

Abstract
The generation of all blood cells depends on the ability of hematopoietic stem cells (HSCs) for self-renewal and multilineage differentiation. We show here that the transcription factor Gfi1 is expressed in HSCs and in more mature cells such as common lymphoid progenitors (CLPs) and granulo/monocytic progenitors, but is absent in common myeloid progenitors and megakaryocyte/erythroid progenitors. When Gfi1 is deleted in mice, HSC frequencies are significantly reduced and CLPs all but disappear from the bone marrow. This specific requirement of Gfi1 for the maintenance of HSC numbers is cell autonomous. Transplantation of Gfi1-deficient bone marrow results in a compromised radioprotection and lower numbers of colony forming units in the spleen of wild-type recipients. Strikingly, Gfi1−/− bone marrow cells are severely impaired in competitive long-term reconstituting abilities after transplantation and show a surprisingly high proportion of actively cycling HSCs, suggesting that Gfi1 restrains proliferation of HSCs and thereby regulates their self-renewal and long-term engraftment abilities.
Keywords: cell cycle, proliferation, self-renewal, stem cells, zinc-finger transcription factor
Introduction
Multilineage hematopoiesis is maintained by a pool of hematopoietic stem cells (HSCs). To sustain the production of blood cells throughout the lifetime of an individual, HSCs are capable of self-renewal to maintain the HSC pool and have the ability for multilineage differentiation (Weissman, 2000). HSCs comprise phenotypically and functionally defined long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs) and multipotent progenitors (MPPs) (Adolfsson et al, 2001; Christensen and Weissman, 2001; Guenechea et al, 2001). This pool of cells can give rise to a series of intermediate lineage committed progenitors such as common lymphoid progenitors (CLPs) and common myeloid progenitor (CMPs) that are able to further generate the different hematopoietic lineages (Kondo et al, 1997; Akashi et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001).
The decisions of HSCs for self-renewal or for differentiation are strictly controlled by both extrinsic and intrinsic mechanisms. Recently, the introduction of mutant alleles in mice by gene targeting has shed light on positive and negative regulators for hematopoiesis (Bertoncello and Williams, 2001). A number of cytokines and their receptors are involved in the regulation of HSC self-renewal and differentiation such as stem cell factor (SCF) and its receptor c-Kit but also the c-Mpl the receptor for thrombopoietin and the flt-2 and flt-3 ligands and their receptors (Broudy, 1997; Kimura et al, 1998; Metcalf, 1998). More recently, the Notch (Karanu et al, 2000; Varnum-Finney et al, 2000; Stier et al, 2002) and Wnt (Reya et al, 2003; Willert et al, 2003) signaling pathways were found to regulate stem cell self-renewal. And transcription factors such as Ikaros (Nichogiannopoulou et al, 1999) and Bmi-1 (Lessard et al, 2003; Park et al, 2003) and cell cycle regulators like the G1-specific inhibitor p21 (Cheng et al, 2000) have been implicated as key regulatory components to maintain the ability of HSCs for self-renewal.
The gene locus encoding the Gfi1 protein was discovered in a screen for Moloney murine leukemia virus (MoMuLV) proviral integration site in NB2 rat lymphoma cells and in T-lymphoid tumors that were elicited by MoMuLV infection (Gilks et al, 1993; Schmidt et al, 1996; Zörnig et al, 1996; Scheijen et al, 1997). The Gfi1 gene encodes a 55-kDa nuclear transcription factor, which harbors six carboxy-terminal C2–H2 zinc-finger domains and a characteristic N-terminal 20-amino-acid stretch termed ‘SNAG' domain, which is well conserved between Gfi1 and the proteins Snail and Slug (Grimes et al, 1996; Zweidler-Mckay et al, 1996). Reporter gene experiments suggested a transcriptional repressor activity of Gfi1 that depends on the DNA binding activity and on intact SNAG domain (Grimes et al, 1996; Zweidler-Mckay et al, 1996). An alternative activity of Gfi1 has been discovered through its interaction with PIAS (protein inhibitor of activated STAT) 3, which is an inhibitor of signal transducers and activators of transcription (STAT) 3 suggesting a role of Gfi1 in a set of specific cytokine signaling pathways (Rödel et al, 2000).
Early studies revealed a remarkably high expression level of Gfi1 in thymic lymphoid cells and a key role of Gfi1 in lymphomagenesis and lymphopoiesis (Gilks et al, 1993; Grimes et al, 1996; Schmidt et al, 1998a). Constitutive Gfi1 expression can relieve peripheral mature T cells from a requirement of IL-2 and sustains proliferation of IL-2-dependent cells in the absence of the cytokine (Grimes et al, 1996; Zörnig et al, 1996), indicating a positive role of Gfi1 in IL-2-dependent cell cycle progression of T cells. Gfi1 can act as a dominant oncogene when overexpressed, and cooperates strongly with other oncoproteins as Pim-1 (a cytoplasmic serine/threonine kinase) or Myc (an HLH-LZ transcription factor) in accelerating progression of T-cell lymphomagenesis (Zörnig et al, 1996; Scheijen et al, 1997; Schmidt et al, 1998a, 1998b). Moreover, Gfi1 regulates IL-4/STAT6-dependent Th2 cell proliferation (Zhu et al, 2002) and IL-6/STAT3-mediated proliferative responses to antigenic stimulation (Rödel et al, 2000).
Later studies showed that Gfi1 is also present in granulocytes and in activated macrophages (Karsunky et al, 2002b) and in distinct areas of the nervous system, most prominently in inner ear hair cells (Wallis et al, 2003). In the immune system, ablation of Gfi1 by gene targeting in mice caused defects in early T-cell maturation (Yücel et al, 2003) and led to severe neutropenia and a profound monocytosis (Karsunky et al, 2002b; Hock et al, 2003). These findings underscored a pivotal role of Gfi1 in T-cell differentiation as was expected, but also demonstrated that Gfi1 regulates myelopoiesis. Specifically, the absence of mature neutrophils and the accumulation of immature myelo-monocytic cells in Gfi1−/− mice in the bone marrow and peripheral blood suggested that differentiation processes from granulo-monocytic precursors are tightly regulated by Gfi1.
The importance of Gfi1 in hematopoiesis, which was revealed by the studies of Gfi1-deficient mice, prompted us to investigate in more detail the expression pattern of Gfi1 in HSCs and progenitor cells. Since Gfi1−/− mice show multilineage defects (Karsunky et al, 2002b; Hock et al, 2003; Yücel et al, 2003), it was of interest to know whether Gfi1 only acts on lineage committed hematopoietic cells or whether Gfi1 controls hematopoiesis at a more primitive level, for instance by regulating the activity of HSCs or progenitor cells. By using different mouse mutants, we can show that Gfi1 is differentially expressed in HSCs and subsets of hematopoietic progenitor cells, and that a loss of Gfi1 results in a significant alteration of stem and precursor cell frequencies. Through a series of functional assays, we present evidence that Gfi1 is an important factor in maintaining the abilities of HSCs for self-renewal, multilineage differentiation and efficient reconstitution of hematopoiesis in transplanted hosts by restricting the proliferation of HSCs.
Results
Expression of Gfi1 in adult mouse bone marrow HSCs and hematopoietic progenitors
To measure Gfi1 expression in different hematopoietic lineages at different differentiation stages during hematopoiesis, we used a ‘knock-in' mouse mutant in which the Gfi1 coding region had been replaced by the gene encoding green fluorescent protein (GFP) by homologous recombination (Yücel et al, 2004). In this mouse mutant, the GFP gene is expressed instead of the endogenous Gfi1 allele under the transcriptional control of the regulatory elements of the Gfi1 gene. Gfi1:GFP heterozygous knock-in mice (Gfi1GFP/+) were phenotypically indistinguishable from their wild-type (WT) littermates and from the previously described animals (Karsunky et al, 2002b; Yücel et al, 2003) that carry a neo resistance marker gene in the Gfi1 locus disrupting one Gfi1 allele (Gfi1+/−).
