

Abstract
IκB Kinase (IKK)α is required for activation of an alternative NF-κB signaling pathway based on processing of the NF-κB2/p100 precursor protein, which associates with RelB in the cytoplasm. This pathway, which activates RelB:p52 dimers, is required for induction of several chemokine genes needed for organization of secondary lymphoid organs. We investigated the basis for the IKKα dependence of the induction of these genes in response to engagement of the lymphotoxin β receptor (LTβR). Using chromatin immunoprecipitation, we found that the promoters of organogenic chemokine genes are recognized by RelB:p52 dimers and not by RelA:p50 dimers, the ubiquitous target for the classical NF-κB signaling pathway. We identified in the IKKα-dependent promoters a novel type of NF-κB-binding site that is preferentially recognized by RelB:p52 dimers. This site links induction of organogenic chemokines and other important regulatory molecules to activation of the alternative pathway.
Keywords: alternate NF-κB signaling pathway, IKKα, LTβ, NF-κB binding site, stromal cells
Introduction
The classical NF-κB signaling pathway, which is activated by proinflammatory cytokines and pathogen-associated molecular patterns (PAMPs), depends on inducible degradation of specific inhibitors, IκBs, which retain different NF-κB dimers in the cytoplasm (Ghosh and Karin, 2002). This pathway is largely dependent on IKKβ, a component of a complex that also contains the IKKα catalytic subunit and the IKKγ/NEMO regulatory subunit (Rothwarf and Karin, 1999). In this pathway, IKKβ phosphorylates IκBs at N-terminal sites to trigger their ubiquitin-dependent degradation and induce nuclear entry of NF-κB dimers (Karin and Ben-Neriah, 2000). Recently, a second NF-κB activation pathway based on regulated processing of the NF-κB2/p100 precursor protein was identified (Senftleben et al, 2001; Xiao et al, 2001). NF-κB2/p100 consists of an N-terminal Rel homology domain (RHD), common to all NF-κB proteins, and an inhibitory IκB-like C-terminal domain (Ghosh et al, 1998). The presence of the latter prevents nuclear translocation of p100 and its partners.
IKKα and IKKβ activate at least a dozen NF-κB dimers, composed of five subunits (Ghosh and Karin, 2002). While the mechanisms of NF-κB activation are well understood (Ghosh and Karin, 2002), the generation of biological specificity by this complex system is more enigmatic (Pomerantz and Baltimore, 2002). Mouse mutagenesis experiments indicate that IKKβ activates the classical NF-κB pathway, represented by RelA:p50 dimers, in response to stimuli such as tumor necrosis factor (TNF)α (Li et al, 1999; Chen et al, 2003). The mechanisms by which IKKα regulates cytokine-induced gene expression are more obscure and controversial (Israel, 2003). In vivo analysis revealed that IKKα activates an alternative NF-κB pathway based on processing of NF-κB2/p100 and release of RelB:p52 dimers in response to LTα/β trimers (Dejardin et al, 2002) and other TNF family members (Claudio et al, 2002; Kayagaki et al, 2002). This pathway is required for secondary lymphoid organogenesis and induction of genes involved in this process, but has no apparent role in TNFα-induced functions (Senftleben et al, 2001; Dejardin et al, 2002). We have used mice in which IKKα was rendered inactivateable (Cao et al, 2001) to study the mechanism responsible for selective gene induction by the alternative NF-κB signaling pathway. Using primary cultures of splenic stromal cells and bone marrow-derived myeloid dendritic cells (BMDCs) as experimental systems, we found that generation of gene induction specificity by IKKα depends on selective activation of RelB:p52 dimers, which recognize a unique type of the NF-κB-binding site.
Results
Reconstitution of lethally irradiated mice with Ikkα−/− fetal liver hematopoietic progenitors revealed a role for IKKα in late B-cell maturation, splenic organization and germinal center (GC) formation (Kaisho et al, 2001; Senftleben et al, 2001). However, embryonic lethality precludes the use of Ikkα−/− mice to identify functions for IKKα in other cell types involved in spleen development and organization. Homozygous knockin mice expressing an IKKα variant that cannot be activated (IkkαAA/AA mice) are viable, yet show defects in lymphoid organogenesis and GC formation (Senftleben et al, 2001). Using an antibody against FDC-M2, a follicular dendritic cell (FDC) marker, we found that IkkαAA/AA mice lack mature FDCs (Figure 1A). To identify the cells in which IKKα acts, reciprocal bone marrow chimeras were generated using IkkαAA/AA and WT mice. At 6 weeks after adoptive transfer, mice were challenged with a T-cell-dependent antigen, sheep red blood cells (SRBC), and killed 7 days later. Using an antibody against CD35, another FDC marker, we examined the formation of mature FDCs. FDC maturation was impaired in IkkαAA/AA recipients reconstituted with WT bone marrow, whereas a mature FDC network formed in WT recipients reconstituted with IkkαAA/AA bone marrow (Figure 1B). These results suggest that IKKα acts in stromal cells of the spleen to induce maturation of FDCs, which are thought to be derived from mesenchymal stromal cells (Fu and Chaplin, 1999).
Figure 1.
