

Abstract
Accumulation of mutant proteins into misfolded species and aggregates is characteristic for diverse neurodegenerative diseases including the polyglutamine diseases. While several studies have suggested that polyglutamine protein aggregates impair the ubiquitin–proteasome system, the molecular mechanisms underlying the interaction between polyglutamine proteins and the proteasome have remained elusive. In this study, we use fluorescence live-cell imaging to demonstrate that the proteasome is sequestered irreversibly within aggregates of overexpressed N-terminal mutant Huntingtin fragment or simple polyglutamine expansion proteins. Moreover, by direct targeting of polyglutamine proteins for proteasomal degradation, we observe incomplete degradation of these substrates both in vitro and in vivo. Thus, our data reveal that intrinsic properties of the polyglutamine proteins prevent their efficient degradation and clearance. Additionally, fluorescence resonance energy transfer is detected between the proteasome and aggregated polyglutamine proteins indicative of a close and stable interaction. We propose that polyglutamine-containing proteins are kinetically trapped within proteasomes, which could explain their deleterious effects on cellular function over time.
Keywords: FLIP, FRAP, FRET, polyglutamine proteins, proteasome
Introduction
The formation of protein aggregates or inclusions in select neurons is a hallmark of neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and the polyglutamine disorders. Expansion of polyglutamine repeats in otherwise unrelated proteins is responsible for at least nine inherited disorders including Huntington's disease, spinocerebellar ataxias 1,2,3,6,7,17, spinal and bulbar muscular atrophy, and dentatorubral and pallidoluysian atrophy (Zoghbi and Orr, 2000; Nakamura et al, 2001). Although the role of polyglutamine aggregates in pathogenesis is still unclear, aggregates have been shown to contain critical cellular components such as the transcription factors TATA binding protein (TBP) and CREB binding protein (CBP), cytoskeletal proteins, molecular chaperones such as Hsc70, Hsp70, Hdj-1, and Hdj-2, as well as ubiquitin and proteasome components (Davies et al, 1997; Cummings et al, 1998; Perez et al, 1998; Kazantsev et al, 1999; Jana et al, 2001; Suhr et al, 2001). As components of the ubiquitin–proteasome pathway colocalize with the aggregates, a question that remains is why polyglutamine proteins are not efficiently eliminated by proteasomal degradation. It has been reported that cells expressing mutant ataxin-1 or Huntingtin exhibit decreased proteasome activity and that degradation of the artificial proteasomal substrate Ub-GFP is impaired in cells overexpressing a fragment of mutant Huntingtin or a folding mutant of cystic fibrosis transmembrane conductance regulator (CFTR) (Cummings et al, 1999; Bence et al, 2001; Jana et al, 2001). Moreover, prevention of polyglutamine oligomerization by Congo red ameliorated polyglutamine-induced decrease in proteasome activity, suggesting that the inhibitory effect of polyglutamine proteins is due to their self-association properties (Sanchez et al, 2003). While these studies suggest that aggregated polyglutamine proteins are inhibitory to the proteasome, recent dynamic image analysis indicates that the proteasome associates only transiently with mutant ataxin-1-containing aggregates (Stenoien et al, 2002). In this study, we show that protein aggregates, initiated by the expression of N-terminal mutant Huntingtin fragment or simple polyglutamine expansion proteins, associate irreversibly with the proteasome via direct interaction. As degradation-tagged polyglutamine proteins are also incompletely degraded by the proteasome in vitro and in vivo, our data reveal that polyglutamine-containing proteins directly interfere with proteasome function.
Results
To monitor the effect of Huntingtin protein (Htt) on localization and mobility of the proteasome, we employed a green fluorescent protein (GFP) fusion of the 20S proteasome β-subunit LMP2. LMP2-GFP serves as an excellent monitor of the assembled proteasome, as it has previously been shown to be quantitatively incorporated into active proteasomes (Reits et al, 1997). LMP2-GFP exhibited a diffuse cytoplasmic and nuclear localization in HeLa cells that was unaltered when coexpressed with soluble N-terminal Htt fragment containing a stretch of 23 glutamines (Htt-Q23; Figure 1A, upper panel). However, when LMP2-GFP was coexpressed with Htt-Q65, the proteasome was partially redistributed into discrete cytoplasmic and nuclear foci that colocalized with Htt-Q65 aggregates (Figure 1A, upper panel). The relocalization of LMP2-GFP is not due to different expression levels of the Htt proteins nor did the Htt proteins affect the expression level of LMP2-GFP (Supplementary Figure S1). Similar redistribution of LMP2-GFP was observed upon coexpression with Flag-tagged 81 glutamines (Flag-Q81; Figure 1A, upper panel), showing that the intrinsic properties of the extended polyglutamine stretch are sufficient to mediate the effect observed on LMP2-GFP distribution. These results are not a consequence of transient overexpression of LMP2-GFP, as expression of Flag-Q81 alone also resulted in relocalization of endogenous proteasomes to aggregates (Figure 1A, lower panel). These observations are in accordance with previous reports on accumulation of endogenous proteasome to aggregates of mutant Htt in cell lines or in R6/1 transgenic mice (Jana et al, 2001; Waelter et al, 2001).
Figure 1.