We analyzed Gfi1 expression in HSC and early hematopoietic progenitors by measuring the fluorescence of GFP in cells of Gfi1GFP/+ mice. Using multiparameter flow cytometry, cells were isolated with surface marker expression patterns that discriminate among HSCs, CLPs, CMPs and the more lineage-restricted granulo/monocytic progenitors (GMPs) and megakaryocyte/erythroid progenitors (MEPs) according to previously described procedures (Kondo et al, 1997; Akashi et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001). A high intensity of green fluorescence was found in a subset of bone marrow cells defined by the absence of lineage markers and high level expression of Sca-1 and c-Kit (Lin−Sca-1+c-Kit+, LSK population), which contains LT-HSCs, ST-HSCs and MPPs (Adolfsson et al, 2001; Christensen and Weissman, 2001), indicating that the Gfi1 gene is expressed in the entire HSC compartment (Figure 1A). Similarly, significant GFP expression was observed in CLPs (Lin−IL-7Rα+Sca-1lowc-Kitlow cells) (Figure 1B) and in GMPs (Lin−Sca-1−IL-7Rα−c-Kit+CD34+FcγRhigh) (Figure 1C) but not in MEPs (Lin−Sca-1−IL-7Rα−c-Kit+CD34−FcγRlow cells) and CMPs (Lin−Sca-1−IL-7Rα−c-Kit+CD34+FcγRlow cells) (Figure 1C). This expression pattern of Gfi1 in HSCs and progenitors was confirmed by RT–PCR analyses with 2000 double-sorted cells for each population from bone marrow of C57BL/6, Thy1.1 mice (Figure 1D).
Figure 1.
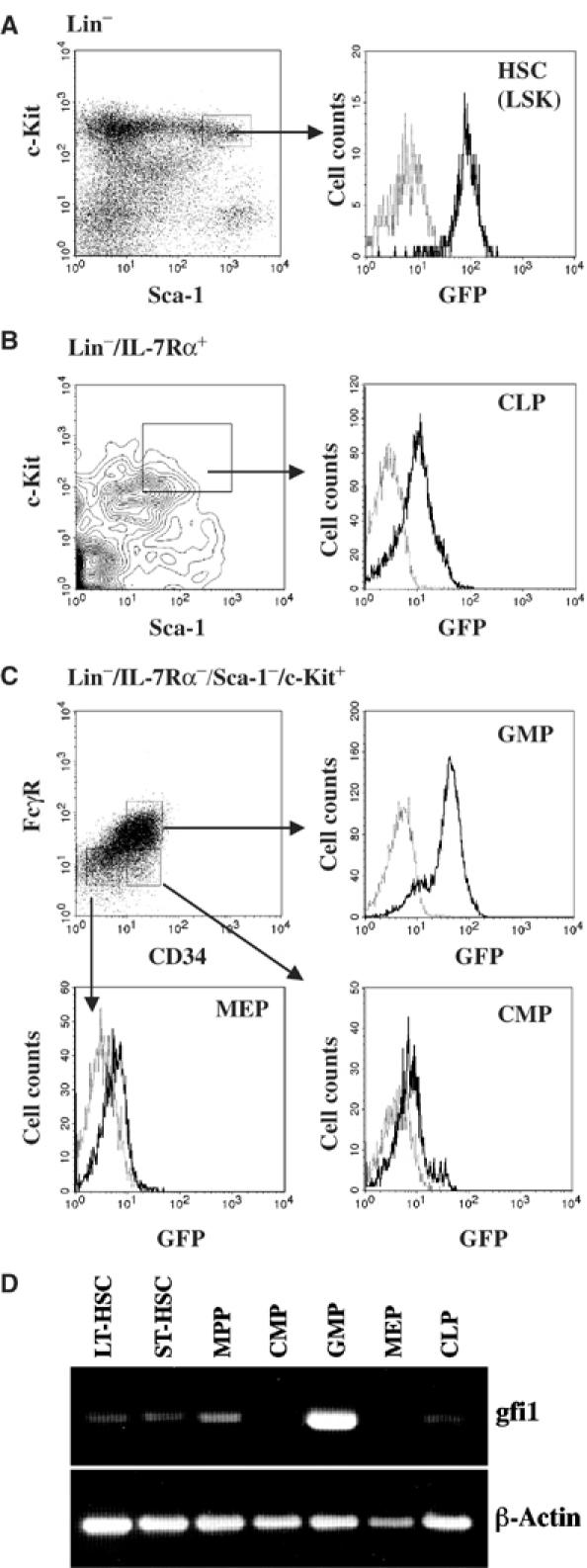
Expression of Gfi1 in adult mouse bone marrow hematopoietic stem cells and subsets of hematopoietic progenitor cells. Total bone marrow cells of WT mice and animals carrying one mutated Gfi1 allele in which part of its coding region was replaced by the reading frame encoding green fluorescent protein (Gfi1:GFP knock-in mice (Gfi1+/GFP); Yücel et al, 2004) were prepared and stained with antibodies against lineage markers (Lin) and the indicated surface markers. The cells were then analyzed by flow cytometry and were gated as previously described (see Materials and methods) to obtain fractions representing HSCs, CLPs, GMPs or CMPs. Electronically gated subsets (left panels) were reanalyzed by measuring green fluorescence (right panels, dark lines). As a control, background green fluorescence of WT cells was monitored (right panels, gray lines). (A–C) Expression of GFP in HSCs (A), CLPs (B) and CMPs, GMPs and MEPs (C) in Gfi1+/GFP mice. (D) Expression analysis by RT–PCR of Gfi1 in LT-HSCs, ST-HSCs, MPPs, CMPs, GMPs, MEPs and CLPs.
Altered frequencies of HSCs and hematopoietic progenitors in Gfi1−/− bone marrow
To investigate whether loss of Gfi1 has any consequences for the cell numbers in the HSC compartment, we compared different subsets of bone marrow cells from Gfi1−/− mice and WT littermates. LSK cells and different LSK subsets defined by the expression of Flt3 were electronically gated (Figure 2A) to distinguish between LT-HSCs and ST-HSCs along previously described procedures (Goodell et al, 1997; Zhao et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001). Loss of Gfi1 was associated with a five- to six-fold decrease of LSK percentages in total bone marrow (Figure 2B). The frequencies of LT-HSCs (Lin−Sca1+c-Kit+CD34− or Lin−Sca1+c-Kit+Flt3−) and ST-HSCs/MPPs (Lin−Sca1+c-Kit+Flt3+) were reduced two- to four-fold and 10-fold, respectively, in Gfi1 null mice (Figure 2B). Strikingly, Lin−Sca1+c-Kit+Flt3high cells were almost completely absent in Gfi1−/− bone marrow (Figure 2B). Since total bone marrow cellularity was not significantly altered in Gfi1−/− mice, the absolute numbers of LSK cells and LSK subsets were reduced in Gfi1-deficient mice to the same degree as the percentages (data not shown).
Figure 2.
