrane Ig on murine B lymphocytes. J Immunol 148: 3021–3027 [PubMed] [Google Scholar]
- Hombach J, Tsubata T, Leclercq L, Stappert H, Reth M (1990) Molecular components of the B-cell antigen receptor complex of the IgM class. Nature 343: 760–762 [DOI] [PubMed] [Google Scholar]
- Huang PS, Davis L, Huber H, Goodhart PJ, Wegrzyn RE, Oliff A, Heimbrook DC (1995) An SH3 domain is required for the mitogenic activity of microinjected phospholipase C-γ 1. FEBS Lett 358: 287–292 [DOI] [PubMed] [Google Scholar]
- Irvin BJ, Williams BL, Nilson AE, Maynor HO, Abraham RT (2000) Pleiotropic contributions of phospholipase C-γ1 (PLC-γ1) to T-cell antigen receptor-mediated signaling: reconstitution studies of a PLC-γ1-deficient Jurkat T-cell line. Mol Cell Biol 20: 9149–9161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JK, Motto DG, Sun Q, Tanemoto M, Turck CW, Peltz GA, Koretzky GA, Findell PR (1995) Molecular cloning of SLP-76, a 76-kDa tyrosine phosphoprotein associated with Grb2 in T cells. J Biol Chem 270: 7029–7032 [DOI] [PubMed] [Google Scholar]
- Ji QS, Winnier GE, Niswender KD, Horstman D, Wisdom R, Magnuson MA, Carpenter G (1997) Essential role of the tyrosine kinase substrate phospholipase C-γ1 in mammalian growth and development. Proc Natl Acad Sci USA 94: 2999–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumaa H, Wollscheid B, Mitterer M, Wienands J, Reth M, Nielsen PJ (1999) Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity 11: 547–554 [DOI] [PubMed] [Google Scholar]
- Kitamura D, Rajewsky K (1992) Targeted disruption of μ chain membrane exon causes loss of heavy-chain allelic exclusion. Nature 356: 154–156 [DOI] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kuhn R, Rajewsky K (1991) A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin μ chain gene. Nature 350: 423–426 [DOI] [PubMed] [Google Scholar]
- Kurosaki T (1999) Genetic analysis of B cell antigen receptor signaling. Annu Rev Immunol 17: 555–592 [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Maeda A, Ishiai M, Hashimoto A, Inabe K, Takata M (2000) Regulation of the phospholipase C-γ2 pathway in B cells. Immunol Rev 176: 19–29 [DOI] [PubMed] [Google Scholar]
- Kurosaki T (2002) Regulation of B-cell signal transduction by adaptor proteins. Nat Rev Immunol 2: 354–363 [DOI] [PubMed] [Google Scholar]
- Lam KP, Kuhn R, Rajewsky K (1997) In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90: 1073–1083 [DOI] [PubMed] [Google Scholar]
- Loder F, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, Carsetti R (1999) B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J Exp Med 190: 75–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin F, Kearney JF (2000) Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity 12: 39–49 [DOI] [PubMed] [Google Scholar]
- Martin F, Kearney JF (2001) B1 cells: similarities and differences with other B cell subsets. Curr Opin Immunol 13: 195–201 [DOI] [PubMed] [Google Scholar]
- Niiro H, Clark EA (2002) Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2: 945–956 [DOI] [PubMed] [Google Scholar]
- Oliver AM, Martin F, Kearney JF (1999) IgMhighCD21high lymphocytes enriched in the splenic marginal zone generate effector cells more rapidly than the bulk of follicular B cells. J Immunol 162: 7198–7207 [PubMed] [Google Scholar]
- Paige CJ, Kincade PW, Ralph P (1981) Independent control of immunoglobulin heavy and light chain expression in a murine pre-B-cell line. Nature 292: 631–633 [DOI] [PubMed] [Google Scholar]
- Pappu R, Cheng AM, Li B, Gong Q, Chiu C, Griffin N, White M, Sleckman BP, Chan AC (1999) Requirement for B cell linker protein (BLNK) in B cell development. Science 286: 1949–1954 [DOI] [PubMed] [Google Scholar]