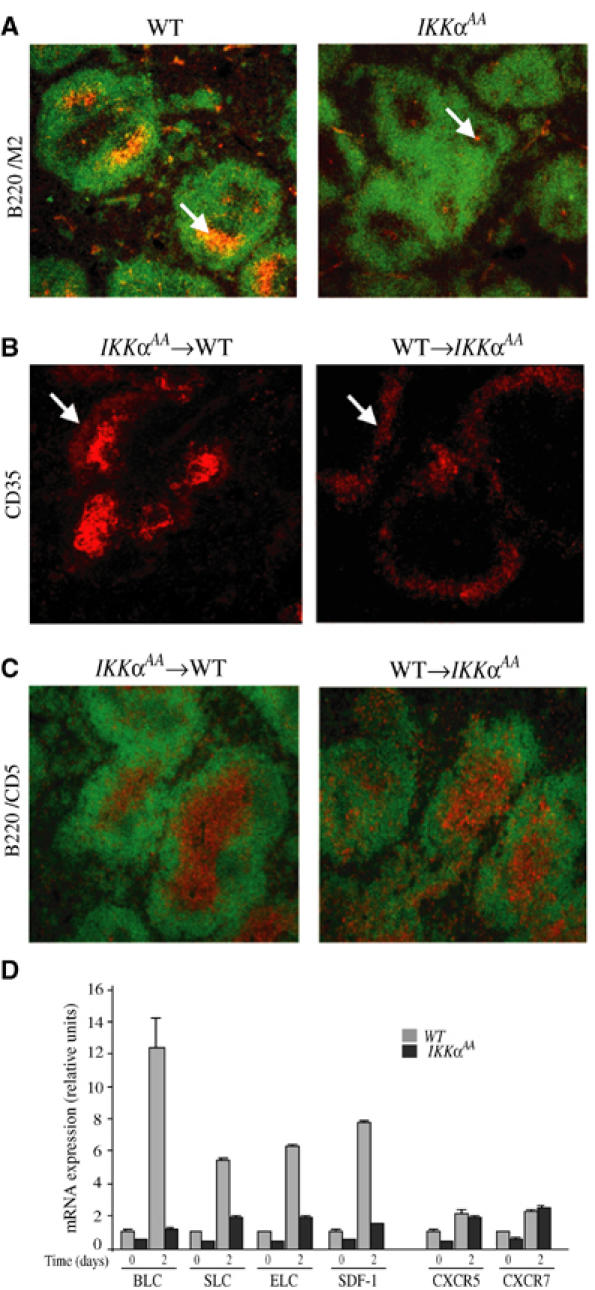
Impaired FDC maturation and chemokine production in stromal cell-derived FDC requires IKKα. (A) Absence of mature FDC network in IkkαAA/AA mice. Cryosections of spleen from WT (n=6) and IkkαAA/AA (n=6) mice, isolated 7 days post-immunization with SRBC, were stained for FDCs (arrows) with FDC-M2 (orange) and anti-B220 (green). (B) Impaired FDC maturation is inherent to the IkkαAA/AA stroma. Lethally irradiated WT (n=6) or IkkαAA/AA (n=6) mice were reconstituted with IkkαAA/AA or WT bone marrow, respectively. Spleens were isolated 7 days after immunization with SRBC, cryosectioned and stained with anti-CD35. An FDC network is present in WT mice reconstituted with IkkαAA/AA bone marrow, while only perifollicular rings of CD35+ immature FDCs are present in IkkαAA/AA mice reconstituted with WT bone marrow. (C) Impaired B/T cell segregation in IkkαAA/AA spleens. Lethally irradiated WT (n=6) or IkkαAA/AA (n=6) mice reconstituted with IkkαAA/AA or WT bone marrow cells were immunized and analyzed as above, using anti-CD5 (to recognize T cells) and anti-B220 (to recognize B cells). Impaired B/T cell segregation is intrinsic to the IkkαAA/AA stroma. (D) Defective chemokine gene expression in IkkαAA/AA spleens. Total splenocytes from naïve and SRBC-immunized (day 2) WT (n=6) and IkkαAA/AA (n=6) mice were isolated. RNA was extracted and analyzed by RT–PCR for expression of mRNAs encoding BLC, SLC, ELC and SDF-1 and two of their receptors (CXCR5, CCR7). The results are averages±s.d. of three independent experiments normalized to the level of cyclophilin mRNA.
Another aspect of spleen development is segregation of B and T lymphocytes to the follicles and the periarterial lymphatic sheath (PALS), respectively. WT chimeras reconstituted with IkkαAA/AA bone marrow, but not IkkαAA/AA mice reconstituted with WT bone marrow, exhibited normal B- and T-cell segregation detected by staining with anti-B220 and anti-CD5 antibodies, respectively (Figure 1C). These results also point to a critical action of IKKα in stromal cells, which, in addition to giving rise to FDCs, control splenic microarchitecture through production of organogenic chemokines that dictate cell migration and localization (Ansel and Cyster, 2001). Critical organogenic chemokines for spleen development include: ELC and SLC, ligands for the chemokine receptor CCR7; BLC, which binds CXCR5 (Forster et al, 1999; Ansel et al, 2000) and SDF-1, which promotes trafficking of both immature and naïve lymphocytes to lymphoid tissues (Kim and Broxmeyer, 1999). Previous work revealed that induction of these chemokines in response to engagement of LTβR is defective in IkkαAA/AA mice (Dejardin et al, 2002). We extended these observations to SRBC immunized mice (Figure 1D). Based on previous experiments, we examined the expression of the different genes 48 h post-immunization. While induction of the mRNAs for BLC, ELC, SLC and SDF-1 was readily detected in WT spleens, these genes were barely induced in the mutant.