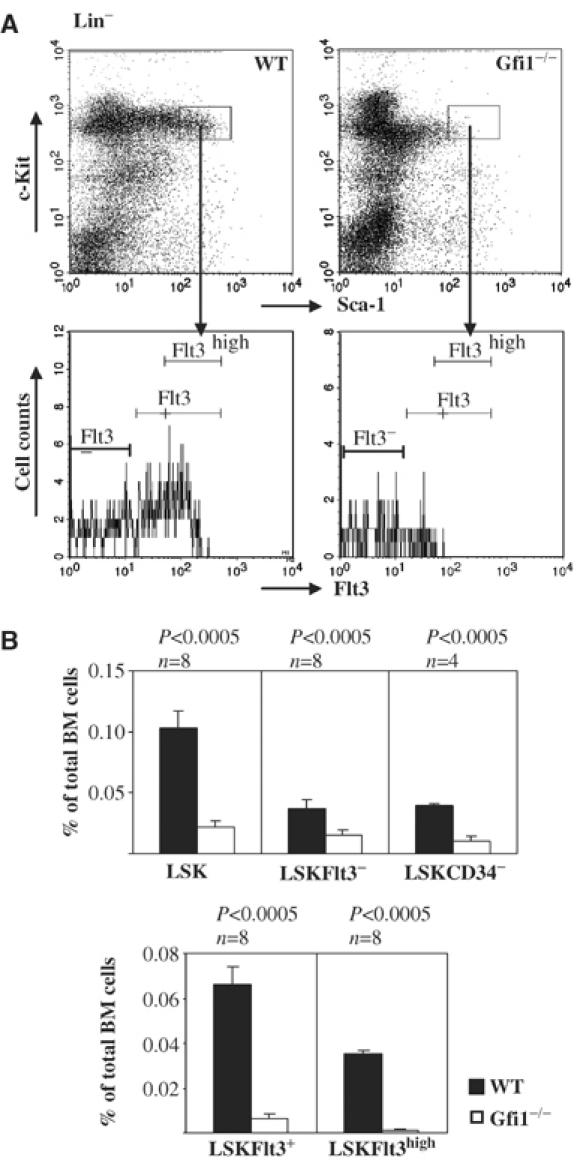
Effects of Gfi1 deletion on the frequencies of HSCs in adult mouse bone marrow. (A) Total bone marrow cells of Gfi1−/− and WT mice were prepared and stained with antibodies against lineage markers (Lin) and the surface markers and receptors Sca-1, c-Kit and Flt3. Lin−Sca-1+c-Kit+ cells (LSK) were analyzed by flow cytometry, gated as previously described to obtain fractions representing HSCs (Akashi et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001) and were reanalyzed for Flt3 expression. (B) The percentages for phenotypically defined HSCs (LSK) and the indicated different HSCs subsets are given with regard to total bone marrow cell numbers. n: number of mice analyzed. P-values were calculated using the Student's t-test.
We also observed that ablation of Gfi1 resulted in a dramatic decrease of CLP (40-fold) and a more mild (two-fold) reduction in CMP frequencies (Figure 3A and B). By contrast, the frequency of GMPs was not reduced in Gfi1-deficient mice, but rather was moderately increased with regard to the Lin−Sca-1−IL-7Rα− c-Kit+ (LISK) population and also with respect to total bone marrow cell numbers compared to the respective WT compartments (Figure 3B). The frequency of MEPs was not altered in Gfi1-deficient mice (Figure 3C). Similar to Gfi1−/− mice, homozygous Gfi1GFP/GFP mice are also unable to produce the Gfi1 protein (Yücel et al, 2004) and present with the same full Gfi1 knockout phenotype. These animals showed the same alterations of HSCs and progenitor frequencies as the original Gfi1 knockout mouse model (data not shown), confirming our findings in a second, independently generated Gfi1 null mouse mutant.
Figure 3.
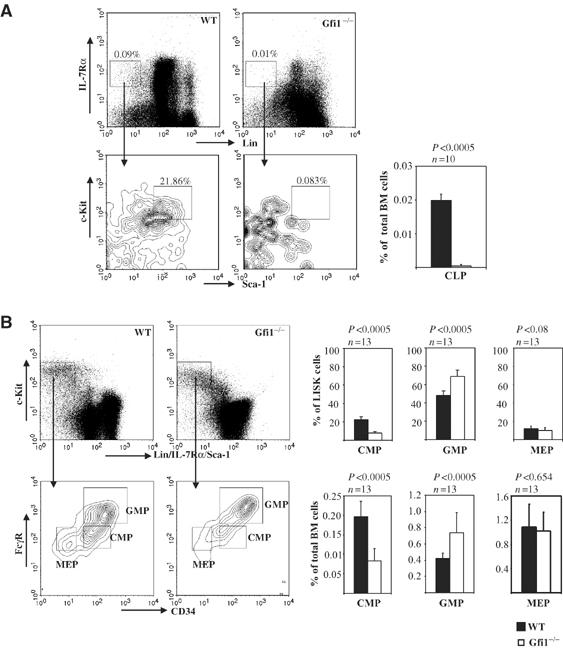
Effects of Gfi1 deletion on the frequencies of hematopoietic progenitors in adult mouse bone marrow. (A) Total bone marrow cells of Gfi1−/− and WT mice were prepared and stained with antibodies against lineage markers (Lin) and the indicated surface markers and receptors. Lin−IL-7Rα+Sca-1+c-Kit+ cells were analyzed by flow cytometry and gated as previously described (Kondo et al, 1997) to obtain fractions representing CLPs. The frequency of CLPs in Gfi1−/− and WT mice is given as percentage of total bone marrow cells. (B) Lin− IL-7Rα− Sca-1−c-Kit+ cells were gated according to their expression of CD34 and FcγR to obtain populations representing CMPs, GMPs and MEPs as described (Akashi et al, 2000). The frequencies of CMPs, GMPs and MEPs in bone marrow of Gfi1−/− or WT adult mice are given as a percentage of total bone marrow cells or as a percentage of the LISK subpopulation. n: number of mice analyzed. P-values were calculated using the Student's t-test. (LISK: Lin−Sca-1−IL-7Rα−c-Kit+.)
Transplantation of Gfi1−/− bone marrow leads to reduced day 12 CFU-S and a compromised short-term radioprotection capacity
The function of HSCs and primitive progenitors can be assessed by in vivo reconstitution assays. It has been demonstrated that the HSCs and primitive progenitors can form colonies in the spleens of lethally irradiated recipients after bone marrow transplantation. About half of day 12 CFU-S (CFU-S12) are derived from the HSC/MPP compartment and the other half are derived from the MEP/CMP populations (Spangrude et al, 1988; Morrison and Weissman, 1994; Nakorn et al, 2002), while the vast majority of day 8 CFU-S (CFU-S8) are derived from MEPs (Nakorn et al, 2002). We tested the ability of Gfi1−/− HSCs and progenitors to form colonies in the spleens in a bone marrow transplantation assay and found that both the numbers and the size of CFU-S12 were significantly reduced in transplanted hosts that received Gfi1−/− bone marrow (204±50.7 and 82±25.6 per 106 bone marrow cells, respectively) (Figure 4A). In contrast, numbers and size of CFU-S8 remained unaltered in irradiated hosts after transplantation of WT or Gfi1−/− bone marrow (174±28.2 and 137.3±15.5 per 106 bone marrow cells, respectively) (Figure 4A). Consistent with the decrease in CFU-S12, we found that the short-term radioprotection capacity of Gfi1−/− bone marrow cells was also compromised. While the transplantation of 2 × 105 WT bone marrow cells led to a complete rescue of lethally irradiated recipients (Figure 4B), the same number of Gfi1−/− bone marrow cells protected less well and about 1/3 of the animals died from the consequences of irradiation within 35 days. However, transplantation of 1 × 106 Gfi1−/− bone marrow cells could provide almost full protection against irradiation (Figure 4B). The results of both experiments suggest functional defects in the progenitor and stem cell compartment of Gfi1−/− bone marrow, in particular in the ST-HSC, MPP and CMP populations.
Figure 4.
