

Abstract
Current standard of care for patients with pediatric acute lymphoblastic leukemia (ALL) is mainly effective, with high remission rates after treatment. However, the genetic perturbations that give rise to this disease remain largely undefined, limiting the ability to address resistant tumors or develop less toxic targeted therapies. Here we report the use of next generation sequencing to interrogate the genetic and pathogenic mechanisms of 240 pediatric ALL cases with their matched remission samples. Commonly mutated genes fell into several categories, including RAS/receptor tyrosine kinases, epigenetic regulators, transcription factors involved in lineage commitment and the p53/cell cycle pathway. Unique recurrent mutational hotspots were observed in epigenetic regulators CREBBP (R1446C/H), WHSC1 (E1099K) and the tyrosine kinase FLT3 (K663R, N676K). The mutant WHSC1 was established as a gain-of-function oncogene, while the epigenetic regulator ARID1A and transcription factor CTCF were functionally identified as potential tumor suppressors. Analysis of 28 diagnosis/relapse trio patients plus 10 relapse cases revealed four evolutionary paths and uncovered the ordering of acquisition of mutations in these patients. This study provides a detailed mutational portrait of pediatric ALL and gives insights into the molecular pathogenesis of this disease.
Introduction
Pediatric ALL is the most common childhood tumor and the leading cause of childhood cancer deaths (1). Unlike adult ALL, most pediatric ALL patients respond well to conventional chemotherapy, and the 5 year overall survival rate can reach 85-90%. However, once the patient develops relapse disease, the outcome became less favorable (2,3). Cytogenetic alterations are frequent, and several molecular markers have been identified to predict prognosis (1): ALL patients with high hyperdiploid chromosomes are usually associated with a better survival, while those with low hypodiploid chromosomes usually have a worse prognosis (1). Nevertheless, a substantial percentage of “good prognosis” patients, based on the above classification, still experience relapse, highlighting the urgent need for a deeper understanding of the molecular features of these patients and identifying those who are at greater risk of relapse.
Recently, mutational profile of several subtypes of pediatric ALL has been interrogated with next generation sequencing (NGS) (4-7). NGS enables an improved understanding of the disease pathogenesis at the nucleotide level, which leads to the discovery of new therapeutic targets and enables a tailored therapeutic regimen for each patient. Herein, a comprehensive mutational profiling was carried out in a large cohort of pediatric ALL patients.
Materials and Methods
Patients’ samples
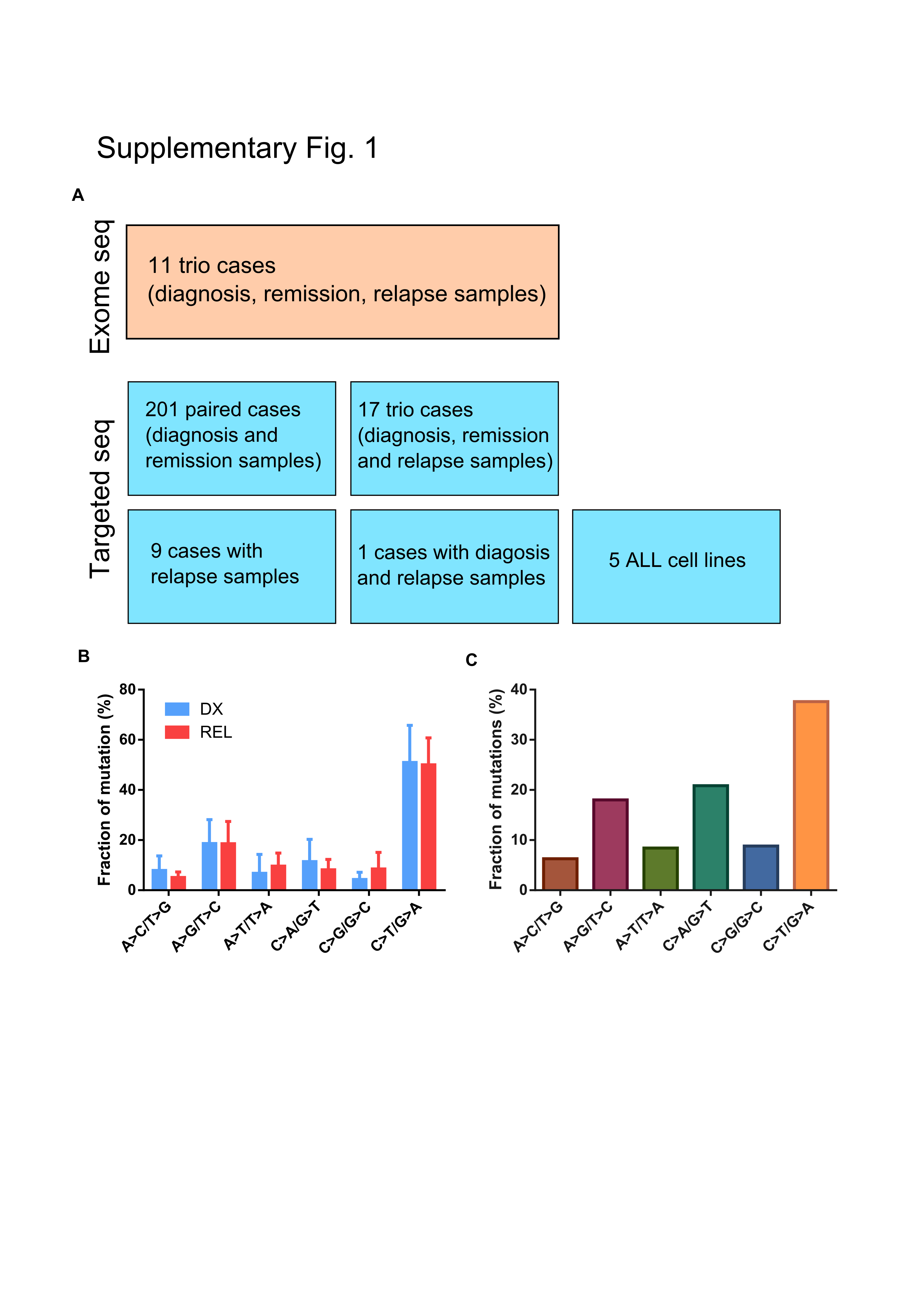
The patients’ samples were collected with informed consent. DNA of pediatric ALL patients (ages 1 to 18) used in this study includes (Supplementary Fig. 1A, Supplementary Tables 1 and 2): 9 complete cases of Asian samples collected from Singapore (including diagnosis, matched remission and relapse samples were cytogenetically analyzed and screened for the presence of either ETV6-RUNX1 or BCR-ABL as part of routine clinical diagnostics); 67 cases of Asian B-ALL patient samples were collected from Taiwan (including 5 complete cases with diagnosis, matched remission and relapse, 59 cases of diagnosis and matched remission, as well as an additional 3 cases with matched remission and relapse); 10 relapse ALL cases collected from MD Anderson Cancer Center. 154 Caucasian pediatric ALL patients samples DNA (with their matched remission control) previously interrogated with SNP-array (8,9) were amplified by whole genome amplification with high fidelity Phi29 (REPLI-g Mini Kit, Qiagen).