The defects shown above are very similar to those exhibited by mice lacking LTβR (Fu and Chaplin, 1999). The majority of LTβR expression is restricted to stromal cells of the spleen; however, BMDCs have also been shown to express LTβR (Browning and French, 2002). We therefore isolated and cultured both of these cell types from WT and IkkαAA/AA mice. Stimulation of WT stromal cells with agonistic anti-LTβR antibody (Dejardin et al, 2002) resulted in four- to six-fold induction of BLC, SDF-1, TNFα, VCAM-1 and IκBα mRNA (Figure 2A). Modest induction of ELC and SLC mRNAs was also observed. Both basal expression and induction of BLC, SDF-1, ELC and SLC mRNAs were defective in IkkαAA/AA stromal cells. Similar defects in expression of these chemokines have been described in RelB−/− and Nfkb2−/− mice (Poljak et al, 1999; Weih et al, 2001). Induction of TNFα, IκBα and VCAM-1 in IkkαAA/AA stromal cells remained intact or was even elevated. The increased expression of VCAM-1 could be related to the defective nuclear entry of RelB in IkkαAA/AA cells (see below), as RelB deficiency was previously found to increase the expression of certain inflammatory genes (Xia et al, 1999). By contrast, very few differences in expression of TNFα-inducible genes were found between WT and IkkαAA/AA stromal cells (Figure 2A). Unlike anti-LTβR, TNFα was a poor inducer of the organogenic chemokines, but was a potent inducer of TNFα, IκBα and VCAM-1.
Figure 2.
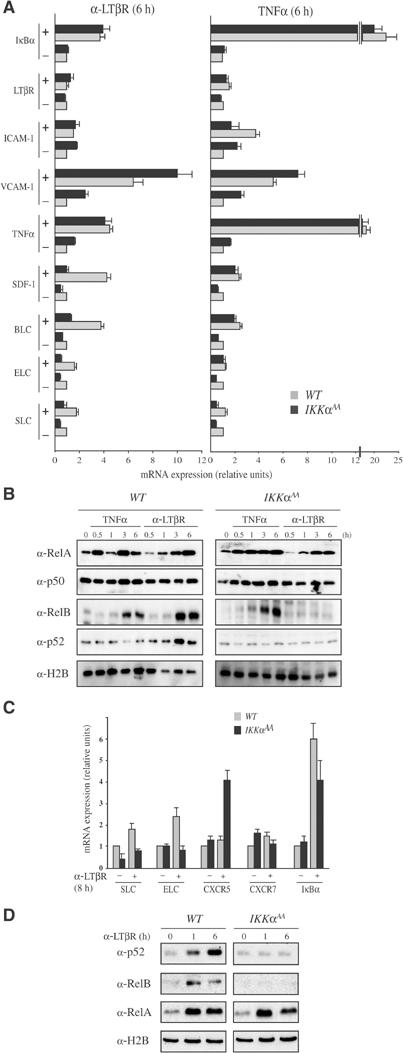
IKKα is required for LTβR-induced RelB:p52 nuclear translocation and chemokine expression in splenic stromal cells and myeloid dendritic cells. (A) IkkαAA/AA stromal cells and (C) BMDC exhibit specific defects in LTβR-induced gene expression. Total RNA was extracted from either WT or IkkαAA/AA stromal cells or BMDC before and after stimulation with 2 μg/ml agonistic anti-LTβR antibody or 20 ng/ml TNFα. Gene expression was analyzed by real-time PCR. Results are averages±s.d. of three independent experiments normalized to the level of cyclophilin mRNA. (B, D) Nuclear translocation of NF-κB proteins. Stromal cells (B) and BMDC (D) were stimulated with either anti-LTβR antibody or TNFα as indicated. At the indicated time points (h), nuclear extracts were prepared and analyzed by immunoblotting for the presence of the indicated NF-κB subunits. The levels of histone H2B were examined to control for loading and proper cell fractionation. Contamination with cytoplasmic proteins was monitored by blotting with anti-actin antibody (not shown).
TNFα induced both rapid and delayed nuclear translocation of RelA in WT and IkkαAA/AA stromal cells (Figure 2B). This response was not considerably different in IkkαAA/AA cells (Figure 2B, right panel). Neither TNFα nor anti-LTβR had a significant effect on the subcellular distribution of p50, as this NF-κB subunit was constitutively nuclear (Figure 2B). Both TNFα and anti-LTβR induced nuclear translocation of RelB in WT cells, but only TNFα was capable of sending RelB to the nucleus of IkkαAA/AA cells (Figure 2B). In either case, the nuclear translocation of RelB is delayed relative to that of RelA. As expected, only anti-LTβR, but not TNFα, stimulated nuclear entry of p52, and this effect was seen only in WT cells (Figure 2B). Similar results with regard to both gene expression and nuclear translocation of NF-κB subunits were observed in BMDCs. In WT BMDCs, LTβR engagement led to induction of SLC, ELC and IκBα mRNA (Figure 2C). However, SLC and ELC were not induced in BMDC from IkkαAA/AA mice. Again, we found that at least one gene, in this case CXCR5, was elevated in mutant cells. Whereas engagement of LTβR resulted in nuclear entry of RelB and p52 in WT BMDCs, this response was defective in IkkαAA/AA cells (Figure 2D). Nuclear translocation of RelA was not affected in IkkαAA/AA cells. These results and the previous genetic analysis of NF-κB2- (Poljak et al, 1999) and RelB- (Weih et al, 2001) deficient mice strongly suggest that Blc, Sdf-1, Elc and Slc gene induction requires RelB:p52 nuclear translocation. Curiously, the induction of RelB and p52 nuclear entry following LTβR engagement was considerably faster in BMDCs than in splenic stromal cells. This is likely to be related to the different origins of these cell types and/or the expression of different levels of LTβR molecules on their surface.
