

Abstract
An interferon-induced endoribonuclease, ribonuclease L (RNase L), is implicated in both the molecular mechanism of action of interferon and the fundamental control of RNA stability in mammalian cells. RNase L is catalytically active only after binding to an unusual activator molecule containing a 5′-phosphorylated 2′,5′-linked oligoadenylate (2-5A), in the N-terminal half. Here, we report the crystal structure of the N-terminal ankyrin repeat domain (ANK) of human RNase L complexed with the activator 2-5A. This is the first structural view of an ankyrin repeat structure directly interacting with a nucleic acid, rather than with a protein. The ANK domain folds into eight ankyrin repeat elements and forms an extended curved structure with a concave surface. The 2-5A molecule is accommodated at a concave site and directly interacts with ankyrin repeats 2–4. Interestingly, two structurally equivalent 2-5A binding motifs are found at repeats 2 and 4. The structural basis for 2-5A recognition by ANK is essential for designing stable 2-5As with a high likelihood of activating RNase L.
Keywords: 2-5A system, ankyrin repeat, crystal structure, drug design, interferon
Introduction
In mammals, viral infections initiate an innate immune response predominantly mediated by type I interferons (IFNs) (Sen, 2001). Type I IFNs regulate the transcription of a number of genes that inhibit or block viral replication by diverse mechanisms (Stark et al, 1998). There are three well-established antiviral pathways for IFN action: these three pathways are associated with the double-stranded RNA (dsRNA)-dependent protein kinase PKR (Williams, 1995), the Mx proteins (Pavlovic and Staeheli, 1991), and the 5′-triphosphorylated, 2′,5′-phosphodiester-linked oligoadenylate (2-5A) system (Player and Torrence, 1998).
In the 2-5A system, treatment of cells with IFN activates genes encoding several 2′,5′-linked oligoadenylate synthetases (OASs) (Chebath et al, 1987) and a single gene encoding ribonuclease L (RNase L) (Zhou et al, 1993). The OASs are activated by binding to dsRNA (Hovanessian et al, 1977), a frequent byproduct of viral infection. The activated OASs generate 2-5A from ATP. The crystal structure of porcine OAS1 was recently reported (Hartmann et al, 2003). The mechanisms of its activation induced by dsRNA and its synthesis of 2-5A were discussed based on its structure and structure-based mutagenesis studies, although the structure contained neither activator nor substrate analogs. RNase L is known to be activated by binding to 2-5A, changing from an inactive monomer to a catalytically active homodimer (Dong and Silverman, 1995). The activated RNase L cleaves RNA containing dyads of UU, UA, AU, AA, and UG (Floyd-Smith et al, 1981; Wreschner et al, 1981). The RNA degradation inhibits protein synthesis and thus inhibits viral replication.
RNase L is an unusual ribonuclease because it requires the activator molecule 2-5A to catalyze the hydrolysis of single-stranded RNA. 2-5A is itself very unusual, consisting of a type of oligoadenylates with 2′,5′ internucleotide linkages, in contrast to the typical 3′,5′ linkages found in RNA and DNA. 2-5A is unstable in cells due to the activities of phosphodiesterases and phosphatases. Early studies revealed the requirement of at least two 5′-phosphoryl groups and at least three 2′,5′-linked adenylyl residues for the efficient activation of murine RNase L (Kerr and Brown, 1978). Thereafter, it was shown that only a single 5′-phosphoryl group on the trimer of 2′,5′-linked oligoadenylate was required for the full activation of human RNase L (Haugh et al, 1983; Yoshimura et al, 2002).
The human form of RNase L is a 741-amino-acid protein with a molecular mass of 83 543 Da (Zhou et al, 1993). RNase L consists of three domains, namely the N-terminal ankyrin repeat domain, the protein kinase homology domain, and the C-terminal ribonuclease domain. The N-terminal ankyrin repeat domain, a region containing nine ankyrin-like macromolecular recognition repeats (the ninth ankyrin repeat is incomplete), is responsible for 2-5A binding, and the C-terminal domain is responsible for catalytic activity (Dong and Silverman, 1997; Nakanishi et al, 2004). The binding of 2-5A to RNase L is thought to induce a conformation change in the enzyme or result in the unmasking of an interaction domain, permitting dimerization and activation of RNase L.
Recently, the enzyme was proposed as a candidate risk factor for hereditary prostate cancer (Casey et al, 2002; Xiang et al, 2003). Among the naturally occurring mutants of RNase L that have been examined, only the Arg462Gln variant showed low RNase activity, and this variant was also shown to be significantly associated with prostate cancer. Therefore, elucidation of all of the amino-acid residues that might influence RNase L activity (i.e., 2-5A binding, dimerization, and catalysis) remains necessary.
Here, we present the crystal structure at 1.8 Å resolution of the N-terminal ankyrin repeat domain of human RNase L complexed with 2-5A. To our knowledge, this is the first structural view of an ankyrin repeat structure directly interacting with a nucleic acid, rather than with a protein. The crystal structure of the ANK/2-5A complex clearly shows that the bound 2-5A molecule directly interacts with ankyrin repeats 2–4. We have re-evaluated the structure–function relationship studies of RNase L and 2-5A in terms of the present crystal structure analysis. The structural basis for 2-5A recognition by ANK is essential for designing stable 2-5As with a high likelihood of activating RNase L.
Results
Structure determination
The crystallization trials have to date not successfully obtained the crystals of full-length human RNase L, as only a minute amount of the full-length recombinant human RNase L has been obtainable; however, crystals of the N-terminal ankyrin repeat domain (ANK) of human RNase L (Figure 1A) were successfully obtained. Addition of 2-5A to the ANK solution was essential for obtaining crystals. For this study, a 2-5A trimer with 5′-monophosphate (p5′(A2′p5′)2A) was used for crystallization (Figure 1B). We determined the crystal structure of the ANK/2-5A complex by the molecular replacement (MR) method using the coordinate of a ‘consensus ankyrin repeat protein' (Kohl et al, 2003) (PDB code: 1MJ0) as the template for constructing a search model, and we refined the resulting model to an R-factor of 0.202 (Rfree of 0.230) at 1.8 Å resolution. The final model consisted of residues 21–305, 220 water molecules, and one 2-5A molecule. The refinement statistics are summarized in Table I.
Figure 1.
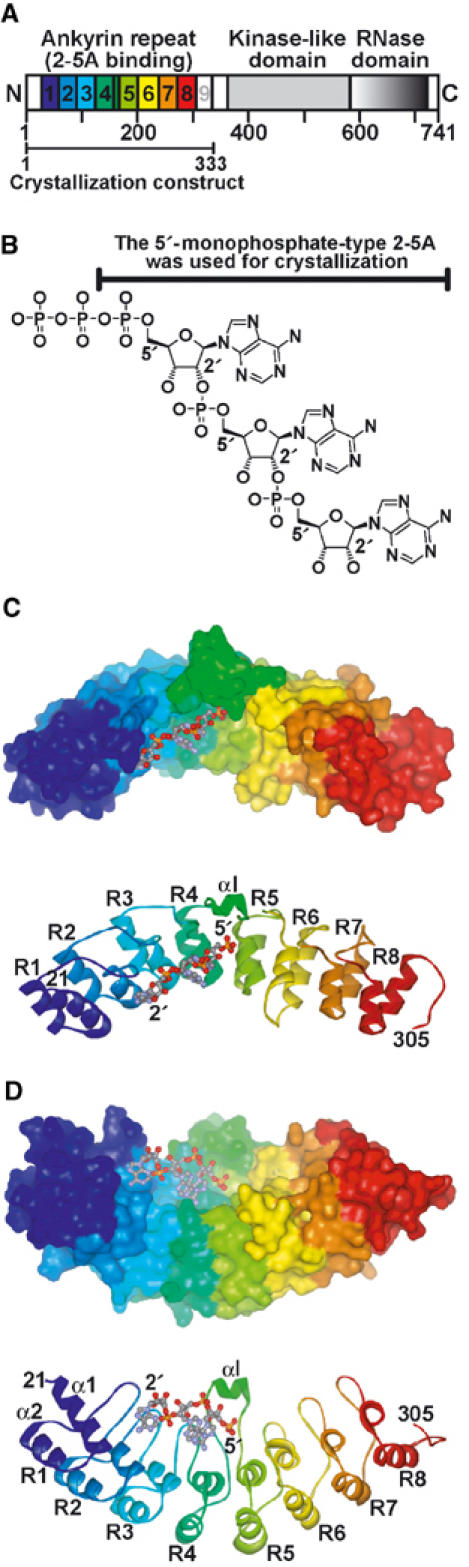
Crystal structure of ANK complexed with 2-5A. (A) Structural and functional domains of RNase L. Ankyrin repeats are shown starting with blue at repeat 1 and ending with red at repeat 8. (B) Structure of the predominant trimeric species of 2-5A ((pp)p(A2′p5′)2A) (Kerr and Brown, 1978). (C, D) Surface (top) and ribbon (bottom) representations of the ANK/2-5A complex. Ankyrin repeats (R1–R8) are shown as in (A). The bound 2-5A molecule is shown as a ball-and-stick model. The view in (D) was obtained by rotating the view in (C) by 90°.