Exome/targeted sequencing and mutation analysis
Exome sequencing and targeted capture libraries (Supplementary Tables 3) were prepared according to recommendations in either the Agilent XT or XT2 kit. DNA was sheared to 250-300 bp fragments using the Covaris machine, end repaired and ligated with adapters. PCR was performed and hybridized with either the human exome or targeted region beads. Captured products were amplified by PCR and sequenced with Illumina Hiseq 2000. Illumina paired-end reads were aligned to NCBI build 37 using Novoalign, and somatic variants were called using Mutect (for single nucleotide variants, SNVs, LOD ≥ 6.3), VarScan (for SNVs and Indels, mutant supported by at least 6 reads, min-coverage ≥10, somatic-p-value ≤0.07, min-tumor-variant-freq ≥0.10, normal-variant-freq ≤0.05) and Pindel [for Indels, only indels that were less than 60bp long were retained (except for FLT3, all the candidate mutations of FLT3 gene were retained for further analysis of potential internal tandem duplication (ITD) event), mutant supported by at least 6 reads and at least a 5% VAF]. For FLT3-ITD detection, the Bam files were further analyzed using Pindel as described previously (10). All the candidate mutations were filtered with dbSNP131 [latest versions of the dbSNP database were not utilized as it contains some well characterized cancer mutations. For example, KRAS G12D mutation was annotated as SNP rs121913529 in dbSNP137 and dbSNP138 (10)]; allele >0.1% in 1000 genome sequencing project or ESP5400 database and repetitive and low complexity regions (genomic SuperDups track, UCSC Genome Browser). Variants that appeared more than once in our in-house panel of normal controls database of 200+ samples were also removed. The sciClone R package was used to analyze clonal evolution. Read counts of each SNV were retrieved using bam-readcount (https://github.com/genome/bam-readcount/) followed by VAF calculation. Mutational diagrams were generated using the proteinPainter (http://explorepcgp.org/proteinPainter).
Cell culture
ALL cell lines NALM-6, RS4;11, REH, SUP-B15 and SEM cells were obtained from DSMZ and authenticated by short tandem repeat (STR) DNA profiling in Dec 2014. Cells were cultured at low passage in RPMI 1640 medium with 10% fetal bovine serum (FBS) and 1% PenStrep. HEK293FT (for generating high-titer lentivirus, obtained from Invitrogen) was growth in DMEM medium with 10% FBS.
Methylcellulose colony formation assay
Methylcellulose colony assays were performed using MethoCult™ H4230 (STEMCELL Technologies). 1000 cells (Nalm6 and REH) or 3000 cells (RS4;11) were resuspended in 80 μl RPMI medium and 320 μl Methocult medium, and cultured in 24-well plates. After 7-14 days, colonies were stained with either gentian violet or MTT dye (1:10 diluted) and counted.
Western blot analysis
SDS-PAGE and Western blot analysis was performed according to standard protocol, using the following primary antibodies: WHSC1 (Santa Cruz, sc-365627), ARID1A (Millipore, 04-080), c-MYC (Cell Signaling, 9402S), CTCF (Santa Cruz, sc-271474) and GAPDH (Cell Signaling, 2118S).
shRNA and CRISPR-Cas9 sgRNA
shRNAs were generated using the lentiviral construct pLKO.1 according to the standard protocol described in the website http://www.addgene.org/tools/protocols/plko/. CRISPR-Cas9 sgRNAs were generated based on the pLENTI-CRISPRv2 according to the procedure described in the website http://www.addgene.org/crispr/zhang/. The shRNA and CRISPR-Cas9 sgRNA sequences are listed in Supplementary Table 4. Real-time PCR was performed to examine the knockdown effect of shRNA and the primers used for the experiment are listed in Supplementary Table 5. PCR and Sanger sequencing were performed to examine the indel induced by CRISPR-Cas9. The primers used in the PCR sequencing are listed in Supplementary Table 6.
Lentivirus infection and stable cell lines
Lentivirus particles were generated using the packaging vector as described previously (11). Plasmids were co-transfected into 293FT (293T cells optimized to generate high titer of lentivirus). Virus was collected 48 hours after transfection and passed through a 0.45 μm filter. B-ALL cells were infected with spin-down infection using 1,400 rpm for 2 hours, twice spaced by 24 hours. Stable cell lines were selected using puromycin (1μg/ml).
Xenograft experiments
Three million ALL cells were suspended in 100 μl RPMI medium and injected into both flanks of 6-8 weeks old NSG mice. The cell mass was harvested (21 days after the injection), photographed and the weight of each mass was recorded.
Luciferase assay
Dual luciferase assay was carried out using Dual-Luciferase® Reporter Assay System (Promega). Luciferase activities were measured 72 h after transfection of 293FT cells either with or without pLenti-ARID1A (a gift from Ie-Ming Shih, Addgene plasmid # 39478), together with a MYC activity reporter pMyc4ElbLuc (a gift from Roger Davis, Addgene plasmid # 53246). The luciferase activity was normalized with the co-transfected Renilla luciferase activity.
