

Abstract
Chemotaxis requires localized F-actin polymerization at the site of the plasma membrane closest to the chemoattractant source, a process controlled by Rac/Cdc42 GTPases. We identify Dictyostelium RacB as an essential mediator of this process. RacB is activated upon chemoattractant stimulation, exhibiting biphasic kinetics paralleling F-actin polymerization. racB null cells have strong chemotaxis and morphogenesis defects and a severely reduced chemoattractant-mediated F-actin polymerization and PAKc activation. RacB activation is partly controlled by the PI3K pathway. pi3k1/2 null cells and wild-type cells treated with LY294002 exhibit a significantly reduced second peak of RacB activation, which is linked to pseudopod extension, whereas a PTEN hypomorph exhibits elevated RacB activation. We identify a RacGEF, RacGEF1, which has specificity for RacB in vitro. racgef1 null cells exhibit reduced RacB activation and cells expressing mutant RacGEF1 proteins display chemotaxis and morphogenesis defects. RacGEF1 localizes to sites of F-actin polymerization. Inhibition of this localization reduces RacB activation, suggesting a feedback loop from RacB via F-actin polymerization to RacGEF1. Our findings provide a critical linkage between chemoattractant stimulation, F-actin polymerization, and chemotaxis in Dictyostelium.
Keywords: chemotaxis, Dictyostelium, morphogenesis, Rac, PI3K
Introduction
Chemotaxis involves leading edge protrusion through localized F-actin polymerization and retraction of the cell's posterior via myosin II assembly and F-actin/myosin II contraction (Chung et al, 2001b; Devreotes and Janetopoulos, 2003; Pollard and Borisy, 2003). F-actin polymerization is initiated by newly formed barbed ends, which are generated by ADF/cofilin severing existing filaments or the Arp2/3 complex forming new branch points. The activity of the Arp2/3 complex is controlled by WASP family members, which are Cdc42 and Rac effectors (Raftopoulou and Hall, 2004).
In Dictyostelium cells and leukocytes, the directionality of cell movement is mediated partially through the lipid kinase PI3K (Iijima et al, 2002; Merlot and Firtel, 2003). PI3K activation occurs preferentially at the leading edge of chemotaxing cells and recruits proteins containing a PH domain (such as Akt/PKB and CRAC), which bind to the PI3K products PI(3,4,5)P3 and PI(3,4,)P2. Abrogation of the function of the appropriate PI3K isoforms in leukocytes and Dictyostelium cells causes defects in directional sensing (Iijima et al, 2002; Merlot and Firtel, 2003). Deletion or overexpression of PTEN, a phosphoinositide 3-phosphatase that downregulates the PI3K pathway by dephosphorylating PI(3,4,5)P3 and PI(3,4,)P2 at the third position, in Dictyostelium cells results in unregulated or reduced PI(3,4,5)P3 function, respectively (Funamoto et al, 2002; Iijima and Devreotes, 2002). PI3K is recruited to the part of the plasma membrane closest to the chemoattractant source and plays an instructive role in leading edge formation (Funamoto et al, 2002). Studies in neutrophils revealed that leading edge function is coordinately controlled through localized PI3K activation and Gβγ-dependent recruitment of PAK1 (a Rac and Cdc42 effector) and PIXα, which are required for chemoattractant-mediated activation of Cdc42 (Li et al, 2003; Xu et al, 2003).
The Rho family of small GTPases are key regulators of the actin/myosin cytoskeleton during chemotaxis (Raftopoulou and Hall, 2004). Studies using dominant-negative and constitutively active forms of RhoA, Cdc42, and Rac1 in mammalian fibroblasts indicate these proteins control lamellipod and uropod function and stress fiber and filopod formation by regulating F-actin polymerization and myosin contractility. In Dictyostelium, 15 genes encode Rho family members, of which Rac1a/b/c, RacF1/F2, and RacB fall into the Rac subfamily (Rivero and Somesh, 2003). No Rho subfamily members nor a true Cdc42 have been identified. As in other systems, studies using dominant-negative and constitutively active forms of Rac family members in Dictyostelium have linked Rac family members to F-actin polymerization and actin/myosin cytoskeleton regulation (Rivero and Somesh, 2003). Disruption of one of the two genes encoding Dictyostelium RhoGDIs causes growth defects, moderate pinocytosis defects, contractile vacuole system defects, and reduced chemoattractant-mediated F-actin polymerization (Rivero et al, 2002). Gene disruption of DRG/DdRacGAP1, a multidomain protein carrying a Rac1GEF (GDP/GTP exchange factor), causes reduced chemoattractant-mediated F-actin polymerization and chemotaxis defects (Knetsch et al, 2001). Disruption of the gene encoding RacF1, which associates with areas of cell–cell contact, macropinosomes, and phagosomes, causes no observable phenotypes, presumably because RacF1 and RacF2 are redundant (Rivero et al, 1999).
Overexpression studies using wild-type, dominant-negative, and constitutively active forms of Rac proteins have been essential in elucidating the function of these proteins, but the correspondence between the results of such studies and endogenous processes is often uncertain. Dominant-negative forms of Rac block the activation of endogenous Rac proteins by inhibiting the function of upstream guanine nucleotide exchange factors (GEFs). However, overexpression of dominant-negative Rac isoforms can potentially inactivate RacGEFs in addition to the cognate GEF. A particular RacGEF may activate more than one Rac, depending on the physiological stimuli, and these Rac isoforms could be blocked by a single dominant-negative Rac. Likewise, interpretation of experiments using constitutively active Rac proteins or overexpressing wild-type Racs, which often have similar effects, is limited by the potential for activating nonphysiological downstream effectors due to mass action. Although the activation or inhibition of a pathway by a constitutively active or dominant-negative Rac has often been interpreted as demonstrating that the pathway is activated by that Rac in vivo, this conclusion can be too simplistic.
Previous work unequivocally implicates Dictyostelium Rac proteins in the control of chemotaxis. Due to the limitations discussed above, the role of individual Rac family members has been difficult to assess. To identify the most relevant members for further study, we measured the yeast two-hybrid interaction of 15 Rac family members with the CRIB domains of two PAKs that are important for chemotaxis. This screen identified RacB, which has been implicated in F-actin responses (Lee et al, 2003), as the Rac with the strongest interaction with both CRIB domains. Using gene knock-in techniques to express wild-type levels of an epitope-tagged form of RacB from its endogenous promoter, we found that RacB activity is stimulated in response to chemoattractant stimulation, with two peaks of activation that correspond to the two peaks of chemoattractant-mediated F-actin polymerization. racB null cells exhibit an ∼60–70% loss of both F-actin peaks and a loss of chemoattractant-mediated PAKc activation. We find that the second peak of RacB activation, correlating to F-actin assembly during pseudopod extension, is dependent on the PI3K pathway (Chen et al, 2003), linking PI3K to F-actin polymerization through RacB. We identify a RacBGEF (RacGEF1) and demonstrate that RacGEF1 is required for ∼50% of RacB activation in vivo. Gene knockouts and domain-function studies with RacGEF1 corroborate an in vivo function of RacGEF1 in directly controlling RacB activity, F-actin polymerization, and downstream chemotaxis and morphogenesis.
Results
RacB is activated by chemoattractant stimulation
To identify which Rac family members are involved in controlling chemotaxis, we employed a two-hybrid assay in which a constitutively active form of each Dictyostelium Rac was tested for interaction with the CRIB domains of Dictyostelium PAKc and PAKa. As shown in Table I and Figure 1A, constitutively active RacB (RacBCA) strongly interacts with both CRIB domains, but dominant-negative RacB does not. Constitutively active, but not dominant-negative, Rac1 family members (only one dominant-negative tested) as well as RacA have weaker interactions, and RacCCA has significantly weaker interactions compared to RacB. No other Dictyostelium Rac scored positive. Constitutively active human Cdc42 and Rac1 but not RhoA had a strong interaction. We confirmed the specificity of the interaction of RacB with the CRIB domains in a pull-down assay (Figure 1B). We therefore focused further studies on RacB. We find that the interaction is GTP-dependent, and GST-RacB has a higher affinity for MBP-PAKa-CRIB than for MBP-PAKc-CRIB. We also find that the CRIB domain of Dictyostelium WASP interacts. A complete analysis with the WASP CRIB was not performed.