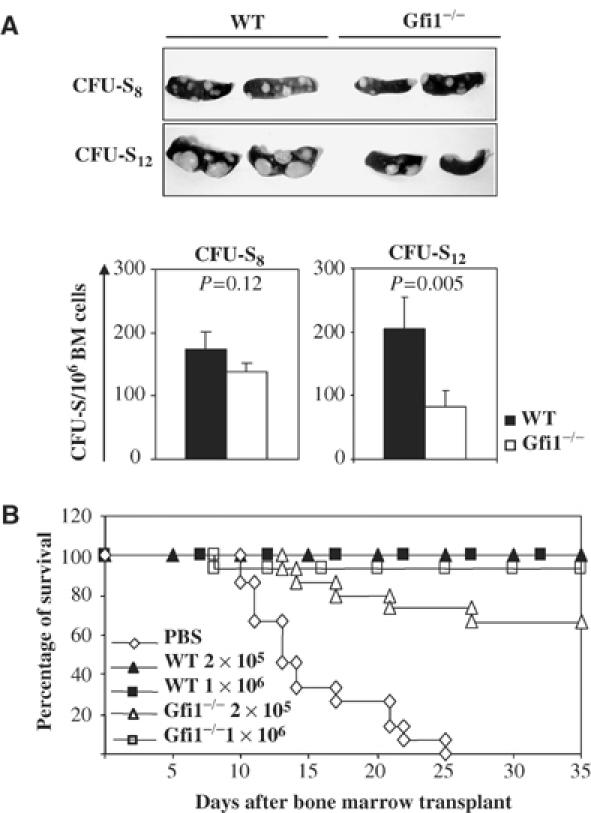
Number of primitive progenitors is reduced and short-term radioprotection ability is moderately compromised in Gfi1-deficient mice. (A) For CFU-S, 5 × 104 Wt or Gfi1−/− bone marrow cells were injected into lethally irradiated (9.6 Gy) recipients (10 mice for each group). Mice were killed 8 or 12 days later and their spleens were fixed in Tellesniczky's fixative for macroscopic examination. Spleen colonies are shown; the size and numbers of day 12 CFU-S but not day 8 CFU-S colonies were significantly reduced when Gfi1-deficient bone marrow was transplanted. (B) Survival graph of lethally irradiated mice (9.6 Gy) reconstituted with the indicated amounts of WT or Gfi1−/− bone marrow cells, or PBS as control as indicated (15 mice per group).
Decreased long-term and competitive reconstitution capacity of Gfi1−/− HSCs
Since the Gfi1−/− bone marrow cells exhibited moderate decrease of radioprotection ability in the primary recipients, it was of interest to determine whether a defect in Gfi1-deficient HSCs would be more predominant upon further proliferative stress, for instance upon transplantation to secondary recipients. To test this, 1 × 106 bone marrow cells from WT or Gfi1−/− mice were transplanted into lethally irradiated WT mice. After 3 months, secondary transplantations were performed with 1 × 106 bone marrow cells from primary recipients, which had previously received either 1 × 106 WT or Gfi1−/− bone marrow cells. Within 6 months, four of 10 secondary recipients that received bone marrow from primary recipients transplanted with Gfi1−/− cells died. In contrast, all secondary recipients injected with cells from primary recipients previously transplanted with WT bone marrow survived this 6-month period. This suggested that a lack of Gfi1 is associated with a defect of the repopulation ability of HSCs.
To further determine the long-term reconstituting abilities of Gfi1-deficient bone marrow cells, WT or Gfi1−/− bone marrow cells (both CD45.2+) were mixed at a 1:1 ratio with competitor CD45.1+ bone marrow cells. The mixtures were transplanted into irradiated CD45.1+ recipients and myeloid and lymphoid reconstitution was measured by FACS analysis of peripheral blood over a period of 22 weeks. WT bone marrow cells successfully reconstituted both myeloid and lymphoid lineages in irradiated recipients, whereas Gfi1−/− bone marrow cells showed a dramatically decreased capacity to reconstitute myeloid or lymphoid hematopoiesis (Figure 5A, upper panels). The possibility that the above defect is simply due to the reduction of phenotypically defined HSCs in Gfi1−/− bone marrow could be excluded because a 10:1 mixture of Gfi1−/− bone marrow with CD45.1 competitor cells, which provided a large excess of phenotypically defined Gfi1−/− HSCs, also failed to compete with CD45.1 cells to reconstitute hematopoiesis in recipient mice in both myeloid and lymphoid lineages (Figure 5A, lower panels). Consistent with these results, a dramatic, 60-fold reduction of long-term culture initiating cell (LTC-IC) numbers was observed in cultures started with Gfi1−/− bone marrow compared to WT bone marrow (data not shown). Thus, the loss of Gfi1 not only correlates with a dramatic decrease of phenotypically defined HSCs (see Figure 2) but is also associated with a clear and significant reduction of functionally defined HSCs.
Figure 5.
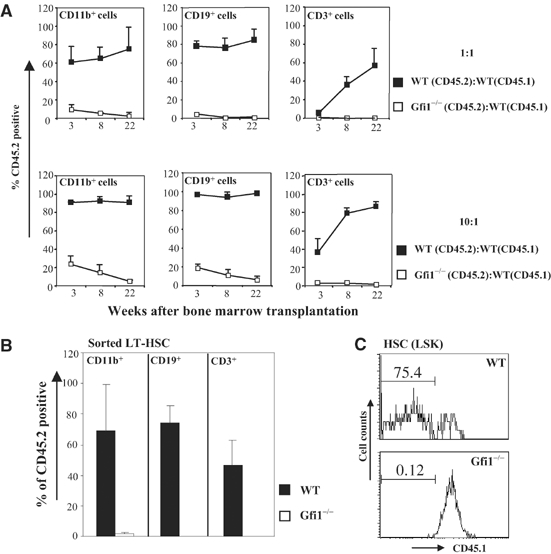
Gfi1-deficient HSCs are defective in their reconstituting ability. (A) Competitive repopulation assay with WT and Gfi1−/− bone marrow cells. In all, 2 × 105 WT or Gfi1−/− bone marrow cells (CD45.2+) were mixed with the same number of competitor CD45.1+ bone marrow cells and were injected into lethally irradiated CD45.1+ mice (n=4). Peripheral blood was analyzed at various times after reconstitution for the presence of WT or Gfi1−/− bone marrow (CD45.2+)-derived myeloid, B-lymphoid and T-lymphoid cells (upper panels marked 1:1). The same experiments were performed using 2 × 106 WT or Gfi1−/− bone marrow cells (CD45.2+) mixed with 2 × 105 competitor CD45.1+ bone marrow cells (n=4, lower panels marked 10:1). (B) A total of 500 sorted WT or Gfi1−/− LT-HSCs (CD45.2+) were mixed with 2 × 105 competitor CD45.1+ bone marrow cells and injected into lethally irradiated CD 45.1+ mice (n=4). Peripheral blood was analyzed 22 weeks after reconstitution for the presence of WT or Gfi1−/− LT-HSC-derived myeloid (CD11b+), B-lymphoid (CD19+) and T-lymphoid cells (CD3+). (C) At 6 months after reconstitution, bone marrow cells from mice described in (B) were analyzed to measure the percentage of LSK cells derived from transplanted WT or Gfi1−/− LT-HSCs. Representative diagrams show the contribution of donor-derived HSCs (CD45.1− LSK) in WT and Gfi1−/− LT-HSCs transplanted recipients (75.4 or 0.12%, respectively).
To exclude the influences of the altered lineage profiles (monocytosis, neutropenia and lymphocytopenia) or a simple dilution effect in Gfi1−/− bone marrow, 500 sorted WT or Gfi1−/− LT-HSCs (CD45.2+Lin−Sca1+c-Kit+Flt3−) were mixed with 2 × 105 competitor CD45.1+ bone marrow cells and competitive transplantation assay was performed. Gfi1−/− HSCs were unable to foster the outgrowth of significant numbers of myeloid or lymphoid cells in irradiated hosts up to 22 weeks after transplantation (Figure 5B). Moreover, sorted Gfi1−/− LT-HSCs were also unable to give rise to any LSK cells upon transplantation in lethally irradiated recipients, whereas 500 sorted WT cells could indeed generate significant numbers of LSK cells in transplanted recipients (Figure 5C). This direct determination of HSC ratios in reconstituted animals strongly suggests that the competitive disadvantage of Gfi1−/− bone marrow cells starts at the level of HSCs.