Results
Mutational landscape of pediatric-ALL
To gain a better understanding of the landscape of somatic mutations in ALL, we performed whole exome and targeted sequencing of 240 pediatric ALL patients (Supplementary Fig. 1A, Supplementary Tables 1 and 2). For discovery cohort, we selected 11 trios (diagnosis/complete-remission/relapse) cases who, based on traditional cytogenetic classification, should have responded well to conventional chemotherapy but unfortunately relapsed. These samples were interrogated for somatic alterations by whole exome sequencing. Mutational spectrum analysis revealed that C>T transitions were the dominant mutational event, which is likely caused by a spontaneous deamination process (Supplementary Fig. 1B). An increase of the mutational burden at relapse was noted in some patients. However, in contrast to the previous observation in AML which showed frequent transversions in relapse samples (12), relapse ALL had a similar mutational spectrum at diagnosis and relapse. This may be explained by the decreased DNA damage of the chemotherapeutic drugs administered for ALL (L-asparaginase, vincristine and prednisolone) compared with daunorubicin used in AML treatment.
We next extended our study to a larger patient cohort of 229 additional paired remission/diagnosis or relapse samples as well as 5 B-cell ALL cell lines. A total of 560 genes, including those found in the discovery cohort plus selected leukemia related genes (Supplementary Table 3) were examined. Similar to the discovery cohort, the mutational signature was again dominated by C>T transition events (Supplementary Fig. 1C). The most prominent altered genes among the entire cohort were involved in RAS-RTK signaling, epigenetic regulators, transcriptional factors and p53/cell cycle pathway (Fig. 1).
Figure 1. Mutational landscapes of 240 ALL patients.

Heatmap diagram showing the mutational landscape of ALL patients. Patients are labeled on the horizontal axis and mutant genes on the vertical axis.
RAS-RTK signaling
The most frequently mutated genes were members of RAS signaling. Besides the well know hotspot mutations [G12 (NRAS, n=21; KRAS, n=12), G13 (NRAS, n= 15; KRAS, n=11) and Q61 (NRAS, n=16; KRAS n=1)], novel mutational sites were also identified for KRAS: A146T/P (n=4), K117N/T (n=4) and V14I (n=1) (Fig. 2A). These non-canonical KRAS mutations often occur in colon cancer (13) (Supplementary Table 7), suggesting a similar oncogenic gain-of-function mutation. A novel in-frame insertion in the G13 of KRAS was noted in one patient. Remarkably, we found mutant KRAS/NRAS were not mutually exclusive in ALL. Fifteen patients harbored multiple RAS mutations in the same leukemic sample. For example, in patient GE145, NRAS G13D (mutated/wild-type reads: 49/228) and KRAS G12D (mutated/wild-type reads: 11/61) were detected at diagnosis; however, at relapse, only the subclone harboring the NRAS G13D (mutated/wild-type reads: 121/120) emerged as the dominant clone (Fig. 2B). For patients having multiple RAS mutations in the same RAS gene and the mutations were close enough to be covered by the same sequencing read pair (i.e. KRAS G12D/G13D), phasing analysis was performed. The results indicated that all of these mutations occurred in a mutually exclusive manner (Figs. 2C, D), reflecting a parallel branching hierarchy instead of a linear acquisition path of these mutations in different subclones (intratumor heterogeneity (14-16)). In addition, loss-of-function mutations of the RAS signaling negative regulator (NF1) occurred in 7 patients (1 stop-gains and 3 frame-shift mutations, Fig. 2A).
Figure 2. Recurrent mutations in RAS-RTK pathway.

A, schematic of the mutations in NRAS, KRAS and NF1. B, comparison of mutational allele frequency (VAF) of two cases harboring different RAS mutations at diagnosis and relapse. C, mutational heatmap of the RAS-RTK pathway genes. D, mutation phasing analysis depicting the mutually-exclusive pattern of RAS mutations in the leukemic cells of the same patient, four representative cases are shown. Sequencing reads are displayed at each locus as a gray bar. Mutations found in the reads (nucleotide differs from the reference sequence) are indicated and highlighted. E, F, schematic diagrams showing mutations of PTPN11 and FLT3, respectively.
PTPN11 encodes a phosphatase that modulates signaling from upstream receptor tyrosine kinases and the RAS genes. High frequency missense mutations clustered in two small regions of the gene, the first cluster located in the SH2 domain included the canonical hotspot A72T/V (6 cases) and E76K/V (6 cases); the second cluster was located in the tyrosine-phosphatase catalytic domain (G503R/V, Fig. 2E). Most of these mutations have been recurrently found in a variety of other cancers (Supplementary Table 8), and the hotspot mutational pattern suggests a potential gain-of-function of these mutations.
FLT3 codes for a cell surface tyrosine kinase receptor which plays a key role in hematopoietic cell growth and survival. The well-appreciated activating hotspot mutations in the kinase domain (D835Y/Y842C) and several novel recurrent mutationswere identified (Fig. 2F). For example, we found FLT3 N676K in two patients, this mutation was recently shown to be a gain-of-function lesion by demonstrating that the mutant protein confers growth of BaF3 cells in growth factor depleted media (17). In addition, missense mutations were also identified in several unreported/unstudied positions, including region either close or within the juxtamembrane (JXM) domain. This region may be involved in the regulation of FLT3 dimerization and self-activation [including K663R (3 cases), V592D (2 cases) and V491L (2 cases); these same mutations were noted in COSMIC/TCGA databases (Supplementary Table 9), implicating a potential oncogenic gain-of-function]. Five patients have in-frame indel of FLT3 gene (two of them occurred in exon 14 within the JXM domain, and three are occurred in exon 20 within the kinase domain, Fig. 2F). No FLT3-ITD mutations were detected in the entire cohort.
Mutation of other cytokine receptors included PDGFRA (3 cases), KIT (3 case), IL7R (2 cases) and CSF3R (2 cases). Downstream of these kinase receptors are the Janus kinase family members which are also mutated: JAK1 (2 cases), JAK2 (2 cases) and JAK3 (1 case). A previously reported hotspot, R683S of JAK2 which occurred in 18-28% of Down-syndrome ALL (18) as well as high risk pediatric ALL (19), was found in one patient. In addition, CBL and CBLB, two negative regulators of endocytosis and degradation of RTK were mutated in four samples.