Table 1.
Yeast two-hybrid analysis of interactions of the PAKc and PAKa CRIB domains with constitutively active and dominant-negative Rho family GTPases
| Rho GTPase | Interaction |
|
|---|---|---|
| PAKa | PAKc | |
| Rac1a Q61L | +++ | +++ |
| Rac1a T17N | − | − |
| Rac1b G12V | +++ | +++ |
| Rac1c G12V | +++ | +++ |
| RacA G12V | +++ | +++ |
| RacA S17N | − | − |
| RacB G12V | ++++ | +++++ |
| RacB Q61L | ++++ | +++++ |
| RacB T17N | − | − |
| RacC G15V | − | + |
| RacC T20N | − | − |
| RacD G17V | − | − |
| RacE G20V | − | − |
| RacF1 G12V | − | − |
| RacF2 G12V | − | − |
| RacG G12V | − | − |
| RacH M13V | − | − |
| RacI S14V | − | − |
| RacJ D18V | − | − |
| RacL G12V | − | − |
| HsRac1 G12V | +++ | +++ |
| HsRac1 T17N | − | − |
| HsCdc42 G12V | +++ | +++ |
| HsCdc42 T17N | − | − |
| HsRhoA G14V | − | − |
Figure 1.

RacB interactions. (A) Two-hybrid interactions between the CRIB domains of PAKa and PAKc and constitutively active and dominant-negative forms of Dictyostelium and human Rho family members. β-gal staining of colonies is shown. (B) The MBP-fused CRIBs of PAKa and PAKc were used in a pull-down assay with GST-RacB preloaded with or without GTP-γ-S (left panel). The right panel shows a similar experiment using the GST-CRIB domain of Dictyostelium WASP and myc-RacB. The bound proteins were analyzed by immunoblot assay with the anti-GST or anti-myc antibody.
Chemoattractant-stimulated F-actin polymerization exhibits a biphasic curve (Hall et al, 1988; Chen et al, 2003). The first peak is very fast, occurring at 5 s, followed by a rapid decrease in F-actin levels. The second peak is significantly broader and lower, with levels ∼30% those of the initial peak and at maximum at 30–60 s. The first peak correlates with the initial cringe reaction in which the cells round up and produce a uniform F-actin cortex. The second peak corresponds to the emergence of pseudopodia and cell movement.
If RacB is important in controlling F-actin assembly, then the kinetics of RacB activation should parallel those of F-actin polymerization. To test this hypothesis, we established a RacB activation assay based on that of Benard et al (1999). To follow RacB in the assay, we employed a myc-tagged RacB protein. As overexpression of wild-type RacB results in excessive F-actin polymerization (Lee et al, 2003), we used a knock-in construct (Supplementary Figure S1) and replaced the endogenous RacB gene with a gene encoding the myc-tagged RacB, which is under the control of the endogenous RacB promoter. We confirmed the knock-in by PCR and Western blot analyses (Figure 2B). The kinetics of RacB activation (RacB-GTP bound to GST-PAKa-CRIB) parallel those of F-actin polymerization with a sharp first peak and a broad second peak (Figure 2B and C). Moreover, the relative magnitudes of these two peaks mirror those of F-actin polymerization.
Figure 2.
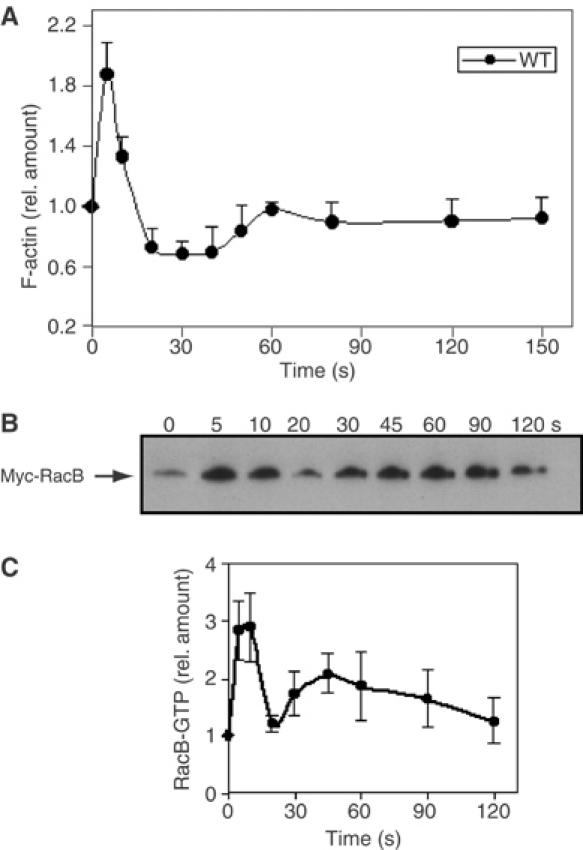
(A) Kinetics of F-actin polymerization in the Triton-insoluble, cytoskeletal fraction of wild-type cells. (B) RacB-GTP proteins in wild-type RacB knock-in cells stimulated with cAMP for the indicated time were separated with glutathione–Sepharose beads using the GST-CRIB of PAKa and analyzed by immunoblot assay (see Materials and methods). The values in these and the other experiments are averages based on five separate experiments.
RacB is important for chemoattractant-mediated F-actin polymerization, myosin II assembly, and PAKc activation
To examine the importance of RacB in chemoattractant-mediated F-actin assembly, we created a racB null mutation via homologous recombination, which we confirmed by Southern (Supplementary Figure S1) and RNA blot analyses (Figure 3A). racB null cells have a reduced basal level of F-actin and a 60–70% reduction in both peaks of chemoattractant-mediated F-actin polymerization, indicating that RacB is required for much, but not all, of the response (Figure 3B). This implies that one or more of the other Racs (see Discussion) and/or a Rac-independent mechanism mediate the remainder of F-actin polymerization.
Figure 3.
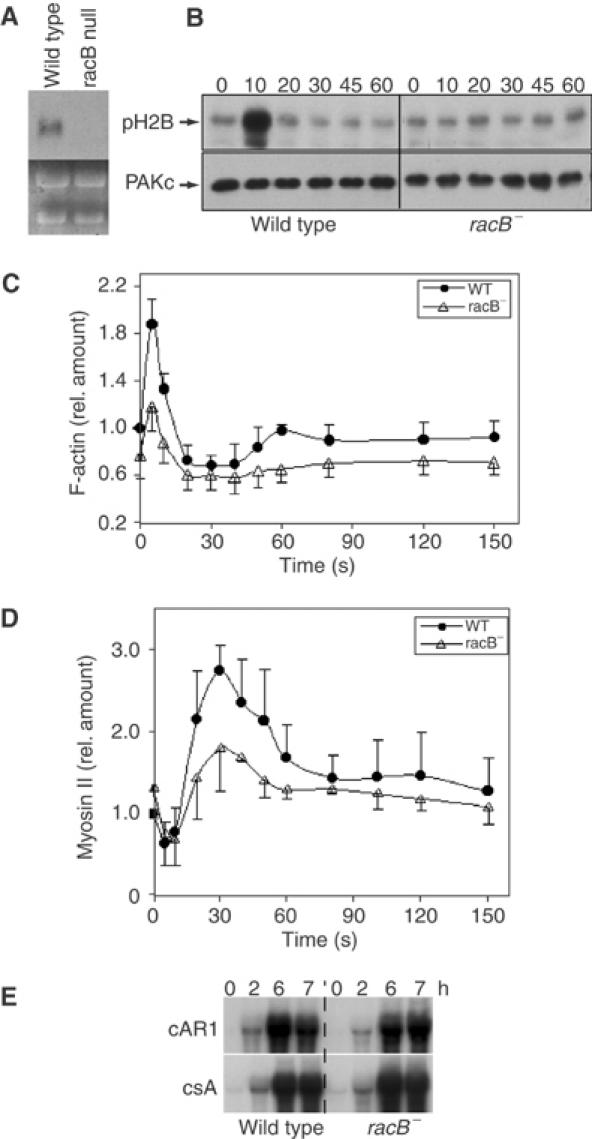
Requirement of RacB for chemoattractant-mediated PAKc activation, F-actin polymerization, and myosin II assembly. (A) RNA blot of wild-type and racB null cells probing for the RacB coding region (Supplementary Figure S1). The lower portion of the blot depicts the rRNA and shows equal loading. (B) cAMP-stimulated kinase activity of myc-PAKc expressed in wild-type and racB null cells. Aggregation-competent cells were stimulated with cAMP and kinase activity was measured as described in Materials and methods. The lower panel depicts the Western blot probed with the anti-myc antibody and indicates the amount of PAKc in each immunoprecipitate. pH2B is 32P-labeled phosphorylated Histone 2B. (C, D) Kinetics of F-actin polymerization and myosin II assembly, respectively, in the Triton-insoluble, cytoskeletal fraction of wild-type and racB null cells.