Table 1.
Data collection and refinement statistics of ANK
| Data collection statistics | |
| X-ray source | PF-AR NW12 |
| Wavelength (Å) | 0.978 |
| Resolution range (outer shell) (Å) | 40–1.8 (1.9–1.8) |
| Observed reflections (no sigma cutoff) | 154 126 |
| Unique reflections | 35 922 |
| Multiplicity | 4.3 (3.7) |
| Mean 〈I/σ(I)〉 | 5.5 (2.1) |
| B-factor (Wilson plot) (Å2) | 27.3 |
| Rsym (%) | 8.4 (33.8) |
| Completeness (%) | 99.7 (99.5) |
| Refinement statistics | |
| Resolution range (Å) | 40–1.8 |
| No. of reflections | |
| Working set | 34 084 |
| Test set | 1791 |
| R-factor | 0.202 |
| Free R-factor | 0.230 |
| No. of protein atomsa (average B-factors (Å2)) | 2184 (28.3) |
| No. of water molecules (average B-factors (Å2)) | 220 (37.0) |
| No. of 2-5A atoms (average B-factors (Å2)) | 67 (23.5) |
| R.m.s. deviations | |
| Bond distances (Å) | 0.008 |
| Bond angles (deg) | 1.119 |
| B-factors related by main-chain bonds (Å2) | 0.669 |
| B-factors related by side-chain bonds (Å2) | 1.825 |
| Ramachandran plot | |
| Most favored (%) | 92.6 |
| Additional allowed (%) | 7.4 |
| aHis tag, N-terminal residues (1–20), and C-terminal residues (306–333) are disordered. | |
Overall fold of ANK
ANK folds into eight ankyrin repeat elements and forms an extended curved structure with a groove running across the long concave surface (Figure 1C and D). Previous primary structure analysis suggested that RNase L had nine ankyrin repeats, but the ninth ankyrin repeat is incomplete (Hassel et al, 1993). However, this prediction for RNase L differs from the present crystal structure of ANK, which consists of eight ankyrin repeats (R1–R8 in Figure 1C and D). Residues 306–333, corresponding to the incomplete ninth repeat (Hassel et al, 1993), are disordered. As in other ankyrin repeat proteins (Sedwick and Smerdon, 1999), each repeat is formed by ∼33 amino-acid residues and consists of pairs of antiparallel α-helices stacked side by side and which are connected by a series of intervening β-hairpin motifs. In general, the structure is shaped similar to a cupped hand (Jacobs and Harrison, 1998). There is a noticeable curvature across the ‘palm', such that the surface created by the β-hairpins (fingers) and the α1 ‘inner' helices is concave, whereas that formed by the α2 ‘outer' helices, that is, the back of the cupped hand, is convex (Figure 1D). The 2-5A molecule fits in the concavity and directly interacts with ankyrin repeats 2–4.
Ankyrin repeats of RNase L
A structure-based sequence alignment of the eight ankyrin repeats of RNase L is shown in Figure 2. Definition of the ankyrin repeat consensus is based on the exon boundaries of the ankyrin gene (Lux et al, 1990). This definition separates the two β-strands of the β-hairpin structure and incorporates the most commonly occurring nonconserved elements within the repeating unit (Sedwick and Smerdon, 1999); however, the insertion helix (αI: residues 159–164) of ANK is incorporated between R4 and R5 (Figures 1C, D, and 2). We therefore consider the ankyrin repeat motif of RNase L to be defined as a strand–helix–loop–helix–loop–strand (βααβ) structure.
Figure 2.
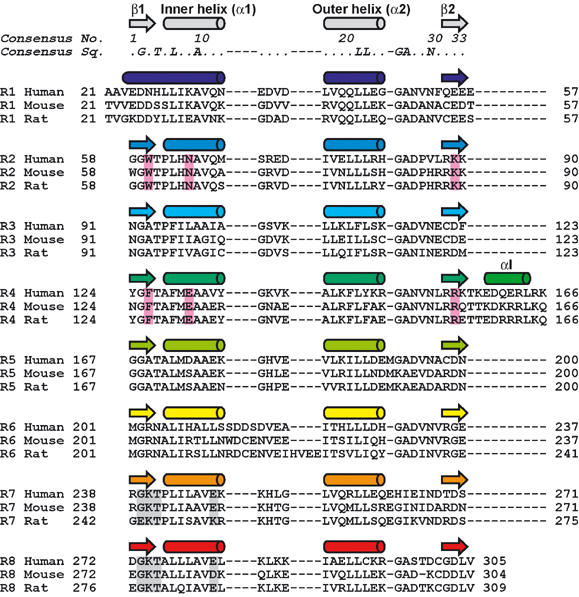
Structure-based sequence alignment of the eight ankyrin repeats (R1–R8) of human, mouse, and rat RNase Ls. Secondary structural elements are shown on top and are colored as in Figure 1. The 2-5A binding motifs, equivalently found in repeats 2 and 4, are boxed in pink. The two GKT motifs are boxed in gray.
Residue Gly(2) in β1 is invariant across all repeats (Figure 2), except for R1. Hereafter, the consensus ankyrin repeat numbering will be shown in italics and parentheses. This glycine residue allows a sharp turn within the β-hairpin structure. Since R1 lacks β1, Gly(2) is not conserved in R1. Highly conserved hydrophobic residues (Leu(6) and Ala(9) from the inner helix α1, and Leu(21) and Leu(22) from the outer helix α2) are involved in packing interactions within and between repeats, giving rise to a continuous hydrophobic core. As previously noted (Jacobs and Harrison, 1998), the inner helix α1 contains hydrophobic residues (Ala(9) and Ala/Val(10)) smaller than those in the outer helix α2 (Leu(21) and Leu(22)), causing the stack of ankyrin repeats to curve toward the inner helix α1. The signature Thr(4)–Ala/Pro(5)–Leu(6)–X(7) motif characteristic of ankyrin repeats forms a tight turn that initiates the inner helix α1. Because the inner helix α1 starts at four residues upstream from Thr(4) in R1, Thr(4) is not conserved in R1. Residues Gly(25) and Ala(26) allow a sharp turn at the end of the outer helix α2. The side chain of the highly conserved polar residue Asn(29) is involved in hydrogen bonds with the carbonyl oxygen atom of Gly(25) and the imino nitrogen atom of X(27) from the next repeat.
