

Abstract
The Escherichia coli twin-arginine protein transport (Tat) system is a molecular machine dedicated to the translocation of fully folded substrate proteins across the energy-transducing inner membrane. Complex cofactor-containing Tat substrates, such as the model (NiFe) hydrogenase-2 and trimethylamine N-oxide reductase (TorA) systems, acquire their redox cofactors prior to export from the cell and require to be correctly assembled before transport can proceed. It is likely, therefore, that cellular mechanisms exist to prevent premature export of immature substrates. Using a combination of genetic and biochemical approaches including gene knockouts, signal peptide swapping, complementation, and site-directed mutagenesis, we highlight here this crucial ‘proofreading' or ‘quality control' activity in operation during assembly of complex endogenous Tat substrates. Our experiments successfully uncouple the Tat transport and cofactor-insertion activities of the TorA-specific chaperone TorD and demonstrate unequivocally that TorD recognises the TorA twin-arginine signal peptide. It is proposed that some Tat signal peptides operate in tandem with cognate binding chaperones to orchestrate the assembly and transport of complex enzymes.
Keywords: chaperones, hydrogenase, Tat export pathway, TMAO reductase, twin-arginine signal peptide
Introduction
Targeting of proteins to their sites of physiological function is an important feature of all biological systems. In bacteria, the generation of energy by electron transfer chains involves cofactor-containing enzymes often embedded in the cytoplasmic membrane or located entirely on the extracytoplasmic side. Thus, bacterial growth and survival in many environments depends upon the controlled targeting and transport of redox enzymes to and across this membrane. Studies of Escherichia coli have established that a subset of cofactor-containing exported proteins are synthesised with N-terminal signal peptides containing an SRR × FLK ‘twin-arginine' amino-acid sequence motif (reviewed by Berks et al, 2003). Preproteins bearing twin-arginine signal peptides are translocated post-translationally across the cytoplasmic membrane by the twin-arginine transport (Tat) system (Berks et al, 2003). Biochemical studies have shown the integral membrane proteins TatA, TatB, and TatC to form the core components of the E. coli Tat system. The TatBC unit is believed to form the twin-arginine signal peptide recognition module (Alami et al, 2003), while TatA forms a very large oligomeric ring structure presumed to be the protein-conducting channel itself (Berks et al, 2003). Most remarkably, substrates of the Tat translocase are required to be fully folded before successful translocation can occur (e.g. DeLisa et al, 2003). Key Tat-dependent components of the respiratory chain, (NiFe) hydrogenase and trimethylamine N-oxide (TMAO) reductase, have been shown to acquire their redox cofactors, and even oligomerise, prior to the transport event (Santini et al, 1998; Rodrigue et al, 1999). Recently, however, a number of cofactor-less Tat substrates have been uncovered in E. coli (e.g. Bernhardt and de Boer, 2003), implicating the Tat pathway as a more general export route specifically evolved for folded substrates.
Tat transport is the ultimate point-of-no-return in periplasmic enzyme biosynthesis. It is important that export of cofactor-containing, and in many cases multimeric, Tat-substrate proteins is not performed or attempted until all assembly processes are complete. Mechanisms are likely to exist, therefore, to coordinate the cofactor-insertion and export processes, to prevent wasteful export of immature substrates, or to curb competition between immature and mature proteins for the transporter. Such ‘proofreading' of Tat substrate protein ‘maturity' may operate at different levels on the export pathway. There is good evidence that the Tat translocase cannot transport unfolded proteins and this has been interpreted as a ‘sensing' of substrate folded state by the Tat translocase itself (DeLisa et al, 2003). We demonstrate here, however, that transport of complex Tat substrates can be regulated at an earlier stage on the Tat pathway by dedicated cytoplasmic chaperones that recognise specific twin-arginine signal peptides. Swapping of twin-arginine signal peptides between the unrelated hydrogenase-2 and the TMAO reductase (TorA) systems established the first practicable assay for chaperone-mediated proofreading activity, and we provide evidence that TorD is a twin-arginine signal peptide-binding protein.
Results and discussion
A specific chaperone and the native signal peptide are required for hydrogenase-2 assembly
E. coli (NiFe) hydrogenase-2 is encoded by the hybOABCDEFG operon and the core enzyme consists of a membrane-bound heterodimer of an Fe–S cluster-binding subunit (HybO), together with a partner subunit that binds the Ni–Fe active site cofactor (HybC). The HybOC dimer is targeted to the Tat translocon as a pre-formed unit by a twin-arginine signal peptide located on only one subunit (HybO) (Rodrigue et al, 1999). The HybO Tat signal peptide is typical of all twin-arginine signals in that it contains a polar ‘n-region' preceding the highly conserved twin-arginine motif, which itself is followed by a relatively hydrophobic ‘h-region' and punctuated by a more polar (often positively charged) ‘c-region' that contains the signal peptidase-I AxA cleavage site. Following successful transport, the HybO Tat signal is cleaved off and plays no other role in enzyme structure or function. HybC is also proteolytically processed during the assembly process, but at the C-terminus prior to transport and immediately following the cofactor-loading event (reviewed by Vignais and Colbeau, 2004). HybC itself contains no obvious targeting signals. Following Tat transport, the HybOC catalytic dimer is anchored at the periplasmic side of the cytoplasmic membrane by a single transmembrane segment located at the C-terminus of HybO (Hatzixanthis et al, 2003).
Biosynthesis of the core HybOC dimer requires careful co-ordination of cofactor loading and subunit recruitment before protein transport can proceed. It is crucial that the signal-bearing HybO subunit is not exported before Fe–S cluster insertion is complete or the HybC partner subunit has docked. A recent two-hybrid study implicated the HybE accessory protein as a key player in this ‘proofreading' or ‘quality control' process (Dubini and Sargent, 2003).
Hydrogenase-2 activity can be specifically measured in whole cells using the redox-dye benzyl viologen (BV) as an artificial electron acceptor (Figure 1A): activity is abolished in a strain lacking only the hydrogenase-2 Tat-dependent subunit HybO (Figure 1A). A ΔhybE strain (FTD673) demonstrates very low hydrogenase-2 activity (Figure 1A). Western analysis revealed that HybO and HybC were synthesised at normal levels in the ΔhybE background (Figure 1B); however, while the HybO subunit could be visualised in the membrane fraction (Figure 1B), HybC was no longer tightly associated and was instead located in the cytoplasm (Figure 1B). Targeting of HybO in the ΔhybE strain remained Tat-dependent since co-inactivation of the Tat translocase resulted in the loss of the HybO antigen from the membrane fraction (Figure 1B) and indeed completely destabilised the HybO protein. Note that HybC was C-terminally processed in the ΔhybE strain, indicating that the nickel cofactor had been incorporated (Figure 1C).
Figure 1.