Dictyostelium PAKc is required for proper chemotaxis and its kinase activity is rapidly and transiently activated in response to chemoattractant stimulation (Lee et al, submitted for publication). We found that point mutations in the PAKc CRIB domain that abrogate binding to RacB-GTP also abrogate chemoattractant-mediated PAKc activation, suggesting that PAKc activates Rac-GTP binding. When we examined PAKc activation in racB null cells, no activation was observed (Figure 3C), implying that PAKc activation is mediated by RacB. This is consistent with RacB-GTP being the Dictyostelium Rac protein with the strongest interaction with the PAKc CRIB domain.
In response to chemoattractant stimulation, there is a small, rapid, transient decrease in myosin II assembly followed by an ∼3-fold increase in assembled myosin II levels, which corresponds to the retraction of the cell's posterior during chemotaxis (Steimle et al, 2001). Myosin II assembly requires PAKa and is regulated, in part, by Akt/PKB phosphorylation (Chung et al, 2001a). Myosin II assembly is reduced by ∼50% in racB null cells compared to wild-type cells, indicating that RacB is required for this process (Figure 3D).
Responsiveness to cAMP is developmentally regulated. To insure that the observed defects were not due to a general reduced ability to respond to cAMP stimulation, we examined the expression of two genes (the receptor cAR1 and the cell adhesion protein csA) whose expression is regulated by cAMP (Aubry and Firtel, 1999). Both genes are expressed normally in racB null cells (Figure 3E).
RacB is required for proper chemotaxis
RacB's requirement for part of chemoattractant-mediated F-actin assembly and myosin II polymerization suggests that racB null cells would be defective in chemotaxis. As shown in Figure 4 and Table II, racB null cells exhibit chemotaxis defects. Wild-type cells have an average speed of ∼9 μm/min and a high directionality, whereas racB null cells have a speed of only ∼3.9 μm/min. However, racB null cells exhibit only a small loss of polarity (increased roundness) and directionality compared to wild-type cells. Thus, RacB is required for speed of movement but is not essential for directionality. The reduced speed and polarity are consistent with defects in F-actin polymerization and myosin II assembly.
Figure 4.
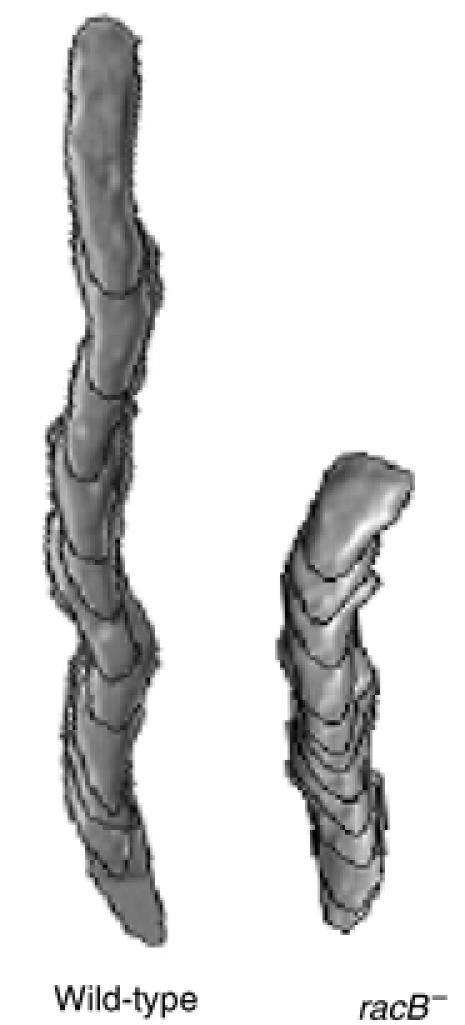
Computer-assisted analysis of chemotaxis (DIAS) of wild-type and racB null cells. The figure depicts the overlapping DIAS image analysis of chemotaxing cells. Overlapping images were captured at 1-min intervals. Cells for analysis were chosen randomly and the paths examined represent a 10-min interval taken from the middle of the chemotaxis movie. The image illustrates the shape, total distance moved, and directionality of movement.
Table 2.
DIAS analysis of chemotaxis
| Speed (μm/min) | Directionality | Directional change (deg) | Roundness (%) | |
|---|---|---|---|---|
| Wild type | 9.16±0.45 | 0.89±0.05 | 17.7±3.7 | 55.8±1.5 |
| racB null | 3.88±0.77 | 0.76±0.08 | 23.1±4.6 | 76.1±3.2 |
| gef1 null | 8.23±1.62 | 0.77±0.07 | 24.2±7.6 | 55.3±2.0 |
| GEF1OE | 5.19±0.18 | 0.80±0.07 | 22.7±4.3 | 68.3±5.6 |
| GEF1ΔN | 3.34±0.25 | 0.25±0.07 | 61.5±2.4 | 59.1±4.3 |
| GEF1ΔIQ | 4.13±0.28 | 0.53±0.05 | 47.2±0.7 | 55.8±0.58 |
| GEF1ΔPH | 3.41±0.38 | 0.59±0.12 | 41.7±8.1 | 62.3±1.2 |
| GEF1ΔIQ/ΔPH | 4.61±0.18 | 0.64±0.04 | 38.9±3.8 | 56.5±4.7 |
| Numbers are mean±s.d. Speed indicates speed of cell's centroid movement along the total path. Direction change is a relative measure of the number and frequency of turns the cell makes. Larger numbers indicate more turns and less efficient chemotaxis. Directionality is a measure of the linearity of the pathway. Cells moving in a straight line to the needle have a directionality of 1.00. Roundness is an indication of the polarity of the cells. Larger numbers indicate that the cells are more round and less polarized. | ||||
Regulation of RacB activation by the PI3K pathway
PI3K is required for proper directional movement in Dictyostelium and neutrophils (see Introduction). pi3k1/2 null cells, a double knockout of the Class I PI3Ks, PI3K1 and PI3K2, exhibit a small decrease in the first (5 s) peak and a significant loss of the second, broader peak that has been linked to pseudopod protrusion (Funamoto et al, 2001; Chen et al, 2003). Loss of PTEN activity, in contrast, leads to increased F-actin polymerization during the second peak (Iijima and Devreotes, 2002). To examine the role of the PI3K pathway in RacB activation, we replaced the endogenous racB gene with myc-RacB in pi3k1/2 null and PTEN hypomorphic strains (Funamoto et al, 2002; Iijima and Devreotes, 2002) as described for wild-type cells. The levels of RacB mRNA and myc-tagged RacB protein were elevated in pi3k1/2 null cells, compared to those in wild-type cells (Figure 5A and C; the developmental expression of RacB mRNA is shown in Figure 5B). This finding was confirmed in several independent pi3k1/2 null strains. The same elevated level of endogenous RacB mRNA was observed in pi3k1/2 null strains carrying the wild-type (untagged) endogenous RacB gene (data not shown). In the PTEN hypomorphic strain, we observed the opposite effects: the levels of myc-RacB mRNA and protein were reduced compared to those in wild-type cells (Figure 5A and C).
Figure 5.