Several insertions are found within and between repeats. Met191 and Glu262 are inserted after the outer helix α2 within R5 and R7, respectively. Each of these insertions allows for a substitution of the Gly(25)–Ala(26) residues in R5 or R7, which enable a sharp turn at the end of α2. Four residues (Ser213–Asp214–Asp215–Ser216) are inserted after the inner helix α1 within R6. The insertion of two acidic residues would be expected to affect the electrostatic properties of the surface. Glu57 is inserted between R1 and R2. A total of 10 residues (Thr157 to Lys166), including the insertion helix αI (residues 159–164), are inserted between R4 and R5. These insertions also exist for mouse and rat RNase Ls.
2-5A binding mode
ANK is the first representative of an ankyrin repeat protein for which the structure has been solved in the form of a nucleotide (rather than a protein) complex. Here, the electron density, shown at 1.8 Å resolution, corresponding to the bound 2-5A molecule is very well defined (Figure 3A). Thus, we were able to unequivocally determine the conformations of 2-5A and the surrounding amino-acid residues, as well as the location of water molecules. As shown in Figure 1C and D, only one 2-5A molecule is bound to the concavity of ANK. Since ANK is responsible for 2-5A binding in RNase L (Dong and Silverman, 1997), the present crystal structure is consistent with previous observations demonstrating that the dimerization of RNase L requires the binding of one 2-5A molecule per RNase L monomer and that the enzymatic activity of RNase L is also maximized at a 1:1 2-5A:RNase L stoichiometry (Cole et al, 1996). As shown in Figure 3A, the bound 2-5A molecule adopts an extended conformation in which the first and second adenine rings (Ade1 and Ade2, respectively) are in an anti conformation, whereas the third adenine ring (Ade3) is in a syn conformation. The first and second ribose rings have 3E (C3′-endo) puckering, whereas the third ribose has 4E (C4′-exo) puckering (IUPAC-IUB, 1983). The 2-5A molecule is accommodated in the concavity and directly interacts with ankyrin repeats 2–4 (Figure 1C and D). In the present crystal structure analysis, we used a 2-5A trimer with 5′-monophosphate (p5′(A2′p5′)2A) for crystallization (Figure 1B). The 2-5A molecule is thus considered to be a 2′,5′-linked trimer of 5′-AMP.
Figure 3.
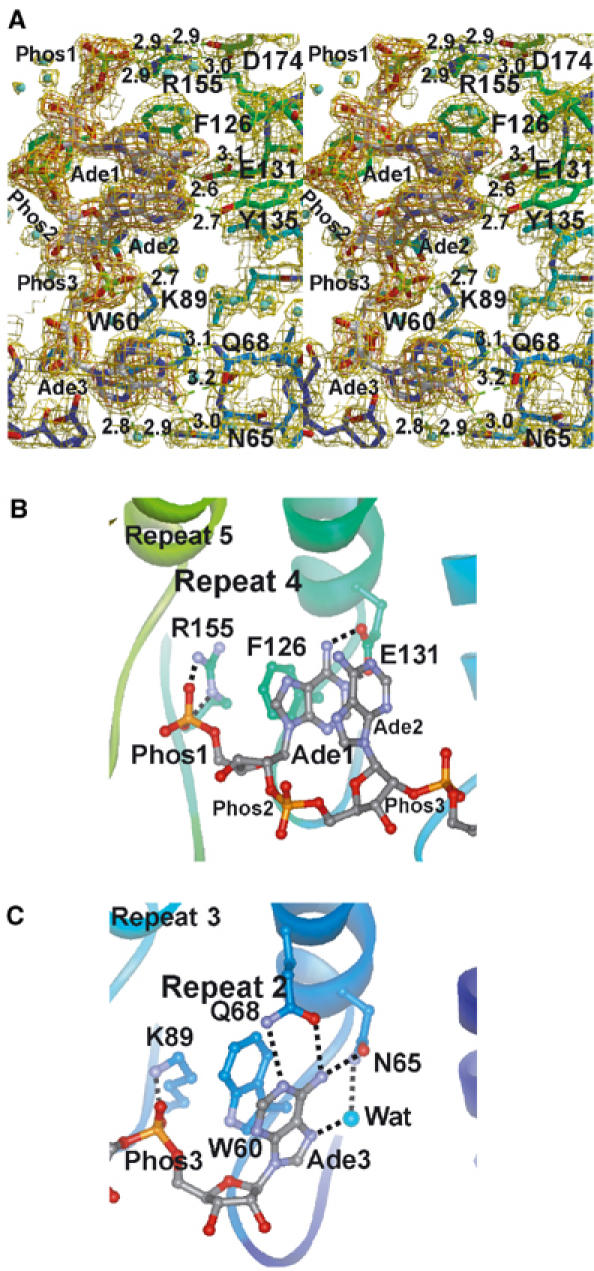
Recognition of 2-5A by ANK. (A) Stereodiagram showing the mode of 2-5A binding to ANK. The carbon atoms of ANK are colored as in Figure 1, and those of the bound 2-5A molecule are shown in white. Possible hydrogen bonds or salt bridges are indicated by dashed lines, and distances (in Å) are given. The refined model is superimposed on the weighted 2∣Fo∣–∣Fc∣ map (1.5σ, yellow) and the ∣Fo∣–∣Fc∣ omit map of 2-5A (4.5σ, orange), calculated at 1.8 Å resolution. Bound water molecules are shown as spheres (cyan). (B, C) Recognition of the first and third AMP moieties of 2-5A by repeats 4 and 2, respectively, of ANK.
The first AMP moiety of the 2-5A directly interacts with the fourth repeat (R4) of ANK (Figure 3A and B). The 5′-phosphate group of the first AMP (Phos1) forms bifurcated salt bridges with the side chain of Arg155. The side chain of Arg155 is fixed by bifurcated salt bridges with the side chain of Asp174 (Figure 3A). The adenine ring of the first AMP (Ade1) is stacked between the side chain of Phe126 and the adenine ring of the second AMP (Ade2), and is fixed by bifurcated hydrogen bonds with the side chain of Glu131, that is, OE1(Glu131)—N6(Ade1) and OE2(Glu131)—N1(Ade1). The side chain of Phe126 also stacks with the guanidino group of the side chain of Arg155, forming a quadruplex (Arg155–Phe126–Ade1–Ade2) of stacking interactions (Figure 3B).
The second AMP moiety of 2-5A interacts only slightly with ANK. The 5′-phosphate group of the second AMP (Phos2) is exposed to solvent, and no direct interactions are found between Phos2 and the surface of ANK. The adenine ring of the second AMP (Ade2) is stacked with Ade1 as described above, and is fixed by a single hydrogen bond with the side chain of Tyr135, that is, OH(Tyr135)—N1(Ade2). The 3′-OH group of the second AMP is involved in a hydrogen bond network and is fixed on the protein surface via water molecules. The second AMP appears to be rather weakly fixed on the ANK surface relative to the two ends of the 2-5A molecule.
The third AMP moiety of 2-5A directly interacts with the second repeat (R2) of ANK (Figure 3A and C). The 5′-phosphate group of the third AMP (Phos3) forms a salt bridge with the side chain of Lys89. The adenine ring of the third AMP (Ade3) is stacked with the side chain of Trp60, and is fixed by a hydrogen bond network involving the side chains of Gln68 (OE1(Gln68)—N6(Ade3) and NE2(Gln68)—N1(Ade3)) and Asn65 (OD1(Asn65)—N6(Ade3)), as well as a water molecule (O(Wat)—N7(Ade3) and O(Wat)—ND2(Asn65)). The side chain of Trp60 also stacks with the CD–CE–NZ bonds of the side chain of Lys89, forming a triplex (Lys89–Trp60–Ade3) of stacking interactions (Figure 3C). Unlike Ade1 and Ade2, Ade3 adopts a syn conformation. The 2′- and 3′-OH groups of the third AMP do not interact with either the protein surface or with water molecules.