To address whether the IKKα-dependent genes are in fact direct targets for RelB-containing dimers and whether they are also recognized by RelA-containing dimers, we performed chromatin immunoprecipitation (ChIP) experiments (Saccani and Natoli, 2002). In splenic stromal cells, anti-LTβR induced efficient recruitment of RelB, but not RelA, to the Blc and Sdf-1 promoters (Figure 3A), which encode the two organogenic chemokines that are most efficiently expressed by these cells (Cyster, 2003). As previously shown, recruitment of NF-κB subunits to promoter DNA may be detected at earlier time points than the ones revealed by immunoblot analysis of nuclear translocation, due to the increased sensitivity of the ChIP assay (Saccani et al, 2001). Anti-LTβR-induced recruitment of RelB to target gene promoters was abolished in IkkαAA/AA cells. However, TNFα-induced RelB promoter recruitment, which was slower and weaker than the response elicited by anti-LTβR, was not affected by the IkkαAA mutation (Figure 3A). The response to TNFα may depend on the formation of RelB:p50 dimers. As a control, we analyzed the same immunoprecipitates for the presence of the Tnfα 0and Vcam1 promoter regions. We found efficient precipitation of both promoter fragments by anti-RelA antibodies, but a weak signal was obtained with anti-RelB (Figure 3A). Recruitment of either Rel protein to these promoters was not IKKα-dependent. Importantly, recruitment of Pol II to the Blc and Sdf-1 promoters correlated with recruitment of RelB and was seen only in anti-LTβR stimulated WT cells (Figure 3A).
Figure 3.
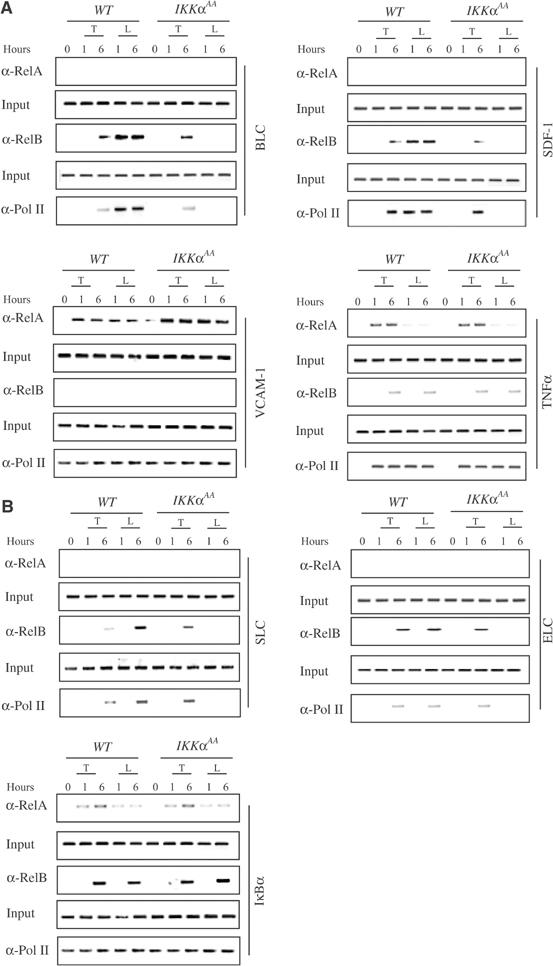
IKKα is required for recruitment of RelB to the Blc, Sdf-1, Elc and Slc promoters. Primary cultures of stromal cells (A) and BMDC (B) from WT and IkkαAA/AA mice were left unstimulated or stimulated with TNFα (T) or anti-LTβR (L). At the indicated time points (h), the cells were collected and recruitment of RelA, RelB and the large subunit of RNA polymerase (Pol II) to the indicated promoter regions was examined by ChIP experiments.
As mentioned above, splenic stromal cells are the major source of production of BLC and SDF, while BMDCs are a major source of ELC and SLC (Cyster, 2003). Therefore, in BMDCs, we examined recruitment of the different NF-κB subunits in response to LTβ signaling to the Elc and Slc promoters. Treatment with anti-LTβR induced efficient recruitment of RelB, but not RelA, to the Elc and Slc promoters (Figure 3B). No recruitment of RelA was observed. By contrast, both RelB and RelA were recruited to the IκBα promoter in response to either TNFα or anti-LTβR, but neither response was IKKα-dependent (Figure 3B). As observed for RelB, the LTβR-induced recruitment of Pol II to the Slc and Elc promoters was IKKα-dependent (Figure 3B).
Selective recruitment of RelB-containing NF-κB dimers to the Blc, Sdf-1, Elc and Slc promoters could reflect, previously unknown, intrinsic differences in sequence selectivity between RelB- and RelA-containing dimers. To examine this possibility, we analyzed binding of NF-κB proteins to the Blc and Elc promoters. In this experiment, we used truncated recombinant NF-κB proteins to generate NF-κB dimers of known composition. All of the proteins used in these experiments were fully characterized and even crystallized (Ghosh et al, 1995; Chen et al, 1999; G Ghosh, unpublished data). Several 32P-labeled probes were derived from the 700 base pair (bp) proximal region (−688 to +12) of the Blc promoter, contained within the ChIP primer set (Figure 4A). One of the probes, spanning positions −191 to −20, exhibited strong binding to recombinant RelB:p52 and weak binding to RelA:p50 dimers (data not shown). Several other probes (from −770 to −460, −460 to −380 and −380 to −150, as well as from −770 to −980) did not detectably bind either dimer (data not shown). To narrow down the sequence responsible for RelB:p52 binding, we generated a shorter probe (Probe 1) covering the region from −191 to −64. This probe exhibited very strong binding to recombinant RelB:p52 and only weak binding to RelA:p50 (Figure 4B). On the other hand, the RelA:p50 and RelB:p52 dimers exhibited little differences in their ability to bind a consensus κB probe, whereas a 200 bp probe (Probe 2) derived from the far 5′ upstream region (−1900 to −1700) of the Blc gene was preferentially recognized by RelA:p50 (data not shown). Probe 1 (−191 to −64) contained only one potential NF-κB-binding site. We synthesized two overlapping smaller probes containing this site (Figure 4C) and used them to examine binding of RelA:p50, RelB:p52, as well as RelB:p50. Both probes, which contained the sequence 5′-GGGAGATTTG-3′, were efficiently recognized by RelB:p52 and only weakly by RelA:p50 (Figure 4B; data not shown). Binding of RelB:p50 to these probes was barely detectable. In all cases, the detected protein–DNA complexes were specific, as indicated by competition experiments (data not shown).