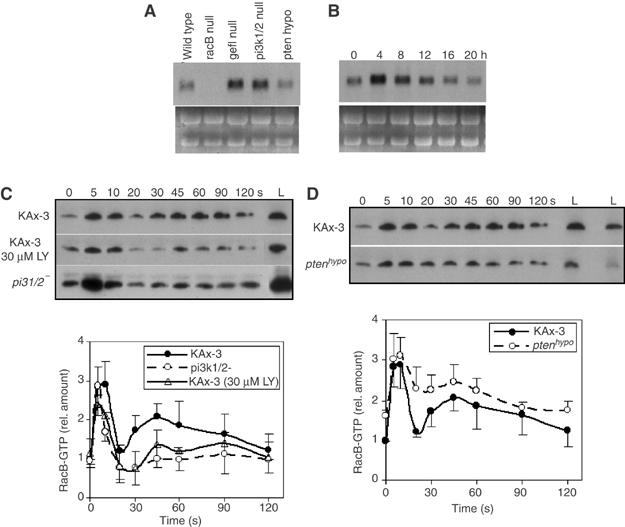
Regulation of RacB by the PI3K pathway. (A) RNA blot of total RNA isolated from vegetative wild-type, racB null, racgef1 null, pi3k1/2 null, and PTEN hypomorphic cells. (B) Developmental RNA blot of RacB RNA. Total cell RNA was isolated from developing wild-type cells plated on non-nutrient agar at the indicated times. 0, vegetatively growing cells; 4 h; early/mid aggregation; 8 h, mound stage; 12 h, tipped aggregates; 20 h, mature fruiting body. (Bush et al (1993) found RacB transcripts present at the same level in vegetative and 4 h starved cells.) (C, D) RacB-GTP in stimulated wild-type cells, wild-type cells pretreated with LY294002, and pi3k1/2 null cells (C) or wild-type and PTEN hypomorphic cells (D). Lanes labeled ‘L' show the levels of total myc-RacB in the same volume of each lysate. The amount of RacB-GTP was quantified by densitometry of developed Western blot films in four independent experiments (lower panel). The wild-type level at 0 s was set at 1.0. The levels of RacB-GTP in a specific mutant strain were normalized to RacB expression in wild-type cells by dividing the total amount of RacB in the mutant by that of wild-type cells. Error bars indicate standard deviation.
We then examined RacB activation in pi3k1/2 null and PTEN hypomorphic cells and compared the resulting data to those obtained with wild-type cells. As the levels of expression of RacB protein are different in all three strains, we normalized the amount of RacB-GTP binding to GST-PAKa-CRIB by dividing the amount of RacB-GTP by the level of myc-RacB protein in each strain compared to the level of myc-RacB protein in wild-type cells. For plotting RacB activation curves, the level of RacB-GTP in unstimulated wild-type cells was set at 1. We observed a strong RacB first activation peak in pi3k1/2 null cells. The fraction of total activated RacB protein was similar to that observed in wild-type cells (a three-fold increase; the basal levels of RacB-GTP in unstimulated cells were proportional to the expression level of the RacB protein). In contrast, the second activation peak was significantly reduced (Figure 5C). To control for a pleiotropic effect of the pi3k1/2 null genetic background on RacB activation, we measured RacB activation in wild-type cells pretreated with the PI3K inhibitor LY294002, which blocks >90% of stimulated Akt/PKB activity (Meili et al, 1999). In these cells, the levels of total myc-RacB protein are the same as in untreated cells (Figure 5C). After chemoattractant stimulation, we observe a slight reduction in the first RacB activation peak and a significant reduction in the second peak, although the level of activation is slightly higher than that observed in pi3k1/2 null cells. In pten hypomorphic cells, we observe an increased fraction of the total RacB in the GTP-bound form in unstimulated cells. Upon chemoattractant stimulation, there is a rapid increase in the level of RacB-GTP with a peak at 5 s, as is observed for wild-type cells; in contrast to wild-type or pi3k1/2 null cells, there is only a slight drop between 10 and 20 s (Figure 5C) followed by a rise at ∼45 s and a subsequent slow decrease. This shallow drop between the first and second peaks is similar to observations for F-actin polymerization in pten null cells.
Dictyostelium RacGEF1 is required for maximal chemoattractant stimulation of RacB
To further examine the pathway leading to RacB activation, we undertook a bioinformatic search for potential RhoGEFs by identifying proteins with linked DH and PH domains. We identified >20 members of this protein family in the Dictyostelium genome sequence database (‘dictyBase', http://www.dictybase.org/). Figure 6A and B depicts a map and the derived amino-acid sequence of one of these, Dictyostelium RacGEF1. To determine whether RacGEF1 plays a part in RacB activation, we created racgef1 null cells (Supplementary Figure S2) and examined their RacB activity. racgef1 null cells exhibit a significant reduction in both RacB activation peaks, suggesting that RacGEF1 mediates part of RacB activation in vivo (Figure 6C). racgef1 null cells normally induce cAR1 and csA mRNA, indicating that they can normally regulate some cAMP-mediated pathways (Figure 6D).
Figure 6.
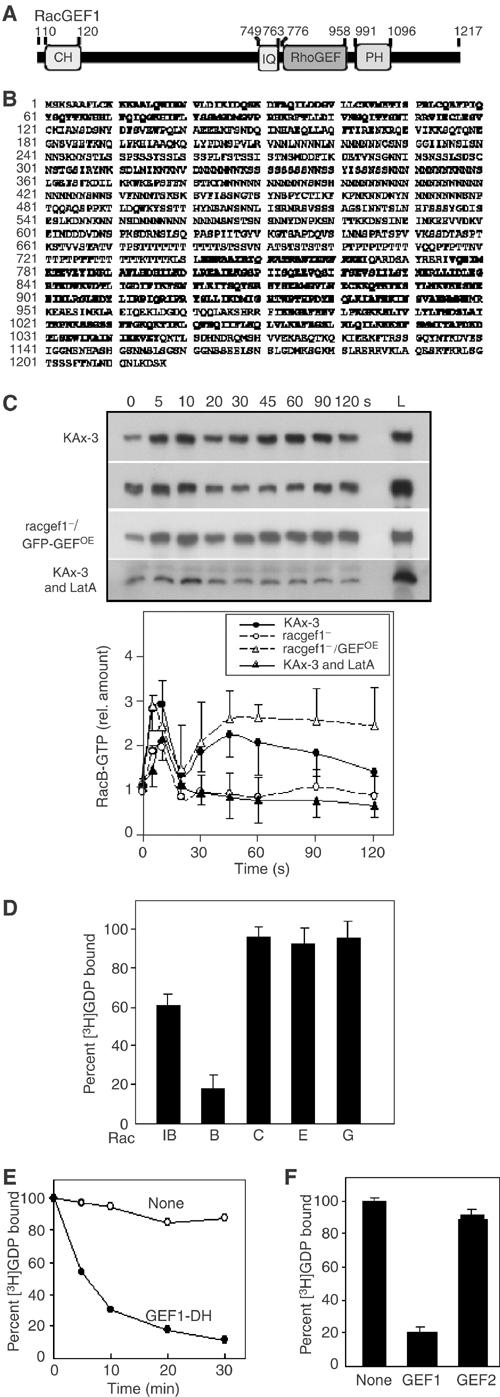
RacGEF structure and function. (A, B) Domain structure and derived amino-acid sequence of RacGEF1. The CH, IQ, RhoGEF (or DH), and PH domains are in bold face. (C) RacB-GTP in the stimulated wild-type cells, racgef1 null and overexpressing cells, and wild-type cells treated with LatA (upper panel). Lane L indicates the levels of total myc-RacB in the same volume of each lysate. RacB-GTP levels were determined by densitometry of developed Western blot films in at least three independent experiments (lower panel). (D) Comparison of RhoGEF domain abilities of RacGEF1 to catalyze GDP/GTP guanine nucleotide exchange on Rac1B, RacB, RacC, RacE, and RacG. (E) Time course of RhoGEF domain activity of RacGEF1 to catalyze GDP/GTP guanine nucleotide exchange on RacB. (F) Comparison of RhoGEF domain abilities of RacGEF1 and RacGEF2 to catalyze GDP/GTP guanine nucleotide exchange on RacB.
To examine the function of RacGEF1 at a biochemical level, the recombinant RacGEF1 DH (catalytic) domain was assayed for its ability to stimulate guanine nucleotide exchange of Dictyostelium Rac1B, RacB, RacC, RacE, and RacG. These Racs were chosen because they have been linked to F-actin-mediated responses (Rivero et al, 2002). The DH (catalytic) domain of RacGEF1 has the highest activity against RacB among the Rac proteins tested (Figure 6E and F). Rac1B is a weaker substrate and no exchange activity was observed for RacC, RacE, or RacG. To test whether any Dictyostelium RacGEF might exhibit exchange activity against RacB, we assayed the DH domain of a second Dictyostelium RacGEF, RacGEF2. Figure 6G indicates that RacGEF2 does not exhibit exchange activity against RacB in our assays. The degree of specificity we observed biochemically makes it likely that RacB is an in vivo RacGEF1 substrate as suggested by our observation of reduced RacB activation in racgef1 null cells.