Epigenetic regulators
Members of the histone methyltransferase MLL family were frequently mutated. MLL (KMT2A) is a well-recognized leukemia related gene, which has been extensively characterized in hematological malignancy. The role of the other MLL family members is not fully established, although exome sequencing studies recently found a large number of mutations occurring in the MLL2 (KMT2D) and MLL3 (KMT2C) genes in a variety of cancers, including pediatric leukemia (20) and more than 80% follicular lymphoma (21). Recurrent mutations of MLL, MLL2, MLL3 genes were noted in our cohort. Among them, MLL2 displayed a higher ratio of inactivating mutation (6 stop-gain and 3 frameshift indel mutations, Fig. 1), suggesting a loss-of-function in a potential tumor suppressor.
Conspicuously, hotspot mutations were identified in histone H3K36 methyltransferase WHSC1. Mutation E1099K located in the SET domain, was identified in 11 patients as well as two of the 5 ALL cell lines that we sequenced (RS4;11, SEM) (Figs. 3A, B). Data mining of the literature as well as CCLE/TCGA database further revealed that another 4 ALL cell lines, one multiple myeloma cell line, mantle cell lymphoma (22), other lymphoid malignancies (23,24) as well as a few colorectal, thyroid, lung and ovarian cancer samples harbored the same mutation, suggesting that this gain-of-function WHSC1 mutation occurred predominantly in B-cell malignancies, but also appear at a lower frequency in solid tumors (Supplementary Table 10). In multiple myeloma, frequent translocation of the WHSC1 [t(4:14)] downstream to the IgG promoter often leads to aberrantly high expression of WHSC1 which contribute to the transformation process (25). In line with these findings, elevated WHSC1 mRNA was found in B-ALL samples and cell lines compare to normal B-cells (Fig. 3C). In addition, in silico analysis of two independent ALL cohorts consistently showed that high expression of WHSC1 was correlated with a higher risk of relapse (data retrieved from GSE11877, Children's Oncology Group Study 9906 for 207 high risk pediatric B-precursor ALL (26,27), Fig. 3D) and shorter remission period (data retrieved from GSE4698, 60 relapse pediatric ALL (28), Fig. 3E). Furthermore, stable silencing of E1099K mutant WHSC1 in RS4;11 cells by either lentiviral shRNA or CRISPR-Cas9 guide RNA (sgRNA) (Fig. 3F) markedly reduced clonogenic growth both in vitro (Figs. 3G, H) and in vivo (Figs. 3I, J). Taken together, these findings underscore the critical role of WHSC1 in lymphoid malignancies, highlighting the need for effective inhibitors of this enzyme.
Figure 3. WHSC1 is a unique oncogene in B-ALL.

A, mutational diagram illustrating the mutations of WHSC1. B, representative Sanger sequence tracings that show somatic E1099K mutation in 4 patients (upper and middle panel, diagnosis and the matched remission sample, respectively) and 2 ALL cell lines (RS4;11, SEM, lower panel). C, elevation of WHSC1 mRNA in B-ALL samples or B-ALL cell lines compared with normal B-cells (data were retrieved from GSE48558). ****, p<0.0001. D, patients whose ALL cells at diagnosis had higher levels of WHSC1, also had a higher likelihood to undergo relapse (GSE11877, 207 Pediatric ALL. Censored: Patients in clinical remission, or with a second malignancy, or with a toxic death as a first event were censored at the date of last contact). **, p<0.01. E, aberrant WHSC1 expression was associated with high risk leukemia and early occurrence of relapse: very early relapse (within 18 months after initial diagnosis); early relapse (>18 months after initial diagnosis but <6 months after cessation of frontline treatment); late relapse (>6 months after cessation of frontline treatment). Data were retrieved from GSE4698 (60 childhood ALL (28)). **, p<0.01. F, western blot shows the silencing effect of CRISPR-Cas9 sgRNA targeting WHSC1. G, H, silencing of WHSC1 by either shRNA or CRISPR-Cas9 guide RNAs markedly reduced the clonogenic growth of B-ALL cells RS4;11 (cell line carrying WHSC1 E1099K mutation) in methylcellulose assay. Non-target, Non-target shRNA Control. EV, empty vector control. **, p<0.01, ***, p<0.001. I, J, silencing of WHSC1 impairs in vivo cell growth of RS4;11. *, p<0.05.
Two highly related histone/non-histone acetyltransferases, CREBBP and EP300, were also prominently mutated in our cohort. Mutations of CREBBP predominantly occurred in the acetyltransferase domain, particularly in the hotspot R1446C/H (Figs. 4A, B). Remarkably, the same hotspot was previously found in pediatric ALL, as well as a variety of other cancers including melanoma, bladder, liver and lung cancers (Supplementary Table 11, Figs. 4C). Despite the hotspot mutation usually associated with gain-of-function, recent studies in B-cell lymphoma (29) and relapsed ALL (30,31) revealed a tumor suppressor role of both genes, implying that this hotspot mutation may act in a dominant-negative fashion. Congruent with this notion, frequent deletions of CREBBP locus were observed (Fig. 4D). ALL cases with lower expression of CREBBP in their leukemic cells at diagnosis have a significantly shorter remission period (data retrieved from GSE18497, which includes 41 cases of diagnosis and relapse matched ALL (32), Fig. 4E), an inferior overall survival (data retrieved from GSE11877, Fig. 4F), and associated with higher number of peripheral white blood cell count (WBC) at diagnosis (data retrieved from GSE11877, Fig. 4G). Further evidence to support the notion that CREBBP may behave as a tumor suppressor arises from the study of Rubinstein-Taybi syndrome that frequently display mutations of the CREBBP gene (33). These individuals have an increased risk of developing cancers including leukemia. Notably, although an analogous hotspot (D1399N/Y, Supplementary Table 12) mutation was found in EP300 by our in silico analysis (corresponded to the R1446C/H of CREBBP), only scatter mutations occurred at other sites in our ALL cohort (Fig. 4D).
Figure 4. CREBBP and EP300 are frequently mutated in ALL.