Defects of Gfi1−/− HSCs and progenitors are cell autonomous
To verify that Gfi1-deficient HSCs have an intrinsic defect and to exclude influences of a potentially defective bone marrow microenvironment in Gfi1−/− mice, an excess of Gfi1-deficient bone marrow cells (CD45.2+) were transplanted into lethally irradiated WT hosts (CD45.1+). The phenotype of Gfi1-deficient mice with regard to frequencies of stem cells and progenitors, or previously reported hallmarks (neutropenia, accumulation of monocytic cells and reduced numbers of thymocytes; Karsunky et al, 2002b; Hock et al, 2003; Yücel et al, 2003) could be exactly reproduced in the transplanted hosts (Supplementary Figure 1), suggesting that the reported hematopoietic defects in Gfi1−/− mice are cell autonomous. Conversely, CD45.1+ WT bone marrow cells were transplanted into lethally irradiated Gfi1-deficient mice (CD45.2+) to test whether Gfi1-deficient mice have an intact bone marrow microenvironment. Normal numbers of HSCs, progenitors, neutrophils and thymocytes were found in the host 4 months after transplantation (Supplementary Figure 2). This indicated that stroma cells and the microenvironment in Gfi1-deficient bone marrow are functional and lack any detectable defects, which also supports the notion that the defects of Gfi1−/− HSCs and progenitors are cell autonomous.
Altered in vivo proliferation kinetics of HSCs in the absence of Gfi1
Since Gfi1 had been implicated in cell cycle regulation (Karsunky et al, 2002a), we tested whether a proliferation defect is manifest in Gfi1−/− HSCs. To this end, we performed in vivo BrdU labeling experiments to determine the proliferative history of LSK cells. We observed that the percentage of cells entering the cell cycle is significantly higher in the Gfi1−/− HSC compartment than in WT LSK cells (Figure 6A). To determine whether enhanced BrdU incorporation in Gfi1−/− LSK cells correlates with an increase of the proportion of HSC in cell cycle, we used a combination of DNA and RNA staining with the dyes Hoechst and pyronin as previously described (Cheshier et al, 1999). Clearly, the absence of Gfi1 was associated with a significant loss of LSK cells in G0 phase and a significantly higher percentage of Gfi1−/− LSK cells in G2/S/M phases when compared to WT LSKs (Figure 6B and C). To test whether this is due to an altered expression of the previously described Gfi1 target genes E2F5, E2F6 or p21cip1/waf (Duan and Horwitz, 2003) that are associated with cell cycle regulation, we sorted LSK cells from WT and Gfi1−/− mice and assessed the expression of these genes by quantitative real-time PCR. Neither E2F5, E2F6 nor p21cip1/waf1 mRNA levels showed a noticeable difference between WT and Gfi1 null cells (Figure 7A). In contrast however, we found that whole bone marrow cells from Gfi1-deficient mice contain significantly higher levels of E2F5 and E2F6 protein than WT cells and almost completely lack expression of the G1-specific negative cell cycle regulator p21cip1/waf1 whereas p27kip1 was expressed at comparable levels in both WT and Gfi1−/− cells (Figure 7B).
Figure 6.
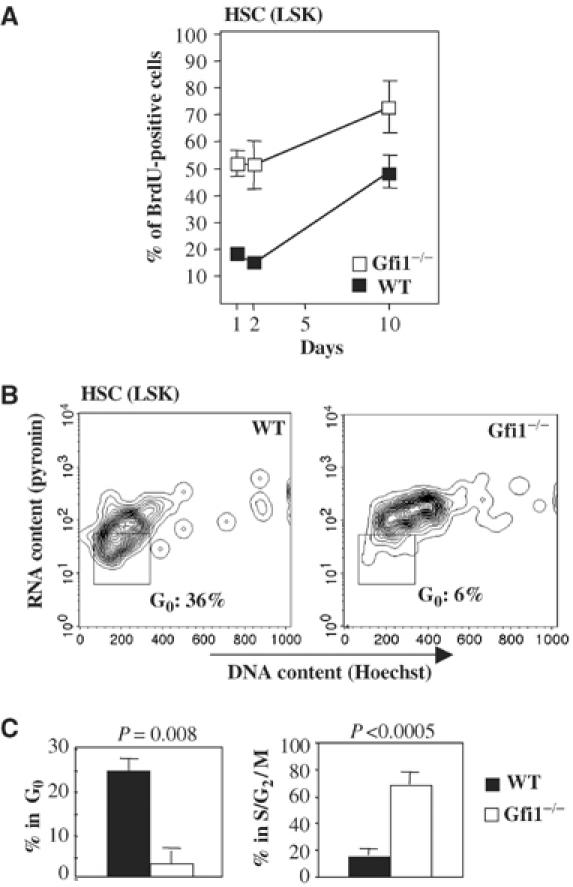
Cell cycle phase distribution of HSCs between G0/G1 and S/G2/M phases is altered in Gfi1−/− mice. (A) LSK cells were labeled with BrdU over the indicated time period. Gfi1−/− LSK cells show a significantly higher incorporation of BrdU than LSK cells from WT mice. (B) Bone marrow cells were stained to identify LSK cells and with a combination of Hoechst (DNA) and pyroninY (RNA). LSK cells were analyzed for incorporation of DNA and RNA dyes to assess the proportions of HSCs in the G0 and the S/G2/M phases of the cell cycle. The absence of Gfi1 correlated with a drastic loss of HSCs (LSK cells) in G0 phase. (C) Percentages of WT and Gfi1−/− HSCs (LSK cells) in different cell cycle phases. Given are average values with standard deviations from four WT and four Gfi1−/− animals.
Figure 7.
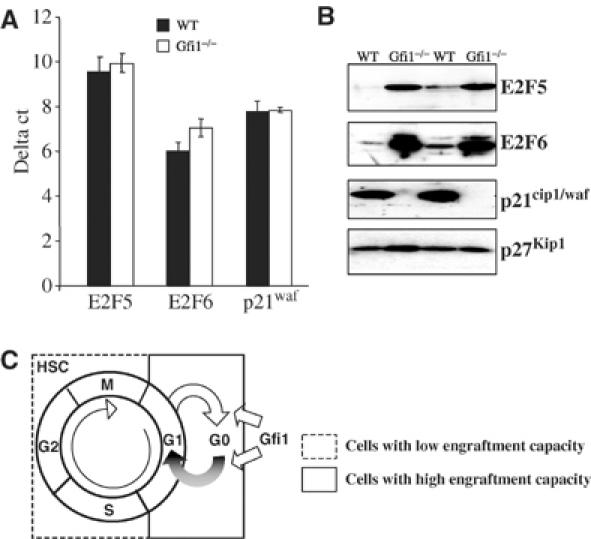
Altered expression of cell cycle regulators in bone marrow of Gfi1−/− mice. (A) Quantification of mRNA expression level of E2F5, E2F6 and p21waf/cip in sorted HSCs (LSK cells) by real-time PCR. Shown is a representative result for two independent experiments with two independent sets of mice. (B) Protein expression levels of E2F5, E2F6, p21cip1/waf1 and p27kip1 in WT or Gfi1−/− bone marrow in two independent sets of mice (total number of animals analyzed: WT, n=4; Gfi1−/−, n=4). (C) Proposed model for a role of Gfi1 in HSCs. A high capacity for long-term engraftment and self-renewal is confined to HSCs in G0/G1 phase but not to those in S/G2/M phase. By controlling the levels of cell cycle regulators, Gfi1 restricts HSC proliferation and helps to maintain the distribution of HSCs between cell cycle phases. The absence of Gfi1 results in a drastic loss of HSCs in G0 phase and, as a consequence, their capacity for long-term engraftment and self-renewal is reduced.
Discussion
In the present study, we show that Gfi1 is expressed in HSCs, CLPs and GMPs, but is absent in CMPs and MEPs. This correlates well with our findings that a loss of Gfi1 affects the frequencies of HSCs and progenitors, in particular ST- and LT-HSCs and CLPs but leaves MEPs unaltered and suggested a role of Gfi1 in regulating the development of certain, distinct myeloid and lymphoid progenitor cell populations and HSCs. In particular, the differential expression of Gfi1 in CLPs versus CMPs and in GMPs versus MEPs suggests that presence or absence of Gfi1 is critical for lineage decision processes between myeloid and lymphoid lineages and at a later stage between granulocytic and monocytic lineages.