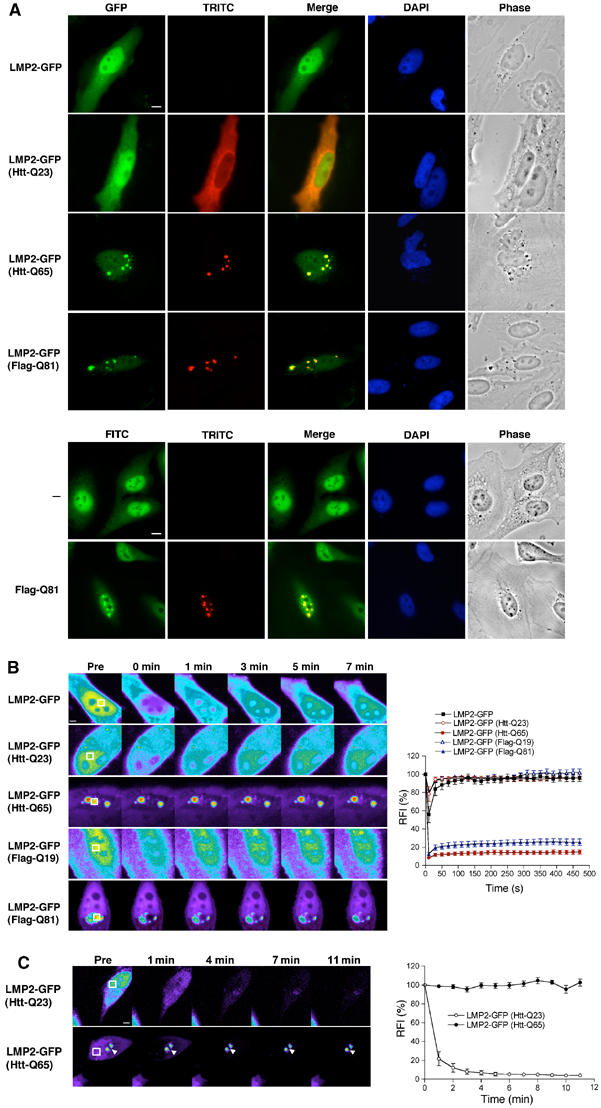
The proteasome is stably associated with aggregates of mutant Huntingtin and simple polyglutamine expansion proteins. (A) Colocalization of the proteasome with Htt-Q65 or Flag-Q81 aggregates. (Upper panel) HeLa cells were transfected with constructs encoding LMP2-GFP together with Htt-Q23, Htt-Q65, or Flag-Q81 as indicated. The Htt proteins and Flag-Q81 were detected with the HP-1 antibody and an antibody against the Flag epitope, respectively, followed by a TRITC-conjugated secondary antibody. Cells expressing LMP2-GFP (GFP, green) and Htt proteins or Flag-Q81 (TRITC, red) were visualized by fluorescence microscopy and phase contrast microscopy (Phase). Colocalization was illustrated by merging GFP and TRITC images (Merge). DNA was stained with DAPI (DAPI). (Lower panel) Localization of endogenous proteasome in the absence or presence of Flag-Q81 expression was detected using an anti-20S proteasome antibody followed by an FITC-conjugated secondary antibody (FITC, green). Scale bar represents 10 μm. (B) FRAP analysis of LMP2-GFP. Cells were imaged before photobleaching (Pre) of the defined area (white box) and at the indicated times after photobleaching. Note that the intensity of the images is scaled differently between the samples. Scale bar represents 5 μm. Quantitative FRAP analysis of soluble or aggregate-associated LMP2-GFP is shown in the graph in the right panel. The relative fluorescence intensity (RFI) was determined for each time point and is represented as the average±s.e.m. of 5–8 cells. (C) FLIP analysis of LMP2-GFP. Single scan images were obtained before (Pre) and at the indicated times between consecutive bleach pulses of the boxed area (white box). Scale bar represents 5 μm. The RFI was determined for each time point and is represented as the average±s.e.m. of five cells (right panel).
To examine whether proteasomes are dynamic components of mutant Htt or simple polyglutamine expansion protein aggregates, we performed fluorescence recovery after photobleaching (FRAP) (Lippincott-Schwartz et al, 2001). When a defined region in the nucleus of a cell expressing LMP2-GFP was photobleached, no discernable bleached zone could be detected and there was a decrease in fluorescence intensity throughout the nucleus indicative of highly mobile LMP2-GFP (Figure 1B). Quantitative analysis of LMP2-GFP fluorescence demonstrated an immediate recovery from photobleaching with a mobile fraction of 98.5±2.6% (Figure 1B, Table I). Similar mobility was detected for cytoplasmic LMP2-GFP (data not shown). Coexpression of LMP2-GFP with Htt-Q23 or Flag-Q19 had no effect on the mobility of the proteasome (Figure 1B, Table I). In contrast, LMP2-GFP associated with Htt-Q65 or Flag-Q81 aggregates displayed no fluorescence recovery and the mobile fraction was reduced to 7.4±4.9 and 15.9±5.9%, respectively (Figure 1B, Table I). Similar results were obtained whether the entire LMP2-GFP signal associated with Htt-Q65 aggregates or a fraction of the associated LMP2-GFP was photobleached (data not shown). By comparison, the mobility of LMP2-GFP outside an aggregate was not reduced (Supplementary Figure S2). Note that the images are scaled differently between the samples to avoid saturation of the LMP2-GFP fluorescence intensity associated with Htt-Q65 or Flag-Q81 aggregates and that a fraction of soluble LMP2-GFP is present in these samples (Supplementary Figure S2). The immobility of LMP2-GFP associated with a polyglutamine aggregate reflects the properties of Q82-YFP aggregates (Figure 2C) (Kim et al, 2002). Moreover, the rapid recovery of LMP2-GFP fluorescence in cells coexpressing proteins with shorter polyglutamine stretches is similar to the recovery of soluble Q19-YFP (Figure 2C).
Table 1.
LMP2-GFP mobility
| Protein | Mobile fraction (%) |
|---|---|
| LMP2-GFP | 98.5±2.6 |
| LMP2-GFP (Htt-Q23) | 97.8±1.9 |
| LMP2-GFP (Htt-Q65 aggregate) | 7.4±4.9 |
| LMP2-GFP (Flag-Q19) | 98.5±1.3 |
| LMP2-GFP (Flag-Q81 aggregate) | 15.9±5.9 |
| The mobile fraction is based on the FRAP analysis in Figure 1B. | |
Figure 2b.
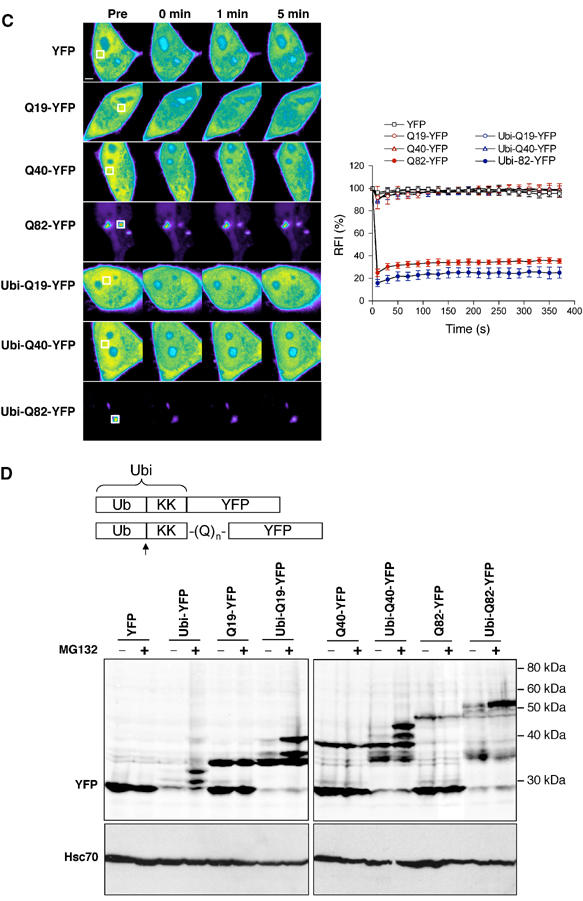
(C) FRAP analysis of untagged and degradation-tagged polyglutamine-YFP proteins. Cells were imaged before photobleaching (Pre) of the defined area (white box) and at the indicated times after photobleaching. Scale bar represents 5 μm. Quantitative FRAP analysis of the respective polyglutamine-YFP proteins is shown in the graph. The RFI was determined for each time point and is represented as the average±s.e.m. of 3–5 cells. (D) Schematic representation of the degradation-tagged YFP fusion proteins. The Ubi consists of a ubiquitin molecule (Ub) followed by a 40-amino-acid lysine-containing linker region. Ubiquitin cleavage is indicated by an arrow. (Upper panel) Western blot analysis of HeLa cells expressing untagged or degradation-tagged YFP fusion proteins in the absence or presence of MG132 (10 μM). (Lower panel) Hsc70 was used as a loading control.