A, Sanger sequencing chromatograms showing the hotspot R1446 mutation of CREBBP (arrowhead). B, Diagram showing the sites of mutations in CREBBP and EP300. C, TCGA pan cancer sequencing analysis identified mutational hotspot in the HAT domain of CREBBP and EP300. D, CREBBP loci is frequently deleted in ALL patients. Each blue color bar indicates a deletion event in one individual. Two cases with focal deletion of CREBBP were highlighted with a red rectangle. Data were retrieved from Tumorscape ALL patients SNP-Array database. E, The lower CREBBP expression in the leukemic cells at diagnosis is associated with early relapse in the relapse ALL cohort GSE18497. The patients are separated into “high” (upper 50%, n=20) and “low” (lower 50%, n=21) groups based on the CREBBP expression of their leukemic cells at diagnosis. F, Kaplan-Meier plot of overall survival: comparison of cases with high versus low expression of CREBBP in 207 ALL patients (GSE11877). “High” and “low” ALL cohorts are defined as upper and lower 50% of expression of CREBBP in their diagnosis leukemic cells. P value was calculated by log-rank test. G, Box plots for peripheral white blood cell counts (WBC) in individuals whose leukemic cells at diagnosis expressed either higher (upper 50%) or lower (lower 50%) level of CREBBP. **, p<0.01.
Mutations of chromatin remodeling genes (SWI/SNF complex) have recently been identified in a number of cancers, particularly the ARID family member ARID1A and ARID2. Consistent with their tumor suppressor role, somatic mutations (Fig. 5A) and copy number deletions of both genes were found (Fig. 5B). Silencing of ARID1A in ALL cell lines by lentiviral shRNA result in upregulation of the pro-growth regulator c-MYC (Fig. 5C), while force expression of ARID1A reduced c-MYC luciferase reporter activity (Fig. 5D). This observation suggests that ARID1A may be involved in the c-MYC pathway and it modulated the ALL cell proliferation. Congruently, silencing of ARID1A by either shRNA or CRISPR-Cas9 sgRNA resulted in enhanced clonogenic growth in methylcellulose colony forming assay (Figs. 5E, F).
Figure 5. Mutation of chromatin remodeling (SWI/SNF complex) genes.

A, diagrams depict the mutations of ARID1A, ARID2, SMARAC4 and ATRX identified in this study. B, deletion of ARID1A (upper panel) or ARID2 (lower panel) in ALL samples. Each blue bar indicates a deletional event in an ALL patient. Data were retrieved from Tumorscape SNP-array database. C, western blots show shRNA silencing of ARID1A in RS4;11, Nalm6 and REH. Ctrl, Non-target shRNA control. D, luciferase assay stimulated by c-MYC activity in 293FT cells (control) versus forced expression of ARID1A (pLenti-ARID1A, see Supplementary Materials and Methods). Mean ± SD, n=3. *, p<0.05. E, F, silencing ARID1A enhances in vitro clonogenic growth of Nalm6 and RS4;11 in methylcellulose assays. Mean ± SD, n=3. **, p<0.01, ***, p<0.001.
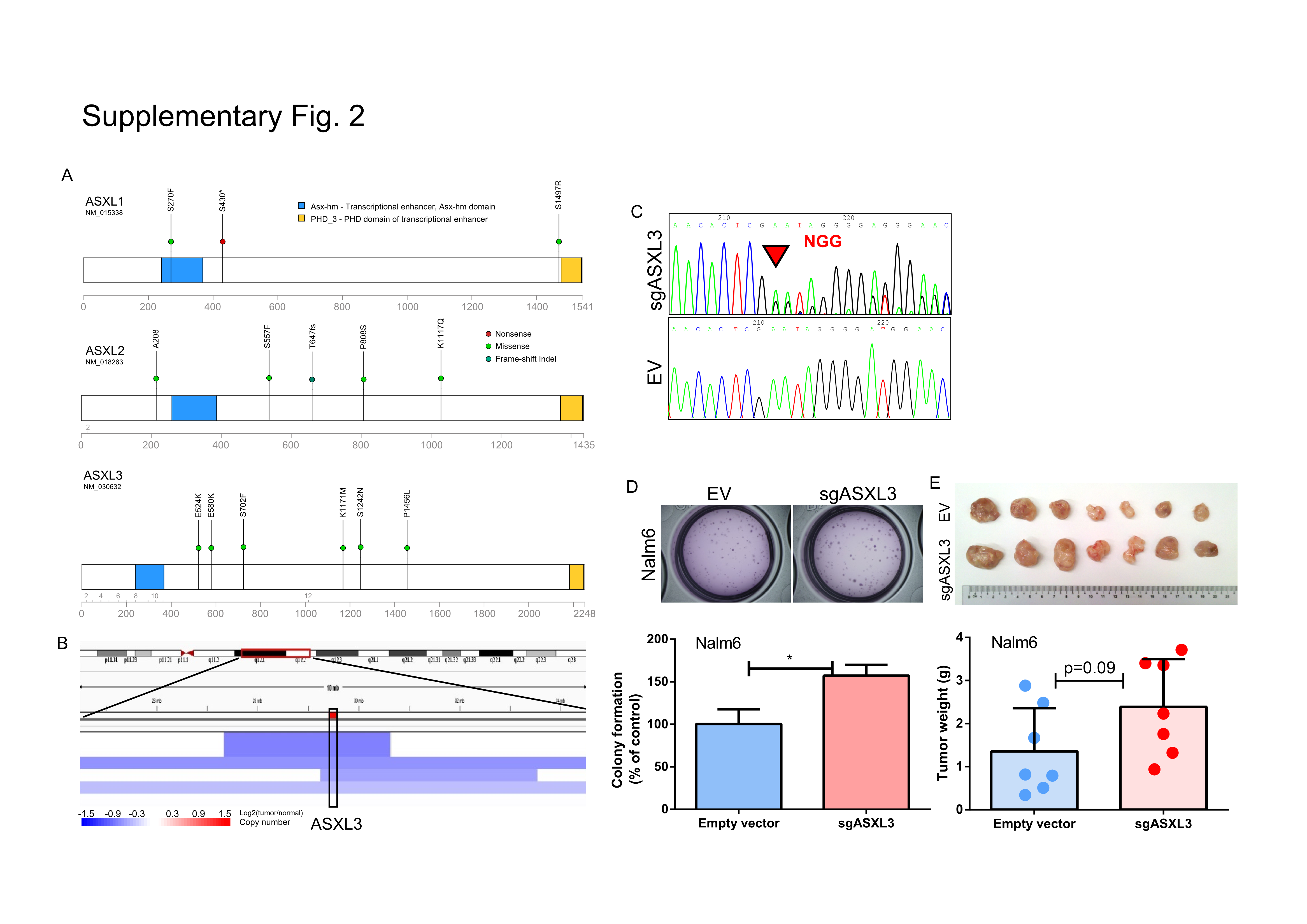
Besides the well-known leukemia gene ASXL1, mutations were also found in the two less studied family members (ASXL2, ASXL3, Supplementary Fig. 2). Mutations of epigenetic regulators were also found in the polycomb complex (EZH2, EED, SUZ12), chromatin/nucleosome structure modifying proteins (CHD2, CHD3, CHD4), TET family proteins [TET1 (4 cases), TET2 (5 cases)] and histone modification proteins (HDAC1, SIRT1, BCOR, BRD8, lysine demethylase PHF2/KDM6A and histone acetyltransferase KAT6B).