If RacGEF1 acts as an exchange factor for RacB in vivo, racgef1 null cells should have decreased F-actin polymerization. racgef1 null cells exhibit an ∼30% reduction in chemoattractant-mediated F-actin assembly (Figure 7A). The magnitude of this decrease, compared to the ∼60–70% decrease found in racB null cells, is consistent with a loss of 50% of RacB activation in racgef1 null cells.
Figure 7.
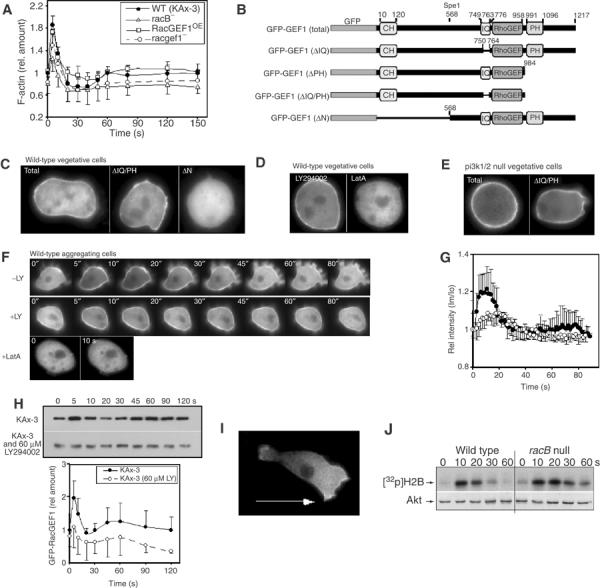
Subcellular localization of GFP-RacGEF1. (A) F-actin profiles of wild-type, racB null, and racgef1 null and overexpressing cells. (B) Maps of GFP-RacGEF1 wild-type and mutant constructs. (C–E) Subcellular localization in vegetative cells of GFP-RacGEF1, GFP-RacGEF1ΔIQ/PH, and GFP-RacGEF1ΔN (C); GFP-RacGEF1 in the presence of LY294002 or LatA (D); GFP-RacGEF1 and RacGEF1ΔIQ/PH in pi3k1/2 null cells (E). (F) Translocation of GFP-RacGEF1 was imaged after stimulation with cAMP in the presence or absence of LY294002 or LatA (pretreated with 60 μM LY294002 or 2 μM LatA for 30 min). (G) Quantitation of the results of untreated and LY294002-treated cells depicted in (F) (see Supplementary data for a detailed protocol). The fluorescence intensity of membrane-localized GFP fusion protein was quantitated using the linescan module of Metamorph software. Im/Io is plotted as a measure of the fluorescence intensity at a point on the membrane relative to that at the cytoplasm. (H) RacB activation in wild-type (Figure 2) and LY294002-treated cells. (J) Akt activation in wild-type and racB null cells.
To understand possible modes of regulation of RacGEF1 activity, we used N-terminally tagged GFP-RacGEF1 (Figure 7B) expressed in myc-RacB knock-in, racgef1 null cells from the Act15 promoter (we observed the same results when it was expressed in wild-type cells; data not shown) to examine RacGEF1's subcellular localization. We expect that this led to a significant overexpression of RacGEF1 because the levels of GFP-RacGEF1 transcripts are elevated compared to endogenous mRNA (Supplementary Figure S3). However, when RacB activation was examined, the level of RacB-GTP in unstimulated racgef1 null/GFP-RacGEF1OE cells was similar to that of wild-type cells, suggesting that overexpression of RacGEF1 does not affect basal RacGEF1 activity or that RacB GAPs compensate. Expression of GFP-RacGEF1 in racgef1 null cells complemented the decrease in the first peak of RacB activation (Figure 6C). As in wild-type cells, the first peak is followed by a dip in RacB-GTP levels and a second peak. In the RacGEF1-overexpressing cells, the dip after the first peak was less and the second peak was elevated and extended. The RacGEF1-overexpressing cell F-actin polymerization profile is similar to that of RacB activation: basal levels and the first peak are similar to those of wild-type cells, but the drop after the first peak is less and the second peak is elevated (Figure 7A).
In vegetative cells, a portion of RacGEF1 is associated with the cortex (Figure 7C). This localization requires the N-terminal portion of the protein that includes the CH domain but does not require the IQ or PH domains (Figure 7B and C). A similar localization is observed in pi3k1/2 null cells or wild-type cells treated with LY294002 (Figure 7D and E). In unstimulated aggregation-competent cells (cells competent to chemotax to cAMP) pretreated with caffeine to inhibit endogenous signaling, GFP-RacGEF1 is predominantly cytosolic with a low basal level associated with the cortex (Figure 7F) but rapidly and transiently translocates to the cortex with a biphasic translocation profile. The first peak, as determined by the change in GFP fluorescence at the cortex, is at ∼5–12 s after stimulation, similar to that of RacB activation (Figure 7G). A second, much lower peak occurs at ∼40–75 s, corresponds to the second peak of RacB activation, and has a visible pseudopod extension (Figure 7F). We obtained similar results when we quantified cortically associated RacGEF1 biochemically (Figure 7H). We asked whether this translocation is dependent on PI3K activity. In aggregation-competent cells, there is a slight increase in cortically localized GFP-RacGEF1 in cells treated with LY294002 compared to untreated cells (Figure 7F). Upon stimulation, RacGEF1 translocates to the cortex, but the fraction of RacGEF1 that translocates is reduced compared to untreated cells (Figure 7F–H), indicating that PI3K plays a role in RacGEF1 cortical localization. In chemotaxing cells, RacGEF1 is preferentially localized along the anterior cortical area and at the posterior of the cell, both of which are regions of F-actin localization and polymerization (Figure 7I).
Because the RacB/RacGEF1 pathway controls chemoattractant-mediated F-actin assembly, we examined whether the F-actin cortex plays a part in RacGEF1 localization. Pretreatment of cells with latrunculin A (LatA), which induces disassembly of the F-actin cytoskeleton, causes a loss of RacGEF1 cortical localization and abrogates chemoattractant-stimulated RacGEF1 cortical localization (Figure 7E–G). Together with the deletion analysis results, these data suggest that RacGEF1 translocates to the plasma membrane through its N-terminal region containing the CH domain (and possible catalytic domain) in a response that requires F-actin assembly.
One prediction from these results is that treatment of cells with LatA to disrupt the F-actin cytoskeleton will reduce chemoattractant-mediated RacB activation. Pretreatment of cells with LatA causes an ∼50% reduction of the first peak of RacB activation and a loss of the second peak, suggesting that F-actin-mediated RacGEF1 translocation plays a key role in RacB activation (Figure 6C).
Models have been proposed for chemotaxing neutrophils in which the activations of PI3K and F-actin are in a positive feedback loop controlled through Rac family GTPases (Wang et al, 2002; Weiner et al, 2002). To examine a component of this pathway in Dictyostelium, we tested whether PKB activation is altered in racB null cells. racB null cells have a slightly longer activation profile than wild-type cells (Figure 7J). There is no evidence that the level of PKB activation is dependent on RacB activation (see Discussion).
RacB and RacGEF1 are required for proper morphogenesis
When Dictyostelium cells are starved and plated on a non-nutrient agar surface, a developmental program is induced. Individual cells aggregate to form a mound of ∼5 × 104 cells at ∼7 h, a process mediated by chemotaxis to cAMP (Aubry and Firtel, 1999; Iijima et al, 2002). Cell-type differentiation and morphogenesis ensue with the formation of a tipped aggregate. Prestalk cells preferentially chemotax to the apical tip of the mound, which elongates and falls over to form a migrating slug or pseudoplasmodium. Culmination follows, resulting in the formation of a mature fruiting body.