We then investigated whether LMP2-GFP was released from the Htt-Q65 aggregates using fluorescence loss in photobleaching (FLIP) (Lippincott-Schwartz et al, 2001). The fluorescence intensity of LMP2-GFP in an Htt-Q65 aggregate was monitored, while a region outside the aggregate, but within the same cell, was continuously photobleached. If LMP2-GFP is released from the Htt-Q65 aggregates, we would expect to observe a decrease in the fluorescence intensity of the aggregate-associated LMP2-GFP. However, FLIP analysis revealed no decrease in the fluorescence intensity of LMP2-GFP in Htt-Q65 aggregates (Figure 1C), whereas the fluorescence intensity of LMP2-GFP coexpressed with Htt-Q23 was reduced by ∼80% within the first minute of photobleaching (Figure 1C). These data indicate that the LMP2-GFP molecules associated with a polyglutamine aggregate are tightly bound with a slow off rate. Together, the FRAP and FLIP analyses demonstrate that the proteasome is sequestered irreversibly within aggregates formed by the expression of Htt-Q65 or Flag-Q81. This irreversible sequestration of the proteasome in Htt-Q65 and Flag-Q81 aggregates is similar to the previously reported association between Htt aggregates and glutamine-rich proteins such as CBP and TBP, but different from the dynamic, substrate-like transient interaction with Hsp70 (Chai et al, 2002; Kim et al, 2002).
What is the basis of sequestration of the proteasome by mutant Htt or simple polyglutamine proteins? To examine whether direct targeting of polyglutamine proteins for proteasomal degradation would affect their association with the proteasome, we generated proteasome substrates by employing the N-end rule pathway (Varshavsky, 1996). A ubiquitin moiety was attached to the N-terminus of a simple polyglutamine protein via a 40-amino-acid linker region containing two lysines (Ubi). The ubiquitin moiety is rapidly cleaved off by endogenous ubiquitin hydrolases resulting in a new destabilizing N-terminus that promotes ubiquitination of internal lysine residues leading to degradation by the proteasome from the N-terminus (Gonda et al, 1989; Varshavsky, 1996). FRAP analysis showed that LMP2-GFP was diffusely localized and highly mobile when coexpressed with Ubi-Q16-Flag and in foci of restricted mobility when coexpressed with Ubi-Q78-Flag (Figure 2A). Therefore, we conclude that the degradation tag did not affect the dynamics of proteasome association with polyglutamine proteins.
Figure 2a.
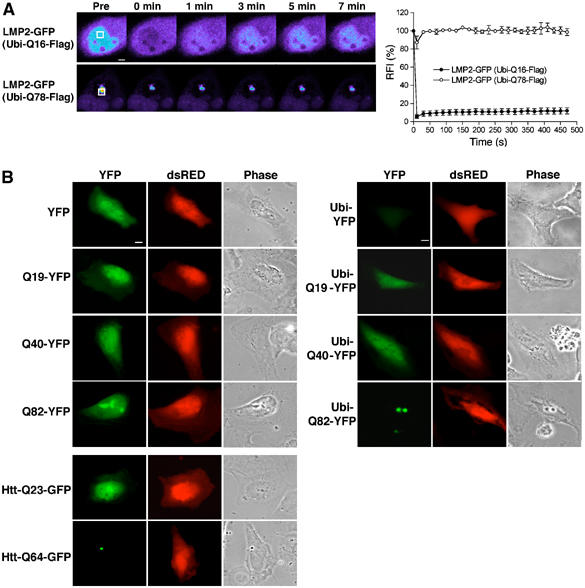
Degradation-tagged simple polyglutamine proteins are detected in vivo. (A) FRAP analysis of LMP2-GFP coexpressed with Ubi-Flag-Q16 or Ubi-Flag-Q78. Cells were imaged before photobleaching (Pre) of the defined area (white box) and at the indicated times after photobleaching. Scale bar represents 5 μm. Quantitative FRAP analysis of soluble or aggregate-associated LMP2-GFP is shown in the graph in the right panel. RFI was determined for each time point and is represented as the average±s.e.m. of four cells. (B) Localization of untagged or degradation-tagged simple polyglutamine-YFP proteins. HeLa cells were transfected with constructs encoding YFP, Q19-YFP, Q40-YFP, Q82-YFP, their degradation-tagged counterparts (Ubi-), Htt-Q23-GFP, and Htt-Q64-GFP, as indicated. Coexpression of dsRED protein was used to identify transfected cells. Cells expressing YFP fusion proteins (YFP, pseudo-colored in green) and dsRED were visualized by fluorescence microscopy and phase contrast microscopy (Phase). Scale bar represents 10 μm.
One expects degradation-tagged polyglutamine proteins to be degraded completely, provided that the polyglutamine stretch itself does not interfere with proteasome activity, and thereby prevent formation of aggregates. To address this, we tested the effect of the degradation tag on polyglutamine-YFP proteins in our cell system. In accordance with earlier studies (Scherzinger et al, 1997), polyglutamine fusion proteins exhibit enhanced solubility resulting in soluble Q40-YFP and a low percentage (5%) of cells showing Q82-YFP aggregates, as shown by fluorescence microscopy (Figure 2B, left panel) and FRAP analysis (Figure 2C). In contrast to the nondegradation-tagged YFP fusion proteins, Ubi-YFP was barely detected in the transfected cells (Figure 2B, right panel, Supplementary Figure S3). However, a fluorescent signal was observed in cells transfected with Ubi-Q19-YFP, Ubi-Q40-YFP, or Ubi-Q82-YFP (Figure 2B, right panel, Supplementary Figure S3), suggesting that introduction of a polyglutamine stretch between the degradation tag and the YFP prevents efficient degradation by the proteasome. Ubi-Q82-YFP was visualized in aggregates, similar to Htt-Q64-GFP, and the number of transfected cells containing aggregates increased to 34% (Figure 2B). FRAP analysis demonstrated that Ubi-Q82-YFP was immobilized in the aggregates, whereas the diffusely expressed Ubi-Q19-YFP and Ubi-Q40 were mobile (Figure 2C). In accordance with the fluorescence microscopy data, Western blot analysis demonstrated that the amount of Ubi-YFP was significantly lower than for YFP (Figure 2D). As expected, no full-length Ubi-YFP of 40 kDa was detected, as the N-terminal ubiquitin molecule is rapidly cleaved off in the cell. Treatment of cells with the proteasome inhibitor MG132 resulted in an accumulation of N-terminal deubiquitinated Ubi-YFP species, establishing that degradation of Ubi-YFP is proteasome dependent (Figure 2D). In comparison to the levels of Ubi-YFP, increased levels of Ubi-Q19-YFP, Ubi-Q40-YFP, and Ubi-Q82-YFP were observed (Figure 2D). In the case of Ubi-Q19-YFP, the major species exhibited a mobility in SDS–PAGE that corresponded to untagged Q19-YFP (Figure 2D). In addition, higher molecular weight bands of Ubi-Q19-YFP were detected. These species most likely correspond to partially degraded N-terminal deubiquitinated Ubi-Q19-YFP, as we do not detect a band of ∼13 kDa higher molecular weight than that of Q19-YFP, which would correspond to the full-length Ubi-Q19-YFP. It is worth noting that all of the polyglutamine-containing fusion proteins exhibit mobilities on SDS–PAGE that are different from their calculated molecular weights. In the Ubi-Q40-YFP and the Ubi-Q82-YFP samples, bands migrating with molecular weight sizes similar, higher, and lower than in the corresponding untagged Q40-YFP and Q82-YFP samples were observed (Figure 2D). Similar to Ubi-Q19-YFP, no full-length Ubi-Q40-YFP and Ubi-Q82-YFP were detected. Treatment with MG132 resulted in an accumulation of the higher molecular weight species of all degradation-tagged polyglutamine-YFP proteins, while no increased accumulation of the corresponding untagged polyglutamine-YFP proteins was observed (Figure 2D). These results reveal an accumulation of partially degraded species of the various degradation-tagged polyglutamine-YFP fusion proteins in the cell. Taken together, our data show that direct targeting of polyglutamine-YFP proteins for proteasomal degradation is neither sufficient to provide efficient clearance nor to prevent aggregate formation in vivo.
