

Abstract
MutL assists the mismatch recognition protein MutS to initiate and coordinate mismatch repair in species ranging from bacteria to humans. The MutL N-terminal ATPase domain is highly conserved, but the C-terminal region shares little sequence similarity among MutL homologs. We report here the crystal structure of the Escherichia coli MutL C-terminal dimerization domain and the likelihood of its conservation among MutL homologs. A 100-residue proline-rich linker between the ATPase and dimerization domains, which generates a large central cavity in MutL dimers, tolerates sequence substitutions and deletions of one-third of its length with no functional consequences in vivo or in vitro. Along the surface of the central cavity, residues essential for DNA binding are located in both the N- and C-terminal domains. Each domain of MutL interacts with UvrD helicase and is required for activating the helicase activity. The DNA-binding capacity of MutL is correlated with the level of UvrD activation. A model of how MutL utilizes its ATPase and DNA-binding activities to mediate mismatch-dependent activation of MutH endonuclease and UvrD helicase is proposed.
Keywords: ATPase, dimerization, DNA binding, MutL, UvrD
Introduction
MutS and MutL are conserved from bacteria to humans and initiate DNA mismatch repair to remove replication errors (Modrich and Lahue, 1996). MutS recognizes mispaired or unpaired bases in a DNA duplex and, in the presence of ATP, recruits MutL to form a MutS–MutL-mismatch DNA signaling complex for repair (Grilley et al, 1989; Habraken et al, 1998; Schofield et al, 2001; Junop et al, 2003). In Escherichia coli, MutS and MutL together activate the latent endonuclease MutH to nick an error-containing daughter strand, which is transiently unmethylated, either 5′ or 3′ to the mismatch site (Modrich and Lahue, 1996). They then recruit the UvrD helicase and appropriate exonucleases (3′ → 5′or 5′ → 3′) to remove the daughter strand from the nick to beyond the mismatch site (Modrich and Lahue, 1996; Burdett et al, 2001). In eukaryotes and bacteria that lack endonuclease MutH homologs, the 3′ hydroxyl group of the daughter strand is believed to be the initiation site for mismatch repair, to which MutS and MutL homologs recruit and load exonuclease (Genschel et al, 2002). Both MutS and MutL are ATPases, and mutations that impair either ATP binding or ATP hydrolysis by MutS or MutL abolish the mismatch repair process (Iaccarino et al, 1998; Hall et al, 2002; Drotschmann et al, 2002b; Junop et al, 2003). In addition, MutS and MutL proteins play roles in mitotic and meiotic DNA recombination and in DNA damage-induced cell death (Borts et al, 2000; Harfe and Jinks-Robertson, 2000; Li, 2003). Mutations in human MutS or MutL homologs have been implicated in predisposition to hereditary nonpolyposis colorectal cancers (HNPCC) and other sporadic cancers (Peltomaki, 2003).
Prokaryotic MutL proteins are homodimeric, but in eukaryotes multiple MutL homologs, including MLH1, PMS1, PMS2 and MLH3, combine to form heterodimers (Li and Modrich, 1995; Raschle et al, 1999; Lipkin et al, 2000). Crystallographic and biochemical studies have shown that MutL proteins contain an N-terminal ATPase region and a C-terminal dimerization region (Ban and Yang, 1998; Ban et al, 1999). The ATPase region is conserved among all MutL homologs and shares four sequence motifs with other GHKL (for Gyrase, Hsp90, histidine kinase and MutL) ATPase/kinase superfamily members (Dutta and Inouye, 2000). Large conformational changes including association and dissociation of the N-terminal ATPase region occur during the ATP hydrolysis cycle (Ban et al, 1999). In the presence of ATP or the nonhydrolyzable ATP analog AMPPNP, the ATPase fragment is fully folded and dimeric. In the absence of nucleotide, it is monomeric and ∼60 out of the 330 residues in the ATPase fragment are disordered (Ban and Yang, 1998). MutL also possesses a nonspecific DNA-binding activity. In the presence of DNA, especially ssDNA, both KM and kcat of the MutL ATPase activity are increased, which results in a redistribution of various forms of MutL due to a decrease of the MutL–ATP complex and increase of apoprotein and possibly MutL–ADP complex (Ban et al, 1999; Junop et al, 2003). Conformational changes of MutL have been suggested to enable the MutS–MutL-mismatch DNA complex to recruit different downstream effectors at different stages of mismatch repair (Junop et al, 2003).
In the presence of AMPPMP, MutL is able to activate MutH endonuclease without MutS or a mismatch site (Ban and Yang, 1998), although such activation is physiologically undesirable and presumably suppressed in vivo by the low protein concentrations. A physical interaction between the N-terminal ATPase domain of MutL and MutH has been detected by protein crosslinking (Ban and Yang, 1998), and AMPPNP enables the interaction of the two proteins and activation of the endonuclease activity.
MutL also activates the UvrD helicase to unwind duplex DNA with a free end, but activation of UvrD on a nicked circular DNA substrate requires a MutS–MutL-mismatch complex (Dao and Modrich, 1998; Yamaguchi et al, 1998). The C-terminal 218 residues (residues 398–615) of MutL were identified to interact with UvrD based on two-hybrid analyses and pull-down assays (Hall et al, 1998). Similar approaches also found that the same C-terminal region of MutL interacted with MutH (Hall and Matson, 1999). Since activation of MutH is ATP-dependent and the ATPase activity resides in the N-terminal and not in the C-terminal region of MutL (Ban and Yang, 1998), physical contacts identified by two-hybrid analyses may not fully correspond to the functional interactions. How MutL functionally interacts with UvrD remains to be determined.
The MutL C-terminal region is essential for homo- or hetero-dimerization of MutL and its homologs (Ban and Yang, 1998; Wu et al, 2003). However, the amino-acid sequence of the C-terminal region is highly divergent among MutL homologs. In fact, no sequence or structural conservation has been described. We report here the crystal structure of the E. coli MutL dimerization domain and demonstrate the structural and functional conservation among MutL homologs beyond the ATPase domain. Based on the crystal structures of the N- and C-terminal fragments of MutL and its ATP hydrolysis and DNA-binding activities, we construct a model of full-length MutL and propose a mechanism by which MutL mediates mismatch-dependent activation of MutH and UvrD.
Results and discussion
Identification of the MutL dimerization domain
Secondary structure prediction for E. coli MutL protein by PsiPred (Jones, 1999) indicates that secondary structures are clustered in the N-terminal 335 (1–335 aa) and the C-terminal 177 residues (439–615 aa). Residues from 336 to 438 are predicted to form random coils. Thrombin digestion of MutL results in two fragments, the N-terminal 350 residues and the C-terminal 265 residues (351–615 aa, LC30). The N-terminal fragment, LN40, contains the ATPase activity and crystal structures reveal that only the first 331 residues are ordered (Ban and Yang, 1998; Ban et al, 1999). Our attempts to crystallize the proteolytically generated C-terminal fragment failed to produce crystals. We therefore subcloned and overproduced three shorter C-terminal fragments of MutL, which contain residues 394–615, 415–615 and 432–615 and are named LC24, LC22 and LC20 according to their molecular weights. Each purified fragment forms dimers as does LC30 (Figure 1A and B). Limited trypsin digestion, which does not reduce the size of LC20, converts the longer fragment LC24 to a uniquely digested product with a molecular weight similar to that of LC20 (Figure 1C). We suspect that residues 432–615 encompass the minimal folded region that is essential for dimerization and that the 100 residues between 332 and 431 form an extended linker, as suggested by secondary structure predictions.
Figure 1.
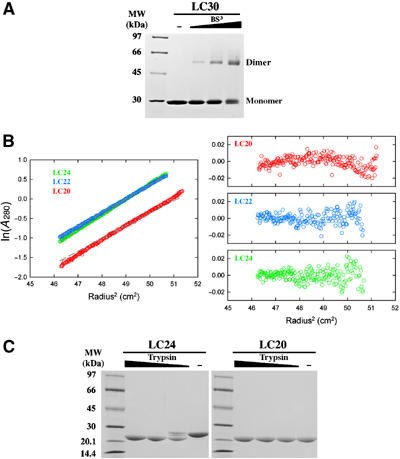
Dimerization domain of E. coli MutL. (A) A Coomassie-blue-stained SDS gel of LC30 dimers crosslinked by 0.11, 0.33 and 1 mM BS3. (B) Sedimentation equilibrium analysis of LC24, LC22 and LC20. Profiles of the three proteins (color coded) at 12 000 r.p.m. and 4°C are plotted on the left. The gray dashed lines represent expected values for the corresponding dimers. Residuals for each protein are shown on the right. (C) Trypsin digestion of LC24 and LC20. In all, 9 μl of 0.5 mg/ml LC24 or LC20 was digested by 1 μl of 0.01, 0.03 or 0.1 mg/ml trypsin in the protein storage buffer for 1 h at 22°C. The digestion products were separated by SDS–PAGE and stained with Coomassie blue.