The alterations of progenitor frequencies in Gfi1−/− mice were confirmed by a series of functional assays such as spleen colony forming assay (CFU-S) and radioprotection assays in which Gfi1 null cells showed a clear deficit compared to WT cells. In addition, a loss of CLPs and unaltered numbers of MEPs in Gfi1−/− mice is consistent with the previously reported phenotypes of lineage-committed cells, in particular with the reduction of thymocytes and peripheral T and B cells and with normal counts of red blood cells and platelets in these animals (Karsunky et al, 2002b; Hock et al, 2003; Yücel et al, 2003), endorsing the notion that Gfi1 is required for multilineage hematopoiesis from early stem cells and progenitors to late committed cells.
Gfi1−/− bone marrow cells could provide short-term and long-term reconstitution of hematopoiesis when transplanted into lethally irradiated recipients without competitive cells and in large excess. However, secondary transplantation revealed deficiencies in this ability that were not detected when WT bone marrow was serially transplanted, suggesting that Gfi1−/− stem cells are defective. Indeed, when measured against WT bone marrow competitor cells, a severe impairment of long-term repopulating activities of Gfi1−/− bone marrow cells became apparent. Most importantly, despite a two- to four-fold decrease in phenotypically defined LT-HSC, that is, by the expression of surface markers, in adult mouse bone marrow, the reduction of functionally defined HSCs was found to be more than 30- to 40-fold in Gfi1−/− mice as estimated by competitive transplantation assays. This severe impairment of the long-term competitive repopulating activity of Gfi1−/− bone marrow cells was observed in several competitive transplantation experiments using total bone marrow cells at different ratios of test cells and competitors and was also evident when sorted, phenotypically defined HSCs were used. These clear and concordant results of both types of experiments strongly suggest that ablation of Gfi1 results in functionally defective HSCs.
Since a defective bone marrow microenvironment in Gfi1 null mice has been excluded by transplantation of WT cells into Gfi1 null recipients (see Supplementary Figures 1 and 2), the observed defects in Gfi1−/− HSCs could be a result of an altered or perturbed homing, a faulty generation of more mature progenitors, or an impairment of self-renewal. First, a homing defect in Gfi1-deficient bone marrow cells seems unlikely since the severe reduction of LTC-IC numbers generated from Gfi1−/− bone marrow cells in culture is not consistent with a homing defect, otherwise a reduction in LTC-IC numbers would not be expected. Second, loss of Gfi1 did not alter the expression of adhesion molecules important for HSC homing such as CXCR4, VLA-4 and VLA-5 in Gfi1−/− LSK populations (Supplementary Figure 3). Third, the early engraftment of Gfi1−/− bone marrow cells that was seen at 3 weeks after transplantation was followed by a gradual decrease of Gfi1−/− progeny, which also suggested that the severe impairment of long-term reconstituting abilities of Gfi1−/− HSCs is not a result of a homing defect, but occurs at the upper level of the hematopoietic hierarchy in Gfi1−/− mice.
Since Gfi1−/− HSCs can generate both myeloid and lymphoid hematopoiesis in the recipients when they were transplanted without competitive cells, Gfi1−/− HSCs do not lack multilineage differentiation potential. Furthermore, the impairment of Gfi1−/− HSCs to competitively reconstitute myeloid cells and lymphoid cells was not only found in peripheral blood but also in bone marrow (data not shown), indicating that the release of Gfi1−/− mature hematopoietic cells from bone marrow to peripheral blood is not perturbed. Importantly, when competitive transplantation experiments were performed with sorted LT-HSCs, a restoration of LSK cells was found in mice reconstituted with WT HSCs, whereas mice reconstituted with Gfi1−/− HSCs failed to generate any Gfi1−/− LSK progeny, which directly indicates that Gfi1−/− HSCs have an impairment of their self-renewal ability. In addition to this, the reduction in long-term radioprotection capacity of Gfi1−/− bone marrow cells upon secondary bone marrow transplantation also supports a role of Gfi1 in HSC self-renewal, at least in response to hematological stress caused by lethal irradiation and serial transplantation.
Of critical importance for the self-renewal and long-term repopulating ability of HSCs is a tight regulation of cell cycle progression. It has been shown that LT-HSCs are asynchronously dividing, repeatedly entering and leaving the cell cycle with a constant fraction in G0 phase under steady-state hematopoietic conditions (Bradford et al, 1997; Cheshier et al, 1999). Impeding HSC proliferation can lead to a decreased repopulating ability (Björnsson et al, 2003; Park et al, 2003). On the other hand, the enhancement of HSC proliferation does not automatically result in a subsequent expansion of self-renewing HSCs, but results in an exhaustion of HSCs (Cheng et al, 2000) and a dramatically decreased long-term engraftment capacity (Fleming et al, 1993; Peters et al, 1996; Orschell-Traycoff et al, 2000). Furthermore, transplantation studies revealed that the ability of HSCs for engraftment in foreign hosts directly correlates with their cell cycle phase position and transit of HSCs through different phases of the cell cycle is accompanied by a significant shift in gene expression that not only affects homing but also other properties of HSCs (Lambert et al, 2003). Therefore, it is crucial for an organism to maintain a pool of HSCs in G0 to assure a potent, self-renewable source of cells that can support lifelong multilineage hematopoiesis.
Earlier studies showed that Gfi1 can be a positive mediator of T-cell proliferation in particular upon T-cell receptor stimulation (Zörnig et al, 1996; Karsunky et al, 2002a), and that Gfi1 can cooperate with positive cell cycle regulators and oncoproteins in T-cell lymphomagenesis (Schmidt et al, 1998a, 1998b). By contrast, our results presented here suggest that the functional defects of Gfi1−/− HSCs are not caused by a lower proliferative ability, but may in contrast be due to a shift of a significant proportion of HSCs from G0 into the cell cycle. Thus, in HSCs, Gfi1 rather seems to dampen cell cycle progression—an entirely different role than in T cells. This can only be explained by proposing that Gfi1 functions differently in different cell types, which is conceivable since antigenic stimulation in T cells for instance triggers other signaling pathways than those that are active in stem cells or hematopoietic progenitor cells. The transcript levels of the previously identified Gfi1 target genes E2F5, E2F6 and p21cip1/waf1 (Duan and Horwitz, 2003) that could be associated with the regulation of cell cycle progression in myeloid cells or T cells were indeed not altered in Gfi1 null LSK cells, and it is thus likely that these targets are not directly regulated by Gfi1 in stem cells. However, E2F5 and E2F6 protein levels are highly upregulated and, surprisingly, p21cip1/waf1 protein is undetectable in Gfi1-deficient bone marrow. This suggests that the genes encoding E2F5, E2F6 and p21cip1/waf1 are not directly regulated by Gfi1 on a transcriptional level but that Gfi1 may be able to influence their protein levels by an indirect and yet unidentified mechanism. The protein levels of the negative G1-specific cell cycle regulator, p27Kip1, in the same bone marrow samples from WT and Gfi1−/− mice remained unchanged, which supports the notion that the alterations of E2F5, E2F6 and p21cip1/waf protein levels are a specific effect associated with the loss of Gfi1 and not a mere consequence of an altered cell cycle distribution in Gfi1 null bone marrow.
Whereas the role of E2F5 in cell cycle regulation seems less clear, it is known that ectopic expression of E2F6 leads to accumulation of cells in S phase (Trimarchi and Lees, 2002) and it has been documented that a loss of p21cip1/waf1 causes increased cell cycling and stem cell exhaustion and as a consequence leads to impaired self-renewal of HSCs (Cheng et al, 2000). Moreover, E2F6 is able to form complexes with the oncoprotein Bmi-1, which is a member of the polycomb group and an essential factor for the self-renewal of HSCs and leukemic stem cells (Lessard and Sauvageau, 2003; Park et al, 2003). Therefore, it is likely that the high levels of E2F5, E2F6 and the lack of p21cip1/waf1 in Gfi1−/− HSCs cause a shift from G0 to the cell cycle leading to exhaustion of the HSC pool and a lower percentage of stem cells with high self-renewal potential similar to the phenotype observed in p21cip1/waf1-deficient mice (Cheng et al, 2000) (Figure 7C). Hence, our data support the interpretation that Gfi1 controls self-renewal and repopulation abilities of HSCs by restraining their proliferative potential and by maintaining a constant proportion of HSCs in G0 (Figure 7C).