To investigate whether the intrinsic properties of the degradation-tagged polyglutamine YFP proteins affect proteasome function, we compared their degradation rates in vitro. Radioactive fusion proteins were synthesized by coupledin vitro transcription and translation, and degradation by the proteasome was measured in rabbit reticulocyte lysates. As expected, Ubi-YFP was rapidly degraded with ∼35% of the protein remaining after 30 min (Figure 3). The disappearance of Ubi-Q19-YFP, Ubi-Q40-YFP, and Ubi-Q82-YFP followed similar kinetics as for Ubi-YFP. However, analysis of Ubi-polyglutamine-YFP time-course samples by SDS–PAGE revealed the appearance of stable intermediates of smaller size, whereas degradation of Ubi-YFP did not result in the accumulation of a specific degradation product (Figure 3, left panel). These species are likely products of incomplete degradation, as addition of RNase A, cycloheximide, and an excess of nonradioactive methionine did not prevent their formation (data not shown). Moreover, addition of MG132 to the lysates prevented accumulation of the degradation end products, showing that the appearance of these species is dependent on proteasome function (Supplementary Figure S4). Addition of MG132 did not block removal of the N-terminal ubiquitin molecule nor polyubiquitination of the lysine-containing linker region in front of YFP or polyglutamine-YFP (Supplementary Figure S4). In the case of Ubi-Q19-YFP and Ubi-Q40-YFP, the degradation end product appeared to be of a discrete molecular size, while a heterogeneous population of degradation products was detected for Ubi-Q82-YFP suggesting that the block in degradation is not designated by a certain glutamine residue or length of polyglutamine (Figure 3, left panel). Analysis of the degradation end products indicated molecular sizes corresponding to the predicted size of the respective polyglutamine expansion-YFP (Figure 3, lower right panel). A direct comparison of the SDS–PAGE migration of the degradation-tagged polyglutamine-YFP proteins with the corresponding untagged polyglutamine-YFP proteins revealed that the degradation end products of Ubi-Q19-YFP and Ubi-Q40-YFP, but not of Ubi-Q82-YFP, were of similar molecular weight sizes as the untagged polyglutamine proteins (Supplementary Figure S5). No significant difference in the amount of accumulated degradation end products of the Ubi-polyglutamine-YFP proteins was detected (Figure 3, graph).
Figure 3.

Polyglutamine-YFP proteins are incompletely degraded by the proteasome in vitro. Proteasome degradation of 35S-labeled Ubi-(Q)n-YFP proteins in reticulocyte lysate. (Left panel) The T lane in the autoradiograms contains the untreated Ubi-tagged YFP fusion proteins (indicated by arrowheads). Upon preincubation of substrate protein in ATP-depleted reticulocyte lysate, the N-terminal ubiquitin moiety has been removed (arrows). Degradation was initiated by addition of ATP and aliquots were collected at the indicated time points. Quantification of the autoradiograms is shown in the upper right panel. The amount of remaining substrate proteins, including their ubiquitinated forms (circle), and the degradation end product (squares) is plotted as the mean±s.e.m. of 3–4 independent experiments. The lower right panel shows an autoradiogram of Ubi-YFP after 30 min of degradation and the degradation end products of Ubi-(Q)n-YFP after 150 min.
These studies show that the progression of substrate degradation is blocked or markedly delayed when the proteasome encounters a protein with a polyglutamine stretch. In agreement with our in vitro data, the appearance of similar degradation products of the Ubi-polyglutamine-YFP proteins was detected in vivo (Figures 2D and 3). The existence of multiple degradation products in vivo could be explained by the asynchronous degradation of Ubi-polyglutamine-YFP proteins in the cells. It seems likely that the polyglutamine proteins are, over time, degraded in vivo, as Ubi-Q40-YFP and Ubi-Q82-YFP are at least partially degraded (Figure 2D). In addition, reports have shown that suppression of mutant Htt expression results in disappearance of aggregates (Yamamoto et al, 2000; Martin-Aparicio et al, 2001).
Our results suggest that the polyglutamine proteins could be trapped within the proteasome, but it is also possible that the partially degraded products are released. To investigate whether we could detect an interaction between the proteasome and polyglutamine-containing proteins in vivo, we employed acceptor photobleaching to reveal whether fluorescence resonance energy transfer (FRET) (Lippincott-Schwartz et al, 2001; Berney and Danuser, 2003) occurs. If the CFP donor and YFP acceptor fluorophores are separated by less than 100 Å, FRET will occur and is detected as an increase in the CFP intensity when YFP is photobleached. As a positive control for FRET, we used cells expressing aggregates formed by Q82-YFP and Q82-CFP or cells expressing a chimeric CFP-YFP fusion protein (Kim et al, 2002). Upon acceptor photobleaching, we detected an increase in the donor fluorescence and a transfer efficiency of 0.28±0.06 and 0.17±0.03 for Q82-CFP/Q82-YFP aggregates and CFP-YFP, respectively (Figure 4A, data not shown). Higher FRET efficiency from the polyglutamine aggregates than from the CFP-YFP chimera is in agreement with a previous report (Kim et al, 2002). No FRET signal was detected in cells coexpressing CFP and YFP (0.01±0.01). In cells expressing LMP2-CFP and Ubi-Q82-YFP, we consistently detected a FRET efficiency of 0.28±0.07 (Figure 4A). Similar results were also observed when LMP2-CFP was associated with Htt-Q78-YFP (0.14±0.05) or Q82-YFP aggregates (Figure 4A, data not shown). However, we did not detect FRET between LMP2-CFP and soluble Ubi-Q82-YFP, Htt-Q78-YFP, Q82-YFP, Ubi-Q19-YFP, Htt-Q23-YFP, or Q19-YFP (Figure 4A, data not shown).
Figure 4a.