Figure 4.
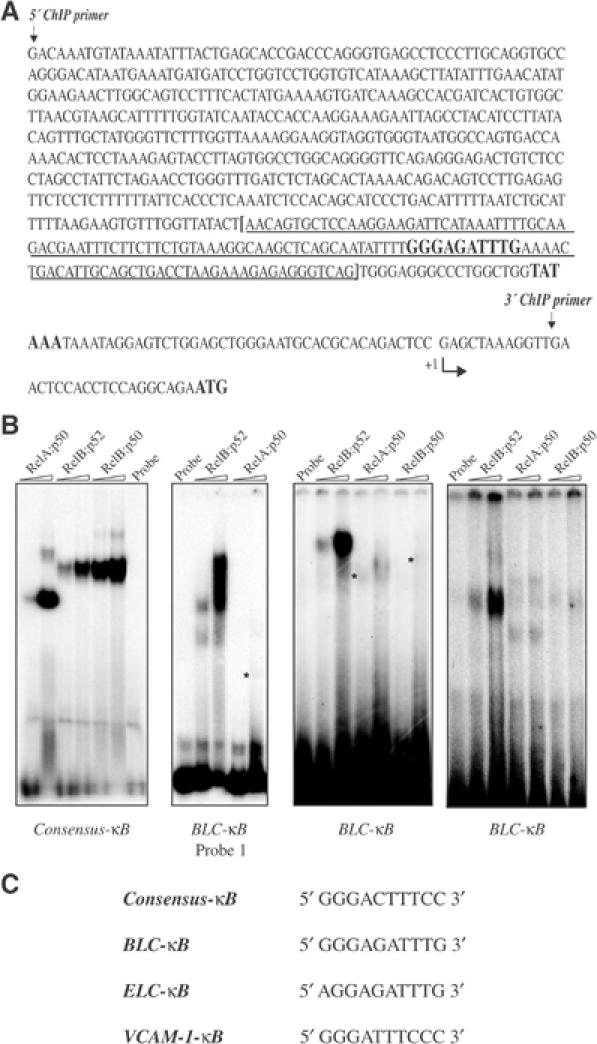
The Blc and Elc promoters contain a unique κB site that is selectively recognized by RelB:p52 dimers. (A) The sequence of the 700 bp region, covering the proximal Blc promoter, contained within the ChIP primer set. The RelB-selective κB site and the TATA box are highlighted. The sequence contained within Probe 1 is indicated by the brackets and is underlined. (B) DNA-binding analysis. The different probes were incubated with two different amounts (250 and 500 ng) of the indicated NF-κB dimers and DNA binding was analyzed by EMSA. Note that the NF-κB subunits are not the full-length proteins, thus giving rise to complexes with different electrophoretic mobilities. (C) The sequences of the different κB sites used in these experiments.
To identify whether nother IKKα-dependent chemokine genes contain a similar sequence, we used the Trafac server (Jegga et al, 2002), which identifies ortholog conserved transcription factor binding sites, to examine the human and rodent Elc genes. The putative binding sites were first identified using the MatInspector program (Professional Version 4.3, 2000) that utilizes a database of eukaryotic transcription factor binding sites (Jegga et al, 2002). This procedure identified a sequence very similar to the Blc-κB site at positions −64 to −50 of the Elc genes (Figure 4C). This site, termed the Elc-κB site, was also preferentially recognized by RelB:p52 dimers (Figure 4B).
We next used MEFs, which, unlike the related stromal cells, are amenable to transfection (Bebien M, unpublished results), to examine the function of the RelB:p52-specific sites. Stimulation of WT MEFs with either TNFα or α-LTβR induced DNA-binding activities recognized by the consensus κB site (Figure 5A). Using the Blc-κB and Elc-κB sites as probes, we detected induced DNA-binding activity only in WT MEFs stimulated with anti-LTβR (Figure 5A). This activity was not induced in Ikkα−/− MEFs. Similar results were obtained in BMDCs analyzed with the Elc-κB probe (Figure 5B). Next, we cloned three copies of either the consensus κB site, the Blc-κB site or an inactive version of the latter (mBlc-κB), upstream to a minimal SV40 promoter driving a luciferase reporter, and transfected the constructs into WT and Ikkα−/− MEFs. The consensus κB site conferred inducibility by either TNFα or anti-LTβR, whereas the Blc-κB site conferred an efficient response to anti-LTβR but only a weak response to TNFα, and the mutated Blc-κB site was inactive (Figure 5C). While the consensus κB site was equally active in WT and Ikkα−/− MEFs, the Blc-κB site did not confer anti-LTβR responsiveness in Ikkα−/− MEFs (Figure 5C). Using the intact Blc promoter fused to a luciferase reporter, we found efficient induction by anti-LTβR in WT but not in Ikkα−/− MEFs. This response was dependent on the integrity of the Blc-κB site and even its conversion to a consensus κB site attenuated the response to anti-LTβR (Figure 5C). The Elc promoter also exhibited preferential activation by anti-LTβR that was IKKα-dependent.
Figure 5.