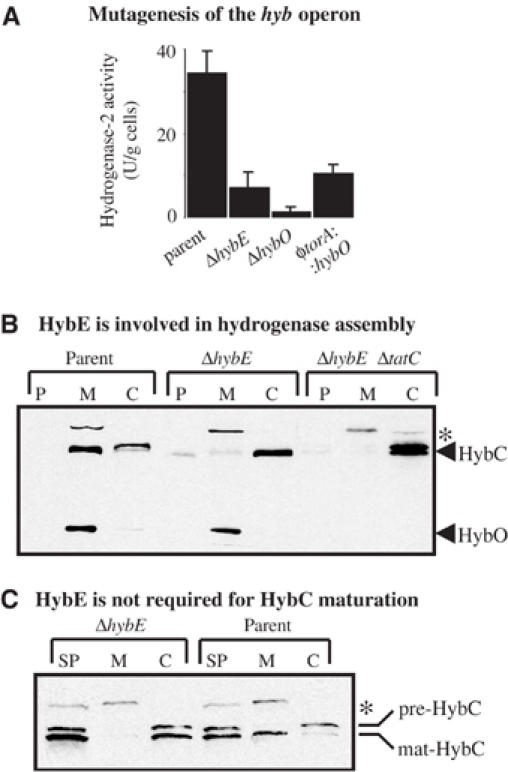
HybE is required for hydrogenase-2 assembly and activity. (A) Hydrogenase-2 activities of mutant strains. Strains MC4100 (‘parent'), FTD673 (‘ΔhybE'), RJ608 (‘ΔhybO'), and RJ603 (‘φtorA∷hybO'), which produce a HybO protein bearing the TorA signal peptide, were grown anaerobically in CR medium containing glycerol and fumarate. Washed whole cells were assayed for hydrogen∷BV oxidoreductase activity with units as μmol BV reduced/min/g cells. (B) Western blot analysis of the core hydrogenase-2 αβ dimer HybOC. Strains MC4100 (‘parent'), FTD673 (‘ΔhybE'), and RJ503 (‘ΔhybE, ΔtatC') were cultured anaerobically in CR medium supplemented with glycerol and fumarate. Cells were fractionated into periplasm (P), total membranes (M), and cytoplasm (C) proteins, separated by SDS–PAGE (14% w/v acrylamide), blotted, and challenged with an antihydrogenase-2 serum. The location of the hydrogenase-2 61 kDa α-subunit (HybC) and 35 kDa β-subunit (HybO) are indicated. (C) Western analysis of the hydrogenase-2 α-subunit HybC. Strains MC4100 (‘parent') and FTD673 (‘ΔhybE') were cultured in CR medium supplemented with glycerol and fumarate. Sphaeroplasts were prepared from whole cells (SP) and further fractionated into membrane (M) and cytoplasm (C). Proteins were separated by SDS–PAGE (10% w/v acrylamide), blotted, and challenged with an antihydrogenase-2 serum. The location of the precursor form of the α-subunit (‘pre-HybC') and the C-terminally processed mature form (‘mat-HybC') are indicated. The asterisks denote a nonspecific immunoreactive band.
These data point to defects in proofreading activity on the hydrogenase-2 assembly pathway rather than protein targeting per se. The normally tight control of assembly and transport has apparently been lost, resulting in premature Tat-dependent targeting of HybO before HybC attachment. The most obvious route to preventing protein export while enabling cofactor insertion and subunit recruitment to proceed would be to mask the HybO signal peptide itself. Taken together with our previous two-hybrid study in which HybE was shown to recognise specifically the signal-bearing HybO precursor (Dubini and Sargent, 2003), these data suggest HybE has a role in coordinating the assembly and export of hydrogenase-2.
In order to investigate further the inter-relationships between E. coli twin-arginine signal peptides and their cognate passenger proteins, we designed a fusion protein. An E. coli strain (RJ603) was constructed in which a signal peptide with a fundamentally different overall primary structure to (NiFe) hydrogenase systems had been swapped for the Tat signal peptide from HybO (Figure 2A). The coding region for the well-characterised E. coli TMAO reductase (TorA) twin-arginine signal peptide (Figure 2A) was precisely fused to the hybO gene at the native hyb locus on the chromosome, thus generating a φtorA∷hybO chimera. Substitution of the Tat signal peptide of the dimeric Fe–S/nickel protein HybOC with that of the monomeric molybdoprotein TorA resulted in very low levels of periplasmic hydrogenase-2 activity (Figure 1A). In order to corroborate these findings, we repeated the experiment in a hybA strain that has enhanced hydrogenase-2-dependent hydrogen∷BV oxidoreductase activity (Dubini et al, 2002). Again, swapping of the HybO signal peptide for that of TorA in the ΔhybA background significantly reduced hydrogenase-2 activity (Figure 2B). Western analysis of the φtorA∷hybO, ΔhybA strain showed very little HybC present in the membrane, while its HybO partner was targeted correctly in a Tat-dependent manner (Figure 2D).
Figure 2.
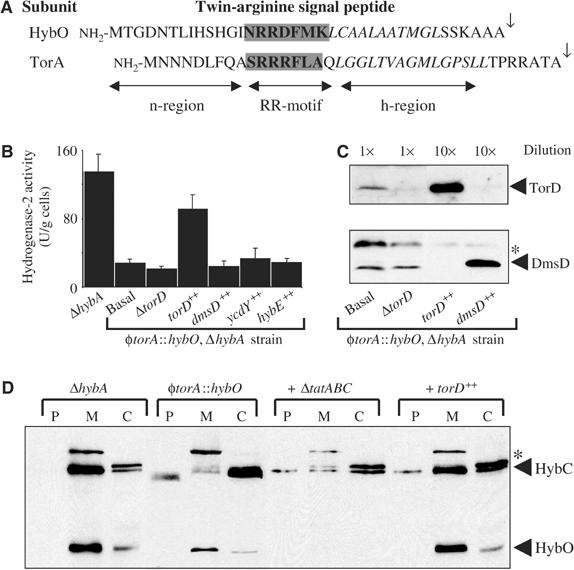
Signal-swapping implicates TorD as a signal peptide chaperone. (A) Amino-acid sequences of the twin-arginine signal peptides from the E. coli hydrogenase-2 β-subunit (HybO) and the TMAO reductase (TorA). The twin-arginine motifs are boxed, the hydrophobic h-regions are shown in italics, and the signal peptidase-I cleavage sites are indicated by the arrows. (B) Hydrogenase-2 activities in mutant strains. Strains RJ606 (‘ΔhybA'), RJ607 φtorA∷hybO, ΔhybA, that produces a HybO protein bearing the TorA signal peptide, and RJ607-D (‘ΔtorD') were grown anaerobically in CR medium containing glycerol and fumarate. In addition, RJ607 was transformed with the pSU series of plasmids that constitutively overproduce TorD (‘torD++'), DmsD (‘dmsD++'), YcdY (‘ycdY++'), or HybE (‘hybE++'), and are grown under identical conditions. Washed whole cells were assayed for hydrogen∷BV oxidoreductase activity with units as μmol BV reduced/min/g cells. (C) Western analysis of TorD and DmsD. Strains RJ607 (φtorA∷hybO, ΔhybA), RJ607-D (‘ΔtorD'), and RJ607 transformed with plasmids overexpressing either torD (‘torD++') or dmsD (‘dmsD++') were cultured anaerobically in CR medium supplemented with glycerol and fumarate. Cells were harvested and resuspended to a concentration of 100 mg (wet weight)/ml (‘1 × '), and a sample diluted to 10 mg/ml (‘10 × '). Identical volumes of protein samples were separated by SDS–PAGE (14% w/v acrylamide), blotted, and challenged with either anti-TorD (top panel) or anti-DmsD (bottom panel) serum at 1:10 000 dilution. (D) Western analysis of the hydrogenase-2 αβ-dimer HybOC. Strains RJ606 (‘ΔhybA'), RJ607 (‘φtorA∷hybO'), RJ607-T (φtorA∷hybO, ΔhybA, ΔtatABC∷KanR; ‘+ ΔtatABC') and RJ607 following transformation with plasmid pSU-torD overexpressing torD (labelled ‘+ torD++') were cultured anaerobically in the presence of glycerol and fumarate. Cells were fractionated in periplasm (P), total membranes (M), and cytoplasm (C), proteins separated by SDS–PAGE (14% w/v acrylamide), blotted, and challenged with an anti-hydrogenase-2 serum. The location of the hydrogenase-2 α-subunit (HybC) and β-subunit (HybO) are indicated. The asterisks denotes a nonspecific immunoreactive band.
TorD operates in tandem with the TorA twin-arginine signal peptide
Clearly, native Tat signal peptides have more far-reaching roles than simply guiding their cognate passenger proteins to the Tat translocon. One possibility is that swapping of the hydrogenase-2 signal peptide has the knock-on effect of also removing any specific signal-binding chaperones, for example, HybE, from the biosynthetic process. Indeed, while the loss of HybE has a slightly more marked effect on hydrogenase activity than the signal swap (Figure 1), probably because this protein may contact HybC and other biosynthetic factors as well as HybO during hydrogenase assembly (Dubini and Sargent, 2003), it is notable that the ΔhybE mutant and the φtorA∷hybO phenotypes are broadly similar. In order to explore this hypothesis further, we considered the possibility that TorA might also employ a signal-binding specific chaperone during its biosynthesis.