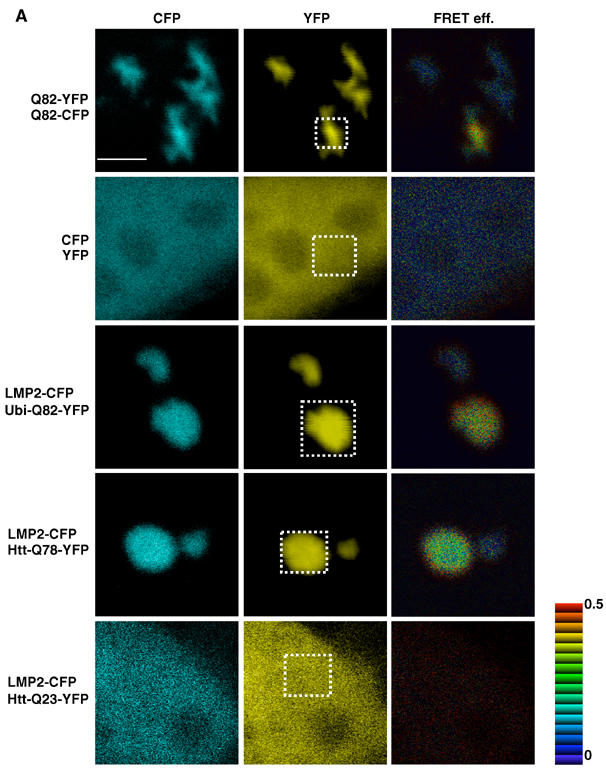
FRET occurs between proteasome and polyglutamine proteins in the aggregates. (A) Acceptor photobleaching experiments were performed using LMP2-CFP as the donor and YFP fused to Ubi-Q82, Htt-Q78, or Htt-Q23 as the acceptor fluorophores. Cells coexpressing aggregated Q82-CFP/Q82-YFP or soluble CFP and YFP were used as positive and negative controls, respectively. Each panel consists of the CFP, YFP, and the FRET efficiency image (FRET eff.) of transiently transfected HeLa cells. The YFP and CFP images were taken before photobleaching of the YFP fusion proteins in the defined area (white box). The FRET efficiency is indicated by the pseudo-color scale next to FRET efficiency image. Scale bar represent 5 μm.
To further confirm our results obtained by the acceptor photobleaching approach, we used a complementary methodology and measured the emission of the acceptor by donor excitation (Figure 4B). Using this approach, we detected FRET between LMP2-YFP and aggregated Htt-Q78-CFP or Q82-CFP, but not between LMP2-YFP and soluble Htt-Q78-CFP, Q82-CFP, or Htt-Q23-CFP (Figure 4B). Moreover, we did not detect FRET in cells expressing aggregated Q82-CFP and YFP (Figure 4B), showing that the FRET signal observed between LMP2 and aggregated polyglutamine proteins is due to a specific and close association between these proteins. Note that the lack of detectable FRET between soluble polyglutamine-containing proteins and LMP2 could be due to a technical limitation; hence, these experiments do not exclude that an interaction could occur between soluble polyglutamine-containing proteins and LMP2.
Figure 4b.
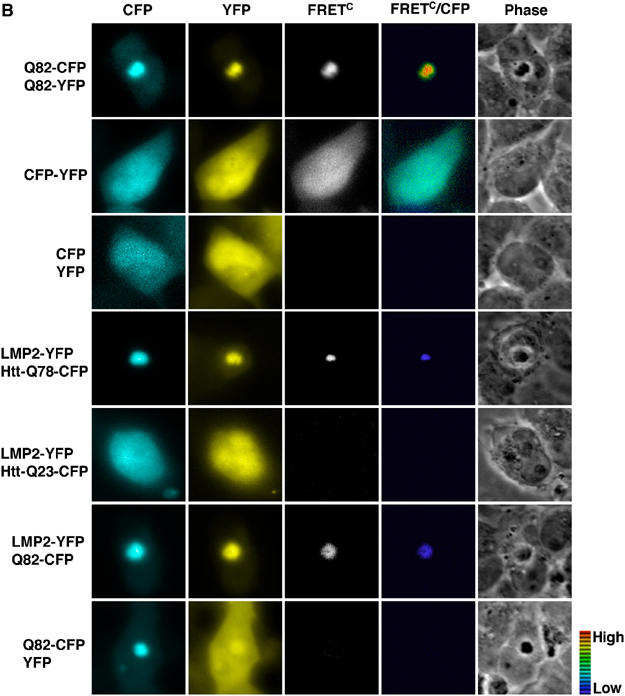
(B) To measure the emission of the acceptor by donor excitation, experiments were performed using LMP2-YFP as the acceptor fluorophore and Htt-Q78-CFP, Htt-Q23-CFP, and Q82-CFP as the donor fluorophores. Cells expressing aggregated Q82-CFP/Q82-YFP or a CFP-YFP chimera were used as positive controls, while cells expressing CFP and YFP were used as a negative control. Each panel consists of CFP, YFP, corrected FRET (FRETC), FRETC/CFP ratio, and phase images. The pseudo-color scale indicates the FRETC/CFP ratio, ranging from 0 to 2, in which violet color indicates low FRET and red indicates high FRET. No FRET is indicated by black.
Discussion
Our studies on the dynamics of the proteasome in cells overexpressing N-terminal mutant Htt or simple polyglutamine expansion proteins show an irreversible sequestration of the proteasome. Additionally, we provide evidence that polyglutamine-containing proteins are incompletely degraded, even when directly targeted for proteasomal degradation, in vitro and in vivo. By FRET analysis, we detect a stable association between the proteasome and the polyglutamine-containing proteins within the aggregates. Taken together, our data suggest that mutant Huntingtin and simple polyglutamine expansion proteins are likely kinetically trapped within the proteasome as partially degraded substrates rather than rapidly released and freely diffusing. Our studies do not exclude that some of the partially degraded polyglutamine-containing proteins are released from the proteasome.
It was recently reported (Venkatraman et al, 2004) that purified eukaryotic proteasomes, which were in an open-gated state either by addition of detergent or by using a mutant yeast proteasome, were unable to digest nonpolyubiquitinated peptides containing 10–30 glutamines. The authors suggested that these polyglutamine peptides would be released from the proteasome. The authors further speculated that longer polyglutamine stretches, as found in polyglutamine diseases, could fail to exit from the proteasome and thereby interfere with proteasome function, a suggestion that is in agreement with our observation. However, unlike their study, we demonstrate here that expanded polyglutamine-YFP fusion proteins that are targeted for degradation via the endogenous ubiquitin–proteasome pathway are at least partially degraded in HeLa cells and rabbit reticulocyte lysates (Figures 2D and 3). For example, we detected a major fragment of degradation-tagged Q40-YFP and Q82-YFP of the same molecular size as untagged Q19-YFP in HeLa cells. The accumulation of these species is decreased upon treatment with the proteasome inhibitor MG132. Taken together, our observations establish that polyglutamine proteins can undergo at least partial proteasomal degradation. Further studies will be needed to establish whether the amino-acid sequences or protein domains surrounding the polyglutamine tract influence the degradation of the polyglutamine disease proteins. Our results corroborate previous reports showing that suppression of mutant Htt expression results in disappearance of aggregates in a conditional mouse model of Huntington's disease (HD94) as well as in HD94 primary striatal neurons (Yamamoto et al, 2000; Martin-Aparicio et al, 2001). These results suggest that polyglutamine proteins could engage the proteasome machinery for longer periods than other proteasome substrates and would therefore indirectly diminish proteasome activity.