Transcription factors and TP53/cell cycle pathway
A number of alterations of transcription factors essential for hematopoietic and lymphoid differentiation were noted, including the lineage regulator PAX5 (3 missense, 3 indels) and ETV6 (7 cases, 5 were frameshift indel and 1 was a splice site mutations). Frequent deletions of PAX5 and ETV6 loci have been previously reported in ALL (8,34,35). In addition, mutations were also found in other lineage transcription factors (IKZF2, IKZF3, EBF1), WT1, RUNX family member [RUNX2 (n=4), RUNX1 (n=1)], ERG1 (n=3) and GATA1/3 (1 case each).
CTCF is a transcription factor involved in many cellular processes, including transcriptional regulation, insulator activity, and regulation of chromatin architecture. Forced expression of CTCF induces growth arrest of B-cells (36). Mutations of CTCF often occur in the conserved R residue of the zinc finger domain, or lead to a functional inactivation (2 frame-shift, 2 stop-gain and 1 splice-site mutation, Supplementary Fig. 3A). Focal deletions of the CTCF locus were observed in pediatric ALL (Supplementary Fig. 3B, data retrieved from Tumorscape, Q value<0.0001). In addition, expression levels of CTCF transcript correlated remarkably with patient prognosis: significantly lower CTCF mRNA was found in the ALL samples of patients with positive MRD and inferior survival outcome (data retrieved from GSE11877, Children's Oncology Group Study 9906 for 207 high-risk pediatric B-precursor ALL (26,27), Supplementary Figs. 3C, D), as well as higher number of peripheral WBC in their diagnosis specimens (Supplementary Fig. 3E). Consistent with its potential tumor suppressor role, stable silencing of CTCF by CRISPR-Cas9 sgRNA in RS4;11 (Supplementary Figs. 3F, G) resulted in increased clonogenic growth (Supplementary Figs. 3H, I).
Somatic mutations of genes involved in the p53 pathway occurred in 18 patients, including TP53, ATM and the kinases that regulate p53 activities (HIPK1, HIPK2). Germline TP53 pathogenic variants were found in 2 patients (R248Q and R278H, Supplementary Fig. 4). The B-cell transcription factor BACH2 (suppressing B-cell transformation via activation of p53) was also mutated in 5 samples. Moreover, a stop-gain as well as a frameshift mutation were found in CDKN2A (the most frequently deleted gene in ALL (8,34)).
NF-κB pathway and splicing factors
Mutations were acquired in genes involved in the NF-κB pathway, including CARD8/CARD11, LRBA, TLR5 and NLRP3/NLRP8. Genes involved in TLR-MYD88-NF-κB pathway are usually activated during pathogen invasion causing an expansion of antibody producing B-cells. Frequent mutation of this pathway has been noted in multiple myeloma and B-cell lymphomas (37). ALL cells may hijack this pathway to gain a growth advantage.
Splicing factors are emerging as a novel class of cancer genes (38) especially in myelodysplastic syndrome (MDS) and chronic lymphocytic leukemia (CLL). Several splicing factors were mutated at a low frequency in our samples, including ZRSR2, SF3B1, SF3B2 and SF3B3. Mutation of SF3B1 at K666N and R625H are recurrent hotspots in MDS (38) and CLL; they were detected in two patients (Supplementary Table 13). Overall, these data suggest that disruption of the RNA splicing machinery contributes to ALL pathogenesis in only a small fraction of patients.
Clonal evolution and relapse
Analysis of the mutations shared between diagnosis and relapse of 28 Trios, as well as 10 relapse-ALL suggest four different clonal evolutionary models (3,14): relapse evolves directly from the dominant clone at diagnosis (Fig. 6A I); relapse evolves from a minor subclone at diagnosis (Fig. 6A II); relapse evolves from ancestral clones prior to the dominant diagnosis clone (Fig. 6A III); or development of a genetically distinct leukemia (Fig. 6A IV). Only one patient followed either models I or IV, most of the cases corresponded to models II or III, indicating that either the malignant or pre-malignant cells survived chemotherapy resulting in relapse (3,14).
Figure 6. Clonal architecture and clonal evolution of ALL.

A, different clonal evolutionary paths inferred from the sequencing data. HSC, Hematopoietic stem cells. B, four representative cases showing the evolution of relapse (REL) ALL from a subclone at diagnosis (DX). C, clonal architecture and evolution of ALL patient SG007. Variant allele frequency (VAF) of mutations of ALL at diagnosis and relapse (upper left panel). Mean VAF mutational clusters (C1, C2, C3, C4) in the primary and relapse ALL samples (upper right panel). Lower panel: schematic diagram illustrating the clonal evolution of ALL cells of patient SG007.
Further scrutiny of the genes that are concordantly mutated at diagnosis and relapse revealed recurrent mutation of three genes (CREBBP, NRAS and PTPN11) in more than one patient. Mutation of CREBBP was found in three trio cases (mutations present at both diagnosis/relapse) and in a relapse sample. Previous studies suggested that CREBBP modulates expression of the glucocorticoid responsive gene (30) and regulates glucocorticoid resistance (3). NRAS and PTPN11 are often present as subclonal drivers (VAF 0.2-0.3); and each are present at diagnosis/relapse specimens of two patients. These mutations carrying clones survived chemotherapy and became the dominant clone at relapse (VAF 0.4-0.5) (Fig. 6B). However, how important are the contribution of mutation of these two genes (NRAS and PTPN11) to ALL clonal survival during chemotherapy remains an open question as we also noted that many mutant NRAS carrying clones were eliminated by chemotherapy and disappeared at relapse.