Interestingly, the 2-5A binding residues in the R4 and R2 of ANK are located at the structurally equivalent position of the ankyrin repeat (Figure 2, boxed in pink) and these residues play a functionally equivalent role. The side chains of Arg155 in R4 and Lys89 in R2 form salt bridges with Phos1 and Phos3, respectively (Figure 3B and C). The side chains of Phe126 in R4 and Trp60 in R2 stack with Ade1 and Ade3, respectively. Furthermore, a quadruplex (Arg155–Phe126–Ade1–Ade2) and a triplex (Lys89–Trp60–Ade3) of stacking interactions are observed at R4 and R2, respectively. The side chains of Glu131 in R4 and Asn65 in R2 form hydrogen bonds with Ade1 and Ade3, respectively. It should be noted here that these 2-5A binding residues are completely conserved among human, mouse, and rat RNase Ls (Figure 2).
Two insertions are found around the 2-5A binding region of ANK. One of these insertions is Glu57, which is inserted between R1 and R2. This insertion extends a finger between R1 and R2, which constitutes a wall of the third AMP binding site (Figure 1D). The other insertion is the insertion helix αI (residues 159–164), which is inserted between R4 and R5. This helix constitutes the bottom of the first AMP binding site (Figure 1D).
The eight ankyrin repeats of ANK are kinked at the middle
As shown in Figure 1C and D, the eight ankyrin repeats of ANK adopt an extremely curved structure. To obtain insight into the curved structure of ANK, we superposed repeats 1 and 2 of other ankyrin repeat proteins on those of ANK (Figure 4). In general, a longer ankyrin repeat protein has a more concave structure than a shorter ankyrin repeat protein does; for example, the structure of ankyrin (Michaely et al, 2002), which consists of 12 ankyrin repeats, is more concave than those of Bcl-3 (Michel et al, 2001) and gankyrin (Krzywda et al, 2004), which each consist of seven ankyrin repeats (Figure 4). However, it is of note that the degree of curvature across the eight ankyrin repeats of ANK is more remarkable than that of the 12 ankyrin repeats of ankyrin in two directions (Figure 4).
Figure 4.
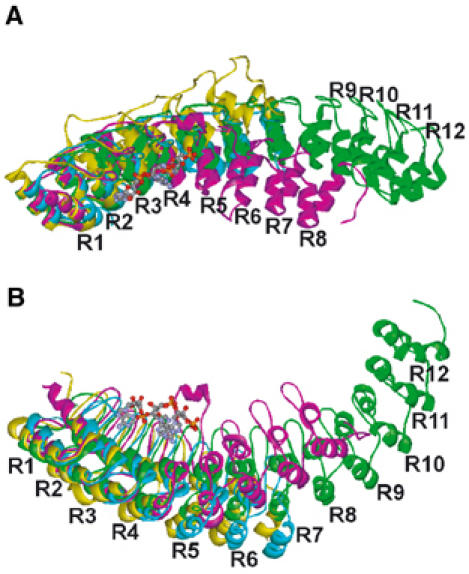
The kinked structure of ANK. (A, B) Superposition of repeats 1 and 2 of human gankyrin (cyan, PDB ID: 1UOH), human Bcl-3 (yellow, 1K1A), and human ankyrin (green, 1N11) on those of ANK (magenta, 1WDY). The views in (A) and (B) are the same as those shown in Figure 1C and D, respectively.
As shown in Figure 4, the eight ankyrin repeats of ANK are clearly kinked at R4 and R5 (15 and 11° in Figure 4A and B, respectively, as compared with ankyrin). A total of 10 residues (Thr157 to Lys166), including the insertion helix αI (residues 159–164), are inserted between R4 and R5 (Figure 2). This insertion of a longer polypeptide chain enables more bending. In addition, the more bulky side chains of Phe143 and Tyr145, instead of Leu residues, are found in the outer helix α2 of R4 (Figure 2). These arrangements appear to result in the wider separation of the outer helices α2 of R3, R4, and R5.
The relationship between the higher degree of curvature of ANK and the 2-5A binding is still unclear, as we have not yet obtained the structure of ANK in the absence of 2-5A. However, the results of time-resolved fluorescence measurements and protease protection assays have indicated that the conformation of ANK does indeed change upon 2-5A binding (M Nakanishi et al, unpublished data). Therefore, we suggest here that the higher degree of curvature of ANK is due to 2-5A binding.
Discussion
2-5A binding domain
Extensive efforts employing various techniques have been directed toward functional analyses of RNase L. As summarized in Figure 5A, we can re-evaluate these results in terms of the newly established crystal structure of the ANK/2-5A complex. The results of the N-terminal truncations of RNase L (Dong and Silverman, 1997) are completely consistent with the present crystal structure analysis showing that the 2-5A molecule directly interacts with ankyrin repeats 2–4 (Figure 1C and D). Truncation of the 23 N-terminal amino acids preceding the first ankyrin repeat (R1), which are disordered in the present crystal structure and are not involved in interactions with 2-5A, does not result in the loss of 2-5A binding activity (NΔN(24–741)). Other N-terminal truncations, that is, mutants NΔR1(57–741), NΔR1-2(91–741), NΔR1-3(124–741), and NΔR1-6(238–741), lacking 1, 2, 3, and 6 ankyrin repeats, respectively, do show a loss of 2-5A binding activity. Although R1 is not directly involved in 2-5A binding, it constitutes a wall of the third AMP moiety of the 2-5A binding site (Figure 1D). Moreover, results showing the C-terminal truncation of RNase L (CΔC(1–335)) (Dong and Silverman, 1997) are also consistent with the present crystal structure analysis demonstrating that the N-terminal ankyrin repeat domain of RNase L is sufficient for 2-5A binding activity.
Figure 5.
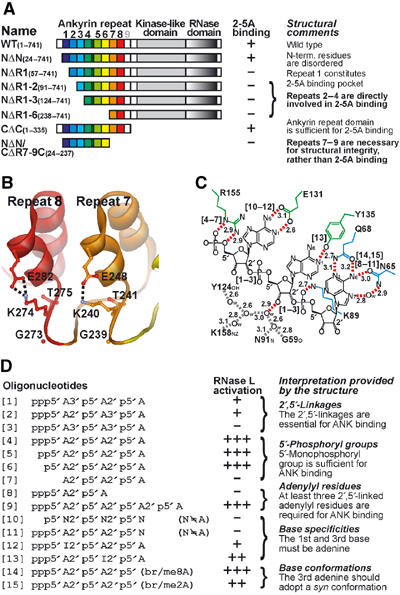
Re-evaluation of previous structure–function relationship studies of ANK and 2-5As in terms of the present crystal structure analysis. (A) Biochemical studies of ANK truncation mutants (Dong and Silverman, 1997). (B) The two GKT motifs in repeats 7 and 8. The side chains of Lys240 and Lys274 are involved in an intrarepeat salt bridge (dashed lines) with the side chains of Glu248 and Glu282, respectively, rather than in 2-5A binding. (C) Schematic representation of the 2-5A binding mode. The side chains of ANK are colored as in Figure 1. Hydrogen bonds or salt bridges and a hydrogen bond network of water molecules are indicated by red and gray dashed lines, respectively, and distances (in Å) are given. Aromatic residues, Trp60 and Phe126, involved in the stacking interactions with 2-5A (seeFigure 3B and C) have been omitted for clarity. Water molecules are shown as ‘Ow'. The [n] indicates important interactions for oligonucleotide [n] in (D). (D) Chemical studies of 2-5A and its analogs (Player and Torrence, 1998). The relative activities of 2-5A analogs, as compared with canonical 2-5A [4], for RNase L activation are shown as ‘+++' (1/2∼), ‘++' (1/20∼1/2), ‘+' (1/200∼1/20), and ‘−' (∼1/200).