It is intriguing to consider that even the presence of Q19 could have detrimental effects on protein degradation. These results suggest that even wild-type Htt may be less efficiently degraded than a nonpolyglutamine-containing protein. However, unlike a short polyglutamine stretch, the presence of the expanded polyglutamine stretch associated with the proteasome could generate additional surfaces for molecular interaction with aggregation-prone proteins, thus promoting recruitment to the aggregates. The inability to efficiently dispose of polyglutamine proteins would likely have deleterious effects on cellular function, as, over time, we would predict the appearance and accumulation of Htt intermediates, oligomers, and aggregates. Moreover, one would predict that kinetic trapping of polyglutamine-containing proteins within the proteasome may interfere with degradation of other cellular substrates of the proteasome. Indeed, it has been shown that expression of N-terminal fragment of mutant Htt results in accumulation of endogenous p53 and the artificial proteasomal substrate GFPU in mammalian cell lines (Bence et al, 2001; Jana et al, 2001) and that striatal cells established from full-length Htt-Q111 knock-in mice have elevated p53 levels (Trettel et al, 2000). Our rationale for expressing the N-terminal fragment of Htt rather than fullprotein is based on observations that full-length Htt is proteolyzed in vivo releasing polyglutamine-containing N-terminal fragments, and that the N-terminal fragments, but not full-length Htt, are found in aggregates from brain tissue of HD patients and an inducible neuronal cell system (DiFiglia et al, 1997; Lunkes et al, 2002). As aggregates in the brain of HD patients have been shown to be ubiquitin positive (DiFiglia et al, 1997) and aggregates found in transgenic N-terminal Htt fragment mice contain both ubiquitin and the proteasome (Jana et al, 2001), our findings that the proteasome is impaired by N-terminal Htt or simple polyglutamine proteins might well correspond to the cellular events associated with polyglutamine diseases.
The differences between our results on N-terminal Htt and previous studies on ataxin-1 with regard to the fate of the proteasome suggest that differences in molecular interactions of aggregates formed by different types of polyglutamine proteins can have distinct biophysical properties. For example, it has been demonstrated using FRAP and FLIP experiments that CBP is irreversibly sequestered in aggregates formed by mutant full-length ataxin-3 or N-terminal Htt fragment, while CBP is mobile in aggregates formed by mutant full-length ataxin-1 (Chai et al, 2002). These observations are in agreement with our results showing irreversible sequestration of the proteasome in aggregates of N-terminal Htt fragment or simple polyglutamine expansion proteins as well as with the results showing a dynamic interaction between the proteasome and mutant ataxin-1 aggregates (Stenoien et al, 2002). Furthermore, it has been shown that aggregates of mutant ataxin-1 are dynamic with both fast- and slow-exchanging ataxin-1 (Stenoien et al, 2002), while aggregates of ataxin-3 or N-terminal Htt fragment are static. The difference in dynamics may be due to intrinsic properties of the aggregating species. For example, expression of ataxin-1 with only two or 30 glutamines, which is within the wild-type range, resulted in the appearance of aggregates (Stenoien et al, 2002), revealing that ataxin-1 aggregate formation is not dependent solely on the expansion of the polyglutamine stretch, but additionally due to expression levels.
Variation in expression levels alone, however, does not explain why the various polyglutamine disease proteins display heterogenous protein association properties and it is becoming clear that polyglutamine protein context and post-translational modifications are important. Recent studies demonstrated that the toxic properties of ataxin-1 depend both on the expansion of the polyglutamine tract and nuclear phosphorylation at serine 776, as expression of an unphosphorylated, but nuclear-localized, ataxin-1(Q82)A776 did not result in toxicity (Emamian et al, 2003). Additionally, it was established that phosphorylation of serine 776 enables binding to the regulatory protein14-3-3, in a polyglutamine length-dependent manner, resulting in enhanced stability of ataxin-1 (Chen et al, 2003). In the case of SBMA, a gender-specific polyglutamine disease, the subcellular localization of mutant androgen receptor appears to be the pathogenic factor. Mouse models of SBMA have demonstrated that ligand-dependent nuclear translocation of the mutant androgen receptor is required for gender-related pathogenesis (Katsuno et al, 2002; Sopher et al, 2004), which explains why most female carriers are usually asymptomatic. Furthermore, an enhanced association between mutant androgen receptors and CBP was detected (Sopher et al, 2004). Thus, it appears that in addition to the expansion of the polyglutamine tract, the protein context, post-translational modifications, and/or subcellular localization result in differential protein associating properties of the polyglutamine disease proteins.
We propose that a detailed understanding of the molecular interactions between polyglutamine proteins and their associating proteins, such as demonstrated here for the proteasome, provides a basis to understand how different disease-associated aggregation-prone proteins can cause substantially different outcomes.
Materials and methods
Constructs
The pcDNA3-LMP2-GFP, pcDNA3-Htt-Q65, pcDNA3-Flag-Q81, pcDNA3-Flag-Q19, pEYFP-N1-Q82, pEYFP-N1-Q19, pECFP-N1-Q82, and pECFP-pEYFP constructs were previously described (Reits et al, 1997; Kim et al, 2002). The pcDNA3-Htt-Q64-GFP and pcDNA-Htt-Q23-GFP constructs were provided by M MacDonald (Harvard University). pECFP-N1-LMP2 and pEYFP-N1-LMP2 were generated by PCR amplifying LMP2 without its stop codon from pcDNA3-LMP2-GFP and inserting into EcoRI/BamHI-digested pECFP-N1 and pEYFP-N1, respectively (BD Bioscience). The 18 nucleotides between the LMP2 and ECFP were deleted using the QuikChange site-directed mutagenesis kit (Stratagene). The pcDNA3-Htt-Q23 construct was generated by subcloning HindIII-digested Htt-Q23 into pcDNA3. pEYFP-N1-Ubi-YFP and pEYFP-N1-Ubi-Qn-YFP were generated by PCR amplifying and inserting XhoI/EcoRI sites into the Ub-R-eK (Ubi) region of pGEM-3Zf(+)-Ub-R-eK-HA102 (Lee et al, 2001) and subcloning into the corresponding pEYFP-N1, pEYFP-N1-Q19, -Q40, or -Q82 construct. pcDNA3-Ubi-Q16-Flag and pcDNA3-Ubi-Q78-Flag were generated by PCR amplifying and inserting the Flag sequence into the 3′ end of the Ubi-Qn region of pEYFP-N1-Ubi-Qn-YFP, and subcloning into the XhoI/XbaI sites of pcDNA3. pGEM-3Zf(+)-Ubi-YFP was generated by PCR amplifying and inserting NcoI/HindIII sites into the YFP region of pEYFP-N1, and subcloning into NcoI/HindIII-digested pGEM-3Zf(+)-Ub-R-eK-HA102 (HA102 is cut out). pGEM-3Zf(+)-Ubi-Qn-YFP was generated by PCR amplifying and inserting AflIII/HindIII sites into the Qn-YFP region of pEYFP-N1-Qn-YFP, and subcloning into NcoI/HindIII-digested pGEM-3Zf(+)-Ub-R-eK-HA102. pGEM-3Zf(+)-YFP and pGEM-3Zf(+)-Qn-YFP were generated by PCR amplifying and inserting EcoRI/HindIII sites into the YFP or Qn-YFP regions of pEYFP-N1 and pEYFP-N1-Q19, -Q40, and -Q82, and subcloning into EcoRI/HindIII-digested pGEM-3Zf(+)-Ub-R-eK-HA102. Ribosomal binding site was introduced into these plasmids using the QuikChange site-directed mutagenesis kit with the primer 5′ccggtcgccaccattaaagaggagaaattaact atggtgagcaagg3′. pTRE2hyg-Htt-Q78-YFP, pTRE2hyg-Htt-Q23-YFP, pTRE2hyg-Htt-Q78-CFP, and pTRE2hyg-Htt-Q23-CFP were generated by insertion of Htt-Qn and YFP or CFP into the BamHI/PvuII and PvuII/MluI sites, respectively, of pTRE2hyg (BD Biosciences). All constructs were verified by sequencing.