Crystal structure of LC20
LC20 crystallized in space group P4322, and these crystals diffracted X-rays to 2.1 Å (Materials and methods). The crystal structure has been determined using the Multiwavelength Anomalous Diffraction (MAD) method with selenomethionine substitution (Table I) (Hendrickson et al, 1990). In the refined crystal structure, each asymmetric unit contains a V-shaped LC20 dimer related by a noncrystallographic dyad axis (Figure 2A). Each LC20 subunit (residues 432–615) can be divided into two subdomains. The external (Ex) subdomain, which contains residues 432–474 and 570–615, forms the outer arm of the ‘V', and the internal (In) subdomain, which consists of residues 475–569, is responsible for dimerization. The Ex subdomain consists of a four-stranded antiparallel β-sheet (β1, β2, β3 and β8), a layer of three helices (αA, αE, and αF), of which αF contains only five residues, and a C-terminal helix (αG) protruding from the Ex subdomain into solvent (Figure 2A and B). The In subdomain is made of a three-stranded antiparallel β-sheet (β4, β5, β6 and β7, with β4 and β5 almost continuous except for a 120° sharp bend) and a layer of three α helices (αB, αC and αD). Two closely packed antiparallel helices αA (residues 460–473) and αD (residues 554–566) tie the Ex and In subdomains into one continuous structural entity (Figure 2A). A search for structural homologues by DALI (Holm and Sander, 1993) finds no close relatives of LC20.
Table 1.
Data collection and refinement statistics
| Space group: P4322 | (a=b=96.0 Å, c=140.2 Å; 2 molecules in each asymmetric unit) | ||
|---|---|---|---|
| Wavelength (Å) | 0.9840 (remote) | 0.9793 (edge) | 0.9790 (peak) |
| Resolution (Å)a | 20–2.1 (2.14–2.1) | 20–2.1 (2.14–2.1) | 20–2.1 (2.14–2.1) |
| Completeness (%)a | 95.0 (97.7) | 95.0 (98.1) | 95.0 (97.7) |
| Redundancy | 5.5 | 5.6 | 5.3 |
| Rmerge (%)a,b | 6.9 (48.1) | 7.5 (50.8) | 7.5 (48.8) |
| I/σ(I) | 22.2 (2.5) | 20.9 (2.2) | 20.0 (2.1) |
| SOLVE (15–3.0 Å) | Figure-of-merit=0.403 | ||
| Refinement | |||
| Resolution (Å) | 20–2.1 | ||
| Reflections work set | 33081 | ||
| Reflections test set | 3678 | ||
| Atoms refined | 3144 (292 water) | ||
| R (Rfree) (%)c | 23.6 (27.8) | ||
| R.m.s.d. in bonds (Å) | 0.008 | ||
| R.m.s.d. in angles (deg) | 1.45 | ||
| Mean B values (Å) |
54.1 |
|
|
| aData in the highest resolution shell are shown in parentheses. | |||
| bRmerge=∑h∑i ∣Ihi−〈Ih〉∣/∑〈Ih〉, where Ihi is the intensity of the ith observation of reflection h, and 〈Ih〉 is the average intensity of redundant measurements of the h reflections. | |||
| cR-value=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣, where Fo and Fc are the observed and calculated structure-factor amplitudes. Rfree is monitored with 3678 reflections excluded from refinement. | |||
Figure 2.
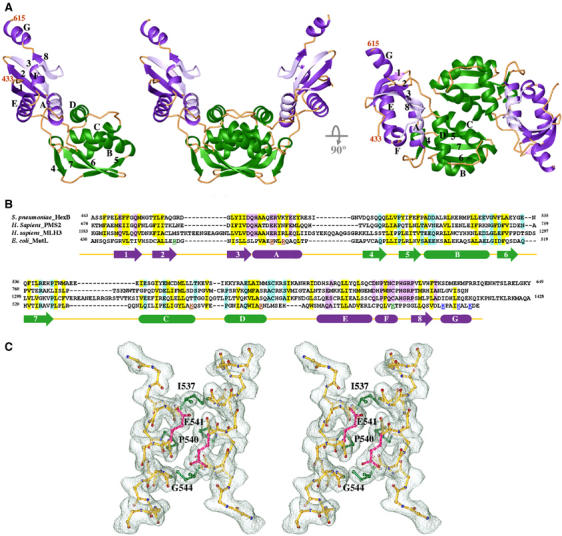
Crystal structure of LC20. (A) Ribbon diagrams of one LC20 subunit and two orthogonal views of an LC20 dimer. The Ex and In subdomains are shown in purple and green, respectively. Secondary structures are labeled in one subunit. (B) Sequence alignment of the C-terminal dimerization domains of Streptococcus pneumoniae HexB, human PMS2 and MLH3, and E. coli MutL. Secondary structures of LC20 that are predicted to also exist in the other three proteins are shown as arrows (β-strand) and boxes (α-helices). Conserved hydrophobic and polar residues are highlighted in yellow and purple (Ex) or green (In). Residues mutated in P-1, P-2 and P-3 are color coded in red, blue and green, respectively. (C) Stereo view of the pair of αC helices at the dimer interface. A 2Fo−Fc electron density map is superimposed. Side chains of Ile537, Pro540 and the Cα atom of Gly544 are highlighted in green, and side chains of Glu541 in pink. Other side chains of the αC helix are omitted for clarity. Figures 2A, C and 3 were generated using RIBBONS (Carson, 1987).
Dimerization of LC20 occurs between the pair of antiparallel αC helices related by a noncrystallographic dyad axis and involves strand β5, the N-terminus of αB and the C-terminus of αD from both subunits (Figure 2A and C). The total surface area buried upon dimerization is ∼1000 Å2, and the surface complementarity index (Lawrence and Colman, 1993) is 0.644, which is comparable to an antibody and antigen interface. The LC20 dimeric interface contains both hydrophobic and hydrophilic residues. Gln536, Asp541 and Lys548 stabilize the dimer interface by direct and water-mediated polar interactions. The 15-residue αC helix contains Pro540 and Gly544 in the middle, yet the helical axis and pitch appear to be normal (Figure 2C). The Cα of Gly544 makes van der Waals contacts with Asp541, and the carbonyl oxygen of residue 536, which would normally be hydrogen bonded to the amine group of residue 540, is rotated outwards due to Pro540. In addition, Asp541 is buried in the dimer interface and likely protonated due to the pH 4.6 crystallization buffer (Materials and methods). It may remain protonated at the physiological pH because of the hydrophobic environment. Neutral pH does not cause dissociation of LC20 dimers, as LC20 monomers are not detectable even at submicromolar protein concentrations by analytical ultracentrifugation (Figure 1B). The dissociation constants of LC20 and MutL dimers are likely in the nanomolar range.
The only reported missense mutation in the C-terminal region of MutL that results in a dominant mutator phenotype in E. coli is Ala518 to Thr (Aronshtam and Marinus, 1996). All other mutations in the C-terminal region of MutL with a dominant mutator phenotype are due to deletion or truncation. Ala518 is located in a tight turn between β6 and β7. It is conceivable that the A518T mutation destabilizes the LC20 structure and results in a phenotype similar to truncations of the C-terminal domain.
Structure conservation among the MutL homologs
Amino-acid sequence conservation between the C-terminal dimerization regions of MutL homologs has not been reported. A recent BLAST search (www.ncbi.nlm.nih.gov/BLAST/) reveals that MutL homologs in Gram-positive bacteria, for example, HexB, share sequence similarity with human PMS2 and MLH3 in the C-terminal dimerization region (Figure 2B). Pairwise comparison between HexB and PMS2 or HexB and MLH3 gives rise to the E-value of 10−10 or 10−6, respectively, suggesting that the sequence similarity is statistically significant (Altschul and Gish, 1996). Furthermore, secondary structure predictions for PMS2, MLH3 and HexB by PsiPred (Jones, 1999) indicate that these three proteins contain the same order and types of secondary structures as the E. coli LC20 structure (Figure 2B). Although sequence identity between E. coli MutL and its three homologs in the C-terminal dimerization region is as low as 13%, alignment guided by the conserved secondary structure results in a similarity of up to 36% (Figure 2B). Residues essential for formation of the hydrophobic core are conserved among these four MutL homologs. Sequence conservation between each of these four MutL homologs and human MLH1 is hard to detect, but, based on secondary structure predictions, human MLH1 is likely to fold as PMS2 and MLH3, with insertions protruding into the solvent surrounding helix αB. Our sequence alignment and structure predictions are consistent with the dimerization domains reported by Wu et al (2003), but not with the results of yeast two-hybrid assays (Kondo et al, 2001). Based on the potential structural homology, the C-terminal residues 571–862 of human PMS2 and residues 475–756 of human MLH1 have been produced and form stable MLH1–PMS2 heterodimers (our unpublished data).