Before the present crystal structure analysis was conducted, two GKT motifs, Gly239–Lys240–Thr241 in R7 and Gly273–Lys274–Thr275 in R8 (Figure 2, boxed in gray), had been postulated as the 2-5A binding motifs (Zhou et al, 1993; Diaz-Guerra et al, 1999). Substitution of the two lysines with asparagines greatly reduced the affinity for 2-5A (Zhou et al, 1993). Similarly, one mutant (NΔN/CΔR7-9C(24–237)) (Dong and Silverman, 1997), which is truncated at both termini and lacks R7–R9, with which Lys240 and Lys274 are involved, was shown to lose 2-5A binding activity (Figure 5A). These observations, together with the lack of structural information about ANK at the time the observations were made, led to the assumption that the two GKT motifs were the 2-5A binding motifs. However, the present crystal structure has clearly revealed that the side chains of Lys240 and Lys274 are involved in an intrarepeat salt bridge with the side chains of Glu248 and Glu282, respectively (Figure 5B), rather than in 2-5A binding.
Although the structural role of the two GKT motifs remains unclear without additional structural studies, one possible explanation for their role would be that the salt bridges in R7 and R8 contribute to maintaining the folding of the ankyrin repeat domain, and hence to its integrity. Consequently, structural perturbation at R7 and R8 greatly diminishes the 2-5A binding ability of RNase L, even though these regions are not directly involved in 2-5A binding.
Structure–activity relationship of 2-5A and its analogs
Several chemical approaches have produced a considerable amount of information about the atoms or groups of atoms in 2-5A that interact with RNase L. Similar to the re-evaluation of previous functional studies of RNase L described above, we can now re-evaluate previous studies of the structure–activity relationship of 2-5A and its analogs (Torrence et al, 1994; Player and Torrence, 1998) in terms of the present crystal structure (Figure 5C and D).
The most intriguing feature of the 2-5A molecule is the presence of the 2′,5′-linked phosphodiester bonds (Figures 1B and 5C). Lesiak et al (1983) showed that replacing either of the 2′,5′-linkages in a 2-5A trimer, [1] and [2] in Figure 5D, decreased binding by 1 and 2 orders of magnitude; however, substituting both linkages [3] reduced binding drastically. The present structure thus accounts for the structural significance of the two 2′,5′-linkages, based on the assumption that the interactions between R4 and R2 of ANK and the first and third AMP, respectively, of 2-5A (Figures 3B, C, and 5C) must be maintained. Replacing the first 2′,5′-linkage with a 3′,5′-linkage [1] would drastically shift the adenosine moiety of the second AMP and the phosphate of the third AMP outwardly, and then the stacking interaction between Ade1 and Ade2 and the salt bridge between Phos3 and Lys89 would be weakened. Replacing the second 2′,5′-linkage [2] would shift the adenosine moiety of the second AMP inward (or drastically shift Phos3 outward), resulting in a weakening of Ade1–Ade2 stacking and possibly the steric hindrance of Ade2 at the protein surface (or the Phos3–Lys89 salt bridge might be weakened). Replacing both the 2′,5′-linkages [3] additively was found to weaken both the Ade1–Ade2 stacking and the Phos3–Lys89 salt bridge. Since the 2′,5′-linkages orient backbone-inwards toward the bases (Figure 1B), rather than away from the bases as in 3′,5′-linkages, 2-5A with both the 2′,5′-linkages enabled all of the essential interactions between 2-5A and ANK (Figure 5C).
The requirement for the 5′-phosphoryl group is an interesting feature of the 2-5A molecule. It was previously demonstrated that only a single 5′-phosphoryl group on the trimer of a 2′,5′-linked oligoadenylate, [4], [5], [6], and [7] in Figure 5D, is required for the full activation of human RNase L (Kerr and Brown, 1978; Haugh et al, 1983; Yoshimura et al, 2002). The present structure reveals that the 5′-monophosphoryl group is tightly fixed by bifurcated salt bridges with Arg155 (Figures 3B and 5C), which is consistent with the finding that the 5′-monophosphoryl group is sufficient for binding to ANK. It is also of note that there is an extra space in the concavity of ANK (Figure 1D, top) to accommodate the 5′-di or -triphosphoryl group of 2-5A. Since [6], which was used for the present crystal structure analysis to prepare ANK/2-5A complex, was about 40% less effective than [4] (Yoshimura et al, 2002), additional interactions between the β- or γ-phosphate of [4] and ANK may exist.
Studies on the ribose moiety of 2-5A showed that only the 3′-OH of the second AMP moiety of 2-5A is required for the efficient activation of RNase L (Torrence et al, 1988). The present structure demonstrates that the 3′-OH group of the second AMP is involved in the hydrogen bond network and is fixed on the protein surface via water molecules (Figure 5C). Our observations are consistent with those of a previous study suggesting that the 3′-OH group of the central adenosine of trimer 2-5A may form a hydrogen bond with an acceptor in RNase L (Torrence et al, 1994).
The length of the 2-5A species is critical for the activation of RNase L. While the dimer, [8] in Figure 5D, failed to activate RNase L, the longer 2-5A oligomer [9], a 5′-monophosphate type of [9], or the canonical 2-5A trimer [4] maximally activated purified recombinant RNase L at 1 nM in the poly-U degradation assay (Dong et al, 1994). Thus, the trimeric 2-5A and tetrameric 2-5As maximally activated RNase L at the same concentration. This finding is consistent with our obtained structure showing that the Phos3 and Ade3 of the third AMP of 2-5A are involved in binding with ANK (Figures 3C and 5C) and that the dimeric form of 2-5A lacking the third AMP is clearly insufficient for binding. On the other hand, the longer 2-5A oligomers appear to be harmless for interactions with ANK. If there were strong interactions between the fourth AMP of a longer 2-5A and the R1 of ANK (Figure 1C and D), the tetrameric 2-5As described above could be more effective than the trimeric 2-5A. Thus, we assume that the fourth or longer AMP moieties of 2-5A would extend out of ANK and would therefore have no or only limited possibility of interacting with ANK.
As regard the role of the adenine rings in 2-5A, it has been reported that RNase L has drastically lower affinity for cytidine, uridine, or inosine analogs, [10] in Figure 5D, compared to its adenine counterpart [6] (Torrence et al, 1984). Similarly, [11] failed to activate RNase L (Drocourt et al, 1982). Torrence et al (1984) suggested that RNase L interacts with N1 and/or N6 of the adenine ring of 2-5A. As suggested by Torrence et al, the present structure shows an adenine ring-specific recognition that includes both N1 and N6 atoms of the adenine ring at the first and the third AMP moiety of 2-5A by R4 and R2 of ANK, respectively (Figures 3B, C, and 5C). Interestingly, the second adenine ring was less important for the activation of RNase L ([12] and [13]) (Imai et al, 1985). Use of uridine analogs of 2-5A also produced similar results (Kitade et al, 1991a). These results are consistent with our structure showing that the second adenine ring (Ade2) is recognized by only a single hydrogen bond with the nonconserved side chain of Tyr135 (Figures 3A and 5C).
We have previously shown that 2-5A molecules modified at the 8-position (8-bromo or 8-methyl) of Ade3, which is thereby forced into a syn conformation, are more effective activators of RNase L, whereas those modified at the 2-position of Ade3, which is thereby forced into an anti conformation, are less effective ([14] and [15]) (Kitade et al, 1991b, 1998, 2000; Yoshimura et al, 2002). In accord with these results, we demonstrated in the present study that the third adenine ring (Ade3) is in a syn conformation, whereas the first and second adenine rings (Ade1 and Ade2, respectively) of 2-5A are in an anti conformation (Figures 3A and 5C). The syn conformation of the Ade3 of 2-5A is stabilized by a hydrogen bond network involving the side chains of Asn65 and Gln68, and a water molecule (Figures 3C and 5C).