Cell culture and transfection
HeLa cervical carcinoma cells were maintained in DMEM supplemented with 10% fetal bovine serum at 37°C in an atmosphere of 5% CO2/95% air. HeLa Tet-off cells (Clontech) were grown similarly with the addition of 200 μg/ml G418. Transient transfections were performed using Lipofectamine Plus, as described in the protocol provided by the manufacturer (Invitrogen), and the cells were analyzed 24 h after transfection. For FRAP and FLIP analyses, cells were grown on 35 mm glass-bottomed microwell dishes (MatTek Corp., MA), and for immunofluorescence and FRET analysis, on coverslips for 24 h before transfection. For transfection of HeLa cells with pTRE2hyg-Htt-Q78-YFP and pTRE2hyg-Htt-Q23-YFP, the pTet-off plasmid (BD Biosciences) was cotransfected. In experiments using MG132 (Peptide Institute Inc.), MG132 (10 μM) was added for the last 9 h of transfection.
Western blotting
The harvested and counted HeLa cells were resuspended in 5% SDS 1 × SDS–PAGE sample buffer, boiled, and sonicated. Equal numbers of cells were loaded on an SDS–PAGE. YFP fusion proteins were detected by an anti-GFP IRDye™800 antibody (600-132-215, Rockland, 1:2500) and the Odyssey infrared imaging system (LI-COR Biosciences). Anti-Hsc70 antibody (SPA-815, StressGen, 1:1000), anti-Htt antibody HP-1 (provided by M MacDonald, Harvard University, 1:5000), and anti-polyglutamine antibody 1C2 (MAB1574, Chemicon, 1:5000) followed by anti-rat, anti-rabbit, or anti-mouse HRP-conjugated antibodies, respectively, were detected by ECL Western blotting detection kit (Amersham Biosciences).
Immunofluorescence analysis
Transfected HeLa cells were fixed in 4% formaldehyde in 1 × PBS, permeabilized in 0.5% saponin and 0.5% Triton X-100 in 1 × PBS, and blocked for 1 h with 10% FBS and 0.3% Triton X-100 in 1 × PBS at 37°C. The Htt proteins were detected using HP-1 antibody (1:500), which recognizes amino acids 80–113 of the Htt protein (provided by M MacDonald, Harvard University) (Trettel et al, 2000). Flag-Q81 was detected with an antibody against the Flag epitope (M5, Sigma, 1:250) and the proteasome was labeled using an anti-20S antibody (PW8155, Affinity Research Products, 1:250). The primary antibodies were detected using TRITC-conjugated anti-mouse or anti-rabbit antibody (Sigma, 1:100) or FITC-conjugated anti-rabbit antibody (Sigma, 1:100). DNA was stained with DAPI (Sigma). Samples were mounted in Vectashield anti-fading solution (Vector Laboratories). Immunofluorescent samples were examined using a Leica DM-IRE2 inverted deconvolution microscope equipped with a × 40 oil objective lens, and Metamorph imaging software (Universal Imaging Corp.). FITC, Texas red, DAPI, and YFP filter sets (Chroma Technology Corp.) were used for visualization. The images were merged and pseudo-colored using the Metamorph imaging software and Adobe Photoshop 7.0, respectively. The percentage of cells containing aggregates was determined by counting the number of cells with aggregates out of ∼200 transfected cells, as determined by fluorescence.
FRAP and FLIP
For live-cell imaging, transfected HeLa cells were maintained at 37°C for the duration of the experiment. Photobleaching for FRAP and FLIP analyses was performed as described previously (Chen and Huang, 2001), using a Zeiss LSM 510 Meta Axiovert confocal microscope and a × 40 oil objective lens. The 488 and 514 nm laser wavelengths were used in photobleaching of the GFP and YFP fusion proteins, respectively. For FRAP analysis, images were taken at ninth zoom power and an area of 36 μm2 was bleached for 10 s (60 iterations), after which an image was collected every 20 s. For FLIP analysis, a single image was taken at fifth zoom power and an area of 36 μm2 was bleached away from aggregates and images were collected after every 1 min (370 iterations) of photobleaching. Relative fluorescence intensity (RFI) was determined using the equation RFI=(Net/N1t)/(Ne0/N10). Net is the average intensity of the bleached area at a given time point and N1t is the average intensity of nonbleached area at the corresponding times and functions as a control for general photobleaching and background fluorescence. Ne0 and N10 are the average intensity before photobleaching of the bleached and nonbleached areas, respectively. RFI for FLIP analysis was calculated with the above equation, except that Net is the average intensity of a nonbleached area, for example, an aggregate at a given time point, and N1t is the average intensity of a nonbleached area in the neighboring cell at the corresponding time. The mobile fraction (Mf) was determined using Mf=(Nefinal−Ne1)/(Ne0−Ne1) (Lippincott-Schwartz et al, 2001), in which Nefinal is the final intensity of the bleached area after full recovery, Ne1 is the fluorescence just after the bleach, and Ne0 is the fluorescence before photobleaching.
Acceptor photobleaching
For acceptor photobleaching experiments, HeLa cells transiently transfected with the indicated CFP and YFP fusion proteins were fixed and images were taken using a Zeiss LSM 510 Meta Axiovert confocal microscope and a × 63 oil objective lens. CFP was excited by 458 nm laser wavelength and emission detected at 480–520 nm using HFT458/514 and BP480-520 filters, whereas YFP was excited by 514 nm laser wavelength and emission detected at 492–535 nm using HFT458/514 and NFT515 filters. Images were taken at fifth zoom power and the defined YFP region was photobleached using the 514 nm laser wavelength and 100% power. The CFP and YFP images were pseudo-colored using Adobe Photoshop 7.0. The FRET efficiency image was generated in Metamorph by image arithmetic of the two CFP images (prebleach, Db, and postbleach, Db(ab)) using the equation E=(Db(ab)−Db)/Db(ab) (Wouters et al, 1998; Berney and Danuser, 2003). For calculations of the average FRET efficiency (n=4–10), a modification to the above-mentioned equation E=1−(Dbc(ab)/Dbc)(Db/Db(ab)) (Berney and Danuser, 2003) was used to take into account variation in CFP intensity in a control region. Dbc and Dbc(ab) are the donor intensity before and after photobleaching of the acceptor in a control region outside of the photobleached area.