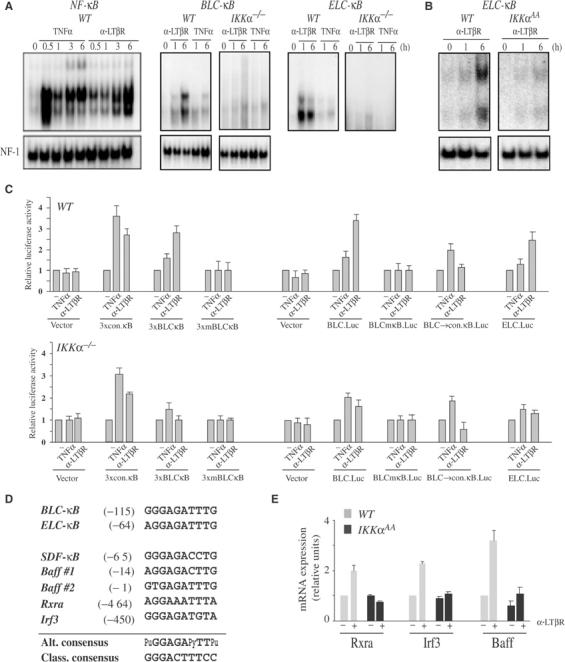
Selective, IKKα-dependent activation of the Blc and Elc promoters by LTβR engagement and IKKα-dependent induction of Rxra, Irf3 and Baff mRNAs. (A, B) Engagement of LTβR selectively induces Blc-κB- and Elc-κB-binding activities. WT and IKKα-defective MEFs (A) and BMDC (B) were left unstimulated or stimulated with either TNFα or anti-LTβR for the indicated times. Nuclear extracts were prepared and incubated with 32P-labeled probes corresponding to the consensus κB site (NF-κB) or the Blc-κB and Elc-κB sites. DNA-binding activity was analyzed by EMSA. NF-1 DNA-binding activity was measured as an internal control. (C) Functional analysis of the different κB sites in the Blc and Elc promoters. Triple repeats of the consensus κB (conκB), Blc-κB and a mutant Blc-κB (mBlc-κB) sites were cloned upstream to a minimal SV40 promoter (pGL3-Promoter vector, Promega). In addition, the Blc (+12 to −688) and Elc (+530 to −320) promoter regions were cloned upstream to a luciferase reporter (pGL3-Basic vector, Promega). To determine the importance of the Blc-κB site, it was converted by site-directed mutagenesis either to an inactive mutant version (mκB) or the consensus κB (conκB) site. The different plasmids were transfected into WT and Ikkα−/− MEFs. After 6 h with TNFα or anti-LTβR, luciferase activity was determined. The results are averages±s.d. of three independent experiments normalized to β-galactosidase activity, produced by a cotransfected β-galactosidase expression vector. (D) Alignment of novel κB sites from the control regions of IKKα-dependent genes. The novel κB sites from the Blc, Elc and Sdf-1 5′ regulatory region were aligned with those identified by computer analysis in the regulatory regions of three other IKKα-dependent genes. These sites form a consensus sequence (Alt. consensus) that, although similar, is distinct from the one associated with the classical NF-κB pathway (Class. consensus). (E) Induction of Baff, Rxra and Irf3 is IKKα-dependent. Expression of the indicated mRNAs was analyzed by real-time PCR as described above, using RNA isolated from nonstimulated and anti-LTβR-stimulated stromal cells (Rxra and Irf3) and BMDCs (Baff) of the indicated genotypes.
To further examine the relevance of the RelB:p52 selective binding site, we conducted a pattern search with two strings, namely AGGAGATTTG (Elc-κB) and GGGAGATTTG (Blc-κB), using the Trafac server and the BlastZ algorithm (http://bio.cse.psu.edu). Closely similar (at least 8/10 identity) sites were detected within 5 kb upstream to the start sites of the Sdf-1 and Baff genes, whose expression is known to be Ikkα-dependent (Dejardin et al, 2002) (Figures 1D, 2A and D; data not shown). We also detected similar and evolutionary conserved sites with the same region of several other genes, whose IKKα dependence was previously unknown (Figure 5D). RT–PCR analysis revealed that two of these genes, Rxra and Irf3, coding for important transcription factors, were induced in stromal cells in response to anti-LTβR in a manner dependent on IKKα (Figure 5E).
Discussion
Two distinct pathways leading to selective activation of RelA:p50 and RelB:p52 dimers, dependent on IKKβ or IKKα, respectively, were identified (Ghosh and Karin, 2002). Each pathway has distinct biological functions (Li et al, 1999; Senftleben et al, 2001; Chen et al, 2003), that could be mediated in part through selective gene activation (Dejardin et al, 2002). How this occurs was previously unknown. We now show in two different cell types, splenic stromal cells and BMDC, that IKKα is required for induction of four genes encoding chemokines critical for spleen organogenesis and maintenance of tissue microarchitecture, because these genes are selectively recognized by RelB-containing dimers, most likely RelB:p52. These genes are preferentially activated by engagement of LTβR and are only weakly induced by TNFα. Whereas the TNFα response is IKKα-independent, the response to LTβR engagement is strictly IKKα-dependent. The latter requires two events. First, RelB:p52 dimers have to enter the nucleus, a process dependent on IKKα-mediated p100 processing (Dejardin et al, 2002; Yilmaz et al, 2003). Second, RelB:p52 dimers are selectively recruited to the IKKα-dependent gene promoter. The selective recruitment of RelB to the Blc and the Elc promoters is likely to depend on a novel κB site, whose consensus sequence (Figure 5D) is distinct from that of the classical κB site. Unlike the classical site, the novel site is preferentially recognized by RelB:p52 dimers. This unique sequence specificity is entirely consistent with sequence differences between the DNA-binding loops of RelA and RelB, but was previously unknown (Ghosh et al, 1995). It is entirely possible, however, that additional factors may contribute to selective IKKα-dependent gene activation and that IKKα may also be responsible in certain cell types for activation of the canonical NF-κB pathway (Cao et al, 2001) or for potentiating its ability to activate transcription (Anest et al, 2003; Israel, 2003; Yamamoto et al, 2003). Nonetheless, an important mechanism responsible for selective gene activation through the IKKα-dependent alternative NF-κB signaling pathway is based on specific recruitment of RelB:p52 dimers to target gene promoters. Sites similar to the RelB:p52 selective κB site were detected in the 5′ regulatory region of three other genes, whose expression was found to be IKKα-dependent (Figure 5D).