In summary, we concluded that the Phos1 and Ade1 of the first AMP moiety, the 3′-OH of the second AMP moiety, and the Phos3 and Ade3 of the third AMP moiety of 2-5A are essential for achieving interactions with ANK. In other words, 2-5A is not a perfect molecule for interacting with RNase L; for example, the 3′-OH group of the first AMP, the Phos2 and Ade2 of the second AMP, and the 2′- and 3′-OH groups of the third AMP could be modified for optimal binding, as described in previous structure–function relationship studies on 2-5A analogs (Torrence et al, 1994; Player and Torrence, 1998). Since our obtained structure revealed that these groups are solvent-exposed, the introduction of a functional group at these positions may enable direct or water molecule-mediated interactions between the group and the protein surface, and may thereby enhance binding and/or activation ability with respect to RNase L.
Surface properties of ANK and the implications for inter- and intramolecular interactions
Several X-ray structures of protein–protein complexes involving ankyrin repeats have been determined (Sedwick and Smerdon, 1999). Although the mechanisms of binding between target molecules and ankyrin repeats are known to vary considerably (aromatic, hydrophobic, hydrophilic, or electrostatic), the binding to target molecules commonly involves contacts with β-hairpin fingers and the surface of the inner helices α1 facing into the concave region of the ankyrin repeat domain. The residues involved in the contact are not conserved in the consensus sequence and are not structurally constrained, and therefore they can be ideally located in order to carry out binding functions (Sedwick and Smerdon, 1999).
Here, we provide the first structural view of the binding of an ankyrin repeat protein to a nonprotein partner. Similar to protein–protein complexes involving ankyrin repeats, the concave region of ANK is involved in 2-5A binding. Since the various structures of nucleic acid binding proteins have shown that the basic surface patch of the proteins is responsible for binding to the exposed phosphate moiety of the target nucleic acid, it is not unreasonable to expect that ANK must possess a basic surface patch for 2-5A binding. Although local basic patches have been found for the binding sites for Phos1 and Phos3 of 2-5A, the majority of the concave region of ANK is covered by acidic patches (Figure 6A). Since the binding mechanism of 2-5A to ANK not only involves electrostatic interactions but also aromatic and hydrophilic interactions (Figure 3), it is not surprising that only a small portion of the concave surface is positively charged. As observed in the ankyrin repeat–protein complexes described above, the residues involved in 2-5A binding are not conserved in the consensus ankyrin repeat sequence (Figure 2, boxed in pink), thus enabling unique 2-5A binding roles.
Figure 6.
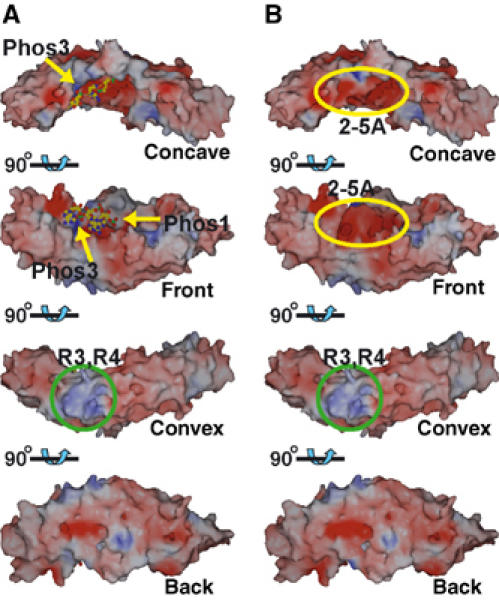
Electrostatic surface potential of ANK. The surfaces are shown in blue and red to indicate positive and negative charges, respectively, from 20 to −20 kT. The view at top is the same as the view provided in Figure 1C. (A) Surface potential of ANK without 2-5A. The bound 2-5A is shown as a ball-and-stick model and has been omitted from the potential calculation. The phosphate binding sites are indicated by yellow arrows. A remarkable basic patch near repeats 3 and 4 is indicated within a green circle. (B) Surface potential of the ANK/2-5A complex. The negative patch arising from bound 2-5A is indicated in a yellow ellipsoid.
The electrostatic surface properties of ANK suggest the potential interactions with the kinase and RNase domains of the full-length RNase L. In the amino-acid sequence of human RNase L, there is a group of negatively charged residues at 451–459 (EDVENEEDE) in the kinase domain, and positively charged residues at 677–684 (KHKKMKLK) in the RNase domain (Zhou et al, 1993). The overall acidic feature of ANK (Figure 6) appears to be favorable for interactions with a basic patch on the other parts of the full-length molecule, and thus acidity may be directly involved in the inhibition/activation mechanism of the positively charged RNase domain. This acidic feature of ANK is furthermore enhanced by 2-5A binding (Figure 6B). The enhancement of the negative charge may be a trigger of dimerization and activation of the full-length RNase L. Although the basic patch around R3 and R4 at the convex region is remarkable (Figure 6), the basic residues in the α2 helices in R3 and R4 are not well conserved among mammalian RNase Ls (Figure 2).
Conclusions
Here, we present the crystal structure of the N-terminal ankyrin repeat domain of human RNase L complexed with activator 2-5A containing unusual 2′,5′ internucleotide linkages. Because the catalytically active form of RNase L is a potent antiviral and anticellular protein, stable derivatives of 2-5A able to penetrate cell membranes might eventually provide novel therapeutic agents for viral infection and tumor development. The structural basis for 2-5A recognition by ANK is essential for designing stable 2-5As with a high likelihood of activating RNase L. Based on the structural information obtained about the ANK/2-5A complex discussed here, the design and synthesis of new 2-5A analogs are currently underway in our laboratory.
It has been reported that the binding of 2-5A to RNase L converts RNase L from its inactive monomer to its catalytically active homodimer. However, the following issues regarding the molecular mechanism of RNase L activity remain unexplained: (1) it is unclear why RNase L is inactive in the absence of 2-5A, and (2) it is unclear how the binding of 2-5A enables RNase L to dimerize and act on target RNA. Although the crystal structure of the ANK/2-5A complex did not provide any definitive answers to the above questions, it clearly revealed the 2-5A binding site of RNase L. Thus, the present structure serves as a starting point for further structure–function relationship studies on RNase L.
Materials and methods
Expression and purification of HisXaANK
A 1.0-kb cDNA encoding residues 1–333 of the N-terminal ankyrin repeat domain of human RNase L (ANK) was PCR-amplified from pKKRNL (Yoshimura et al, 2002) with primers 5′-cgcaggcctatggagagcagggatcataac-3′ and 5′-gcgaagcttttaagcaggagggtg-3′ (underlinings indicate added StuI and HindIII sites, respectively), digested with StuI and HindIII, and then ligated into the corresponding sites of a pQE-30Xa expression vector (pQEHisXaANK). Escherichia coli JM109 harboring pQEHisXaANK was grown in 4 l of LB medium at 303 K. When OD600 reached 0.6, expression was induced with 0.5 mM IPTG. The bacterial cells were collected after an additional 7 h cultivation by centrifugation at 5000 g for 10 min. The cell pellet was resuspended in buffer A (25 mM Tris–HCl (pH 7.5) and 10% glycerol), and then homogenized by sonication disruption on ice. Cell debris was removed from the homogenate by centrifugation at 12 000 g for 15 min at 277 K. The supernatant was loaded onto a SP-Sepharose FF column (bed volume: 40 ml) pre-equilibrated with buffer A. After an extensive wash with buffer A, HisXaANK was eluted from the column with buffer A containing a linear gradient of NaCl, from 0 to 0.8 M, and the fractions containing HisXaANK were pooled. The pooled fractions were combined and then loaded onto a TALON metal affinity column (bed volume: 2 ml) pre-equilibrated with buffer A. Elution was carried out with buffer A containing 150 mM imidazole. The fractions containing HisXaANK were then subjected to gel filtration on a Sephacryl S200 column (bed volume: 180 ml) pre-equilibrated with buffer A. The elution containing HisXaANK was pooled and concentrated with YM-10 ultrafiltration membrane, and stored at 253 K.