In E. coli, TorA is encoded by the torCAD operon, where TorC is a membrane-bound quinol dehydrogenase and torD encodes a cytoplasmic protein (Méjean et al, 1994). In the current literature, TorD has been characterised as a ‘cofactor chaperone' required for efficient molybdopterin guanine dinucleotide (MGD) cofactor insertion into TorA (Pommier et al, 1998; Ilbert et al, 2003, 2004). A crystal structure for a TorD homodimer from Shewanella massilia has recently been described (Tranier et al, 2003), although TorD may also exist as a 22.5 kDa monomer in solution (Tranier et al, 2002). It is clear that TorD makes extensive protein–protein contacts with TorA during the cofactor-loading process (Pommier et al, 1998); however, details of recognition site(s), stoichiometry of binding, or energetic requirements remain ambiguous. Sequence analysis highlighted that TorD is related to an uncharacterised E. coli protein, YcdY, and intriguingly, to the only twin-arginine signal peptide-binding protein described to date, DmsD (Oresnik et al, 2001; Ilbert et al, 2004).
In order to investigate the action of TorD on our φTorA∷HybOC chimera, we introduced a multi-copy plasmid constitutively expressing torD into our φtorA∷hybO, ΔhybA (RJ607) strain (Figure 2B). Very interestingly, the low hydrogenase activity phenotype of RJ607 can be significantly rescued by introduction of our pSU-torD plasmid overproducing TorD (Figure 2B). Furthermore, co-targeting of HybO and HybC was also shown to be restored by co-expression of torD in the φtorA∷hybO strain (Figure 2D). Western analysis suggests that introduction of our pSU-torD plasmid to fumarate-grown cells increases the cellular levels of TorD ∼90 times (Figure 2C). This induction of TorD synthesis suppresses the assembly defects caused by attachment of the TorA signal peptide to hydrogenase-2. The most plausible explanation for these data is that TorD is forced to recognise our engineered φTorA∷HybO chimera and that this event mimics the native hydrogenase-2 proofreading event so closely that assembly is rescued. The basal level of hydrogenase-2 activity in the RJ607 strain (φtorA∷hybO, ΔhybA) is TorD independent, since deletion of the torD gene in this background had no further deleterious effects (Figure 2B).
The restoration of hydrogenase assembly in this system is entirely specific to the TorD protein. Overproduction of the TorD homologs DmsD (confirmed by Western blot in Figure 2C) or YcdY, or the hydrogenase chaperone HybE, in our φtorA∷hybO, ΔhybA background, had no significant effect on hydrogenase activity (Figure 2B). These data are consistent with a proofreading process in which a specific signal-recognising protein regulates export of a Tat substrate until assembly is complete. Since (NiFe) hydrogenase-2 is not at all related to any known molybdoproteins, these data must point very strongly to a previously unidentified role in TorA signal peptide recognition for the TorD protein. This hydrogenase-based assay cannot be biased by the molybdenum cofactor-insertion activity of the TorD protein (e.g. Ilbert et al, 2003). Thus, we have successfully uncoupled a previously submerged Tat proofreading activity from the cofactor-insertion activity, and our assay can be exploited to study the molecular mechanism of proofreading performed by TorD.
Further evidence that TorD binds the TorA signal peptide
In an independent but complementary set of experiments, we took a further genetic approach to assess the physiological role of TorD. Protein–protein interactions can be detected using a bacterial two-hybrid system devised by Karimova et al (1998) based on reconstitution of Bordetella pertussis adenylate cyclase in an E. coli cya mutant. We employed this method to screen a random E. coli genomic library for gene products that could interact with TorD.
TorD was fused to the C-terminus of B. pertussis adenyate cyclase T25 fragment (pT25-TorD) and this fusion was used as ‘bait' in a blind screen of a random library expressing fusions to the N-terminus of B. pertussis adenyate cyclase T18 fragment. We performed six independent rounds of screening and isolated 15 colonies expressing possible interacting partners for TorD. In all, 5/15 isolates harboured identical 1007 bp fragments covering codon 147 of torC through codon 92 of torA (clone 1; Figure 3A). A further 8/15 isolates harboured identical 264 bp fragments covering codon 5–92 of the torA gene (clone 2; Figure 3A). The remaining 2/15 isolates proved to be ‘false positives' containing out-of-frame fusions to small fragments of chromosome.
Figure 3.
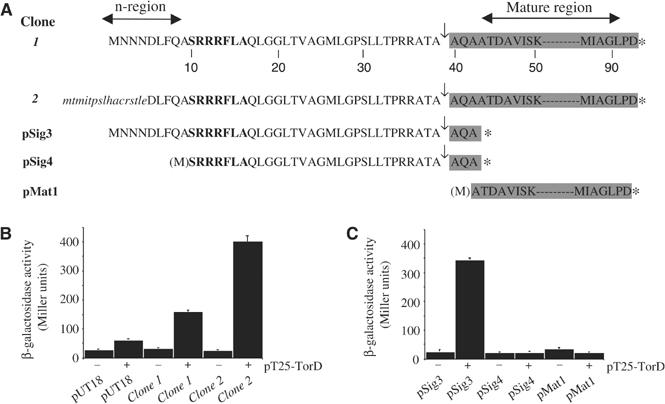
TorD interacts with the TorA signal peptide. (A) Amino-acid sequences of N-terminal TorA fusions to the adenylate cyclase T18 fragment. All TorA-specific residues are shown in upper case and the twin-arginine motif is shown in upper bold case. Residues are numbered from the extreme N-terminus of clone 1. For the clone 2 sequence, residues derived from the lacZ′ gene on the vector are shown in lower case italics. Signal peptidase cleavage sites are indicated by the arrows. Fusion junctions with the T18 fragment are indicated by the asterisks. Sequences corresponding to the mature TorA protein (signal peptide cleaved) are shaded. (B) TorD interacts with the N-terminus of TorA. Interactions were measured between pT25 plasmid (‘−') or pT25-TorD expressing the TorD fusion (‘+') and either pUT18 or the library isolates clone 1 and clone 2 in the reporter strain BTH101. (C) TorD recognises the TorA signal peptide. Interactions were measured between pT25 plasmid (‘−') or pT25-TorD expressing the TorD fusion (‘+') and the engineered fusions to the complete signal peptide, pSig3, the signal peptide lacking n-region, pSig4, and 49 residues of the mature TorA protein, pMat1, in the reporter strain BTH101.
It is striking that, using TorD as bait, 13/15 possible interacting clones expressed in-frame fusions of the TorA N-terminus, including twin-arginine signal peptide, to the T18 fragment. Clone 1 produces a T18 fusion to the intact TorA signal peptide plus 53 amino acids of the mature protein. Clone 2 produces a chimeric T18 protein in which 16 residues derived from the pUT18 vector are fused to the TorA signal (though the first four residues of the n-region are missing) and the identical 53 amino acids from the mature TorA polypeptide (Figure 3A). Subsequent β-galactosidase measurements (reconstitution of adenylate cyclase induces transcription of the lac operon in the reporter strain) confirmed that TorD interacts with both fusions (Figure 3B). However, given the possibility that fusion protein expression levels may differ between clones 1 and 2, and so influence the β-galactosidase activities measured, we decided to deliberately construct N-terminal T18 fusions to TorA using a strategy that would equalise transcription and translation levels as much as possible.