What is the purpose of the functional separation between the two NF-κB signaling pathways? The IKKα-dependent organogenic chemokines optimize adaptive immunity through proper organization of secondary lymphoid organs. By contrast, IKKβ is mostly involved in inflammatory and innate immune responses. Thus, IKKβ-mediated NF-κB signaling is responsible for rapid responses to infection and injury, which require recruitment of immune cells out of lymphoid organs to sites of infection. This response depends on pro-inflammatory chemokines, such as MIP-1, MCP-1 and RANTES, which are induced by the canonical NF-κB signaling pathway (Alcamo et al, 2001). The arrival of antigens to secondary lymphoid tissues from distal sites of infection and their processing, presentation and recognition require coordinated activity of DC, macrophages, T cells and B cells, whose recruitment to secondary lymphoid organs depends on IKKα-regulated organogenic chemokines. Premature expression of such chemokines would compromise the immediate antimicrobial response as it may abort the emigration of immune cells to the periphery. It is, therefore, logical that expression of organogenic chemokines would not be induced through the canonical NF-κB signaling pathway. Consistent with its delayed function in adaptive immunity, activation of the alternative NF-κB signaling pathway is slower than the canonical NF-κB signaling pathway and seems to depend on prior activation of the latter (Dejardin et al, 2002). The dependence of the two pathways on distinct but related protein kinases and transcription factors allows for both functional integration and kinetic separation.
Materials and methods
Primary cell cultures
Stromal cell cultures were established from spleens of WT and IkkαAA/AA mice as described (Skibinski et al, 1998). Spleens were gently ground and released cells cultured in DMEM supplemented with heat-inactivated FCS (Invitrogen, Carlsbad, CA). After 1 week, nonadherent cells were removed, adherent cells were washed twice with PBS and cultured for one more week in DMEM/FCS. The absence of contaminating myeloid and lymphoid cells was verified by flow cytometry (FACSCalibur, Becton Dickinson). Stromal cells are uniformly positive for ICAM-1 (data not shown). BMDCs were cultured as described (Wu and Hwang, 2002).
Adoptive transfers
Bone marrow cells (3–4 × 106 cells per mouse) were isolated from femurs of WT or IkkαAA/AA mice and injected intravenously into lethally irradiated recipients. Mice were H-2 matched and, in the case of IkkαAA/AA, were from the F3–F5 backcross to C57Bl/6. Mice were provided antibiotics in drinking water and killed 6–8 weeks post reconstitution. When indicated, mice were immunized i.p. with SRBC (Colorado Serum Company, Denver, CO) 7 days prior to killing (Poljak et al, 1999).
Immunohistochemical analysis
Cryosections (8–10 μM) of spleen were prepared, dried and fixed with acetone before immunohistochemical analysis (Poljak et al, 1999; Weih et al, 2001). Staining reagents were: FDC-M2 (ImmunoKontact, UK), ICAM-1 (Santa-Cruz Biologicals, CA), B220, and CD35-bio (clone 8C12) (all from BD Pharmingen). Immunecomplexes were detected using species-specific secondary reagents. Sections were viewed by immunofluorescence microscopy (HM505E Microm Inc, Walldorf, Germany) and images captured with a digital camera (Nikon E800 Scope with Spot Diagnostics Digital Camera, AG Heinze Inc., Lake Forest, CA).
Electrophoretic mobility shift assay and immunoblots
Nuclear and cytoplasmic extracts were prepared and analyzed for levels of NF-κB subunits and DNA-binding activity (Bonizzi et al, 1999; Senftleben et al, 2001). Recombinant NF-κB subunits (not full-length proteins) were produced in Escherichia coli and purified as described (Chen et al, 1999). All antibodies and immunoblotting procedures were described (Senftleben et al, 2001).
Real-time PCR analysis and ChIP assay
Real-time PCR was performed using a PE Biosystems 5700 thermocycler following the SyBr Green™ protocol. Briefly, 12 ng of total cDNA, 50 nM of each primer and 1 × SyBr Green™ mix were used in a total volume of 25 μl. All values were standardized to that of cyclophilin mRNA. Primer sequences are available upon request. ChIP assays were as described (Saccani and Natoli, 2002). Polyclonal antibodies to p65 (C-20), RelB (C-19) and Pol II (N-19) were from Santa Cruz. The sequences of the promoter-specific primers (Blc +12 to −688, Sdf-1 +22 to −678, Vcam-1 +30 to −640, Iκbα +20 to −340, Tnfα +20 to −545) and a detailed experimental protocol are available upon request.
Acknowledgments
We thank G Natoli for advice regarding ChIP experiments, M Delhase, L-C Hsu and A Hoffmann for discussions, C Ware and J Browning for generous gifts of agonistic anti-LTβR antibodies and many useful suggestions and discussions, T Kato for oligo synthesis. GB and YC were supported by postdoctoral fellowships from the Human Frontier Science Project (HFSP) and the State of California Breast Cancer Research Program, respectively. This work was supported by NIH grants to MK and to GG. RCR is an awardee of the Hellman Fellows Program for junior faculty. MK is the Frank and Else Schilling American Cancer Society Research Professor.