To understand better the clonal hierarchy and evolutionary process, VAF analysis was performed to trace the potential clonal evolution of each trio samples. One representative clonal path (model II) is shown in Figs. 6C. In this patient, mutations were grouped into three clonal clusters at diagnosis and two clonal clusters at relapse. Chemotherapy eradicated two subclones, which had been present at diagnosis, but not the founder clone; the latter survived, gain mutations and reemerged at relapse.
Discussion
Relapse of pediatric ALL is usually associated with high rates of treatment failure and less favorable outcome (2,3). Recently, several genetic defects have been linked to chemotherapy resistance in relapse ALL, including CREBBP (3,30), NT5C2 (39,40) and PRPS1 (41). In line with these findings, we observed CREBBP (4 cases) and NT5C2 (1 case) mutations in our relapse ALL samples, suggesting that an alternative chemotherapy regimen with different mechanism of action should be adapted for these mutants carrying patients. On the other hand, we identified mutations of genes involved in DNA repair pathway in 8 relapse samples (21%, 8 out of 38 relapse samples, Supplementary Table 14). Mutation of genes involved in either DNA repair or maintenance of genomic stability (i.e., TP53, POLE, MSH6 (2) or SETD2 (2,42)) may result in an elevated mutation rate and enhanced clonal diversification, while extensively diversified subclones may behave as a reservoir facilitating selection of a new adaptive (i.e. drug resistant) clone (43). Nevertheless, we also noticed a few patients with high mutational burden at diagnosis who did not develop a relapse ALL. The possible involvement of DNA repair pathways in disease relapse thus still requires further study.
As an oligoclonal disease, leukemia is generally considered to arise from a single hematopoietic stem cell which then subsequently acquires a series of mutations in a stepwise manner. Models of development from preleukemia to overt-leukemia have been established in ETV6-RUNX1 and hyperdiploid-ALL by comparing leukemia that occurred in monozygotic twins, or backtracking studies of archived neonatal blood samples (44,45). In this model, the initiating ETV6-RUNX1 fusion or hyperdiploidy abnormality was often acquired in utero to generate a preleukemic founder clone, the latter subsequently acquired a series of CNAs/SNVs, and eventually transformed to overt/frank ALL. This model has been supported by recent single cell genotyping based on copy-number variants/FISH (16) or NGS (4). Both approaches showed that the majority of SNVs are acquired after the structural chromosomal change. A similar conclusion was reached by analyzing the sequencing data of our hyperdiploid-ALL patients, as the distribution pattern of the allele burden indicated that the majority of the mutations (located in the gained chromosomes) was acquired after the hyperdiploidy event. In addition, comparing the diagnosis/relapse mutational profile of the same patients adds further evidence to support this model, as hyperdiploidy always persists in ALL relapse patients who had hyperdiploidy at diagnosis, indicating that the hyperdiploidy occurred as an early initiating event.
Recently, several groups of researchers, including ourselves, uncovered the persistence of preleukemic clone at remission in adult AML, by showing the retention of canonical AML mutations with high VAF in remission samples (10,46-48). However, this scenario seems unlikely to be the case in pediatric ALL as our NGS data suggest that none of the somatic mutations persisted in matched remission samples (except for 2 cases with germline TP53 mutations and one case with a CREBBP stop-gain mutation that persisted in remission, which is likely also a germline mutation). Considering that 85-90% of pediatric ALL are cured, this observation probably reflects the eradication of the ALL cells in the majority of patients. To reinforce this concept, we focused on patients who relapsed (which is indicative of the survival of leukemia cells or its ancestral clone). Integrative analysis of copy-number alterations and sequencing data revealed no detectable abnormalities in the remission samples, suggesting that the surviving leukemic ALL clone is present in a very small fraction of the hematopoietic compartment at remission, which is below the detectable threshold of either NGS or SNP-array. This observation is in accordance with an earlier clonal study which applied X-chromosome skewing pattern analysis, and consistently showed the absence of abnormal clonal hematopoiesis at remission in ALL patients (49,50). Collectively, identification of the residue leukemic clone and targeting it at remission may help prevent relapse.
One of the striking observations of this study is the prevalence of non-mutually exclusive pattern of RAS pathway gene mutations in ALL. The traditional view is to consider mutant genes involved in the same pathway as being generally mutually exclusive, and only one mutation is required for each pathway. The frequent co-occurrence of the mutant RAS genes in our study turns this traditional view on its end, raising the interesting question of why ALL would acquire more than one RAS mutation. The most likely explanation is that these mutations are present in different subclones, and that they are mutually exclusive in each subclone. In the entire cohort (15 patients) that harbored more than one RAS mutations in their leukemic cells, 9 patients have more than one mutation in the same RAS gene (usually in codon G12/G13). The latter observation enables us to perform phasing analysis: two mutations that were close enough to be spanned by the same read-pair allowing the determination if the mutations are on either the same or different alleles. Accordingly, we found that all RAS mutations are acquired on different alleles with diverse VAF values. This observation, together with the fact that these ALL cells have a neutral copy-number at the RAS locus, precludes the possibility that these mutations are present on different alleles of the same clone. A similar conclusion was also made concerning a patient with two close-occurring FLT3 mutations. Overall, the above observations highlight the genetic heterogeneity of pediatric ALL. The development and selection of RAS-RTK mutations in different subclones of the same patient's ALL suggest that these mutations follow a branching hierarchy and occurred as a late event. This further underscores their crucial role for the clonal expansion and evolution of ALL.
In summary, we extensively interrogated the mutational landscape of a large cohort of pediatric ALL samples by exome and targeted resequencing. We revealed prominent dysregulation of RAS/RTK signaling and epigenetic regulators in pediatric ALL, providing a therapeutic rationale for the simultaneous targeting of several dysregulated pathways. The study reveals a number of new potential therapeutic targets and provides new insights into the genetic basis of relapse ALL.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We thank Prof Patrick Tan and Dr Zhijiang Zang for generously sharing their relevant facilities.
Grant Support
This research is supported by the Leukemia Lymphoma Society of America, by the National Research Foundation Singapore under its Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/0021/2014) to H.P.Koeffler and administered by the Singapore Ministry of Health's National Medical Research Council (NMRC), the NMRC Centre Grant awarded to National University Cancer Institute of Singapore, the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives. This research is supported by the National Institutes of Health of the USA (R01CA026038-35), as well as the generous donations from the Melamed family and Reuben Yeroushalmi to H.P.Koeffler. This research is also supported by grants from Chang Gung Memorial Hospital (CORPG3C0202), Ministry of Science and Technology of Taiwan (MOST-104-2314-B-182-032-MY3) to L.Y. Shih and Mackay Memorial Hospital (MMH-E-105-09) to D.C. Liang.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122:3407–15. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014;5:3469. doi: 10.1038/ncomms4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. doi: 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat Genet. 2015;47:672–6. doi: 10.1038/ng.3301. [DOI] [PubMed] [Google Scholar]
- 5.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–25. doi: 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, et al. Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray. Blood. 2008;111:776–84. doi: 10.1182/blood-2007-05-088310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawamata N, Ogawa S, Seeger K, Kirschner-Schwabe R, Huynh T, Chen J, et al. Molecular allelokaryotyping of relapsed pediatric acute lymphoblastic leukemia. Int J Oncol. 2009;34:1603–12. doi: 10.3892/ijo_00000290. [DOI] [PubMed] [Google Scholar]
- 10.Sun QY, Ding LW, Tan KT, Chien W, Mayakonda A, Lin DC, et al. Ordering of mutations in acute myeloid leukemia with partial tandem duplication of MLL (MLL-PTD). Leukemia. 2016 doi: 10.1038/leu.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding LW, Sun QY, Lin DC, Chien W, Hattori N, Dong XM, et al. LNK (SH2B3): paradoxical effects in ovarian cancer. Oncogene. 2015;34:1463–74. doi: 10.1038/onc.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–10. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–11. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–7. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- 16.Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–61. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 17.Opatz S, Polzer H, Herold T, Konstandin NP, Ksienzyk B, Zellmeier E, et al. Exome sequencing identifies recurring FLT3 N676K mutations in core-binding factor leukemia. Blood. 2013;122:1761–9. doi: 10.1182/blood-2013-01-476473. [DOI] [PubMed] [Google Scholar]
- 18.Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet. 2008;372:1484–92. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 19.Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2009;106:9414–8. doi: 10.1073/pnas.0811761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630. doi: 10.1038/ncomms4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 2013;110:18250–5. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28:198–201. doi: 10.1038/leu.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45:1386–91. doi: 10.1038/ng.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirabella F, Wu P, Wardell CP, Kaiser MF, Walker BA, Johnson DC, et al. MMSET is the key molecular target in t(4;14) myeloma. Blood Cancer J. 2013;3:e114. doi: 10.1038/bcj.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang H, Chen I-M, Wilson CS, Bedrick EJ, Harvey RC, Atlas SR, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010;115:1394–405. doi: 10.1182/blood-2009-05-218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874–84. doi: 10.1182/blood-2009-08-239681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirschner-Schwabe R, Lottaz C, Todling J, Rhein P, Karawajew L, Eckert C, et al. Expression of late cell cycle genes and an increased proliferative capacity characterize very early relapse of childhood acute lymphoblastic leukemia. Clin Cancer Res. 2006;12:4553–61. doi: 10.1158/1078-0432.CCR-06-0235. [DOI] [PubMed] [Google Scholar]
- 29.Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–95. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–9. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, et al. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012;26:1797–803. doi: 10.1038/leu.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Staal FJ, de Ridder D, Szczepanski T, Schonewille T, van der Linden EC, van Wering ER, et al. Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia. 2010;24:491–9. doi: 10.1038/leu.2009.286. [DOI] [PubMed] [Google Scholar]
- 33.Stef M, Simon D, Mardirossian B, Delrue MA, Burgelin I, Hubert C, et al. Spectrum of CREBBP gene dosage anomalies in Rubinstein-Taybi syndrome patients. Eur J Hum Genet. 2007;15:843–7. doi: 10.1038/sj.ejhg.5201847. [DOI] [PubMed] [Google Scholar]
- 34.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 35.Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453:110–4. doi: 10.1038/nature06866. [DOI] [PubMed] [Google Scholar]
- 36.Qi C-F, Martensson A, Mattioli M, Dalla-Favera R, Lobanenkov VV, Morse HC. CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc Natl Acad Sci U S A. 2003;100:633–38. doi: 10.1073/pnas.0237127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–9. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365:1384–95. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med. 2013;19:368–71. doi: 10.1038/nm.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet. 2013;45:290–4. doi: 10.1038/ng.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, Li H, Bai Y, Kirschner-Schwabe R, Yang JJ, Chen Y, et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat Med. 2015;21:563–71. doi: 10.1038/nm.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landau DA, Carter SL, Getz G, Wu CJ. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia. 2014;28:34–43. doi: 10.1038/leu.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alpar D, Wren D, Ermini L, Mansur MB, van Delft FW, Bateman CM, et al. Clonal origins of ETV6-RUNX1(+) acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia. 2015;29:839–46. doi: 10.1038/leu.2014.322. [DOI] [PubMed] [Google Scholar]
- 45.Bateman CM, Alpar D, Ford AM, Colman SM, Wren D, Morgan M, et al. Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia. 2015;29:58–65. doi: 10.1038/leu.2014.177. [DOI] [PubMed] [Google Scholar]
- 46.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111:2548–53. doi: 10.1073/pnas.1324297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314:811–22. doi: 10.1001/jama.2015.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Busque L, Ilaria R, Jr., Tantravahi R, Weinstein H, Gilliland DG. Clonality analysis of childhood ALL in remission: No evidence of clonal hematopoiesis. Leukemia Research. 1994;18:71–77. doi: 10.1016/0145-2126(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 50.Jowitt SN, Liu Yin JA, Saunders MJ, Lucas GS. Clonal remissions in acute myeloid leukaemia are commonly associated with features of trilineage myelodysplasia during remission. Br J Haematol. 1993;85:698–705. doi: 10.1111/j.1365-2141.1993.tb03211.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.