Because both aggregation and morphogenesis require regulated cell movement, we examined the potential involvement of RacB and RacGEF1 in these processes. racB null cells exhibit a significant developmental delay compared to wild-type cells; only loose aggregates are formed by 10 h and many cells have not entered the aggregate (Figure 8A). These phenotypes are consistent with racB null cells being defective in cell movement. By 24 h, wild-type cells form mature fruiting bodies. racB null cell aggregates break up into smaller aggregates and form morphologically abnormal structures. Even at 24 h, many of the cells have not entered the aggregates. These results indicate that RacB is required for morphogenetic processes in Dictyostelium, consistent with a role of RacB in cell movement.
Figure 8.
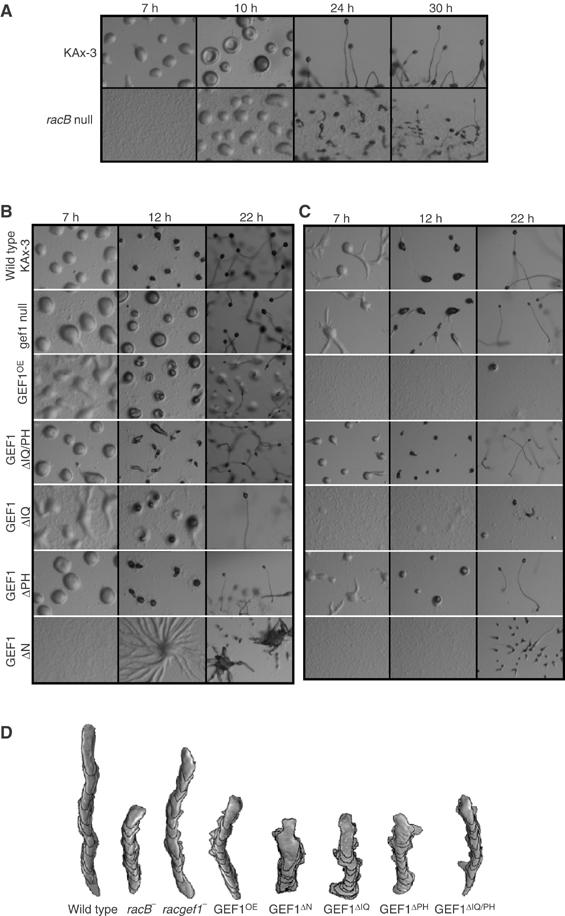
Development on non-nutrient agar plates. (A) racB null cells plated at a density of 1.8 × 106 cells/cm2. (B) Cells expressing mutant RacGEF1 proteins plated at a density of 1.8 × 106 cells/cm2. (C) Cells expressing mutant RacGEF1 proteins plated at a density of 2.4 × 105 cells/cm2. (D) Computer-assisted analysis of chemotaxis (DIAS) of strains examined in (A–C).
Morphogenetic defects were also observed in racgef1 null cells, although these were significantly weaker than those of racB null cells under normal plating conditions (1.8 × 106 cells/cm2; Figure 8B). Like wild-type aggregates, racgef1 null cell aggregates formed by ∼7 h, but a greater fraction of the cells had not entered the aggregate at this time compared to wild-type cells. By 12 h in development, wild-type cells had formed tipped aggregates, whereas the development of racgef1 null cells was delayed and the organisms were still at the tight aggregate stage. By 22 h, both wild-type cells and racgef1 null cells had formed mature fruiting bodies, although some of the racgef1 null organisms had not fully culminated.
The effects of RacGEF1 overexpression were more severe: cells exhibited a delay in aggregation and morphogenesis, with the majority of the mounds not undergoing further morphogenesis (Figure 8B). These cells exhibit a significant reduction in chemotaxis (Figure 8D; Table II), presumably because of a misregulation of RacB activation. Expression of RacGEF1 lacking the N-terminal region (RacGEF1ΔN) resulted in highly delayed aggregation and the formation of extremely large aggregation streams (Figure 8B). These streams formed large multitipped aggregates that produced many extended finger-like structures from the mass of cells. Some of these structures differentiated into mature fruiting bodies. Wild-type cells expressing RacGEF1 lacking the IQ and PH domains or the PH domain alone exhibited fairly normal morphogenesis, whereas deletion of the IQ domain alone led to slightly delayed aggregation and morphogenesis prior to the tipped aggregate stage. All of these strains exhibit reduced chemotaxis speed (Figure 8D; Table II).
Aggregation is mediated by a combination of chemotaxis of cells and cell–cell adhesion (Aubry and Firtel, 1999). Chemotaxis defects can sometimes be masked when the cells are plated at the cell densities used in our developmental analysis because cell–cell contacts help cells coalesce and form aggregates (Meili et al, 1999). At lower cell densities, cells are not touching and therefore cell–cell contacts cannot assist aggregation. Strains with chemotaxis defects often exhibit stronger developmental defects when plated at lower densities. When we plated cells at a lower cell density (2.4 × 105 cells/cm2), many of the RacGEF1-overexpressing strains had stronger morphogenetic phenotypes (Figure 8C). Wild-type cells overexpressing full-length RacGEF1, RacGEF1ΔIQ, or RacGEF1ΔCH have very delayed aggregation and form only a few small aggregates. For RacGEF1- and RacGEF1ΔIQ-expressing cells, once aggregates form, they develop into mature fruiting bodies by 30 h. RacGEF1ΔIQ-expressing cells that aggregate form morphologically abnormal structures similar to those formed when cells are plated at higher cell densities, except that the structures are smaller. The results are consistent with the requirement of RacGEF1/RacB for proper cell movement and morphogenesis during the multicellular stages of Dictyostelium development.
Discussion
RacB involvement in F-actin polymerization and chemotaxis
We have examined the role of Dictyostelium RacB and the exchange factor RacGEF1 in controlling chemotaxis. RacB is activated in response to chemoattractant stimulation and has a bimodal activation profile paralleling that of F-actin polymerization. Our analysis of racB null cells indicates the requirement of RacB for normal F-actin polymerization and chemotaxis. racB null cells have very reduced speed, although directionality is unaffected. Consistent with the reduced speed of chemotaxis, racB null cells have an ∼60–70% decrease in the initial F-actin polymerization peak that corresponds to the initial cringe response (F-actin peak at 5 s) and a further reduction in the second, broader peak of F-actin polymerization associated with pseudopod extension (Hall et al, 1988). Our findings genetically link a specific Dictyostelium Rac protein with chemoattractant-mediated F-actin polymerization and cell movement. Another group previously examined the role of RacB through the expression of constitutively active and dominant-negative forms of the protein (Lee et al, 2003). They reported that cells expressing the dominant-negative form had no morphological changes but they did not examine motility, chemotaxis, or chemoattractant-mediated F-actin polymerization.
The remaining F-actin response in racB null cells indicates that other pathways mediate a portion of chemoattractant-stimulated F-actin polymerization. The remainder of the first and/or second peak of F-actin polymerization could be controlled by one or more other Dictyostelium Rac proteins, such as Rac1, which has been linked to chemotaxis (see Introduction). Alternatively, a Rac-independent mechanism may be involved in mediating F-actin polymerization.
Dictyostelium lacks a true Cdc42 as determined by sequence comparison of the Rac family members identified in the Dictyostelium genome, and it is unclear whether any of the Dictyostelium Rac proteins is its direct functional equivalent. RacB is a possible candidate, since, in addition to its interaction with the CRIB domains of PAKa and PAKc, RacB-GTP interacts with the CRIB domain of WASP, which in mammalian cells preferentially interacts with Cdc42-GTP. We have no direct evidence that RacB mediates WASP or SCAR activity in Dictyostelium.