Crystallization of ANK
We used purified HisXaANK (hereafter simply ANK), containing a factor Xa-digestion site between His tag and ANK, for crystallization without removal of the His tag. 5′-O-monophosphoryladenylyl(2′ → 5′)adenylyl(2′ → 5′)adenosine (p5′(A2′p5′)2A, 2-5A trimer with 5′-monophosphate) was synthesized in our laboratory as described previously (Kitade et al, 1998). Crystallization of ANK was carried out in a way similar to the method described for non-tag ANK (Tanaka et al, in preparation). Briefly, a stock solution of 0.5 mg/ml ANK was concentrated using a Centricon-10 (Millipore), yielding a working solution of 3 mg/ml ANK. The protein solution was mixed with 20 mM 2-5A trimer with 5′-monophosphate dissolved in pure water at a volume ratio of 9:1. Crystallization was carried out at 293 K by the hanging-drop vapor diffusion method. In the best case, a droplet was prepared by mixing equal volumes (1.5+1.5 μl) of the protein solution (2.7 mg/ml ANK and 2 mM 2-5A) described above and a reservoir solution (500 μl) containing 15% (w/v) PEG8000, 0.2 M sodium acetate, and 20% (v/v) glycerol in 0.1 M Hepes buffer at pH 7.5. Rod-shaped crystals with typical dimensions of approximately 0.02 × 0.02 × 0.25 mm3 could be grown in 2 weeks.
Data collection
Since the crystallization conditions of ANK described above contained 20% (v/v) glycerol in reservoir solutions, X-ray data collections could be performed at cryogenic conditions without further addition of a cryoprotectant. The data collection was performed by rotation method at 100 K using an ADSC Q210 CCD detector with synchrotron radiation (λ=0.978 Å at beam line NW12 of the PF-AR). The crystals belong to an orthorhombic space group P212121 with cell dimensions of a=63.20 Å, b=72.83 Å, and c=82.63 Å (α=β=γ=90°). An assumption of one molecule per asymmetric unit leads to an empirically acceptable VM value of 2.38 Å3/Da, corresponding to a solvent content of 48%. The best diffraction data from a native crystal were collected up to 1.8 Å resolution and processed with the program packages DPS (Rossmann and van Beek, 1999) and CCP4 (Collaborative computational project, number 4, 1994). The data collection statistics are summarized in Table I.
Structure determination
Initial phase determination was performed by the MR technique. The coordinate set of ‘consensus ankyrin repeat protein' (Kohl et al, 2003) (PDB code: 1MJ0), having an approximately 15% sequence identity with ANK and consisting of five consensus ankyrin repeats, was used as a template of the search model. The search model was constructed as follows: the ankyrin repeats 1–4 of one coordinate set of 1MJ0 were superimposed on the ankyrin repeats 2–5 of the other coordinate set of 1MJ0. The fifth ankyrin repeat of the former coordinate set is regarded as the additional sixth ankyrin repeat of the latter coordinate set. Thus, a coordinate set of consensus ankyrin repeat protein consisting of six ankyrin repeats was constructed. Crossrotation and translation functions were calculated using the program AMoRe (Navaza, 1994) in the CCP4 suite. After one cycle of simulated-annealing refinement using the program CNS (Brunger et al, 1998) (R-factor of 0.468 and free R-factor of 0.517 in the resolution range 40–1.8 Å), initial model building was automatically carried out using the program ARP/wARP (Lamzin and Wilson, 1993). Without the simulated-annealing refinement before the ARP/wARP model building, the auto model building failed. Manual rebuilding was carried out using the program XtalView (McRee, 1999). Difference electron density maps clearly showed density corresponding to the bound 2-5A trimer with 5′-monophosphate. Crystallographic refinement was performed with the maximum-likelihood procedure of the program REFMAC (Murshudov et al, 1997) in the resolution range of 40–1.8 Å. The stereochemistry of the model was verified using the program PROCHECK (Laskowski et al, 1993). The final model consists of residues 21–305, 220 water molecules, and one 2-5A molecule. His tag, N-terminal residues (1–20), and C-terminal residues (306–333) are disordered. The refinement statistics are summarized in Table I.
Structure analysis
Least-squares comparisons of the molecular models were carried out using the program MATRAS (Kawabata, 2003). Figures were produced using the program ViewerPro (Figures 1C, D, 3B, C, 4, and 5B), the two programs Raster3D (Merritt and Murphy, 1994) and XtalView (McRee, 1999) (Figure 3A), and the program SPOCK (Figure 6).
Accession number
The atomic coordinates have been deposited in the Protein Data Bank with the entry code 1WDY.
Acknowledgments
We thank the late Prof. Y Mitsui for his constant encouragement. We also thank Drs N Matsugaki, N Igarashi, and M Suzuki of Photon Factory, and Drs H Sakai and M Kawamoto of SPring-8 for their help with the data collection at synchrotron facilities. This study was supported in part by the High-Technology Research Center Project (to NT), Grants-in-Aid for Encouragement of Young Scientists No. 12780457 (to NT), Protein 3000 Project (to NT), and Grants-in-Aid for Scientific Research on Priority Area No. 15030237 (to KTN) from MEXT of Japan.