The intact TorA signal peptide, the TorA signal peptide minus the entire n-region, and the region of mature TorA identified in the original screen, were fused separately to the T18 fragment (Figure 3A). The ability of each fusion to be recognised by TorD expressed from pT25-TorD was assessed (Figure 3C). Significant β-galactosidase activity, indicative of strong in vivo protein–protein interaction, was measured only when the clone expressing the full-length TorA signal peptide was co-expressed with the TorD fusion (Figure 3C). No interactions between TorD and either a signal peptide lacking the n-region (Figure 3A and C), or the region of TorA mature protein identified by the random screen (Figure 3A and C), could be detected by this system. These data demonstrate unequivocally that TorD binds directly to the TorA twin-arginine signal peptide. Moreover, this assay suggests that recognition of the TorA signal peptide by TorD is dependent on a completely intact signal n-region. Note that the signal peptide twin-arginine motif itself is so highly conserved between all Tat substrates that this is unlikely to be the principle feature that a TorA-specific chaperone like TorD would recognise. The TorA signal n-region is unique to TorA and may enable TorD to specifically recognise this signal above all others.
Taken together with our studies of the φTorA∷HybOC fusion protein, this work points directly to TorD as a TorA signal peptide-binding protein that operates to harmonise assembly and export of Tat-dependent proteins.
Role of TorD-family proteins in TMAO reductase activity
The twin-arginine signal peptide-binding function of TorD compelled us to re-visit the role of TorD in TorA assembly. Western analysis shows that TorD is a cytoplasmic protein (Figure 4A) and is therefore not part of the periplasmic TMAO reductase system per se. We next constructed single in-frame deletion mutants in the torD and dmsD genes, a torD/dmsD double deletion strain, and a torD/dmsD/ycdY triple deletion mutant. Only strains carrying torD deletions showed any marked effect on periplasmic TMAO reductase activity (Figure 4B), and only the addition of a plasmid expressing TorD was able to rescue the mutant phenotype of the torD/dmsD/ycdY strain (not shown). Our data therefore agree precisely with those of others (Pommier et al, 1998; Ilbert et al, 2004). The loss of TorD causes a significant decrease, but does not abolish, in TorA activity and does not impair targeting of the enzyme to the periplasm. Interestingly, all TorA activity in torD mutants is located in the periplasm (Ilbert et al, 2004), a phenotype unlike that observed for defects in MGD insertion into other enzymes (e.g. Blasco et al, 1998), or Tat transport (e.g. Sargent et al, 1998).
Figure 4.
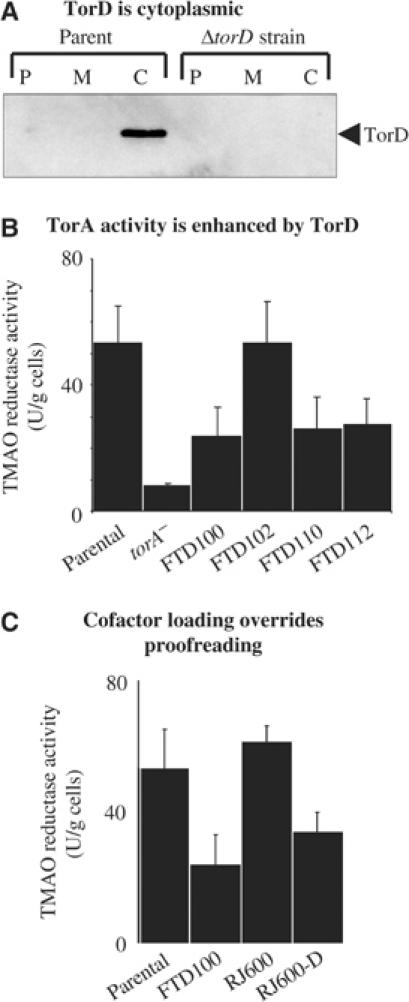
TorD is a cytoplasmic protein with a complex role in TorA assembly. (A) Western analysis of TorD. Strains MC4100 (‘parent') and FTD100 (‘ΔtorD') were cultured anaerobically in CR medium with glycerol and TMAO. Cells were fractionated into periplasm (P), total membranes (M), and cytoplasm (C), proteins separated by SDS–PAGE (14% w/v acrylamide), blotted, and challenged with anti-TorD serum. (B) TMAO reductase activity. Strains MC4100 (‘parent'), LCB628 (torA−), FTD100 (ΔtorD), FTD102 (ΔdmsD), FTD110 (ΔtorD, ΔdmsD), and FTD112 (ΔtorD, ΔdmsD, ΔycdY) were grown anaerobically in CR medium containing glycerol and TMAO. Washed intact cells were assayed for TMAO::BV oxidoreductase activity with units as μmol BV reduced/min/g cells. (C) TMAO reductase activity. Strains MC4100 (‘parent'), FTD100 (ΔtorD), RJ600 (φhybO∷torA) that produces a TorA protein bearing the HybO signal peptide, and RJ600-D (φhybO∷torA, ΔtorD∷KanR) were grown anaerobically in CR medium containing glycerol and TMAO. Whole cells were assayed.
Evidence for a second TorD-binding site on the TorA precursor
In order to test the role of the native twin-arginine signal peptide in TorA enzyme assembly, we next swapped exactly the TorA signal peptide for that of the (NiFe) hydrogenase-2 subunit HybO. The RJ600 strain expressing the φHybO∷TorA chimera displayed periplasmic TMAO reductase activity indistinguishable from that of the parent strain (Figure 4C). The TMAO reductase activity stemming from the φHybO∷TorA enzyme is not being boosted by hydrogenase-specific chaperones since inactivation of either the hya operon, encoding the Tat-dependent hydrogenase-1 (strain RJ601; φhybO∷torA, Δhya) or the hyb operon (strain RJ602; φhybO∷torA, Δhyb), did not modulate TMAO reductase activity (not shown). Furthermore, deletion of torD in the φhybO∷torA strain (yielding strain RJ600-D) resulted in a significant drop in periplasmic TMAO reductase activity—but again of about 50%, precisely that observed for a torD deletion in a strain expressing native torA (Figure 4C). As the TorA signal peptide is not present in these experiments, the phenotype of the φhybO∷torA, ΔtorD (RJ600-D) strain indicates that the reduced TMAO reductase activity observed in other torD strains (Figure 4) is as a result of inefficient MGD insertion rather than a loss of proofreading activity. Thus, when the native TorA/TorD system is studied, the dominant cofactor-insertion activity eclipses the Tat proofreading activity of TorD. Indeed, previous in vitro and in vivo studies of MGD insertion into TMAO reductases had already indicated that the signal peptide was not required for this process (Temple and Rajagopalan, 2000; Ilbert et al, 2004). Given that TorA is a single-subunit enzyme binding a single cofactor, and that transport should be allowed to proceed as soon as cofactor loading is complete, it is perhaps understandable that in the course of evolution the cofactor-insertion role of TorD has taken precedence over the Tat proofreading role. In multi-subunit and multi-cofactor systems like hydrogenase, however, the requirement for an overriding Tat proofreading system remains strong. This again highlights the importance of our unique TorA∷Hydrogenase-2 fusion experiments, which allow the twin-arginine signal peptide-binding proofreading activity of TorD to be completely uncoupled from the overlapping MGD cofactor-loading events. Our data taken together with that of others point towards the existence of at least two TorD-binding sites on the TorA protein: one within the twin-arginine signal peptide and a second within the ‘mature' portion of the protein.
Molecular dissection of TorD activity
We next considered how it was possible that TorD could recognise two different regions of the TorA polypeptide and participate in two different assembly processes. Structural studies of a TorD-family protein have revealed that each protomer consists of two distinct domains (an N-terminal domain and a C-terminal domain) linked by a short conserved ‘hinge' region (Tranier et al, 2003). The TorD protein from S. massilia can be purified as mixtures of stable monomer and dimer (Tranier et al, 2002). Dimerisation is driven by ‘domain swapping' (Tranier et al, 2003) in which the N domain from one protomer interacts with the C domain of the second protomer (Figure 5A). It is not yet clear which forms exist in vivo, or what the functional significance of each form might be. We therefore considered that each domain may play a separate role in TorA assembly.