A model of intact MutL and a large central cavity separating LN40 and LC20
A full-length MutL dimer was modeled assuming that the dyad axes of LN40 and LC20 dimers are collinear (Figure 3A). By placing the C-terminus of LN40 and the N-terminus of LC20 adjacent to each other, the saddle-shaped LN40 is opposite the V-shaped LC20, and the resulting MutL model contains a large central cavity. As the dyad axes relating the N- and C-terminal domains are not necessarily collinear and the linker between the two domains is unstructured, the size and shape of this cavity is likely to vary depending on the relative orientation of LN40 and LC20.
Figure 3.
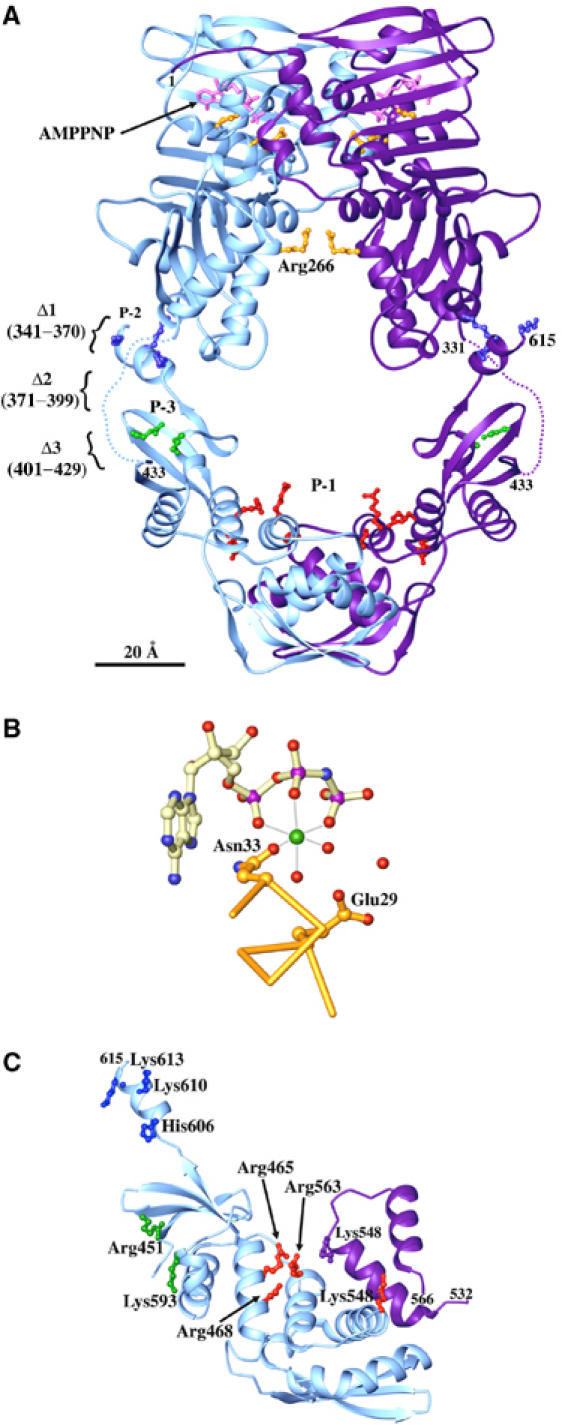
A model of intact MutL. (A) A ribbon diagram of full-length MutL. LN40 and LC20 are placed to share a common dyad axis. For aesthete, separation of the two domains (160 Å) is not drawn to scale. The C-terminus of LN40 (331) and N-terminus of LC20 (433) are linked by a dotted line. AMPPNP (pink), Asn33, Glu29 and Arg266 (gold), residues in the P-1 (red), P-2 (blue) and P-3 (green) patches, and the linker deletion Δ1, Δ2 and Δ3 are shown. (B) Asn33 chelates Mg2+ (green sphere) for ATP binding and Glu29 is the general base for ATP hydrolysis. Water molecules are shown as red spheres. (C) Patch mutations. An LC20 subunit (light blue) is shown with ball-and-stick presentations of the side chains in P-1 (red), P-2 (blue) and P-3 (green). The P-1 patch extends across the dimer interface to the neighboring subunit (purple).
The linker region between the N-terminal ATPase and C-terminal dimerization domains shares no sequence similarity among MutL homologues. In E. coli MutL, it is 100 residue long and dominated by Pro (20%), Ala (17%), Gln (12%) and charged residues (20%). It is predicted to be devoid of secondary structures and is indeed susceptible to protease digestion (Figure 1C). Based on the crystal structure of SMAD4 (Qin et al, 1999), proline-rich regions are often extended and not overly flexible because of the restricted backbone torsion angles of prolines. To examine the functional role of this linker region, we constructed the following seven deletion mutants. The first three mutations sequentially removed 30 residues in the linker, MutLΔ1 (residues 341–370 removed), MutLΔ2 (371–400 removed) and MutLΔ3 (400–429 removed). The next four mutations sequentially remove 10 additional residues from MutLΔ2, MutLΔ4 (40 residues removed, 366–405), MutLΔ5 (50, 361–410), MutLΔ6 (60, 356–415) and MutLΔ7 (70, 351–420).
mutLΔ2, mutLΔ3 and mutLΔ4 mutants behave like wild type in the mismatch repair assays (Table II). Plasmids bearing any one of the three deletion mutants can complement a mutL− strain resulting in mutation rates no more than three-fold of wild-type cells as compared to a 300-fold difference between wild-type and mutL null cells (Table II). Not surprisingly, purified MutLΔ2, MutLΔ3 and MutLΔ4 proteins bind DNA and hydrolyze ATP like native MutL, and undergo conformational changes during the ATPase cycle (Table II and Figure 4). In spite of normal ATPase and DNA-binding activities in vitro, mutLΔ1 is defective in mismatch repair in vivo and has a 30-fold increase in the mutation rate (Table II).
Table 2.
In vivo and in vitro characterization of MutL mutants
| Mutations | Mutation rate (f) and repair efficiency |
ATPase activity |
||
|---|---|---|---|---|
| f (10−8)a | Repair (%)b | KM (μM) | Kcat (min−1) | |
| Wild type | 3.2 (1.7–5.8) | 100 | 31 | 0.13 |
| mutL null | 1070 (913–1410) | 0 | N/A | N/A |
| MutLΔ1 | 105 (69.4–134) | 90.5 | 30 | 0.11 |
| MutLΔ2 | 5.4 (2.3–9.2) | 99.8 | 46 | 0.12 |
| MutLΔ3 | 10 (8.4–12) | 99.4 | 39 | 0.14 |
| MutLΔ4 | 4.4 (2.0–13) | 99.9 | 43 | 0.15 |
| MutLΔ5 | 59 (49–82) | 94.8 | 29 | 0.19 |
| MutLΔ6 | 192 (146–231) | 82.3 | 45 | 0.19 |
| MutLΔ7 | 618 (498–695) | 42.4 | 40 | 0.20 |
| Patch-1 | 282 (168–387) | 73.9 | 53 | 0.10 |
| Patch-2 | 2.1 (0.7–5.7) | 100.1 | 66 | 0.12 |
| Patch-3 | 5.8 (3.8–7.7) | 99.8 | 38 | 0.12 |
| R465E/R468E | 48 (27–56) | 95.8 | N/D | N/D |
| N33A |
518 (383–731) |
51.7 |
N/A |
N/A |
| aThe rpoB mutation frequency (f) per cell was calculated by dividing the median number of mutants by the average number of cells in a series of cultures. Mean value with 95% confidence limit (Dixon and Massey, 1969) in parentheses. | ||||
| bRepair efficiency is defined as (fnull−fn)/(fnull−fwt) × 100, where fnull, fwt and fn are mean values of mutation rates observed from mutL null cells, wild type and a specific mutant strain. | ||||
Figure 4.