- Park DJ, Rho HW, Rhee SG (1991) CD3 stimulation causes phosphorylation of phospholipase C-γ 1 on serine and tyrosine residues in a human T-cell line. Proc Natl Acad Sci USA 88: 5453–5456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL (1994) Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med 180: 1955–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RJ, Stoddart A, Pennycook JL, Huner HO, Furlonger C, Wu GE, Paige CJ (1998) Stromal cell-independent maturation of IL-7-responsive pro-B cells. J Immunol 160: 5886–5897 [PubMed] [Google Scholar]
- Reth M, Wienands J, Schamel WW (2000) An unsolved problem of the clonal selection theory and the model of an oligomeric B-cell antigen receptor. Immunol Rev 176: 10–18 [DOI] [PubMed] [Google Scholar]
- Rhee SG, Bae YS (1997) Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem 272: 15045–15048 [DOI] [PubMed] [Google Scholar]
- Roifman CM, Wang G (1992) Phospholipase C-γ 1 and phospholipase C-γ 2 are substrates of the B cell antigen receptor associated protein tyrosine kinase. Biochem Biophys Res Commun 183: 411–416 [DOI] [PubMed] [Google Scholar]
- Schweighoffer E, Vanes L, Mathiot A, Nakamura T, Tybulewicz VL (2003) Unexpected requirement for ZAP-70 in pre-B cell development and allelic exclusion. Immunity 18: 523–533 [DOI] [PubMed] [Google Scholar]
- Secrist JP, Karnitz L, Abraham RT (1991) T-cell antigen receptor ligation induces tyrosine phosphorylation of phospholipase C-γ 1. J Biol Chem 266: 12135–12139 [PubMed] [Google Scholar]
- Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, Charron J, Datta M, Young F, Stall AM, Alt FW (1992) RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68: 855–867 [DOI] [PubMed] [Google Scholar]
- Smith MR, Liu YL, Kim SR, Bae YS, Kim CG, Kwon KS, Rhee SG, Kung HF (1996) PLCγ1 Src homology domain induces mitogenesis in quiescent NIH 3T3 fibroblasts. Biochem Biophys Res Commun 222: 186–193 [DOI] [PubMed] [Google Scholar]
- Smith MR, Liu YL, Matthews NT, Rhee SG, Sung WK, Kung HF (1994) Phospholipase C-γ 1 can induce DNA synthesis by a mechanism independent of its lipase activity. Proc Natl Acad Sci USA 91: 6554–6558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YW, Jumaa H (2003) LAT links the pre-BCR to calcium signaling. Immunity 19: 295–305 [DOI] [PubMed] [Google Scholar]
- Takata M, Homma Y, Kurosaki T (1995) Requirement of phospholipase C-γ 2 activation in surface immunoglobulin M-induced B cell apoptosis. J Exp Med 182: 907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Boekel E, Melchers F, Rolink A (1995) The status of Ig loci rearrangements in single cells from different stages of B cell development. Int Immunol 7: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, Ihle JN (2000) Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity 13: 25–35 [DOI] [PubMed] [Google Scholar]
- Wen R, Wang D, McKay C, Bunting KD, Marine JC, Vanin EF, Zambetti GP, Korsmeyer SJ, Ihle JN, Cleveland JL (2001) Jak3 selectively regulates Bax and Bcl-2 expression to promote T-cell development. Mol Cell Biol 21: 678–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienands J, Schweikert J, Wollscheid B, Jumaa H, Nielsen PJ, Reth M (1998) SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J Exp Med 188: 791–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Bae SS, Suh PG, Snyder SH (2002) Phospholipase C γ 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415: 541–544 [DOI] [PubMed] [Google Scholar]
- Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, Samelson LE (1998) LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92: 83–92 [DOI] [PubMed] [Google Scholar]
- Zhang W, Sommers CL, Burshtyn DN, Stebbins CC, DeJarnette JB, Trible RP, Grinberg A, Tsay HC, Jacobs HM, Kessler CM, Long EO, Love PE, Samelson LE (1999) Essential role of LAT in T cell development. Immunity 10: 323–332 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material