Filter-based FRET analysis
To measure acceptor emission by donor excitation, transiently transfected HeLa Tet-off cells were fixed and images were taken using a Leica DM-IRE2 inverted deconvolution microscope with × 63 objective. CFP (430ex/470em), YFP (500ex/535em), and FRET (430ex/535em) channel images were taken with the beam splitter 86002v2 JP4 for CFP and YFP, exciter 430/25 and 500/20, emitter 470/30 and 535/30 (Chroma Technology Corp.). The acquired images were analyzed using Metamorph imaging software. As the overlap between donor emission and acceptor excitation spectra contributes to the detection of donor and acceptor fluorescence through the FRET filter set, the images acquired consist of both FRET and non-FRET signals. Therefore, we applied a correction algorithm to each image to generate the corrected FRET images, as previously described (Gordon et al, 1998; Kim et al, 2002). The corrected FRET image was generated using the following equation: FRETC=(FRET-95)−0.46(CFP-95)−0.016(YFP-100)−8. The FRETC/CFP image was then generated by calculating the ratio between FRETC and background corrected CFP.
In vitro proteasome degradation assay
To target proteins to the proteasome by the N-end rule (Varshavsky, 1992), a ubiquitin moiety was attached to the N-terminus of the substrate protein via a 40-amino-acid linker derived from the Escherichia coli Lac repressor. Radioactive substrates were generated from pGEM-3Zf(+)-Ubi-YFP, pGEM-3Zf(+)-Ubi-Qn-YFP, pGEM-3Zf(+)-YFP, and pGEM-3Zf(+)-Qn-YFP using T7 promoter-driven in vitro transcription and translation in [35S]methionine-supplemented E. coli S30 extracts (Promega). The substrate proteins were partially purified by high-speed centrifugation and ammonium sulfate precipitation and resuspended in 40 μl buffer (250 mM Tris–HCl, pH 7.4, 25 mM MgCl2, 25% (v/v) glycerol). Degradation by the proteasome was assayed in rabbit reticulocytes (Green Hectares), as previously described (Lee et al, 2001). A 10 μl portion of the translated protein was added to 35 μl of ATP-depleted reticulocytes supplemented with 1 mM DTT and incubated for 10 min at 30°C to allow removal of the N-terminal ubiquitin. Ubiquitination and degradation were initiated by addition of ATP and an ATP-regenerating system (2 mM ATP, 10 mM creatine phosphate, 0.1 mg/ml creatine phosphokinase, final concentration) and incubation was continued at 30°C. At the indicated time points, 4.8 μl aliquots were transferred to ice-cold 5% trichloroacetic acid (TCA). The TCA-insoluble fractions were analyzed by 12% SDS–PAGE and quantified by electronic autoradiography (InstantImager, Packard). In experiments to inhibit protein synthesis, 5 mM methionine, 40 μg/ml RNase A, and 100 μg/μl cycloheximide were included in the degradation mixture. In experiments using MG132, 100 μM was added to the lysates before the substrate was included in the ATP-depleted reaction mixture.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Acknowledgments
We thank Jacques Neefjes (The Netherlands Cancer Institute) and Marcy MacDonald (Harvard University) for generously sharing reagents, and the Biological Imaging Facilities, Thomas O'Halloran's laboratory, and the Cell Imaging Facilities at the Department of Cell and Molecular Biology, Robert H Lurie Comprehensive Cancer Center at Northwestern University for use of microscopes. We are grateful to members of the Morimoto laboratory for sharing reagents and for their comments on the manuscript. These studies were supported by grants from the National Institute of General Medical Science GM38109, the Huntington Disease Society of America Coalition for the Cure (RIM), and the NIH grant GM63004 (AM). KES was supported by Achievment Reward for College Scientists-Chicago Chapter, KNM by NIH grant T32GM08061, and CIH by Sigrid Jusélius Foundation Fellowship, The Academy of Finland, and a Human Frontier Science Program Organization Long-Term Fellowship.
References
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin–proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Berney C, Danuser G (2003) FRET or no FRET: a quantitative comparison. Biophys J 84: 3992–4010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Shao J, Miller VM, Williams A, Paulson HL (2002) Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci USA 99: 9310–9315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Huang S (2001) Nucleolar components involved in ribosome biogenesis cycle between the nucleolus and nucleoplasm in interphase cells. J Cell Biol 153: 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY (2003) Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113: 457–468 [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19: 148–154 [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY (1999) Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24: 879–892 [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537–548 [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277: 1990–1993 [DOI] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375–387 [DOI] [PubMed] [Google Scholar]
- Gonda DK, Bachmair A, Wunning I, Tobias JW, Lane WS, Varshavsky A (1989) Universality and structure of the N-end rule. J Biol Chem 264: 16700–16712 [PubMed] [Google Scholar]
- Gordon GW, Berry G, Liang XH, Levine B, Herman B (1998) Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J 74: 2702–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana NR, Zemskov EA, Wang G, Nukina N (2001) Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 10: 1049–1059 [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35: 843–854 [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404–11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI (2002) Polyglutamine protein aggregates are dynamic. Nat Cell Biol 4: 826–831 [DOI] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 7: 627–637 [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Snapp E, Kenworthy A (2001) Studying protein dynamics in living cells. Nat Rev Mol Cell Biol 2: 444–456 [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10: 259–269 [DOI] [PubMed] [Google Scholar]
- Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ (2001) Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J Neurosci 21: 8772–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441–1448 [DOI] [PubMed] [Google Scholar]
- Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN (1998) Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol 143: 1457–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reits EA, Benham AM, Plougastel B, Neefjes J, Trowsdale J (1997) Dynamics of proteasome distribution in living cells. EMBO J 16: 6087–6094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Mahlke C, Yuan J (2003) Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421: 373–379 [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549–558 [DOI] [PubMed] [Google Scholar]
- Sopher BL, Thomas PS Jr, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR (2004) Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron 41: 687–699 [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Mielke M, Mancini MA (2002) Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat Cell Biol 4: 806–810 [DOI] [PubMed] [Google Scholar]
- Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH (2001) Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 153: 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 9: 2799–2809 [DOI] [PubMed] [Google Scholar]
- Varshavsky A (1992) The N-end rule. Cell 69: 725–735 [DOI] [PubMed] [Google Scholar]
- Varshavsky A (1996) The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA 93: 12142–12149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14: 95–104 [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12: 1393–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters FS, Bastiaens PI, Wirtz KW, Jovin TM (1998) FRET microscopy demonstrates molecular association of non-specific lipid transfer protein (nsL-TP) with fatty acid oxidation enzymes in peroxisomes. EMBO J 17: 7179–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57–66 [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217–247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5