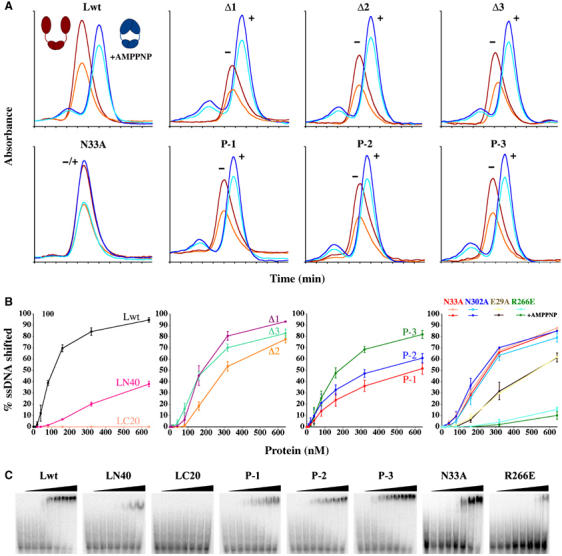
Properties of mutant MutL proteins. (A) Association of the N-terminal ATPase domain upon binding of AMPPNP (+AMPPNP) is monitored by elution profiles from a Sephadex-200 size-exclusion column. Wild-type MutL without AMPPNP is eluted early (red for A280 and orange for A260), since its N-terminal ATPase domains are dissociated as depicted in the red cartoon. When bound to AMPPNP, the ATPase domains become associated (blue cartoon), and the protein is eluted later (dark and light blue for A280 and A260). AMPPNP binding also results in an increased A260 to A280 ratio. For mutant proteins, ‘+' and ‘−' indicate the presence or absence of AMPPNP. Among seven mutant proteins, only N33A fails to bind AMPPNP and does not undergo conformational changes. (B) Electrophoresis mobility shift assays (EMSA) of ssDNA binding by wild-type MutL, LN40 and LC20 (first panel), linker deletion (second panel) and patch-mutant proteins (third panel); ssDNA binding by four MutL mutant proteins with and without AMPPNP (fourth panel). AMPPNP has no effect on the N33A and E29A mutant MutL, and R266E has little DNA-binding activity. (C) EMSA gels of eight MutL proteins shown in (B). DNA binding was assayed in the presence of ATP (first six) or AMPPNP (last two).
Our results agree well with the systematic mutagenesis analyses of yeast MLH1 (Argueso et al, 2003). A missense mutation scan across the entire yeast MLH1 sequence found that mutations that had no effect on either mismatch repair or meiotic DNA recombination were clustered in a span of 70 residues between the N-terminal ATPase and C-terminal dimerization domains. Alterations of the residues immediately N-terminal to this mutation-insensitive region but still within the predicted 150-residue random-coil linker led to functional defects. It appears that the 70-residue mutation-insensitive region of yeast MLH1 corresponds to the E. coli MutL residues deleted in MutLΔ2 and MutLΔ3, and the mutation-sensitive region of yeast MLH1 corresponds to those deleted in MutLΔ1. The native phenotype exhibited by the Δ2, Δ3 and Δ4 mutants indicate that the C-terminal two-thirds of the linker between LN40 and LC20 does not require a specific amino-acid sequence and the linker can be shortened from 100 to 60 residues.
The mutLΔ5, Δ6 and Δ7 mutants with 50, 60 and 70 residues removed, however, have mutation rates increased by 20-, 60- and 200-fold in spite of the normal ATPase activity of the mutant proteins (Table II). Dynamic light-scattering analyses reveal that the diffusion coefficient of wild-type, Δ2 and Δ5 mutant MutL increases monotonously with decreasing linker length (Table III), suggesting that the N- and C-terminal domains are brought closer as the linker is shortened. Assuming that in the presence of AMPPNP and Mg2+ the N- and C-terminal domain are two distinct structural entities, separations of 160, 111 and 76 Å between their centers of mass in wild-type, Δ2 and Δ5 mutant protein, respectively, are required to account for the observed Stokes radii. The diameter of the central cavity in wild-type MutL is thus ∼100 Å, large enough to encircle 2–4 DNA duplexes simultaneously. Additional removal of the linker in the Δ7 mutant protein does not result in further reduction of the protein size in the presence of either EDTA or AMPPNP and Mg2+ (Table III). The 20 residues deleted in Δ7 but present in Δ5 may restrict the linker with local structure, and their removal probably releases the linker from compaction and increases the observed size of the Δ7 mutant protein. The mutator phenotype of mutLΔ7 implies that the large central cavity of MutL play a critical role in mismatch repair.
Table 3.
Dynamic light-scattering analyses of MutL proteins
| Protein | Dso/10−7 cm2 s−1 a | Stokes radius r (Å)b | f / foc | Distance (Å)d |
|---|---|---|---|---|
| WT (AMPPNP) | 3.86±0.08 (11) | 55.5 | 1.62 | 160 |
| Δ2 (AMPPNP) | 4.03±0.06 (24) | 53.2 | 1.58 | 111 |
| Δ5 (AMPPNP) | 4.48±0.08 (25) | 47.8 | 1.44 | 76 |
| Δ7 (AMPPNP) | 4.20±0.04 (14) | 51.0 | 1.54 | 98 |
| WT (EDTA) | 2.48±0.07 (8) | 86.4 | 2.53 | — |
| Δ2 (EDTA) | 2.45±0.04 (13) | 87.5 | 2.60 | — |
| Δ5 (EDTA) | 3.32±0.14 (8) | 64.5 | 1.92 | — |
| Δ7 (EDTA) |
3.12±0.06 (11) |
68.7 |
2.10 |
— |
| aAverage corrected diffusion coefficient based on data collected at angles of 90, 75, 60 and 45°. The number of measurements is shown in parentheses. | ||||
| bStokes radius, r, is calculated as r=kT/6πηDso, where k is Boltzmann's constant and η the viscosity of water at 20.0°C. | ||||
| cThe friction coefficient, f, is calculated as f=kT/Dso, and friction coefficient for a sphere of equivalent mass (M), fo, is given by fo=6πη(3Mv/4πN)1/3, where v is the partial specific volume of the protein and N is Avogadro's number. Deviation of f/fo from 1.0 indicates that the protein molecule is elongated. | ||||
| dLN40 dimer is translated relative to LC20 along a shared dyad axis. The distance between their centers of mass that results in a calculated diffusion coefficient consistent with those measured experimentally is listed. | ||||
LC20 is involved in DNA binding
MutL binds both double- and single-stranded DNA with no sequence specificity (Bende and Grafström, 1991; Mechanic et al, 2000; Drotschmann et al, 2002a). The DNA-binding activity of MutL is detected in the absence of a nucleotide cofactor but enhanced in the presence of AMPPNP (Ban et al, 1999). The crystal structure of an LN40–AMPPNP complex revealed a positively charged groove inside the saddle-shaped LN40 dimer (Figure 3A). Mutation of Arg266 to Glu in the middle of the groove largely abolishes the DNA-binding activity of the full-length MutL (Junop et al, 2003). Although LN40 alone binds DNA, the presence of the C-terminal dimerization region in the full-length MutL greatly enhances DNA binding (Figure 4B). Curiously, DNA binding by LC20 alone is not detected (Figure 4B).
To determine whether LC20 is directly involved in DNA binding, clusters of positively charged residues were identified in LC20 and mutated to glutamate in three patches (Figures 2B and 3). Arg465, Arg468, Lys548 and Arg563 define a large positively charged patch (P-1) on the concave surface of LC20 facing the central cavity; His606, Lys610 and Lys613 (P-2) at the C-terminus and Arg451 and Lys593 (P-3) on the convex surface of LC20 form two additional patches that are not facing the central cavity (Figure 3C). All three patch-mutant proteins behave well in solution and retain normal ATPase activity (Table II). The P-2 and P-3 mutant proteins have reduced DNA-binding activity (Figure 4B), but the corresponding mutants behave like wild type in the mismatch repair assay (Table II), suggesting that DNA binding by the residues in P-2 and P-3 patches is functionally unimportant. In contrast, the P-1 mutation reduces the DNA-binding activity of the mutant protein to a level similar to that of LN40 alone (Figure 4B) and increases the mutation rate of the mutant cell by nearly 100-fold (Table II). Even a double mutation of Arg465 and Arg468 to Glu in the P-1 patch results in a 10-fold increase in the mutation rate (Table II). Since the P-1 mutation does not affect dimerization or the ATPase activity of MutL (Figure 4A and Table II), the impaired mismatch repair probably results from the reduced DNA binding in the central cavity.
Interactions between MutL and UvrD
MutL stimulates the 3′ → 5′ helicase activity of UvrD and, in the presence of a mismatch and MutS, MutL loads UvrD from a nick onto either the nicked or continuous strand, depending on whether the nick is 5′ or 3′ to the mismatch site, so that UvrD unwinds DNA toward the mismatch (Dao and Modrich, 1998; Yamaguchi et al, 1998). MutL may directly interact with UvrD, as suggested by two groups (Hall et al, 1998; Spampinato and Modrich, 2000) or alter DNA structure to enhance the helicase activity. Using potassium permanganate footprinting (McCarthy and Rich, 1991), we find no evidence that MutL distorts DNA. By protein crosslinking, however, we are able to characterize interactions between MutL and UvrD.
The full-length MutL, LN40 and LC30 were studied for interaction with native UvrD in the presence of AMPPNP, a 23 bp duplex DNA with a 20 nt 3′ overhang, or both. As positive controls, dimeric LC30, MutL and UvrD were readily detected by crosslinking with bis-sulfosuccinimidyl suberate (BS3, Pierce) (Figures 1A and 5). Dimeric LN40 was detected at a very low level (Figure 5B) because formation of such dimers requires long incubation of LN40 with AMPPNP (Ban and Yang, 1998) and the crosslinking reported here was initiated immediately after mixing. Mixtures of UvrD and MutL, UvrD and LN40 or UvrD and LC30 each results in unique crosslinked species (Figure 5), indicating that both the N- and C-terminal domains of MutL interact with UvrD. The interaction between LN40 and UvrD depends on the presence of both AMPPNP and DNA (Figure 5B), while the interactions between LC30 and UvrD occur in the absence of nucleotide or DNA (Figure 5C). Two species of crosslinked UvrD and MutL are observed, one in the absence of AMPPNP or DNA and another in the presence of both (Figure 5A). The UvrD and MutL interactions are confirmed by repeated crosslinking experiments with different concentrations of BS3 and with a different crosslinker SPDP (data not shown).
Figure 5.
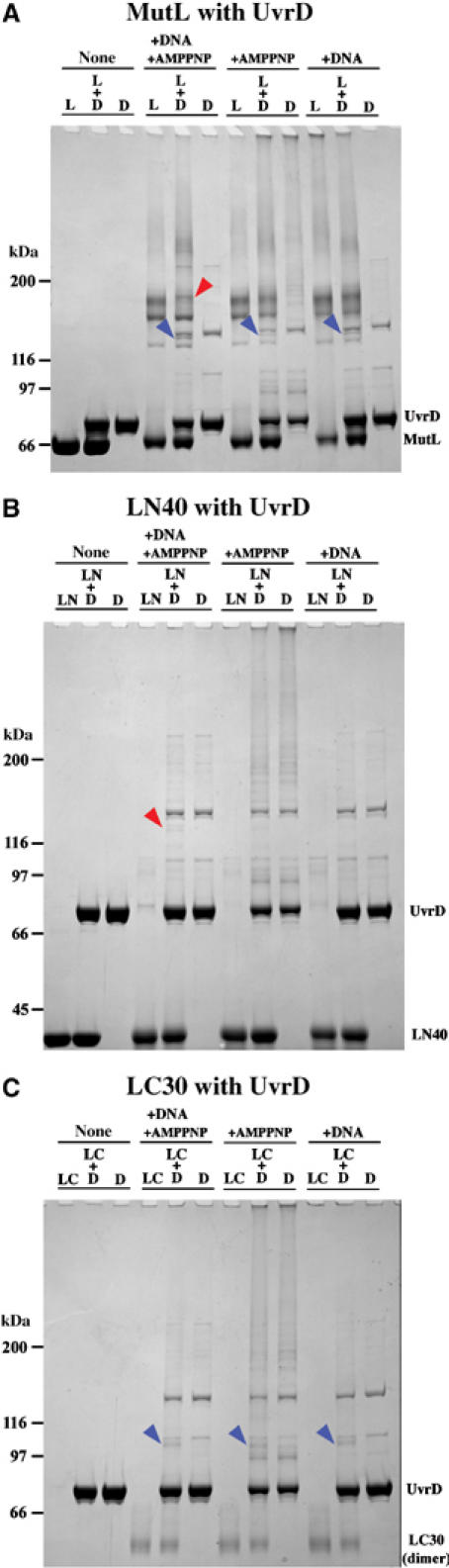
Crosslinking UvrD and MutL by BS3. Coomassie-blue-stained SDS–PAGE gels of UvrD (D) and full-length MutL (L) (A), LN40 (LN) (B), or LC30 (LC) (C) crosslinked by 0.11 mM BS3. The first three lanes are MutL (LN40 or LC30), MutL (LN40 or LC30) mixed with UvrD and UvrD. The following nine lanes are results of crosslinking each set of three protein samples in the presence of AMPPNP and DNA, AMPPNP, or DNA. Molecular-weight standards are indicated. MutL (70 kDa), LN40 (40 kDa) and UvrD (80 kDa), and dimeric LC30 (60 kDa) are labeled. LC30 monomers ran off the gel and are thus absent. The red arrowheads point to the crosslinked bands, whose formation depends on AMPPNP and DNA; the blue arrowheads point to the species crosslinked independent of AMPPNP or DNA.
DNA-dependent activation of UvrD helicase by MutL
MutL stimulates UvrD helicase activity in two distinct modes (Figure 6A). At first, the helicase activity rises steeply with the increase of MutL concentrations and reaches a maximum when the molar ratio of UvrD to MutL is approximately 1:1. This observation and the protein crosslinking data (Figure 5) strongly suggest the formation of a 1:1 MutL–UvrD functional complex. Following this steep rise, the helicase activity continues to rise gradually with further increases of MutL concentrations even when MutL is eight-fold molar excess over UvrD. The slow rise of the helicase activity may be due to nonspecific interactions between MutL and DNA that stabilize the ssDNA product. Unlike the full-length MutL, LN40 or LC24 has little discernable effect on UvrD helicase activity (Figure 6A), despite that they physically interact (Figure 5) (Hall et al, 1998). Not surprisingly, neither the N- nor C-terminal fragments of MutL retain any in vivo mismatch repair activity (Aronshtam and Marinus, 1996).
Figure 6.
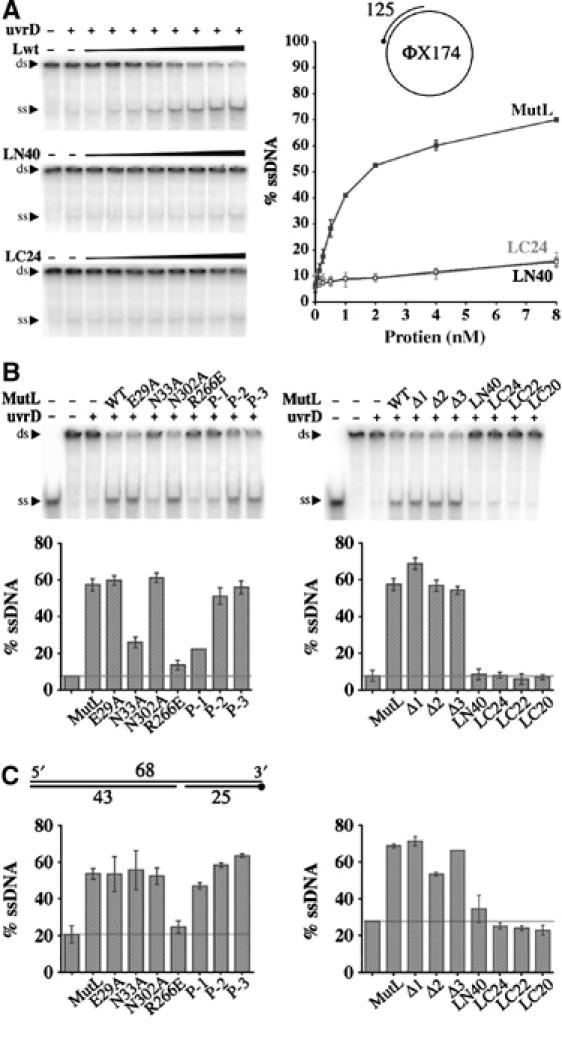
UvrD activation by MutL. (A) Unwinding of 1 nM circular DNA as illustrated in the cartoon by 1 nM UvrD helicase and increasing amount of MutL, LN40 or LC24 is shown on 4.5% TBE polyacrylamide gels (left panels). The amounts of DNA unwound are quantified and plotted on the right panel. (B) Activation of UvrD by mutant MutL proteins to unwind the circular DNA substrate. Both TBE gels and corresponding bar graphs are shown. (C) Activation of UvrD to unwind a linear DNA substrate as diagrammed. The 5′ end of a 32P-labeled strand is marked by a dot. Only the bar graphs are shown. In each bar graph, the left-most column is UvrD (1 nM) alone, to its right 1 nM of wild-type or mutant MutL is added as labeled. A horizontal line marks the amount of DNA unwound by UvrD alone.
We subsequently analyzed whether ATP binding and hydrolysis by MutL are required for UvrD stimulation. A new ATP-binding mutant was constructed replacing Asn33 that chelates Mg2+ and is essential for ATP binding with Ala (Figure 3B). Mutations of the Asn33 equivalent in yeast MutL homologs eliminate ATP binding (Hall et al, 2002). Based on the filter-binding assay (Ban et al, 1999) (data not shown) and gel filtration analyses (Figure 4A), which monitors both ATP binding and ATP-dependent N-terminal association, the E. coli N33A MutL protein does not bind ATP either. With a circular DNA substrate, the ATP-binding-defective N33A mutant protein shows >50% reduction in UvrD stimulation, while the ATP-hydrolysis-defective proteins, E29A and N302A (Junop et al, 2003), are indistinguishable from wild-type MutL in the helicase activity assay (Figure 6B). Interestingly, with a linear DNA duplex, all ATPase mutant proteins stimulate UvrD as efficiently as wild-type MutL (Figure 6C). The linear substrate is much shorter (25 bp) than the circular one (125 bp) and devoid of single-stranded DNA. We suspect that ATP binding enables MutL to either load UvrD more efficiently onto the circular substrate or enhance the processivity of UvrD (∼45 bp in the absence of MutL; Runyon et al, 1993). The absence of detectable defect of the ATP-hydrolysis mutant MutL in the helicase assays may be due to the nonsupercoiled substrates we used and the absence of MutS and mismatch (see later discussion).
In contrast to the subtle defects of the ATPase mutant proteins, MutL proteins defective in DNA binding are remarkably deficient in activating UvrD. The R266E MutL protein, which is the most defective in DNA binding (Figure 4B and C), yet has normal ATPase activity (Ban et al, 1999), is 7–10 times less efficient than wild-type MutL in activating UvrD with linear or circular DNA substrates (Figure 6B and C). Likewise, P-1 mutation in the C-terminal dimerization domain, which results in reduced DNA binding in vitro and increased mutation rate in vivo, leads to a 70% reduction in UvrD activation with the circular DNA substrate and 20% reduction with the linear substrate (Figure 6B and C). R266 and P-1 clearly define two distinct DNA-binding sites of MutL, both facing the central cavity but separated in two domains. The R266 site appears to be generally important for activating UvrD to unwind DNA, and P-1 and the C-terminal domain may play a specific role in recruiting UvrD to circular DNA. We were originally puzzled by the mutator phenotype of the R266E mutant and the ability of the mutant protein to fully activate MutH (Junop et al, 2003). It becomes clear that the defects in DNA binding directly correlate with the reduced ability of MutL to activate UvrD.
A model for mismatch repair in E. coli
Based on previously published data and the new results reported here, we propose a working model for mismatch repair in E. coli (Figure 7). After binding to a mismatch site, MutS recruits MutL in the presence of ATP to form a MutS(ATP)–MutL-mismatch ternary complex (Figure 7, middle left). This complex extends the DNA protection from ∼20 bp by a binary MutS–DNA complex to ∼100 bp (Grilley et al, 1989; Schofield et al, 2001). The extended shape of MutL and multiple DNA-contacting sites facing the central cavity may account for the large span of the DNase I footprint.
Figure 7.
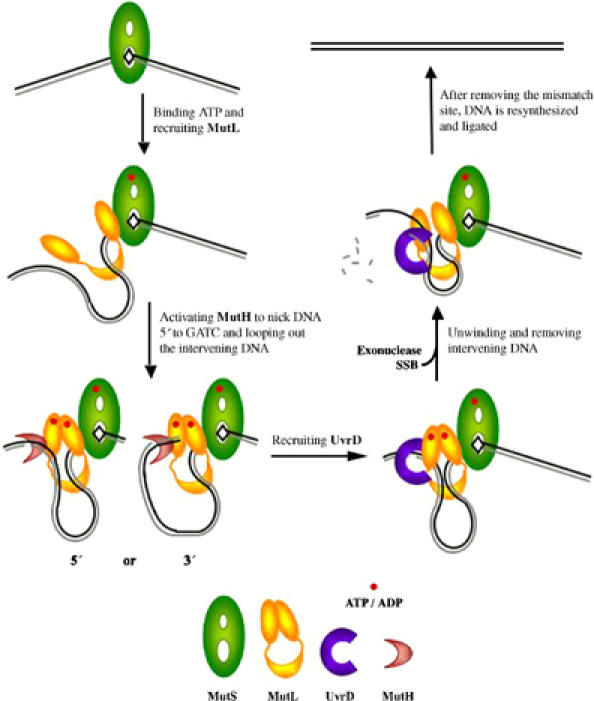
A working hypothesis of MutL-mediated mismatch repair in E. coli. MutS, MutL, MutH and UvrD are shape- and color-coded, and methylated (template) and unmethylated (daughter) DNA strands are shown in black and gray, respectively. After MutS binds to a mismatch site (top left), it recruits MutL in the presence of ATP (shown as a red dot) to form a MutS(ATP)–MutL–DNA ternary complex (middle left). This ternary complex activates MutH to cleave the unmethylated strand at a hemimethylated GATC site. Cleavage either 5′ or 3′ to the mismatch site is possible because both orientations of a GATC site can be accommodated (bottom left). For UvrD activation, MutL contacts DNA near both the mismatch and the nicked GATC sites and loops out the intervening sequence. In the presence of ATP, a ‘closed' MutS–MutL–UvrD–DNA complex is formed (bottom right). ATP hydrolysis by MutL ‘opens' the MutS–MutL–UvrD–DNA complex to release topological constrains and reduce the size of DNA loop as UvrD unwinds the DNA (middle right). Completion of mismatch repair requires DNA re-synthesis and strand ligation (top right).
This MutS(ATP)–MutL-mismatch ternary complex recruits the endonuclease MutH. Mismatch-dependent activation of MutH requires MutL to bind and hydrolyze ATP (Junop et al, 2001). ATP binding by MutL leads to association of the N-terminal ATPase domains, which may establish communication between MutS and MutH and ensure the mismatch repair specificity. ATP hydrolysis is required probably because the hydrolysis product, ADP, is more effective than ATP in enabling MutL to activate MutH (MS Junop and W Yang, unpublished data). Since MutS remains bound to the mismatch site (Junop et al, 2001; Wang and Hays, 2004), to bring MutH into proximity with the MutS(ATP)–MutL-mismatch complex requires looping out DNA between the mismatch and hemimethylated GATC cleavage site (Yang et al, 2000). Due to the asymmetry of MutS–DNA interactions (Lamers et al, 2000; Obmolova et al, 2000), the MutS(ATP)–MutL-mismatch complex is likely to be stereo-specific and directional. Depending on whether the hemimethylated GATC site is 5′ or 3′ to the mismatch site, the DNA loop of the intervening sequence needs to be configured differently to place the scissile bond in the MutH active site (Modrich and Lahue, 1996). Our observation that DNA binding by MutL is not required for MutH activation (Junop et al, 2003) suggests that DNA adjacent to the MutH cleavage site is unconstrained and the DNA substrate can approach MutH from either direction (Figure 7, bottom left).
After the daughter strand is nicked, MutH leaves and UvrD is recruited. The requirement for MutL to interact with both UvrD and DNA substrate (Figure 6) constrains how UvrD is loaded. We propose that, during activation of UvrD, MutL contacts DNA adjacent to the mismatch site as well as where UvrD unwinds the duplex and loops out the intervening sequence in its large central cavity (Figure 7, bottom right). The orientation of UvrD is thus directed by the MutS–MutL–UvrD–DNA complex and not by the polarity of the nick relative to the mismatch site. This proposed model may also apply to eukaryotic mismatch repair. Not only are the structures of MutL homologs conserved, recruiting of Exonuclease I by human MutSα and MutLα to remove DNA from a nick either 5′ or 3′ to the mismatch site is reminiscent of UvrD activation by MutL (Genschel et al, 2002).
Although ATP hydrolysis by MutL is not required for the activation of UvrD in our helicase activity assays (Figure 6), it may play a role in UvrD activation in vivo. ATP binding by MutL, which promotes association of the ATPase domain, may facilitate the formation of a ‘closed' MutS–MutL–UvrD–DNA complex (Figure 7, bottom right) and increase UvrD processivity. ATP hydrolysis by MutL, which dissociates the ATPase domains, may open the MutS–MutL–UvrD–DNA complex to release topological tensions and reduce the size of DNA loop as UvrD unwinds DNA (Figure 7, middle right). The slow ATPase cycle of MutL may maximize the number of base pairs unwound each time when the MutS–MutL–UvrD complex closes on a DNA substrate. To unwind DNA past the mismatch site, UvrD may have to displace MutS and MutL.
Materials and methods
Protein preparation and crystallization
Three mutL C-terminal fragments were cloned into pET15b (Novagen) using the NdeI and BamHI restriction sites to generate pWY1293 (LC24), pWY1294 (LC22) and pWY1295 (LC20). Histidine-tagged LC24, LC22 and LC20 proteins were purified using a Ni-chelating affinity column. His tags were removed by thrombin digestion, and the proteins were further purified over a MonoS column and concentrated to ∼10 mg/ml in 20 mM Tris (pH 8), 150 mM NaCl, 0.1 mM EDTA, 5 mM DTT, 5% glycerol and 3% isopropanol for storage.
Se-Met-labeled LC20 was produced in B834(DE3) cells as described (Hendrickson et al, 1990). Crystals of LC20 were grown using the hanging-drop vapor diffusion method at 4°C against the reservoir containing 1.0–1.5 M NaCl, 60 mM Li2SO4 and 100 mM sodium acetate (pH 4.6) and improved by macro-seeding. For flash freezing, 25% glycerol was added to the mother liquor.
Structure determination and refinement
The LC20 crystals diffracted X-rays to better than 2.1 Å with 0.12° mosaicity. A three-wavelength MAD data set of a Se-Met crystal was collected at X9B in NSLS, Brookhaven National Laboratory. Data were processed using HKL2000 (Otwinowski and Minor, 1997). Four out of six Se sites were found and refined at 2.5 Å resolution using SOLVE (Terwilliger, 2003). Phases were improved by solvent flattening using DM (CCP4, 1994). Structural models were built and refined using CNS and O (Jones et al, 1991; Brünger et al, 1998). The final model contains an LC20 dimer (residues 433–613 and 432–613), 2 Cl−, 1 Na+, 292 water, 2 isopropanol and 2 glycerol molecules. Over 89% of the residues are within the most favored regions in the Ramachandran plot and none in disallowed regions.
Preparation of mutant MutL
MutL mutants were derived from plasmid pTX418 using QuikChange (Stratagene) and designated as pWY1383 (N33A), pWY1368 (MutLΔ1), pWY1369 (MutLΔ2), pWY1370 (MutLΔ3), pWY1404 (P-1), pWY1400 (P-2) and pWY1398 (P-3). pWY1431 (MutLΔ4), pWY1432 (MutLΔ5), pWY1433 (MutLΔ6) and pWY1434 (MutLΔ7) were derived from pWY1369. The coding regions were transferred into pProEXHTb (Life Technologies), which encodes an N-terminal His tag removable by Tev protease. Mutations were verified using DNA sequencer PRISM-310 (ABI). Expression and purification of mutant proteins were carried out similarly to wild-type MutL (Ban and Yang, 1998).
Sedimentation equilibrium analysis
Purified LC24, LC22 and LC20 were dialyzed into 200 mM NaCl, 20 mM Tris (pH 8.0), 4.2 mM 2-mercaptoethanol, 0.1 mM EDTA and 5% (v/v) glycerol. Sedimentation equilibrium experiments were conducted at 4.0°C on a Beckman Optima XL-A analytical ultracentrifuge. Data were acquired at rotor speeds ranging from 8000 to 12 000 rpm. Equilibrium was achieved within 48 h. Data were analyzed in terms of a single ideal solute to obtain the buoyant molecular mass, M(1−vρ). Experimental values of the molecular mass M were determined using densities, ρ, and v as described (Perkins, 1986).
Dynamic light-scattering experiment
Wildtype, Δ2, Δ5 and Δ7 mutant proteins were incubated with AMPPNP and purified by gel filtration in 20 mM Tris (pH 8.0), 0.2 M KCl, 5 mM MgCl2, 1 mM DTT and 0.1 mM EDTA. Half of each sample was treated with 8 mM EDTA to dissociate the N-terminal domains. The translational diffusion coefficient D was measured from autocorrelation analysis of the quasielastically scattered light. Autocorrelation functions were accumulated from 200 μl of ∼0.2 mg/ml protein samples for 1–3 min at 24.0°C using a Brookhaven Instruments BI-9000 AT autocorrelator at angles θ ranging from 90 to 45° and sampling time of 0.1 μs–40 ms. An argon ion laser (Lexel, Model 95) was used in the TEM00 mode at 514.5 nm. Diffusion coefficient, Stokes radius and frictional coefficient were calculated using Brookhaven Instruments analysis software. A series of models with the dimeric LN40 and LC20 separated from 60 to 180 Å along a shared dyad axis were constructed. Theoretical diffusion coefficient and Stokes radius of these models were calculated using HYDROPRO (Garcia de la Torre et al, 2000).
DNA mobility shift assays
MutL binds equally well to a 110 nt ssDNA or 110 bp dsDNA, and ssDNA binding was tested in this study. A 32P 5′-end-labeled single-stranded 110-mer DNA (5 nM) was incubated with 10, 20, 40, 80, 160 and 640 nM MutL proteins (monomer) in 20 mM Tris (pH 8.0), 90 mM KCl, 1 mM DTT, 0.1 mg/ml BSA on ice for 1 h. ATP (1 mM) or AMPPNP with 5 mM MgCl2 was added to the binding buffer when needed. Reaction mixtures (15 μl) were analyzed on 4.5% tris-glycine polyacrylamide gels and quantified using TYPHOON 8600 (Molecular Dynamics). All in vitro assays were repeated at least three times. The mean values with standard deviations are presented.
ATPase activity assays
The ATPase activity of MutL mutants was assayed as described (Ban et al, 1999). Proteins (1.7 μM) were incubated with α-32P-labeled ATP (20 μM–1 mM) in 20 mM Tris (pH 8), 1 mM DTT, 90 mM KCl and 5 mM MgCl2 for 2 h at 22°C or 1 h at 37°C. Reactions were initiated by the addition of α-32P-labeled ATP and terminated by the addition of an equal volume of 50 mM EDTA.
Helicase assay
Native UvrD was overexpressed in BL21 Star™ (DE3)pLysS cells (Invitrogen) at 25°C, purified over heparin and MonoQ columns and stored in 20 mM Tris (pH 8.0), 0.1 mM EDTA, 5 mM DTT, 200 mM NaCl and 20% glycerol. A 68 bp dsDNA substrate containing a G:T mismatch (bold) and a nick at the GATC site (underlined) was prepared by annealing 32P 5′ end-labeled 5′-P-d(ACATGCGGTACCAAGCTTCTCGAGG) with 5′-d(GATCCTTGAATTCCAATAGGCCTGCCCTG GAAATACAGGTTTT) and 5′-d(GAAAACCTGTATTTTCAGGGCAGGCCTAT TGGAATTCAACGATCCTCGAGAAGCTTGGTACCG CATG). φX174 ssDNA was annealed with a 32P 5′ end-labeled 125-mer oligonucleotide. For MutL titration, 1 nM circular DNA substrate and increasing amount of MutL, LN40 or LC24 were mixed in 20 mM Tris (pH 7.5), 50 mM NaCl, 3 mM MgCl2, 3 mM ATP, 4.5 mM β-mercaptoethanol and 0.1 mg/ml bovine serum albumin, and reactions were initiated by addition of 1 nM UvrD, incubated for 15 min at 37°C, and stopped by the addition of an equal volume of 30% glycerol, 0.2% SDS, 5 mM EDTA and 100 μg/μl proteinase K (New England Biolabs). For UvrD activation assay, 1 nM MutL (dimer), 1 nM UvrD and 1 nM circular or linear DNA substrates were used. Reaction time for the linear DNA substrates was 5 min at 22°C. Reaction samples (10 μl) were analyzed on a 10% (linear DNA) or 4.5% (circular DNA) TBE gel and quantified using TYPHOON 8600.
Protein crosslinking by BS3
MutL, LN40 or LC30 (2.5 μM) and 2.5 μM UvrD were incubated with 0.11, 0.33, 1 or 2 mM BS3 in 20 mM Hepes (pH 7.5), 90 mM KCl, 5 mM MgCl2 and 1 mM DTT for 1 h at 22°C. AMPPNP (3 mM) and 7.5 μM dsDNA, which was made by annealing a 23-mer (5′GGACGAGCCGCGCGCTAGCGTCG3′) and a 43-mer (5′CGACGCTAGCGTGCGGCTCGTCCTCA TGGTCATGGTCATCTAG3′), were added as indicated. Reaction products (10 μl) were separated on 4–12% SDS gels and stained with Coomassie blue.
In vivo mismatch repair assay
The E. coli strain CC107 is ara Δ(gpt-lac)5 thi/F′128 lacIZ proA+B+ (Cupples et al, 1990). CC107 mutL:: miniTn10 was constructed by transducing CC107 to Tetr with a P-1 vir lysate grown on strains carrying a miniTn10 insertions in mutL (Miller et al, unpublished data). CC107 mutL was transformed with the plasmid constructs of mutL and plated on LB plus ampicillin (Junop et al, 2003). The frequencies of rifampicin-resistant (Rifr) mutants were determined by plating samples of overnight cultures grown in LB (plus ampicillin if the Ampr-conferring plasmid was used) on plates with 100 μg/ml rifampicin, incubating at 37°C for 20 h, and scoring the Rifr colonies. Dilutions were also plated on LB plates to determine the titer.
Atomic coordinates and structure factors of LC20 have been deposited with the Protein Data Bank (accession code 1X9Z).
Acknowledgments
We thank Dr Z Dauter for synchrotron beam line support, Dr J Hurley for help with MAD data collection, Dr S Matson for the UvrD expression vector, Dr M Junop for sharing unpublished data, Dr D Hinton for help with the KMnO4 foot-printing assay, and Drs R Craigie and D Leahy for reading the manuscript. We also thank one referee for detailed and insightful comments. AG and SR-M have been recipients of post-doctoral fellowship from the Human Frontiers Science Program.
References
- Altschul SF, Gish W (1996) Local alignment statistics. Methods Enzymol 266: 460–480 [DOI] [PubMed] [Google Scholar]
- Argueso JL, Kijas AW, Sarin S, Heck J, Waase M, Alani E (2003) Systematic mutagenesis of the Saccharomyces cerevisiae MLH1 gene reveals distinct roles for Mlh1p in meiotic crossing over and in vegetative and meiotic mismatch repair. Mol Cell Biol 23: 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronshtam A, Marinus MG (1996) Dominant negative mutator mutations in the mutL gene of Escherichia coli. Nucleic Acids Res 24: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban C, Junop M, Yang W (1999) Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97: 85–97 [DOI] [PubMed] [Google Scholar]
- Ban C, Yang W (1998) Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 95: 541–552 [DOI] [PubMed] [Google Scholar]
- Bende SM, Grafström RH (1991) The DNA binding properties of the MutL protein isolated from Escherichia coli. Nucleic Acid Res 19: 1549–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borts RH, Chambers SR, Abdullah MF (2000) The many faces of mismatch repair in meiosis. Mutat Res 451: 129–150 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P (2001) In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci USA 98: 6765–6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M (1987) Ribbon models of macromolecules. J Mol Graphics 5: 103–106 [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cupples CG, Cabrera M, Cruz C, Miller JH (1990) A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics 125: 275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao V, Modrich P (1998) Mismatch-, MutS-, MutL-, and helicase II-dependent unwinding from the single-strand break of an incised heteroduplex. J Biol Chem 273: 9202–9207 [DOI] [PubMed] [Google Scholar]
- Dixon WJ, Massey FJJ (1969) Introduction to Statistical Analysis, p 349. New York: McGraw Hill [Google Scholar]
- Drotschmann K, Hall MC, Shcherbakova PV, Wang H, Erie DA, Brownewell FR, Kool ET, Kunkel TA (2002a) DNA binding properties of the yeast Msh2–Msh6 and Mlh1–Pms1 heterodimers. Biol Chem 383: 969–975 [DOI] [PubMed] [Google Scholar]
- Drotschmann K, Yang W, Kunkel TA (2002b) Evidence for sequential action of two ATPase active sites in yeast Msh2–Msh6. DNA Repair (Amst) 1: 743–753 [DOI] [PubMed] [Google Scholar]
- Dutta R, Inouye M (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25: 24–28 [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre J, Huertas ML, Carrasco B (2000) Calculation of hydrodynamic properties of globular proteins from their atomic level structure. Biophys J 78: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschel J, Bazemore LR, Modrich P (2002) Human exonuclease I is required for 5′ and 3′ mismatch repair. J Biol Chem 277: 13302–13311 [DOI] [PubMed] [Google Scholar]
- Grilley M, Welsh KM, Su S-S, Modrich P (1989) Isolation and characterization of the Escherichia coli mutL gene product. J Biol Chem 264: 1000–1004 [PubMed] [Google Scholar]
- Habraken Y, Sung P, Prakash L, Prakash S (1998) ATP-dependent assembly of a ternary complex consisting of a DNA mismatch and the yeast MSH2–MSH6 and MLH1–PMS1 protein complexes. J Biol Chem 273: 9837–9841 [DOI] [PubMed] [Google Scholar]
- Hall MC, Jordan JR, Matson SW (1998) Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J 17: 1535–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Matson SW (1999) The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J Biol Chem 274: 1306–1312 [DOI] [PubMed] [Google Scholar]
- Hall MC, Shcherbakova PV, Kunkel TA (2002) Differential ATP binding and intrinsic ATP hydrolysis by amino-terminal domains of the yeast Mlh1 and Pms1 proteins. J Biol Chem 277: 3673–3679 [DOI] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S (2000) DNA mismatch repair and genetic instability. Annu Rev Genet 34: 359–399 [DOI] [PubMed] [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM (1990) Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J 9: 1665–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Dali: version 2.0. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Iaccarino I, Marra G, Palombo F, Jiricny J (1998) hMSH2 and hMSH6 play distinct roles in mismatch binding and contribute differently to the ATPase activity of hMutSalpha. EMBO J 17: 2677–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292: 195–202 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW (1991) Improved methods for building models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Junop MS, Obmolova G, Rausch K, Hsieh P, Yang W (2001) Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell 7: 1–12 [DOI] [PubMed] [Google Scholar]
- Junop MS, Yang W, Funchain P, Clendenin W, Miller JH (2003) In vitro and in vivo studies of MutS, MutL and MutH mutants: correlation of mismatch repair and DNA recombination. DNA Repair (Amst) 2: 387–405 [DOI] [PubMed] [Google Scholar]
- Kondo E, Horii A, Fukushige S (2001) The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS2. Nucleic Acids Res 29: 1695–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers MH, Perrakis A, Enzlin JH, Winterwerp HH, de Wind N, Sixma TK (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature 407: 711–717 [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Colman PM (1993) Shape complementarity at protein/protein interfaces. J Mol Biol 234: 946–950 [DOI] [PubMed] [Google Scholar]
- Li GM (2003) DNA mismatch repair and cancer. Front Biosci 8: d997–d1017 [DOI] [PubMed] [Google Scholar]
- Li GM, Modrich P (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci USA 92: 1950–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkin SM, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis AD, Lynch HT, Elliott RM, Collins FS (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet 24: 27–35 [DOI] [PubMed] [Google Scholar]
- McCarthy JG, Rich A (1991) Detection of an unusual distortion in A-tract DNA using KMnO4: effect of temperature and distamycin on the altered conformation. Nucleic Acids Res 19: 3421–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechanic LE, Frankel BA, Matson SW (2000) Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem 275: 38337–38346 [DOI] [PubMed] [Google Scholar]
- Modrich P, Lahue R (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem 65: 101–133 [DOI] [PubMed] [Google Scholar]
- Obmolova G, Ban C, Hsieh P, Yang W (2000) Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407: 703–710 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Peltomaki P (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 21: 1174–1179 [DOI] [PubMed] [Google Scholar]
- Perkins SJ (1986) Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem 15: 169–180 [DOI] [PubMed] [Google Scholar]
- Qin B, Lam SS, Lin K (1999) Crystal structure of a transcriptionally active Smad4 fragment. Struct Fold Des 7: 1493–1503 [DOI] [PubMed] [Google Scholar]
- Raschle M, Marra G, Nystrom-Lahti M, Schar P, Jiricny J (1999) Identification of hMutLb, a heterodimer of hMLH1 and hPMS1. J Biol Chem 274: 32368–32375 [DOI] [PubMed] [Google Scholar]
- Runyon GT, Wong I, Lohman TM (1993) Overexpression, purification, DNA binding, and dimerization of the Escherichia coli uvrD gene product (helicase II). Biochemistry 32: 602–612 [DOI] [PubMed] [Google Scholar]
- Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P (2001) Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem 276: 28291–28299 [DOI] [PubMed] [Google Scholar]
- Spampinato C, Modrich P (2000) The MutL ATPase is required for mismatch repair. J Biol Chem 275: 9863–9869 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2003) SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol 374: 22–37 [DOI] [PubMed] [Google Scholar]
- Wang H, Hays JB (2004) Signaling from DNA mispairs to mismatch-repair excision sites despite intervening blockades. EMBO J 23: 2126–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Platt JL, Cascalho M (2003) Dimerization of MLH1 and PMS2 limits nuclear localization of MutLa. Mol Cell Biol 23: 3320–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Dao V, Modrich P (1998) MutS and MutL activate DNA helicase II in a mismatch-dependent manner. J Biol Chem 273: 9197–9201 [DOI] [PubMed] [Google Scholar]
- Yang W, Junop MS, Ban C, Obmolova G, Hsieh P (2000) DNA mismatch repair: from structure to mechanism. Cold Spring Harb Symp Quant Biol 65: 225–232 [DOI] [PubMed] [Google Scholar]