References
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Casey G, Neville PJ, Plummer SJ, Xiang Y, Krumroy LM, Klein EA, Catalona WJ, Nupponen N, Carpten JD, Trent JM, Silverman RH, Witte JS (2002) RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet 4: 581–583 [DOI] [PubMed] [Google Scholar]
- Chebath J, Benech P, Hovanessian A, Galabru J, Revel M (1987) Four different forms of interferon-treated 2′,5′-oligo(A) synthetase identified by immunoblotting in human cells. J Biol Chem 262: 3852–3857 [PubMed] [Google Scholar]
- Cole JL, Carroll SS, Kuo LC (1996) Stoichiometry of 2′,5′-oligoadenylate-induced dimerization of ribonuclease L. J Biol Chem 271: 3979–3981 [DOI] [PubMed] [Google Scholar]
- Collaborative computational project, number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Diaz-Guerra M, Rivas C, Esteban M (1999) Full activation of RNase L in animal cells requires binding of 2-5A within ankyrin repeats 6 to 9 of this interferon-inducible enzyme. J Interferon Cytokine Res 19: 113–119 [DOI] [PubMed] [Google Scholar]
- Dong B, Silverman RH (1995) 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270: 4133–4137 [DOI] [PubMed] [Google Scholar]
- Dong B, Silverman RH (1997) A bipartite model of 2-5A-dependent RNase L. J Biol Chem 272: 22236–22242 [DOI] [PubMed] [Google Scholar]
- Dong B, Xu L, Zhou A, Hassel BA, Lee X, Torrence PF, Silverman RH (1994) Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J Biol Chem 269: 14153–14158 [PubMed] [Google Scholar]
- Drocourt JL, Dieffenbach CW, Ts'o PO, Justesen J, Thang MN (1982) Structural requirements of (2′-5′) oligoadenylate for protein synthesis inhibition in human fibroblasts. Nucleic Acids Res 10: 2163–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd-Smith G, Slattery E, Lengyel P (1981) Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate-dependent endonuclease. Science 212: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Hartmann R, Justesen J, Sarker SN, Sen GS, Yee VC (2003) Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′-5′-oligoadenylate synthetase. Mol Cell 19: 1173–1185 [DOI] [PubMed] [Google Scholar]
- Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH (1993) A dominant negative mutant of 2-5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J 12: 3297–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugh MC, Cayley PJ, Serafinowska HT, Norman DG, Reese CB, Kerr IM (1983) Analogues and analogue inhibitors of ppp(A2′p)nA. Eur J Biochem 132: 77–84 [DOI] [PubMed] [Google Scholar]
- Hovanessian A, Brown RE, Kerr IM (1977) Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature 268: 537–539 [DOI] [PubMed] [Google Scholar]
- Imai J, Lesiak K, Torrence PF (1985) Respective role of each of the purine N-6 amino groups of 5′-O-triphosphoryladenylyl(2′-5′)adenylyl(2′-5′)adenosine in binding to activation of RNase L. J Biol Chem 260: 1390–1393 [PubMed] [Google Scholar]
- IUPAC-IUB Joint Commission on Biochemical Nomenclature (1983) Abbreviations and symbols for the description of conformations of polynucleotide chains. Eur J Biochem 131: 9–15 [DOI] [PubMed] [Google Scholar]
- Jacobs MD, Harrison SC (1998) Structure of an IκBα/NF-κB complex. Cell 95: 749–758 [DOI] [PubMed] [Google Scholar]
- Kawabata T (2003) MATRAS: a program for protein 3D structure comparison. Nucleic Acids Res 31: 3367–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr IM, Brown RE (1978) pppA2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc Natl Acad Sci USA 75: 256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitade Y, Alster DK, Pabuccuoglu A, Torrence PF (1991a) Uridine analogs of 2′,5′-oligoadenylates: on the biological role of the middle base of 2-5A trimer. Bioorg Chem 19: 283–299 [Google Scholar]
- Kitade Y, Nakata Y, Hirota K, Maki Y, Pabuccuoglu A, Torrence PF (1991b) 8-Methyladenosine-substituted analogues of 2-5A: synthesis and their biological activities. Nucleic Acids Res 19: 4103–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitade Y, Wakana M, Terai S, Tsuboi T, Nakanishi M, Yatome C, Dong B, Silverman RH, Torrence PF (1998) 2-Bromoadenosine-substituted 2′,5′-oligoadenylates modulate binding and activation abilities of human recombinant RNase L. Nucleosides Nucleotides 17: 2323–2333 [Google Scholar]
- Kitade Y, Wakana M, Tsuboi T, Yatome C, Bayly SF, Player MR, Torrence PF (2000) 2-Methyladenosine-substituted 2′,5′-oligoadenylates: conformations, 2-5A binding and catalytic activities with human ribonuclease L. Bioorg Med Chem Lett 10: 329–331 [DOI] [PubMed] [Google Scholar]
- Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A, Grutter MG (2003) Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Natl Acad Sci USA 100: 1700–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywda S, Brzozowski AM, Higashitsuji H, Fujita J, Welchman R, Dawson S, Mayer RJ, Wilkinson AJ (2004) The crystal structure of gankyrin, an oncoprotein found in complexes with cyclin-dependent kinase 4, a 19 S proteasomal ATPase regulator, and the tumor suppressors Rb and p53. J Biol Chem 279: 1541–1545 [DOI] [PubMed] [Google Scholar]
- Lamzin V, Wilson KS (1993) Automated refinement of protein models. Acta Crystallogr D 49: 129–147 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lesiak K, Imai J, Floyd-Smith G, Torrence PF (1983) Biological activities of phosphodiester linkage isomers of 2-5A. J Biol Chem 258: 13082–13088 [PubMed] [Google Scholar]
- Lux SE, John KM, Bennett V (1990) Analysis of cDNA for human erythrocyte ankyrin indicates a repeated structure with homology to tissue-differentiation and cell-cycle control proteins. Nature 344: 36–42 [DOI] [PubMed] [Google Scholar]
- McRee DE (1999) XtalView/Xfit: a versatile program for manipulating atomic coordinates and electron density. J Struct Biol 125: 156–165 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Murphy EP (1994) Raster3D version 2.0: a program for photorealistic molecular graphics. Acta Crystallogr D 50: 869–873 [DOI] [PubMed] [Google Scholar]
- Michaely P, Tomchick DR, Machius M, Anderson RGW (2002) Crystal structure of a 12 ANK repeat stack from human ankyrinR. EMBO J 21: 6387–6396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Soler-Lopez M, Petosa C, Cramer P, Siebenlist U, Muller CW (2001) Crystal structure of the ankyrin repeat domain of Bcl-3: a unique member of the IκB protein family. EMBO J 20: 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Yoshimura A, Ishida N, Ueno Y, Kitade Y (2004) Contribution of Tyr712 and Phe716 to the activity of human RNase L. Eur J Biochem 271: 2737–2744 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated program for molecular replacement. Acta Crystallogr A 50: 157–163 [Google Scholar]
- Pavlovic J, Staeheli P (1991) The antiviral potentials of Mx proteins. J Interferon Res 11: 215–219 [DOI] [PubMed] [Google Scholar]
- Player MR, Torrence PF (1998) The 2-5A system: modulation of viral and cellular processes through acceleration of RNA degradation. Pharmacol Ther 78: 55–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann MG, van Beek CG (1999) Data processing. Acta Crystallogr D 55: 1631–1640 [DOI] [PubMed] [Google Scholar]
- Sedwick SG, Smerdon SJ (1999) The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci 24: 311–316 [DOI] [PubMed] [Google Scholar]
- Sen GC (2001) Viruses and interferons. Annu Rev Microbiol 55: 255–281 [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silvermanm RH, Schreiber RD (1998) How cells respond to interferons. Annu Rev Biochem 67: 227–264 [DOI] [PubMed] [Google Scholar]
- Torrence PF, Brozda D, Alster D, Charubala R, Pfleiderer W (1988) Only one 3′-hydroxyl group of ppp5′A2′p5′A2′p5′A (2-5A) is required for activation of the 2-5A-dependent endonuclease. J Biol Chem 263: 1131–1139 [PubMed] [Google Scholar]
- Torrence PF, Imai J, Lesiak K, Jamoulle J-C, Sawai H (1984) Oligonucleotide structural parameters that influence binding of 5′-O-triphosphoryladenylyl(2′-5′)adenylyl(2′-5′)adenosine to the 5′-O-triphosphoryladenylyl(2′-5′)adenylyl(2′-5′)adenosine-dependent endoribonuclease: chain length, phosphorylation state, and heterocyclic base. J Med Chem 27: 726–733 [DOI] [PubMed] [Google Scholar]
- Torrence PF, Xiao W, Li G, Khamnei S (1994) Development of 2′,5′-oligonucleotides as potential therapeutic agents. Curr Med Chem 1: 176–191 [Google Scholar]
- Williams BRG (1995) The role of the dsRNA-activated kinase, PKR, in signal transduction. Semin Virol 6: 191–202 [Google Scholar]
- Wreschner DH, McCauley JW, Skehel JJ, Kerr IM (1981) Interferon action-sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 289: 414–417 [DOI] [PubMed] [Google Scholar]
- Xiang Y, Wang Z, Murakami J, Plummer S, Klein EA, Carpten JD, Trent JM, Isaacs WB, Casey G, Silverman RH (2003) Effects of RNse L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res 63: 6795–6801 [PubMed] [Google Scholar]
- Yoshimura A, Nakanishi M, Yatome C, Kitade Y (2002) Comparative study on the biological properties of 2′,5′-oligoadenylate derivatives with purified human RNase L expressed in E. coli. J Biochem 132: 643–648 [DOI] [PubMed] [Google Scholar]
- Zhou A, Hassel BA, Silverman RH (1993) Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72: 753–765 [DOI] [PubMed] [Google Scholar]