Regulation of RacB by the PI3K pathway
The PI3K pathway has been linked to directional signaling and the control of F-actin polymerization. Studies indicate that the initial peak of F-actin polymerization is relatively unaffected (no inhibition or a modest 20–30% reduction) in cells treated with the PI3K inhibitor LY294002 or in pi3k1/2 null cells (Funamoto et al, 2001; Chen et al, 2003). However, the second peak of F-actin polymerization appears to be highly regulated by the PI3K pathway. pi3k1/2 null cells or cells treated with LY294002 have an almost complete loss of the second peak of RacB activation and Dictyostelium pten null cells exhibit a significantly elevated and extended second peak of F-actin polymerization with little effect on the first peak (Chen et al, 2003). We show that the PI3K pathway plays a role in RacB activation, suggesting that the effect of the PI3K pathway on F-actin polymerization is mediated, in part, through the regulation of RacB. We find that, compared to the first peak of RacB activation in wild-type cells, the second peak is significantly reduced in pi3k1/2 null cells and highly elevated in pten null cells. pi3k1/2 null cells express a higher level of RacB compared to wild-type cells, making it difficult to draw conclusions on the biological consequences of the reduction of the second RacB activation peak. However, our finding that treatment with LY294002, which does not detectably alter RacB protein levels in the time course of our experiments, also results in a modest reduction of the first and a significant reduction of the second RacB-GTP peak strongly supports a model in which the PI3K pathway directly controls chemoattractant-mediated RacB-GTP levels. No RacGEF like mammalian P-Rex1, which provides a direct linkage between PI3K and Rac activation, has been found in Dictyostelium (Welch et al, 2002).
Interestingly, our experiments identified a compensatory mechanism that controls RacB protein levels in genetic backgrounds that affect 3-phosphoinositide levels. We examined the regulation of RacB expression at the level of mRNA accumulation and protein accumulation by using knock-in technologies to replace the endogenous RacB gene with a tagged form of the protein. In pi3k1/2 null cells, in which the second peak of RacB activation is reduced, there is an increased level of RacB mRNA and RacB protein, whereas pten null cells, in which the second peak of RacB activation is elevated, have reduced RacB protein and mRNA levels. These data suggest that the cells need to regulate the level of RacB protein to prevent toxic levels of RacB-GTP, F-actin, and/or activated forms of other RacB effectors. We presume this compensatory regulatory mechanism at the level of mRNA and protein accumulation exists and is independent of regulatory mechanisms that control RacGEF activation and RacGAP hydrolysis of RacB-GTP. Consistent with this, we discovered that in racgef1 null cells, which exhibit a 50% reduction in RacB activation, there is an apparently compensatory increase in the expression of RacB protein compared to RacB protein levels in wild-type cells. RacGEF1-overexpressing cells, which do not exhibit elevated basal levels of RacB-GTP, show no appreciable change in RacB levels. These findings suggest that the cells monitor basal RacB-GDP or RacB-GTP levels and these levels feed back to control RacB expression.
Regulation and role of RacGEF1
We identified RacGEF1 and demonstrated that it preferentially uses RacB as a substrate for GDP/GTP exchange activity in vitro and is required for ∼50% of RacB activation in vivo. racgef1 null cells exhibit a reduction in chemoattractant-mediated F-actin polymerization that is intermediate to that observed for wild-type cells and racB null cells. RacGEF1 is localized to the cortex in vegetative cells. However, in aggregation-competent cells, RacGEF1 is predominantly cytosolic and translocates to the plasma membrane in response to global chemoattractant stimulation. In chemotaxing cells, RacGEF1 is associated with the leading edge and to a lesser degree with the cell's posterior, sites of F-actin polymerization. RacGEF1 translocation to the cortex is inhibited by LatA, suggesting that RacGEF1 cortical localization is driven by F-actin polymerization. Treatment with LatA reduces RacB activation, suggesting that RacGEF1 must translocate to the cortex in an F-actin-dependent manner to activate RacB. RacGEF1 translocation is also reduced in pi3k1/2 null cells or in wild-type cells treated with LY294002. We suggest that RacB, RacGEF1, and PI3K may be part of a feedback loop in which PI3K stimulates RacB activation by further translocating RacGEF1 to the plasma membrane through its interaction with the F-actin cytoskeleton. RacGEF1 localization may cause additional RacB activation, increasing F-actin polymerization and possibly recruiting additional RacGEF1 to the leading edge.
A positive feedback loop between PI3K and F-actin regulated by Rac GTPases has been implicated in leading edge formation in neutrophils (Wang et al, 2002; Weiner et al, 2002). We examined this indirectly and found little difference in PKB activation between wild-type and racB null cells. In other studies, we found that chemoattractant-mediated PI3K translocation to the cortex is dependent on F-actin polymerization (Sasaki et al, submitted for publication). These findings suggest that an F-actin-dependent feedback loop may exist in Dictyostelium and may be partially controlled by RacB, but the level of PKB activation is not altered. This may be because RacB is required for ∼50% of the first peak of F-actin polymerization. It is possible that PI3K activity is reduced in racB null cells but PKB activity is insensitive to this level of change.
Roles of RacB and RacGEF1 in morphogenesis
Morphogenesis in Dictyostelium is mediated, in part, through the chemotactic movement of prestalk cells within the organism, which depends on the cytoskeleton. Not unexpectedly, racB null cells exhibit morphogenesis defects. racgef1 null cells have a weaker phenotype, presumably because there is only a partial loss of RacB activation and a modest loss of F-actin polymerization in these cells. However, overexpression of wild-type and various deleted forms of RacGEF1 causes a range of morphological phenotypes that are most obvious at lower cell densities in which defects in chemotaxis could not be overcome by cell adhesion mechanisms facilitating aggregation. These data support other studies in which mutants in Rac pathways affect morphogenetic movement of cells (Raftopoulou and Hall, 2004). We expect RacB to be required to activate other effectors such as PAKc and IQGAPs in addition to directly controlling F-actin polymerization.
Our studies on RacB and RacGEF1 provide a better mechanistic understanding of the regulatory pathways controlling F-actin polymerization, cell movement, and morphogenesis in Dictyostelium. The partial dependence of RacB activation on the PI3K pathway and the recruitment of RacGEF1 to the plasma membrane in response to F-actin polymerization link RacB to the PI3K pathway, leading edge formation, localized F-actin polymerization, and pseudopod extension.
Materials and methods
Assays
The GDP dissociation assay was performed as described (Rossman and Campbell, 2000). [3H]GDP-bound Rac proteins were prepared by incubating 12 μM of GST-Rac in 10 mM HEPES buffer (pH 7.5) containing 100 mM NaCl, 7.5 mM EDTA, 15 μM GDP, and 5.5 μM [3H]GDP for 30 min at 23°C, and stabilized by supplementing with 20 mM MgCl2 solution. Nucleotide exchange activity was performed by diluting [3H]GDP-bound Rac to 4 μM in 10 mM HEPES buffer (pH 7.5) containing 4 μM His-GEF(DH) or no GEF, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT, 50 μg/ml BSA, and 100 μM GTP at 23°C. A 30 μl portion of reaction mixture was quenched in 1 ml of ice-cold Tris buffer (pH 7.5) containing 100 mM NaCl and 20 mM MgCl2. [3H]GDP bound to the Rac proteins was determined using a scintillation counter.
The RacB activation assay is a modification of the protocol of Benard et al (1999). The levels of RacB-GTP were measured by affinity precipitation using the MBP- (maltose-binding protein) or GST-CRIB (Cdc42 and Rac interactive binding region) of DdPAKa, DdPAKc, or DdWASP. Log-phase vegetative cells were washed and shaken at a density of 8–9 × 106 cells/ml in Na/K phosphate buffer for 5 h with 30 nM cAMP added every 6 min to obtain cAMP-responsive, aggregation-competent cells. Cells were treated with 1 mM caffeine for 30 min, collected, and resuspended at 4 × 107 cells/ml in Na/K phosphate buffer containing 1 mM caffeine. Samples were stimulated with 1 μM cAMP in a syringe attached to a filter holder. At the indicated times, the cells were disrupted by filtering into 5 × binding buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 100 mM MgCl2, 1 mM DTT, 2.5% Triton X-100) containing aprotinin, leupeptin, and GST-PAK CRIB protein. Glutathione–Sepharose beads were added and incubated for 30 min at 4°C. The beads were washed three times in binding buffer, suspended in sample buffer, and subjected to SDS–PAGE and Western blot analysis with an anti-myc monoclonal antibody. The amount of RacB (RacB-GTP) bound to beads was quantified by densitometry. The wild-type level at 0 s was set at 1.0. The levels of RacB-GTP in the mutant strains were divided by the relative level of myc-RacB protein in that strain compared to the level of myc-RacB protein in wild-type cells. See Supplementary data for a detailed protocol.