Figure 5.
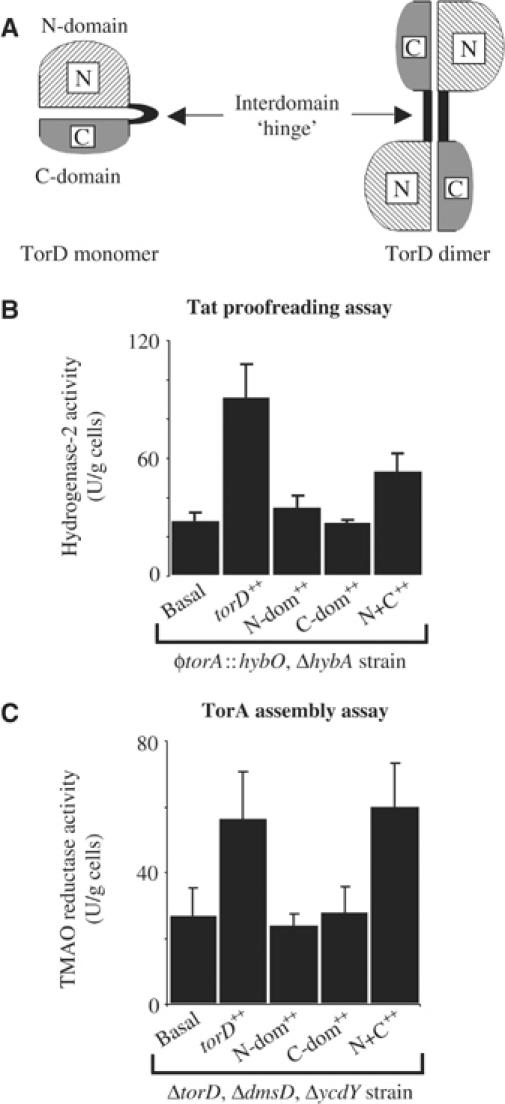
Dissection of TorD activity. (A) Cartoon representation of the predicted structures of TorD-family proteins based on the crystal structure of a TorD dimer from S. massilia. An N-terminal domain (hatched) is separated from a C-terminal domain (shaded) by a short ‘hinge' region. ‘Domain swapping' results in the formation of the TorD homodimer. (B) Hydrogenase-2 activity. Strain RJ607 (φtorA∷hybO, ΔhybA) was grown anaerobically in CR medium containing glycerol and fumarate. In addition, the RJ607 strain was transformed with plasmids that constitutively overproduce TorD from pSU-torD (‘torD++'), the TorD N-terminal domain from pUNI-NDOM (‘N-dom++'), the TorD C-terminal domain from pSU-CDOM (‘C-dom++'), or both separated domains together from plasmids pUNI-NDOM and pSU-CDOM (‘N+C++'), and hydrogenase-2 activity assayed in intact cells. (C) TMAO reductase activity. Strain FTD112 (ΔtorD, ΔdmsD, ΔycdY) was grown anaerobically in CR medium containing glycerol and TMAO. In addition, the FTD112 strain was transformed with plasmids that constitutively overproduce TorD from pSU-torD (‘torD++'), the TorD N-terminal domain from pUNI-NDOM (‘N-dom++'), the TorD C-terminal domain from pSU-CDOM (‘C-dom++'), or both separated domains together from plasmids pUNI-NDOM and pSU-CDOM (‘N+C++'), and TMAO reductase activity assayed in intact cells.
We cloned DNA encoding the TorD N domain (from Methionine-1 through Histidine-125) and C domain (from Methionine-113 through Arginine-199) into our vectors. When expressed separately, neither domain could rescue the mutant phenotypes of either the torD/dmsD/ycdY triple mutant (FTD112) or the φtorA∷hybO, ΔhybA strain (RJ607) (Figure 5C and B, respectively). However, when the separate TorD domains were co-expressed together in the torD/dmsD/ycdY mutant, periplasmic TMAO reductase activity could be restored close to the levels observed in the parent strain (Figure 5C). This clearly indicates that each separate domain is functional and need not be covalently attached in order to enhance the assembly of native TorA. In the case of our hydrogenase-based Tat proofreading assay, co-expression of the separate TorD domains did not significantly rescue the defect in hydrogenase-2 assembly (Figure 5B). Thus, separated TorD domains are essentially incapable of performing a Tat proofreading function (Figure 5B).
We pursued this line of investigation further with a programme of site-directed mutagenesis. Analysis of TorD-family proteins highlighted four conserved polar residues: Serine-95 in the N-terminal domain; Aspartate-124, and Histidine-125 in the interdomain ‘hinge'; and Glutamate-142 in the C-terminal domain (Tranier et al, 2003—E. coli numbering applied), and we separately substituted each with alanine. Western immunoblotting established that the variant TorD proteins were stable and produced at levels equivalent to native TorD in the same system (Figure 6A).
Figure 6.
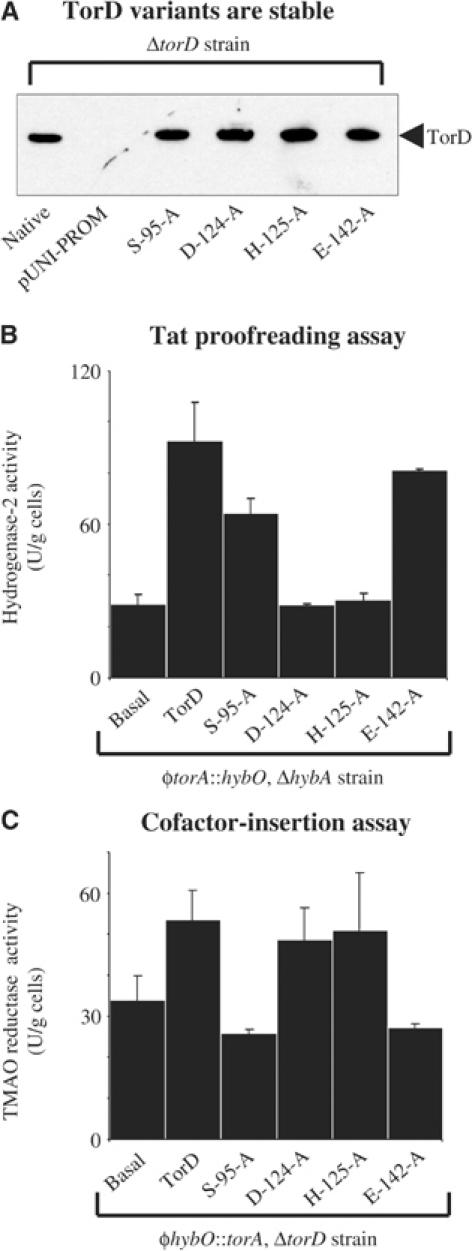
Site-directed mutagenesis of the TorD protein. (A) Expression, production, and relative stability of mutant TorD proteins. The FTD100 strain (ΔtorD) was transformed with the original cloning vector pUNI-PROM (AmpR), pUNI-torD (‘native'), and the four pUNI-torD derivatives expressing the mutant torD genes (as indicated). Cultures were grown anaerobically in CR medium supplemented with glycerol and TMAO. Cell pellets were harvested, washed, and resuspended to 100 mg (wet weight)/ml. Whole-cell proteins were separated by SDS–PAGE (14% w/v acrylamide), blotted, and challenged with anti-TorD. Identical proportions of cellular protein were loaded in each lane. (B) Hydrogenase-2 activity. Strain RJ607 (φtorA∷hybO, ΔhybA) was grown anaerobically in CR medium containing glycerol and fumarate. The RJ607 strain was transformed with plasmids that constitutively overproduce TorD from pUNI-torD (‘torD++') or the four pUNI-torD derivatives expressing the mutant torD genes (as indicated). Whole cells were assayed for hydrogen∷BV oxidoreductase activity with units as μmol BV reduced/min/g cells. (C) TMAO reductase activity. Strain RJ600-D (φhybO∷torA, ΔtorD∷KanR) was grown anaerobically in CR medium containing glycerol and TMAO. The RJ600-D strain was transformed with plasmids that constitutively overproduce TorD from pUNI-torD (‘torD++') or the four pUNI-torD derivatives producing TorD variants (as indicated). Whole cells were assayed for TMAO::BV oxidoreductase activity with units as μmol BV reduced/min/g cells.
In order to study only the MGD cofactor-insertion activities of the mutant proteins without interference from the Tat proofreading activity, we utilised our φhybO∷torA, ΔtorD (RJ600-D) strain that expresses TorA with the HybO signal peptide. Complementation analysis of the φhybO∷torA, ΔtorD (RJ600-D) strain with our TorD S95A, D124A, H125A, and E142A variants revealed that the residues located in the inter-domain hinge (D124 and H125) are not required for efficient cofactor insertion into TorA (Figure 6C). However, N-domain residue S95 and C-domain residue E142 are required to catalyse the enhancement of cofactor insertion (Figure 6C). These data corroborate our experiments with separated TorD domains (Figure 5) and confirm that residues located in both N and C domains are involved in the cofactor-insertion process.
We next employed our hydrogenase-based proofreading assay and introduced the variant TorD proteins to our φtorA∷hybO, ΔhybA (RJ607) strain. In this case, TorD variants D124A and H125A were found to be completely inactive in our proofreading assay (Figure 6B), demonstrating that the hinge region is critical either for TorA signal recognition per se or for a downstream biochemical process normally triggered by signal recognition. Interestingly, the domain-located residues S95 and E142 essential for cofactor loading into TorA (Figure 6C) are dispensable in the TorD-dependent rescue of proofreading activity (Figure 6B). Thus, amino-acid requirements for Tat proofreading by TorD are exactly the opposite to those required for cofactor insertion.
We have highlighted key differences in the biochemical requirements for TorD-dependent Tat proofreading and TorD-dependent cofactor insertion. Signal recognition/proofreading clearly requires an intact TorD protein that would undoubtedly result in placement of the hinge residues (D124 and H125) in the correct orientation to fulfil their function, while domain-located side chains are important in the TorD-dependent cofactor-insertion process. TorD-family proteins can be isolated both in monomeric and dimeric forms, and it remains a possibility that one isoform is predominantly involved in proofreading while the other governs cofactor insertion. In the S. massilia TorD homodimer, the D124 and H125 hinge-located residues seem to be intimately intertwined in the structure (Tranier et al, 2003). The D124 equivalent in S. massilia (D134) is hydrogen-bonded to a histidine sidechain in the opposite protomer (Tranier et al, 2003), perhaps implicating it in dimer formation. This histidine residue is not conserved in the E. coli protein, however. The H125 equivalent in the S. massilia TorD (H135) is buried at the dimer interface and may be hydrogen-bonded to Tyr93 (which is conserved in the E. coli protein). Again, H125 could be implicated in influencing dimer formation; however, both D124 and H125 are part of an extensively intertwined stretch of nearly 30 consecutive sidechains from each protomer (Tranier et al, 2003), and single amino-acid substitutions would therefore not be expected to disrupt this structure. E. coli S95 (also S95 in the S. massilia TorD structure) probably contributes to a ‘polar patch' within the N domain on the surface of the molecule, close to the inter-domain hinge (Tranier et al, 2003). E142 (E151 in S. massilia TorD) is a surface-exposed sidechain in the C-terminal domain (Tranier et al, 2003). Neither S95 nor E142 appear to be intimately involved with TorD dimer formation.
Concluding remarks
In the current work, we have shed light on an aspect of Tat-dependent protein transport that has puzzled researchers from the outset: what prevents premature targeting of cofactor-containing proteins before cofactor loading is complete, and what coordinates export of multi-protein complexes where one protein ‘piggy-backs' on another? A system of ‘proofreading' or ‘quality control' governing these processes has been postulated since the bacterial Tat system was first conceived (e.g. Santini et al, 1998), and here we provide the first experimental evidence for the existence of a wide-ranging chaperone-mediated proofreading mechanism. Previous work had demonstrated that some signal peptides were both organism- and enzyme-specific (Sambasivarao et al, 2000; Blaudeck et al, 2001); however, our work suggests that specialisation can be extended to specific chaperones that operate hand-in-glove with signal peptides harmonising assembly and export processes. A model of how such a system may operate is shown in Figure 7. In E. coli, this system is in operation for TorA (with TorD as chaperone) and hydrogenase-2 (with HybE as chaperone). Biochemical, genetic, and sequence analyses suggest other cofactor-containing and multi-subunit enzymes could utilise similar systems during biosynthesis (Oresnik et al, 2001; Gross and Simon, 2003). Indeed, this work describes an assay that could, in principle, be used to identify signal peptide/chaperone pairs from any system.
Figure 7.
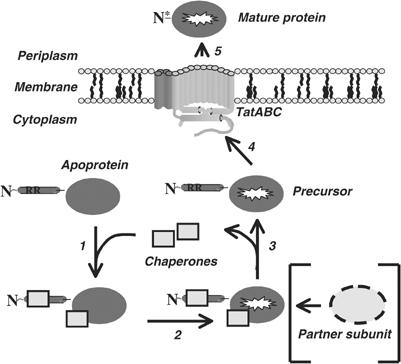
A model for chaperone-mediated proofreading. An apoprotein bearing a twin-arginine signal peptide is released from the ribosome. Step 1: In order to prevent premature export, a proofreading chaperone binds to the twin-arginine signal peptide. In some cases, specific or general chaperones also bind to the mature portion of the protein at this stage. Step 2: Cofactor loading into the mature portion of the protein proceeds. At this stage, binding of partner subunits would also occur, if applicable. Step 3: All chaperones are released. Step 4: Targeting of the precursor to the Tat translocon proceeds. Step 5: Following Tat transport, the signal peptide is cleaved (‘N*') and the mature protein is released to the periplasm. TatABC model is from Palmer et al (2004).
Chaperone-mediated proofreading is an alternative concept of ‘quality control' as previously documented by DeLisa et al (2003). DeLisa et al (2003) demonstrated by utilising alkaline phosphatase fusions that only folded proteins could be transported by the Tat translocase, and postulated that the Tat machinery itself performed a final quality control check before transport (essentially at step 4 in Figure 7). The system of chaperone-mediated proofreading described here would be in operation at a much earlier stage on the Tat pathway, preventing premature targeting and ensuring that only fully assembled substrates are presented to the Tat translocase. Such a system would greatly increase the efficiency of Tat transport by decreasing the incidence of abortive engagements, and reducing competition, with substrates that are not ready for export.
We have shown that the E. coli TorA protein employs a proofreading system involving its cognate signal peptide and a specialised chaperone termed TorD. Most remarkably, the TorA signal peptide/TorD system can be isolated and transposed onto a completely unrelated enzyme, hydrogenase, where it continues to operate almost unabated. Hydrogenases are not at all related to TMAO reductases: the primary structures of the enzymes are unrelated; the subunit composition is dissimilar; the cofactors used are different; and the accessory proteins are not homologous. The fact that the TorA/TorD proofreading system ‘works' for E. coli HybOC suggests that the native hydrogenase-2 system is functionally very similar. This perhaps points to a converging of functionality between unrelated systems rather than a common evolutionary origin. The exchangeability of the TorA/TorD system with the hydrogenase system not only raises questions about the exquisite specificity of recognition (step 1 in Figure 7) but also about what triggers release of the chaperone following assembly (step 3 in Figure 7). It is possible that bound chaperones are simply dislodged by the folding substrate protein, though it should be noted that most chaperone–protein interactions are much more tightly controlled (e.g. Slepenkov and Witt, 2002). This work establishes the existence, and lays the foundation for detailed molecular analysis, of a chaperone-mediated proofreading system operating on the bacterial Tat pathway.
Materials and methods
Bacterial strains and growth conditions
Strains used are based on MC4100 (Table I). Most unmarked deletion mutant strains were constructed by cloning the mutant alleles into pMAK705 (CmR) as previously described (Sargent et al, 1998) and transferring onto the chromosome by the method of Hamilton et al (1989). In short, deletion alleles carry 500–600 bp of DNA upstream of the target gene, linked to a similar amount of downstream DNA to form a plasmid-borne in-frame deletion preserving translation stop signals and Shine–Dalgarno motifs for adjacent sequences. Marked deletions (e.g. KanR) were prepared by cloning resistance cassettes between the upstream and downstream segments of the deletion alleles.
Table 1.
List of E. coli strains featured in this study
| Strain | Relevant genotype | Source |
|---|---|---|
| MC4100 | F-, ΔlacU169, araD139, rpsL150, relA1, ptsF, rbs, flbB5301 | Lab Stocks |
| RJ606 | ΔhybA | This work |
| RJ608 | ΔhybO | This work |
| FTD673 | ΔhybE | This work |
| RJ503 | ΔhybE, ΔtatC::SpecR | This work |
| RJ603 | φtorA::hybO (produces TorA signal peptide on HybO mature protein) | This work |
| RJ607 | φtorA::hybO, ΔhybA | This work |
| RJ607-D | φtorA::hybO, ΔhybA, ΔtorD::KanR | This work |
| RJ607-T | φtorA::hybO, ΔhybA, ΔtatABC::KanR | This work |
| FTD100 | ΔtorD | This work |
| FTD101 | ΔtorD::KanR | This work |
| FTD102 | ΔdmsD | This work |
| FTD110 | ΔtorD, ΔdmsD | This work |
| FTD112 | ΔtorD, ΔdmsD, ΔycdY | This work |
| LCB628 | torC::Ω SpecR | V Méjean |
| RJ600 | φhybO::torA (produces HybO signal peptide on TorA mature protein) | This work |
| RJ600-D | φhybO::torA, ΔtorD::KanR | This work |
| RJ601 | φhybO::torA, Δhya::KanR | This work |
| RJ602 | φhybO::torA, Δhyb::KanR | This work |
| BTH101 | F-, cya99, araD139, galE15, galK16, rpsL1(StrR), hsdR2, mcrA1, mcrB1 | D Ladant |
Strain RJ603 (as MC4100, φtorA∷hybO), in which DNA encoding the TorA signal peptide exactly replaced that encoding the HybO signal peptide within the native hyb operon, was constructed by cloning DNA encoding the TorA signal peptide into pBluescript (Stratagene Europe) to yield plasmid pRAT7. Upstream and downstream sequences flanking the native HybO signal-coding region was incorporated into pRAT7, thus generating a plasmid-borne φtorA∷hybO construct (pRAT46) which was subcloned into pMAK705 before transfer to the chromosome of MC4100. Strain RJ607 (φtorA∷hybO, ΔhybA) was constructed by incorporating the ΔhybA deletion allele used to construct RJ606 (as MC4100, ΔhybA) into RJ603 by the method of Hamilton et al (1989). RJ600 (as MC4100 φhybO∷torA), in which DNA encoding the HybO signal peptide exactly replaced that encoding the TorA signal peptide at the native tor locus, was constructed using identical methodology employed to make RJ603. Strains RJ600-D (φhybO∷torA, ΔtorD∷KanR) and RJ607-D (φtorA∷hybO, ΔhybAΔtorD∷KanR) were generated by P1 transduction (Russell and Sambrook, 2001) of the ΔtorD∷KanR allele from FTD101 into RJ600. Strains RJ601 (φhybO∷torA, Δhya∷KanR) and RJ602 (φhybO∷torA, Δhya∷KanR) were constructed following P1 transduction of the appropriate alleles from HDJ123 (as MC4100, Δhya∷KanR, Δhyb∷KanR, Δhyc∷CmR; A Böck) into RJ600. The tat mutation used to construct RJ607-T (φtorA∷hybO, ΔhybA, ΔtatABC∷KanR) was made using the method of Datsenko and Wanner (2000).
During all genetic manipulations, cultures were grown in LB medium (Russell and Sambrook, 2001) with the appropriate antibiotics. For hydrogenase-2 assays, cultures were grown anaerobically in CR medium (Sargent et al, 1998) containing 0.5% (v/v) glycerol and 0.4% (w/v) fumarate. For TMAO reductase assays, cultures were grown in 0.5% (v/v) glycerol and 0.4% (w/v) TMAO.
Plasmids
Plasmid-expressed chaperone constructs used in this work are driven from the tat promoter to give constitutive transcription (Jack et al, 2001). They carry the tatA Shine–Dalgarno motif engineered close to the BamHI site within the polylinker which, when a BamHI is placed immediately upstream of the target gene AUG by PCR, can be used to align all heterologous start codons exactly with the native tatA AUG codon. The constructs were based on two different vectors:
(a) pSU-PROM (KanR): An engineered 100 bp PCR fragment covering the tatA promoter up to the tatA AUG was digested with EcoRI (5′ end) and BamHI (3′ end) into vector pSU40 (KanR). Plasmids pSU-hybE, pSU-torD, pSU-dmsD, and pSU-ycdY (encoding hybE, torD, dmsD, and ycdY respectively) were constructed following amplification of the respective PCR fragments, digestion with BamHI–XbaI and cloning into pSU-PROM. For site-directed mutagenesis, the pSU-torD plasmid was used as template and the Quickchange (Stratagene) protocol was employed. Four independent mutations were incorporated into torD, resulting in pSU-S95A, pSU-D124A, pSU-H125A, and pSU-E142A in which the codons indicated were replaced by alanine codons. DNA encoding the C-terminal domain of TorD (from Methionine-113 through to the native torD stop codon) was amplified by PCR and cloned as a BamHI–XbaI fragment into pSU-PROM to give pSU-CDOM.
(b) pUNI-PROM (AmpR) was constructed by cloning the identical 100 bp EcoRI–BamHI tatA promoter fragment into plasmid vector pT7.5 (AmpR). The hybE gene, the torD-family genes, and the four site-specific mutants of torD were moved into pUNI-PROM. DNA encoding the N-terminal domain of TorD (from codon Methionine-1 through Histidine-125) and the C-terminal domain of TorD (above) were cloned as BamHI–XbaI fragments into pUNI-PROM to give pUNI-NDOM and pUNI-CDOM, respectively.
Bacterial two-hybrid system
Direct protein interactions were detected using the method of Karimova et al (1998). The torD gene was incorporated downstream of the coding region of the N-terminal T25 fragment of adenylate cyclase on plasmid pT25 (CmR) to give pT25-TorD. An E. coli genomic library was prepared on plasmid pUT18 (AmpR) encoding the C-terminal T18 fragment of adenylate cyclase. MC4100 chromosomal DNA was isolated and partially digested with Sau3AI. Fragments between 0.2 and 2 kbp were extracted from agarose and ligated into BamHI-digested pUT18. Approximately 40 000 independent transformants were harvested and plasmid DNA stored as the Sau3AI library. For library screening, BTH101 was co-transformed with pT25-TorD and the Sau3AI library, and plated onto MacConkey-maltose at 30°C. Red colonies (indicating possible interactions) were selected after 48 h, plasmids isolated, and inserts sequenced.
Derivatives of pUT18 engineered to carry DNA fragments encoding the TorA signal peptide (pSig3 and pSig4) and the TorA mature protein (pMat1) were constructed as follows: DNA encoding the complete TorA signal, the TorA signal minus N-region, and the TorA mature protein from codons 43–92 were cloned as BamHI–XbaI fragments into pUNI-PROM. Clones were then moved onto the pUT18 vector between the HindIII and EcoRI sites using PCR primers that preserved the optimised ribosome-binding site and placed a stop codon immediately upstream of the fragments of interest to prevent read-through translation from the vector lacZ gene. Protein interactions were estimated by measurement of β-galactosidase activity from BTH101 grown to mid-log phase at 30°C as described (Karimova et al, 1998).
Protein methods
During genetic manipulations, strains were grown aerobically in Luria–Bertani (LB) medium (Russell and Sambrook, 2001). For biochemical characterisations, strains were cultured anaerobically in Cohen–Rickenberg (CR) medium containing 0.5% (v/v) glycerol and 0.4% (w/v) fumarate or 0.4% (w/v) TMAO (Sargent et al, 1998). Subcellular fractionation of cell pellets was performed by the lysozyme/EDTA protocol (Hatzixanthis et al, 2003).
Antisera to TorD and DmsD was provided as a service by Sigma-Genosys. Hydrogenase-2 antiserum has been described previously (Sargent et al, 1998).
SDS–PAGE and immunoblotting analyses were by Lämmli (1970) and Towbin et al (1979). Immunoreactive bands were detected by ECL (Amersham) and intensities were estimated using a densitometer and MacBas version 2.0 software. Hydrogen- and TMAO-BV oxidoreductase activities were measured as described (Sargent et al, 1998; Dubini et al, 2002; Stanley et al, 2002). For intact-cell assays, pellets were washed twice in 20 mM Tris–HCl (pH 7.6) and resuspended at 0.1 g cells (wet weight) ml−1.
Acknowledgments
We thank BC Berks, B Gust, R Little, M Hutchings, and M Hicks for technical help and discussions. D Ladant is thanked for providing the two-hybrid system. This work was funded by BBSRC through research grants 88/P11832 and 83/P15018 and by a grant-in-aid to the John Innes Centre. FS and TP are Royal Society University Research Fellows.
References
- Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Müller M (2003) Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell 12: 937–946 [DOI] [PubMed] [Google Scholar]
- Berks BC, Palmer T, Sargent F (2003) The Tat protein translocation pathway and its role in microbial physiology. Adv Microb Physiol 47: 187–254 [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA (2003) The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol 48: 1171–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco F, Dos Santos JP, Magalon A, Frixon C, Guigliarelli B, Santini CL, Giordano G (1998) NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol Microbiol 28: 435–447 [DOI] [PubMed] [Google Scholar]
- Blaudeck N, Sprenger GA, Freudl R, Wiegert T (2001) Specificity of signal peptide recognition in Tat-dependent bacterial protein translocation. J Bacteriol 183: 604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa MP, Tullman D, Georgiou G (2003) Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci USA 100: 6115–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubini A, Pye RL, Jack RL, Palmer T, Sargent F (2002) How bacteria get energy from hydrogen: a genetic analysis of periplasmic hydrogen oxidation in Escherichia coli. Int J Hydrogen Energy 27: 1413–1420 [Google Scholar]
- Dubini A, Sargent F (2003) Assembly of Tat-dependent [NiFe] hydrogenases: identification of precursor-binding accessory proteins. FEBS Lett 549: 141–146 [DOI] [PubMed] [Google Scholar]
- Gross R, Simon J (2003) The hydE gene is essential for the formation of Wolinella succinogenes NiFe-hydrogenase. FEMS Microbiol Lett 227: 197–202 [DOI] [PubMed] [Google Scholar]
- Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR (1989) New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 171: 4617–4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzixanthis K, Palmer T, Sargent F (2003) A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase. Mol Microbiol 49: 1377–1390 [DOI] [PubMed] [Google Scholar]
- Ilbert M, Mejean V, Giudici-Orticoni MT, Samama JP, Iobbi-Nivol C (2003) Involvement of a mate chaperone (TorD) in the maturation pathway of molybdoenzyme TorA. J Biol Chem 278: 28787–28792 [DOI] [PubMed] [Google Scholar]
- Ilbert M, Mejean V, Iobbi-Nivol C (2004) Functional and structural analysis of members of the TorD family, a large chaperone family dedicated to molybdoproteins. Microbiology 150: 935–943 [DOI] [PubMed] [Google Scholar]
- Jack RL, Sargent F, Berks BC, Sawers G, Palmer T (2001) Constitutive expression of the Escherichia coli tat genes indicates an important role for the twin-arginine translocase during aerobic and anaerobic growth. J Bacteriol 183: 1801–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D (1998) A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA 95: 5752–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 277: 680–685 [DOI] [PubMed] [Google Scholar]
- Mejean V, Iobbi-Nivol C, Lepelletier M, Giordano G, Chippaux M, Pascal MC (1994) TMAO anaerobic respiration in Escherichia coli: involvement of the tor operon. Mol Microbiol 11: 1169–1179 [DOI] [PubMed] [Google Scholar]
- Oresnik IJ, Ladner CL, Turner RJ (2001) Identification of a twin-arginine leader-binding protein. Mol Microbiol 40: 323–331 [DOI] [PubMed] [Google Scholar]
- Palmer T, Sargent F, Berks BC (2004) Light traffic: photo-crosslinking a novel transport system. Trends Biochem Sci 29: 55–57 [DOI] [PubMed] [Google Scholar]
- Pommier J, Mejean V, Giordano G, Iobbi-Nivol C (1998) TorD, a cytoplasmic chaperone that interacts with the unfolded trimethylamine N-oxide reductase enzyme (TorA) in Escherichia coli. J Biol Chem 273: 16615–16620 [DOI] [PubMed] [Google Scholar]
- Rodrigue A, Chanal A, Beck K, Müller M, Wu LF (1999) Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial Tat pathway. J Biol Chem 274: 13223–13228 [DOI] [PubMed] [Google Scholar]
- Russell DW, Sambrook J (2001) Molecular Cloning: A Laboratory Manual, 3rd edn. Cold Spring Harbour, NY: Cold Spring Laboratory Press [Google Scholar]
- Sambasivarao D, Turner RJ, Simala-Grant JL, Shaw G, Hu J, Weiner JH (2000) Multiple roles for the twin arginine leader sequence of dimethyl sulfoxide reductase of Escherichia coli. J Biol Chem 275: 22526–22531 [DOI] [PubMed] [Google Scholar]
- Santini CL, Ize B, Chanal A, Müller M, Giordano G, Wu LF (1998) A novel Sec-independent periplasmic protein translocation pathway in Escherichia coli. EMBO J 17: 101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F, Bogsch E, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T (1998) Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J 17: 3640–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slepenkov SV, Witt SN (2002) The unfolding story of the Escherichia coli Hsp70 DnaK: is DnaK a holdase or an unfoldase? Mol Microbiol 45: 1197–1206 [DOI] [PubMed] [Google Scholar]
- Stanley NR, Sargent F, Buchanan G, Shi J, Stewart V, Palmer T, Berks BC (2002) Behaviour of topological marker proteins targeted to the Tat protein transport pathway. Mol Microbiol 43: 1005–1021 [DOI] [PubMed] [Google Scholar]
- Temple CA, Rajagopalan KV (2000) Mechanism of assembly of the bis(molybdopterin guanine dinucleotide)molybdenum cofactor in Rhodobacter sphaeroides dimethyl sulfoxide reductase. J Biol Chem 275: 40202–40210 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranier S, Iobbi-Nivol C, Birck C, Ilbert M, Mortier-Barriere I, Mejean V, Samama JP (2003) A novel protein fold and extreme domain swapping in the dimeric TorD chaperone from Shewanella massilia. Structure 11: 165–174 [DOI] [PubMed] [Google Scholar]
- Tranier S, Mortier-Barriere I, Ilbert M, Birck C, Iobbi-Nivol C, Mejean V, Samama JP (2002) Characterization and multiple molecular forms of TorD from Shewanella massilia, the putative chaperone of the molybdoenzyme TorA. Prot Sci 11: 2148–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais PM, Colbeau A (2004) Molecular biology of microbial hydrogenases. Curr Issues Mol Biol 6: 159–188 [PubMed] [Google Scholar]