References
- Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, Doerschuk CM, Hynes RO, Baltimore D (2001) Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J Immunol 167: 1592–1600 [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IκB kinase-α in NF-κB-dependent gene expression. Nature 423: 659–663 [DOI] [PubMed] [Google Scholar]
- Ansel KM, Cyster JG (2001) Chemokines in lymphopoiesis and lymphoid organ development. Curr Opin Immunol 13: 172–179 [DOI] [PubMed] [Google Scholar]
- Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, Browning JL, Lipp M, Cyster JG (2000) A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 406: 309–314 [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Piette J, Schoonbroodt S, Greimers R, Havard L, Merville MP, Bours V (1999) Reactive oxygen intermediate-dependent NF-κB activation by interleukin-1β requires 5-lipoxygenase or NADPH oxidase activity. Mol Cell Biol 19: 1950–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JL, French LE (2002) Visualization of lymphotoxin-β and lymphotoxin-β receptor expression in mouse embryos. J Immunol 168: 5079–5087 [DOI] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves T, Greten F, Johnson R, Schmidt E, Karin M (2001) IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107: 763–775 [DOI] [PubMed] [Google Scholar]
- Chen FE, Kempiak S, Huang DB, Phelps C, Ghosh G (1999) Construction, expression, purification and functional analysis of recombinant NF-κB p50/p65 heterodimer. Protein Eng 12: 423–428 [DOI] [PubMed] [Google Scholar]
- Chen L-W, Egan L, Li Z-W, Greten FR, Kagnoff MF, Karin M (2003) The two faces of IKK and NF-κB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia–reperfusion. Nat Med 9: 575–581 [DOI] [PubMed] [Google Scholar]
- Claudio E, Brown K, Park S, Wang H, Siebenlist U (2002) BAFF-induced NEMO-independent processing of NF-κB2 in maturing B cells. Nat Immunol 3: 958–965 [DOI] [PubMed] [Google Scholar]
- Cyster JG (2003) Lymphoid organ development and cell migration. Immunol Rev 195: 5–14 [DOI] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li Z-W, Karin M, Ware CF, Green DR (2002) The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity 17: 525–535 [DOI] [PubMed] [Google Scholar]
- Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf ELM (1999) CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99: 23–33 [DOI] [PubMed] [Google Scholar]
- Fu YX, Chaplin DD (1999) Development and maturation of secondary lymphoid tissues. Annu Rev Immunol 17: 399–433 [DOI] [PubMed] [Google Scholar]
- Ghosh G, Vanduyne G, Ghosh S, Sigler PB (1995) Structure of NF-κB p50 homodimer bound to a IκB. Nature 373: 303–310 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell 109: S81–S96 [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Ann Rev Immunol 16: 225–260 [DOI] [PubMed] [Google Scholar]
- Israel A (2003) Signal transduction: a regulator branches out. Nature 423: 596–597 [DOI] [PubMed] [Google Scholar]
- Jegga AG, Sherwood SP, Carman JW, Pinsky AT, Phillips JL, Pestian JP, Aronow BJ (2002) Detection and visualization of compositionally similar cis-regulatory element clusters in orthologous and coordinately controlled genes. Genome Res 12: 1408–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, Akira S (2001) IκB kinase α is essential for mature B cell development and function. J Exp Med 193: 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18: 621–663 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, Grewal IS, Cochran AG, Gordon NC, Yin J, Starovasnik MA, Dixit VM (2002) BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity 17: 515–524 [DOI] [PubMed] [Google Scholar]
- Kim CH, Broxmeyer HE (1999) Chemokines: signal lamps for trafficking of T and B cells for development and effector function. J Leukoc Biol 65: 6–15 [DOI] [PubMed] [Google Scholar]
- Li Q, Van Antwerp D, Mercurio F, Lee K-F, Verma IM (1999) Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 284: 321–325 [DOI] [PubMed] [Google Scholar]
- Poljak L, Carlson L, Cunningham K, Kosco-Vilbois MH, Siebenlist U (1999) Distinct activities of p52/NF-κB required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J Immunol 163: 6581–6588 [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D (2002) Two pathways to NF-κB. Mol Cell 10: 693–695 [DOI] [PubMed] [Google Scholar]
- Rothwarf DM, Karin M (1999) The NF-κB activation pathway: a paradigm in information transfer from membrane to nucleus. Science's STKE, www.stke.org/cgi/content/fullOC_sig trans; 1999/5/re1 [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G (2002) Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 16: 2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G (2001) Two waves of NF-κB recruitment to target promoters. J Exp Med 193: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten F, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun S-C, Karin M (2001) Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 293: 1495–1499 [DOI] [PubMed] [Google Scholar]
- Skibinski G, Skibinska A, Stewart GD, James K (1998) Enhancement of terminal B lymphocyte differentiation in vitro by fibroblast-like stromal cells from human spleen. Eur J Immunol 28: 3940–3948 [DOI] [PubMed] [Google Scholar]
- Weih DS, Yilmaz ZB, Weih F (2001) Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol 167: 1909–1919 [DOI] [PubMed] [Google Scholar]
- Wu MT, Hwang ST (2002) CXCR5-transduced bone marrow-derived dendritic cells traffic to B cell zones of lymph nodes and modify antigen-specific immune responses. J Immunol 168: 5096–5102 [DOI] [PubMed] [Google Scholar]
- Xia Y, Chen S, Wang Y, Mackman N, Ku G, Lo D, Feng L (1999) RelB modulation of IκBα stability as a mechanism of transcription suppression of interleukin-1alpha (IL-1alpha), IL-1beta, and TNFα in fibroblasts. Mol Cell Biol 19: 7688–7696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC (2001) NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell 7: 401–409 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Udit N, Verma I, Prajapati S, Kwak Y-T, Gaynor RB (2003) A nucleosomal role of IKKα is critical for cytokine-induced activation of NF-κB regulated genes. Nature 423: 655–659 [DOI] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F (2003) RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J 22: 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]