However, other mechanisms of Gfi1 action cannot be excluded. For instance, we have previously reported that Gfi1 can enhance STAT3 activity by relieving it from PIAS3 inhibition, an activity of Gfi1 that is independent of DNA binding (Rödel et al, 2000). Thus, it is possible that loss of Gfi1 impairs the ability of STAT3 to efficiently relay signals from cytokine receptors in HSCs that are required to control their proliferation. Such a hypothesis is supported by the finding that retrovirus-mediated overexpression of a dominant-negative STAT3 mutant in HSCs can permanently reduce their in vivo reconstituting ability (Oh and Eaves, 2002). Alternatively, the previously shown function of Gfi1 in early pre-T-cell survival (Yücel et al, 2003) points to a role of Gfi1 in preventing apoptosis in specific cell types. Since we have preliminary evidence that coexpression of a Bcl-2 gene can specifically rescue the loss of short-term HSCs in Gfi1-deficient mice, it will be important to asses this aspect in future experiments to show whether the inhibition of cell death by Gfi1 is involved in the maintenance of normal HSC functions.
Materials and methods
Mice
Gfi1-deficient mice have been previously described (Karsunky et al, 2002b). WT and Gfi1−/− mice were bred and maintained under specific pathogen-free conditions at the animal facility of the Institut für Zellbiologie, Universitätsklinikum Essen in individually ventilated cages. Mice that were used for analyses were healthy 4- to 8-week-old animals from a more than 20 generations backcross with C57BL/6 mice. Gfi1:GFP knock-in mice were generated in a similar way as the Gfi1−/− animals with the exception that a GFP open reading frame was inserted immediately downstream of the Gfi1 translation initiation codon and that the neo cassette was flanked by loxP sites, which allowed its germline deletion upon expression of a Cre recombinase (Yücel et al, 2004).
Flow cytometry analysis and sorting of HSC and progenitors
HSCs and progenitors were analyzed by flow cytometry and sorted from adult mouse bone marrow as described (Kondo et al, 1997; Akashi et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001). In brief, bone marrow cells were obtained by flushing the femurs and were then stained with primary and labeled secondary antibodies. HSCs were defined by the absence of lineage markers (CD3, CD4, CD8, Mac-1, Gr-1, Ter119 and B220) and the expression of Sca-1, c-Kit and Flt3. CLPs were defined by the absence of lineage markers and the expression of Sca-1, c-Kit and IL-7Rα. CMPs, GMPs and MEPS were defined by the absence of lineage markers and different expression levels of Sca-1, c-Kit, IL-7RαCD34 and Fcγ receptor. Cells were analyzed with a FACS Calibur or sorted with a FACSVantageDiVa (Becton Dickinson).
Expression analysis of Gfi1 by RT–PCR
Bone marrow cells were harvested from C57BL/6, Thy1.1 mice and stem cell and progenitor populations were sorted according to previously described criteria (Kondo et al, 1997; Akashi et al, 2000; Adolfsson et al, 2001; Christensen and Weissman, 2001). Total RNA from 2000 double-sorted cells was isolated using TRIzol Reagent (Invitrogen) according to the manufacturer's protocol and 10 μg/ml linear acrylamide (Ambion) was used as a carrier. All RNA samples were treated with DNase1 to avoid genomic DNA contamination and reverse transcribed into cDNA using the SuperScript First Strand Synthesis System with random hexamers according to the manufacturer's protocol. For PCR, 1 μl of cDNA (equivalent to cDNA from 100 cells) and the following primers were used for amplification: Gfi1 5′ CTG CTA CAA GAG GAG GCA TCA-3′, 5′-GAA GCA CAG AAC ACA GGC TCT-3′, β-actin: 5′-ACG AGG CCC AGA GCA AGA GAG G-3′, 5′-AGC CAC CGA TCC ACA CAG AGT A-3′.
In vivo analysis of HSC function: competitive reconstitution
WT or Gfi1−/− bone marrow cells (CD45.2) were mixed with competitor CD45.1+ bone marrow cells at a ratio of 1:1 or 10:1, and injected into CD45.1 recipient mice lethally irradiated with 9.6 Gy (n=4 per group). Reconstitution of donor (CD45.2+) myeloid and lymphoid cells was monitored by staining peripheral blood cells with antibodies against CD45.2, Mac-1 (CD11b), Gr-1, CD3 and CD19. The same experiments were performed using 500 sorted WT or Gfi1−/− Lin−c-Kit+Sca-1+Flt3− cells mixed with 2 × 105 competitor CD45.1 bone marrow cells.
Cell cycle analysis of HSCs
For BrDU incorporation, mice were initially injected intraperitoneally with 1.8 mg of BrdU in 200 μl PBS and then were continuously given BrdU at 1 mg/ml in the drinking water. After different time points, bone marrow cells were harvested and stained for surface markers, and then fixed and stained with FITC-conjugated anti-BrdU antibody using the Cytofix/Cytoperm Kit (BD Biosciences) according to the manufacturer's instructions. Bone marrow cells were stained with Hoechst 33342 and Pyronin Y at 37°C for 45 min as described (Cheshier et al, 1999). Cells then were analyzed by flow cytometry to determine the cell cycle profile of LSK cells.
Real-time quantitative PCR
HSCs were sorted by FACSDiVa™ for quantitative RT–PCR. Approximately 18 000 cells of two sets of mice were collected and Trizol (Invitrogen) was applied using carrier provided by the ExpressArt® mRNA Kit. Quantitative RT–PCR was performed in a 20-μl reaction volume containing 900 nM of each primer, 250 nM TaqMan probe and 1 × TaqMan Universal PCR Master Mix (ABI) according to the manufacturer's instructions. Reaction was monitored in an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). Quantitative RT–PCR was performed on two amounts of cDNA (1/20 and 1/5 of total cDNA). To correct for the amount of cDNA added to any individual reaction, PCR was performed in duplicate. The expression of the gene of interest was calculated relative to the levels of GAPDH mRNA.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Klaus Lennartz for excellent assistance with flow cytometry. We are also indebted to Petra Plessow for excellent animal care and Holger Karsunky and Ulrich Dührsen for critically reading the manuscript. We thank Holger Karsunky for the Gfi1-specific PCR reactions of sorted HSCs and progenitors. This work was supported by the Deutsche Forschungsgemeinschaft, DFG (grant no. 435/10-5, 10-6), the ‘Fonds der chemischen Industrie', the European Community Framework 5 Program and the ‘IFORES Program' of the University of Essen Medical School.
References
- Adolfsson J, Borge OJ, Bryder D, Theilgaard-Monch K, Astrand-Grundstrom I, Sitnicka E, Sasaki Y, Jacobsen SE (2001) Upregulation of Flt3 expression within the bone marrow Lin−Sca1+c-kit+ stem cell compartment is accompanied by loss of self-renewal capacity. Immunity 15: 659–669 [DOI] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, Weissman IL (2000) A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404: 193–197 [DOI] [PubMed] [Google Scholar]
- Bertoncello I, Williams B (2001) Analysis of hematopoietic phenotypes in knockout mouse models. Methods Mol Biol 158: 181–203 [DOI] [PubMed] [Google Scholar]
- Björnsson JM, Larsson N, Brun AC, Magnusson M, Andersson E, Lundström P, Larsson J, Repetowska E, Ehinger M, Humphries RK, Karlsson S (2003) Reduced proliferative capacity of hematopoietic stem cells deficient in Hoxb3 and Hoxb4. Mol Cell Biol 23: 3872–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford GB, Williams B, Rossi R, Bertoncello I (1997) Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp Hematol 25: 445–453 [PubMed] [Google Scholar]
- Broudy VC (1997) Stem cell factor and hematopoiesis. Blood 90: 1345–1364 [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT (2000) Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287: 1804–1808 [DOI] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL (1999) In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA 96: 3120–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JL, Weissman IL (2001) Flk-2 is a marker in hematopoietic stem cell differentiation, a simple method to isolate long-term stem cells. Proc Natl Acad Sci USA 98: 14541–14546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Horwitz M (2003) Targets of the transcriptional repressor oncoprotein Gf11. Proc Natl Acad Sci USA 100: 5932–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming WH, Alpern EJ, Uchida N, Ikuta K, Spangrude GJ, Weissman IL (1993) Functional heterogeneity is associated with the cell cycle status of murine marrow cells. J Cell Biol 122: 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks CB, Bear SE, Grimes HL, Tsichlis PN (1993) Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol 13: 1759–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodell MA, Rosenzweig M, Kim H, Marks DF, De Maria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP (1997) Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med 3: 1337–1345 [DOI] [PubMed] [Google Scholar]
- Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN (1996) The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol 16: 6263–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenechea G, Gan OI, Dorrell C, Dick JE (2001) Distinct classes of human stem cells that differ in proliferative and self-renewal potential. Nat Immunol 2: 75–82 [DOI] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, Orkin SH (2003) Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 18: 109–120 [DOI] [PubMed] [Google Scholar]
- Karanu FN, Murdoch B, Gallacher L, Wu DM, Koremoto M, Sakano S, Bhatia M (2000) The Notch ligand Jagged-1 represents a novel growth factor of human hematopoietic stem cells. J Exp Med 192: 1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsunky H, Mende I, Schmidt T, Möröy T (2002a) High levels of the onco-protein Gfi-1 accelerate T-cell proliferation and inhibit activation induced T-cell death in Jurkat T-cells. Oncogene 21: 1571–1579 [DOI] [PubMed] [Google Scholar]
- Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Dührsen U, Möröy T (2002b) Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet 30: 295–300 [DOI] [PubMed] [Google Scholar]
- Kimura S, Roberts AW, Metcalf D, Alexander WS (1998) Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci USA 95: 1195–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo M, Weissman IL, Akashi K (1997) Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell 91: 661–672 [DOI] [PubMed] [Google Scholar]
- Lambert JF, Liu M, Colvin GA, Dooner M, McAuliffe CI, Becker PS, Forget BG, Weissman SM, Quesenberry PJ (2003) Marrow stem cell shift gene expression and engraftment phenotype with cycle transit. J Exp Med 197: 1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255–260 [DOI] [PubMed] [Google Scholar]
- Metcalf D (1998) Lineage commitment and maturation in hematopoietic cells. The case for extrinsic regulation. Blood 92: 345–352 [PubMed] [Google Scholar]
- Morrison SJ, Weissman IL (1994) The long-term repopulating subsets of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1: 661–673 [DOI] [PubMed] [Google Scholar]
- Nakorn TN, Miyamoto T, Weissman IL (2002) Myeloid erythroid-restricted progenitors are sufficient to confer radioprotection and provide the majority of Day 8 CFU-S. J Clin Invest 109: 1579–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichogiannopoulou A, Trevisan M, Neben S, Friedrich C, Georgopoulos K (1999) Defects in hemopoietic stem cell activity in Ikaros mutant mice. J Exp Med 190: 1201–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh IH, Eaves CJ (2002) Overexpression of a dominant negative form of STAT3 selectively impairs hematopoietic stem cell activity. Oncogene 21: 4778–4787 [DOI] [PubMed] [Google Scholar]
- Orschell-Traycoff CM, Hiatl K, Dagher RN, Rice S, Yoder MC, Srour EF (2000) Homing and engraftment potential of Sca-1+lin− cells fractionated on the basis of adhesion molecule expression and position in cell cycle. Blood 96: 1380–1387 [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF (2003) Bmi-1 is required for maintenance of adult self-renewing heamatopoietic stem cells. Nature 423: 302–305 [DOI] [PubMed] [Google Scholar]
- Peters SO, Kittler ELW, Ramshaw HS, Quesenberry PJ (1996) Ex vivo expansion of murine marrow cells with interleukin-3: interleukin-6: interleukin-11: and stem cell factor leads to impaired engraftment in irradiated hosts. Blood 87: 30–37 [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423: 409–414 [DOI] [PubMed] [Google Scholar]
- Rödel B, Tavassoli K, Karsunky H, Schmidt T, Bachmann M, Schaper F, Heinrich P, Shuai K, Elsasser HP, Möröy T (2000) The zinc finger protein Gfi-1 can enhance STAT3 signaling by interacting with the STAT3 inhibitor PIAS3. EMBO J 19: 5845–5855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheijen B, Jonkers J, Acton D, Berns A (1997) Characterization of pal-1: a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J Virol 71: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Karsunky H, Gau E, Zevnik B, Elsässer HP, Möröy T (1998a) Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene 17: 2661–2667 [DOI] [PubMed] [Google Scholar]
- Schmidt T, Karsunky H, Rödel B, Zevnik B, Elsässer HP, Möröy T (1998b) Evidence implicating Gfi-1 and Pim-1 in pre-T-cell differentiation steps associated with beta-selection. EMBO J 17: 5349–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T, Zörnig M, Beneke R, Möröy T (1996) MoMuLV proviral integrations identified by Sup-F selection in tumors from infected myc/pim bitransgenic mice correlate with activation of the gfi-1 gene. Nucleic Acids Res 24: 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude GJ, Heimfeld S, Weissman IL (1988) Purification and characterization of mouse hematopoietic stem cells. Science 241: 58–62 [DOI] [PubMed] [Google Scholar]
- Stier S, Cheng T, Dombkowski D, Carlesso N, Scadden DT (2002) Notch1 activation increases hematopoietic stem cell self-renewal in vivo and favors lymphoid over myeloid lineage outcome. Blood 99: 2369–2378 [DOI] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20 [DOI] [PubMed] [Google Scholar]
- Varnum-Finney B, Xu L, Brashem-Stein C, Nourigat C, Flowers D, Bakkour S, Pear WS, Bernstein ID (2000) Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat Med 6: 1278–1281 [DOI] [PubMed] [Google Scholar]
- Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, Zoghbi HY, Orkin SH, Bellen HJ (2003) The zinc finger transcription factor Gfi1: implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130: 221–232 [DOI] [PubMed] [Google Scholar]
- Weissman IL (2000) Stem cells, unit of development, unit of regeneration, and its evolution. Cell 100: 157–168 [DOI] [PubMed] [Google Scholar]
- Willert K, Brown D, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR III, Nusse R (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423: 448–452 [DOI] [PubMed] [Google Scholar]
- Yücel R, Karsunky H, Klein-Hitpass L, Möröy T (2003) The transcriptional repressor Gfi1 affects development of early, uncommitted c-Kit+ T-cell progenitors and CD4/CD8 lineage decision in the thymus. J Exp Med 197: 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yücel R, Kosan C, Heyd F, Möröy T (2004) Gfi1:GFP knock-in mutant reveals differential expression and autoregulation of the gene growth factor independence 1 (Gfi1) during lymphocyte development. J Biol Chem, Jul 13 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Zhao Y, Lin Y, Zhan Y, Yang G, Louie J, Harrison DE, Anderson WF (2000) Murine hematopoietic stem cell characterization and its regulation in BM transplantation. Blood 96: 3016–3022 [PubMed] [Google Scholar]
- Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, Tsichlis PN, Paul WE (2002) Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity 16: 733–744 [DOI] [PubMed] [Google Scholar]
- Zörnig M, Schmidt T, Karsunky H, Grzeschiczek A, Möröy T (1996) Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene 12: 1789–1801 [PubMed] [Google Scholar]
- Zweidler-Mckay PA, Grimes HL, Flubacher MM, Tsichlis PN (1996) Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol 16: 4024–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3