Chemotaxis analysis was performed as described previously (Funamoto et al, 2001). Briefly, a small volume of aggregation-competent cells was placed on a 30 mm Petri plate with a hole covered by a glass coverslip. A glass capillary needle filled with 150 μM cAMP solution was positioned to stimulate cells with an Eppendorf micromanupulator and the response of the cells was recorded with a time-lapse video recorder and NIH Image software. Computer analysis was performed using DIAS software. At least five cells from each of at least three independent experiments were analyzed. See Supplementary data for a detailed protocol.
F-actin polymerization and myosin II assembly were assayed as previously described (Steimle et al, 2001). Briefly, caffeine-treated, aggregation-competent cells were stimulated with 10 μM cAMP and at the indicated times, aliquots of cells were lysed by addition of an equal volume of 100 mM MES (pH 6.8) buffer containing 1% Triton X-100, 5 mM EGTA, and 10 mM MgCl2. The cytoskeletal pellet was collected by centrifugation, suspended in 2 × sample buffer, and subjected to SDS–PAGE. Actin and myosin levels were determined by densitometric analysis of scanned Coomassie-stained gels. See Supplementary data for details.
To assay RacGEF1 membrane localization, aggregation-competent cells were resuspended at a density of 1 × 107 cells/ml in Na/K phosphate buffer and were treated with 60 μM LY294002 or DMSO (control) and incubated for 25 min by shaking. At the indicated times after stimulation with 10 μM cAMP, the cells were lysed with an equal volume of 2 × Triton lysis buffer (2 × PBS, 100 mM NaF, 1% Triton X-100, 4 mM EDTA, 2 mM pyrophosphate, 1 mM DTT, and protease inhibitors leupeptin and aprotinin) on ice for 10 min. Triton-resistant cytoskeletal pellets were collected by centrifugation, suspended in 2 × sample buffer, and subjected to SDS–PAGE. GFP-GEF1 protein was determined by Western blotting using Santa Cruz Biotech anti-GFP polyclonal antibody.
Yeast two-hybrid assays were performed as described previously (Lee et al, 1999) or using protocols from the Matchmaker two-hybrid system (Clontech). See Supplementary data for details.
The PKB kinase activity was assayed as previously described (Meili et al, 1999). The PAKc kinase activity assay is a modification of that used for PKB except that myc-tagged PAKc was immunoprecipitated with anti-myc antibody. See Supplementary data for details.
Constructs
The null and knock-in constructs were made using standard approaches (Supplementary figures). All knockout clones were confirmed by Southern blot analysis, and positive racB knock-in clones were confirmed by Western blotting using the anti-myc antibody.
Supplementary Material
Supplementary Materials
Acknowledgments
We thank Sharon Campbell, UNC, for invaluable assistance in establishing the RacGEF activity assays and Gary Bokoch, TSRI, for assisting us in establishing our RacB-GTP binding assay. We thank Ann-Kathrin Meyer for invaluable technical assistance and the Firtel laboratory for helpful suggestions. This work was supported by grants of the DFG (RI 1034/2) and the Köln Fortune program to FR and by USPHS grants to RAF.
References
- Aubry L, Firtel R (1999) Integration of signaling networks that regulate Dictyostelium differentiation. Annu Rev Cell Dev Biol 15: 469–517 [DOI] [PubMed] [Google Scholar]
- Benard V, Bohl BP, Bokoch GM (1999) Characterization of rac and cdc42 activation in chemoattractant-stimulated human neutrophils using a novel assay for active GTPases. J Biol Chem 274: 13198–13204 [DOI] [PubMed] [Google Scholar]
- Bush J, Franek K, Cardelli J (1993) Cloning and characterization of seven novel Dictyostelium discoideum Rac-related genes belonging to the Rho family of GTPases. Gene 136: 61–68 [DOI] [PubMed] [Google Scholar]
- Chen L, Janetopoulos C, Huang YE, Iijima M, Borleis J, Devreotes PN (2003) Two phases of actin polymerization display different dependencies on PI(3,4,5)P3 accumulation and have unique roles during chemotaxis. Mol Biol Cell 14: 5028–5037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C, Potikyan G, Firtel R (2001a) Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol Cell 7: 937–947 [DOI] [PubMed] [Google Scholar]
- Chung CY, Funamoto S, Firtel RA (2001b) Signaling pathways controlling cell polarity and chemotaxis. TIBS 26: 557–566 [DOI] [PubMed] [Google Scholar]
- Devreotes P, Janetopoulos C (2003) Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J Biol Chem 278: 20445–20448 [DOI] [PubMed] [Google Scholar]
- Funamoto S, Meili R, Lee S, Parry L, Firtel R (2002) Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 109: 611–623 [DOI] [PubMed] [Google Scholar]
- Funamoto S, Milan K, Meili R, Firtel R (2001) Role of phosphatidylinositol 3′ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in Dictyostelium. J Cell Biol 153: 795–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AL, Schlein A, Condeelis J (1988) Relationship of pseudopod extension to chemotactic hormone-induced actin polymerization in amoeboid cells. J Cell Biochem 37: 285–299 [DOI] [PubMed] [Google Scholar]
- Iijima M, Devreotes P (2002) Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 109: 599–610 [DOI] [PubMed] [Google Scholar]
- Iijima M, Huang Y, Devreotes P (2002) Temporal and spatial regulation of chemotaxis. Dev Cell 3: 469–478 [DOI] [PubMed] [Google Scholar]
- Knetsch M, Schafers N, Horstmann H, Manstein D (2001) The Dictyostelium Bcr/Ab-related protein DRG regulates both Rac- and Rab-dependent pathways. EMBO J 20: 1620–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Seastone D, Harris E, Cardelli J, Knecht D (2003) RacB regulates cytoskeletal function in Dictyostelium spp. Eukaryot Cell 2: 474–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Parent CA, Insall R, Firtel RA (1999) A novel Ras-interacting protein required for chemotaxis and cyclic adenosine monophosphate signal relay in Dictyostelium. Mol Biol Cell 10: 2829–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, Huang CK, Wu D (2003) Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 114: 215–227 [DOI] [PubMed] [Google Scholar]
- Meili R, Ellsworth C, Lee S, Reddy T, Ma H, Firtel R (1999) Chemoattractant-mediated transient activation and membrane localization of Akt/PKB is required for efficient chemotaxis to cAMP in Dictyostelium. EMBO J 18: 2092–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlot S, Firtel R (2003) Leading the way: directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J Cell Sci 116: 3471–3478 [DOI] [PubMed] [Google Scholar]
- Pollard T, Borisy G (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112: 453–465 [DOI] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265: 23–32 [DOI] [PubMed] [Google Scholar]
- Rivero F, Albrecht R, Dislich H, Bracco E, Graciotti L, Bozzaro S, Noegel A (1999) RacF1, a novel member of the Rho protein family in Dictyostelium discoideum, associates transiently with cell contact areas, macropinosomes, and phagosomes. Mol Biol Cell 10: 1205–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero F, Illenberger D, Somesh B, Dislich H, Adam N, Meyer A (2002) Defects in cytokinesis, actin reorganization and the contractile vacuole in cells deficient in RhoGDI. EMBO J 21: 4539–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero F, Somesh B (2003) Signal transduction pathways regulated by Rho GTPases in Dictyostelium. J Muscle Res Cell Motil 23: 737–749 [DOI] [PubMed] [Google Scholar]
- Rossman KL, Campbell SL (2000) Bacterial expressed DH and DH/PH domains. Methods Enzymol 325: 25–38 [DOI] [PubMed] [Google Scholar]
- Steimle PA, Yumura S, Cote GP, Medley QG, Polyakov MV, Leppert B, Egelhoff TT (2001) Recruitment of a myosin heavy chain kinase to actin-rich protrusions in Dictyostelium. Curr Biol 11: 708–713 [DOI] [PubMed] [Google Scholar]
- Wang F, Herzmark P, Weiner OD, Srinivasan S, Servant G, Bourne HR (2002) Lipid products of PI(3)Ks maintain persistent cell polarity and directed motility in neutrophils. Nat Cell Biol 4: 513–518 [DOI] [PubMed] [Google Scholar]
- Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR (2002) A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol 4: 509–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR (2002) P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell 108: 809–821 [DOI] [PubMed] [Google Scholar]
- Xu J, Wang F, Van Keymeulen A, Herzmark P, Straight A, Kelly K, Takuwa Y, Sugimoto N, Mitchison T, Bourne HR (2003) Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 114: 201–214 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials