

Abstract
Cabezas syndrome is a syndromic form of X-linked intellectual disability primarily characterized by a short stature, hypogonadism and abnormal gait, with other variable features resulting from mutations in the CUL4B gene. Here, we report a clinically undiagnosed 5-year-old male with severe intellectual disability. A genome-first approach using targeted exome sequencing identified a novel nonsense mutation [NM_003588.3:c.2698G>T, p.(Glu900*)] in the last coding exon of CUL4B, thus diagnosing this patient with Cabezas syndrome.
Intellectual disability (ID) affects 1–3% of the general population and is more prevalent in males than in females.1 X-linked ID (XLID) accounts for 5–10% of all types of ID and is the most common cause of ID in males.1 In nonsyndromic XLID, ID is the only clinical feature. In contrast, syndromic ID is associated with various neurological and/or behavioral manifestations and structural anomalies. Thus far, more than 150 XLID disorders have been described, with approximately half of them accounting for over 100 mutated genes.2 However, because of the extensive phenotypic and genetic heterogeneity of XLID disorders, the identification of causal gene mutations on the X chromosome among families with XLID and among sporadic cases of XLID via clinical diagnosis-based approaches remains challenging.3 CUL4B (MIM #300304) is a relatively large gene (913 amino acids) containing 21 coding exons (exons 2–22) encoding the cullin-4B scaffold protein, which forms a ubiquitin–ligase complex. Variants of this gene are a known cause of the Cabezas type of X-linked syndromic ID (MRXSC or Cabezas syndrome, MIM #300354).4–6 A screening of XLID families and patients with ID using targeted panel-based next-generation sequencing (NGS) determined the frequency of CUL4B variants leading to XLID and ID, which were ~2% (8/407)7–3% (8/250)4 and 0.5% (5/986)8–0.9% (1/106),9 respectively. This result implicates CUL4B as one of the most commonly mutated genes with loss-of-function variants in patients with ID, regardless of their mode of inheritance. Patients with Cabezas syndrome display overlapping phenotypes, including short stature, hypogonadism, gait abnormalities and other variable features such as ID, seizures, tremors, behavioral problems, macrocephaly, obesity and various dysmorphic features.7 The presence of these overlapping features other than ID are indicative of the syndrome associated with CUL4B variants,7 and a guideline can be used to identify potential patients with this syndrome (Supplementary Table S1);4 however, the only previously reported consistent feature in affected individuals was ID,4 and at least some of these features are commonly observed among individuals with ID.10 Indeed, some CUL4B-associated XLID cases with no features or partially overlapping features have been reported.9,11 Therefore, diagnosis can be challenging due to the high number of overlapping phenotypes among syndromes.
In addition to the aforementioned CUL4B variants, gross deletions within or around CUL4B have also been observed as disease-causing alterations.12 Therefore, a genome-first approach using NGS that can simultaneously detect single-nucleotide variants and copy number aberrations of candidate ID-associated genes, including CUL4B,13 might be useful for cost- and time-effective screening and the differential diagnosis of CUL4B-associated XLID. Here, we report the first Japanese case of Cabezas syndrome caused by a novel nonsense mutation of CUL4B to be successfully detected by a genome-first approach using targeted exome sequencing.
The patient was a 5-year-old, first-born child of healthy, non-consanguineous Japanese parents with an unremarkable family history. He was delivered at full term with a birth weight of 2,500 g (−1.5 s.d.), body length of 46.5 cm (−1.3 s.d.) and occipitofrontal head circumference of 28.6 cm (−3.4 s.d.). The child displayed feeding difficulty and failure to thrive in newborn infancy. Psychomotor development was delayed beginning from the infantile period and apparently at 5 years of age; he could not sit or crawl and could not speak. He displayed stereotypical patterns of behavior including head banging, licking his hands and bruxism. Brain magnetic resonance imaging showed cortical dysplasia, ventriculomegaly, loss of cerebral white matter and a thin corpus callosum (Figure 1), which were reported to be observed in 33%, 53%, 27% and 20% of XLID cases with CUL4B mutations, respectively.7 At 4 years of age, the patient visited a pediatric neurology department because of frequent complex partial seizures. Normal results were obtained from routine laboratory tests and metabolic surveys. On physical examination, the child showed short stature and relative macrocephaly: his weight, height and occipitofrontal head circumference were 13.0 kg (−2.2 s.d.), 101 cm (−2.0 s.d.) and 52.5 cm (+0.8 s.d.), respectively, at 5 years and 7 months of age. Craniofacial dysmorphic features, including anterior cowlick, broad forehead, sparse eyebrow, prominent eyes, down-slanting palpebral fissures, low-set ears, flat nasal bridge, short nose, short philtrum, high arched palate and a wide mouth with a prominent lower lip were noted. He exhibited a micropenis and small testes. Standard karyotyping using peripheral blood revealed no abnormalities. Although clinical features including cerebral magnetic resonance imaging abnormalities of the patient were retrospectively at least partially consistent with the recently proposed clinical spectrum associated with CUL4B mutations (Supplementary Table S1),7 the patient remained undiagnosed because of the absence of several symptoms including tremors, gait abnormalities and behavioral problems.
Figure 1.
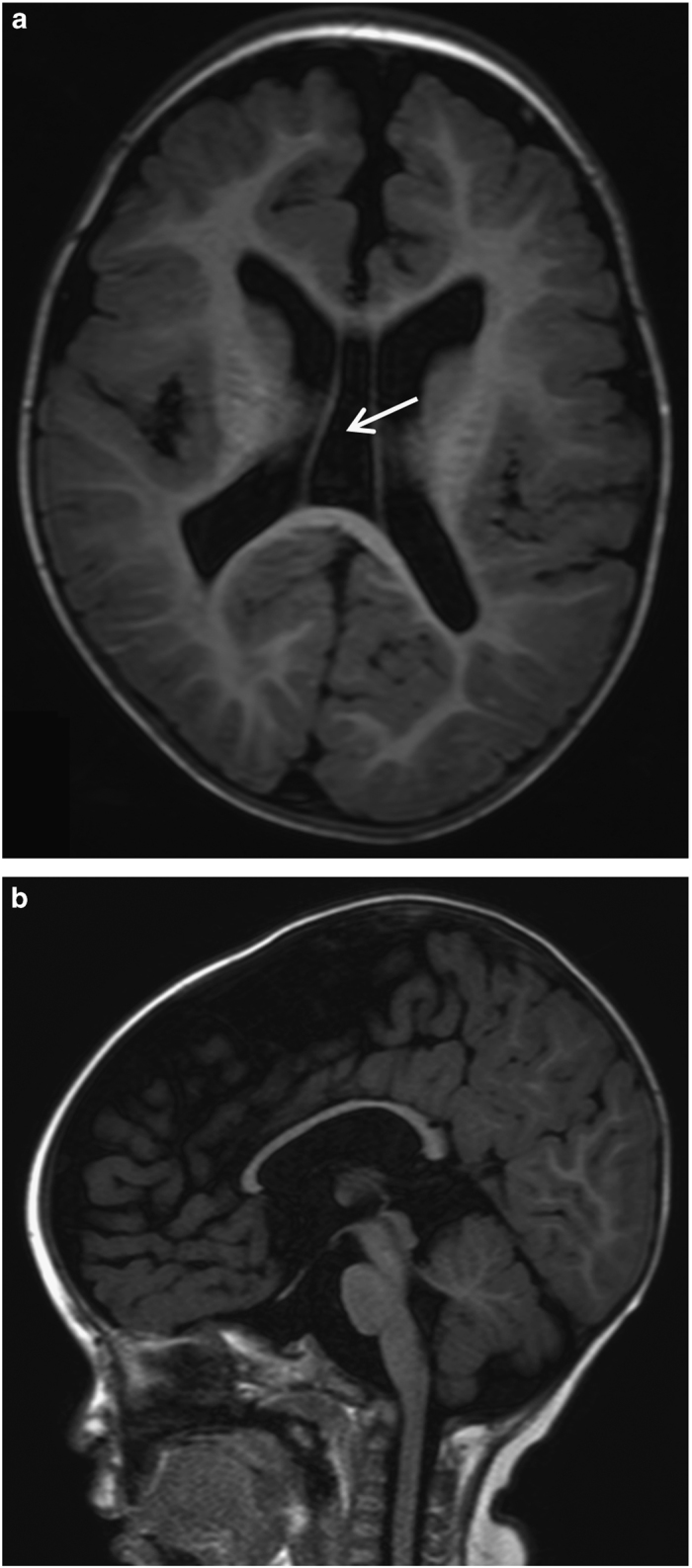
Representative magnetic resonance imaging (MRI) images from the patient at 1 year and 6 months of age. MRI T1 axial (a) and sagittal (b) sections show enlarged lateral ventricle, cavum vergae (arrow), loss of white matter volume and thin corpus callosum.
After informed consent was obtained from the parents, molecular diagnosis was performed using genomic DNA extracted from a blood sample. The study was approved by the ethics committees of Tokushima University and Osaka Medical Center and Research Institute for Maternal and Child Health. To screen known disease-associated genes, we used a TruSight One Sequencing Panel (Illumina, San Diego, CA, USA) to perform NGS with a MiSeq sequencer (Illumina) and our pipeline for NGS data analysis, as described elsewhere,14 with a minor modification due to a software update specific for a bioinformatics pipeline.13 To identify presumably pathogenic single-nucleotide variants, we excluded sequence variants with low-allele frequencies, i.e., >0.01, included in the 1000 Genomes Project database (http://www.1000genomes.org/), NHLBI GO Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/), Human Genetic Variation Database (HGVD, http://www.genome.med.kyoto-u.ac.jp/SnpDB/) and integrative Japanese Genome Variation Database (iJGVD, https://ijgvd.megabank.tohoku.ac.jp/). Copy number aberration analysis was also performed as described elsewhere.13,15 These analyses detected a single-base substitution, c.2698G>T, in the last coding exon (exon 22) of NM_003588.3 (CUL4B_v001), which was confirmed by Sanger sequencing (Figure 2a). Primer sequences are available upon request. This mutation transformed the codon for glutamic acid 900 to a premature stop codon in the protein sequence [NM_003588.3(CUL4B_i001):p.(Glu900*)] (Figure 2a). This mutation is not present in the Human Gene Mutation Database professional 2016.2 (HGMD, http://www.hgmd.org/) or ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and is the first mutation detected in the last exon of CUL4B. Because nonsense mutations within the last exon do not activate nonsense-mediated messenger RNA decay, this mutation yields a stable CUL4B messenger RNA that directs the synthesis of C-terminally truncated polypeptides.16,17 The C-terminal extremity is the most conserved part of the cullin family protein (Figure 2b), and the CULLIN_1 cullin family signature ([LIV]-[KL]-x(2)-[LIVM]-x(2)-L-[IL]-[DEQGVT]-[KRHNQ]-x-[YT]-[LIVM]-x-R-x(5,7)-[FYLV]-x-Y-x-[SAT]; PROSITE entry PS01256, http://prosite.expasy.org/), located between codons 886 and 913 in the CUL4B protein, is one of the consensus patterns within this conserved region. Fourteen amino acids from the C-terminus in this highly conserved amino acid sequence are lost in the truncated CUL4B protein of the aforementioned case (Figure 2b). In re-evaluating the clinical features of the affected patient, together with the clinical spectrum of patients harboring CUL4B mutations (Supplementary Table S1), it appears that there is a correlation between mutations missing 14 amino acids in this region of CUL4B and the presented phenotype. As a result of this molecular diagnosis, the patient was diagnosed with Cabezas syndrome caused by a novel nonsense mutation in CUL4B. The mother was an asymptomatic carrier of the same mutation (Figure 2a).
Figure 2.
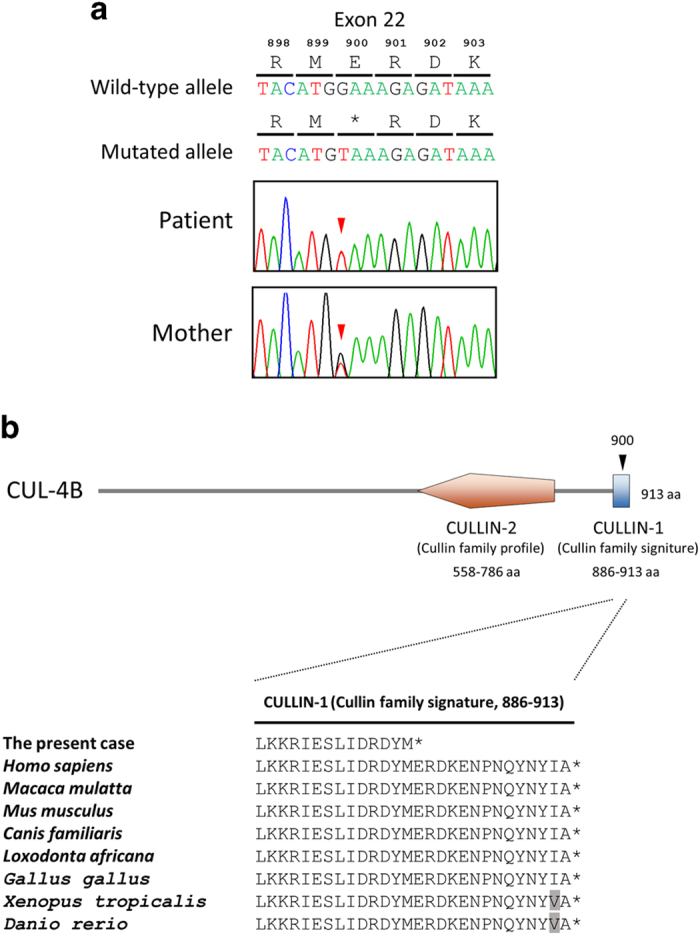
Mutation in CUL4B observed in the present X-linked ID (XLID) case. (a) Partial sequence chromatograms of exon 22 of CUL4B in a patient and his mother. Red arrowheads denote the mutated base. The DNA and corresponding amino acid sequences of the wild-type and mutant CUL4B alleles are also shown. (b) A schematic representation of the CUL4B protein (top) and multiple alignment of the CUL4B amino acid sequences within the CULLIN-1 cullin family signature (bottom). Arrowheads denote the mutated amino acid. Amino acids that did not match those in Homo sapiens are shaded.
CUL4B has been reported to be frequently mutated as detected by targeted exome sequencing analysis investigating individuals with undiagnosed ID and XLID, who previously tested negative for mutations and pathogenic copy number aberrations in a subset of known ID-related genes by Sanger sequencing.9,18 Although the clinical phenotype may be retrospectively consistent with observations in previous reports, a clinical diagnosis of Cabezas syndrome could not be offered even by experienced clinical geneticists because of the lack of certain clinical features. Badura-Stronka et al.10 proposed screening for mutations in CUL4B in males with ID, severe speech impairment and primary intention tremor, but many clinical features of Cabezas syndrome, including a wide range of cerebral malformations in neuroimaging examination, are also common among individuals with ID. Indeed, in most of the reported cases, the clinical diagnosis of Cabezas syndrome was established following the identification of genetic defects in CUL4B,3–5,8,9,11,18 suggesting that Cabezas syndrome is clinically underdiagnosed.12 Therefore, individuals affected with Cabezas syndrome may remain undiagnosed through conventional means of sequencing genes other than CUL4B that are more specific to other ID syndromes evoked by the patient phenotype.9 Molecular diagnosis by this genome-first approach using targeted high-throughput NGS in undiagnosed ID cases may be crucially important for providing appropriate therapeutic options and optimized healthcare in patients through facilitating the differential diagnosis of this syndrome and related diseases involving ID in a cost-effective manner. In addition, a correct diagnosis through this diagnostic approach is useful for establishing recurrence risks and providing appropriate genetic counseling to the family.
Acknowledgments
The authors thank the patient and his family for their participation in this study. Parts of this work were performed at the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University. This work was supported by JSPS KAKENHI grant numbers 26293304 (I.I.), 16K15618 (I.I.) and 15K19620 (T.N.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv).
The authors declare no conflict of interest.
References
- Bassani S, Zapata J, Gerosa L, Moretto E, Murru L, Passafaro M. The neurobiology of X-linked intellectual disability. Neuroscientist 2013; 19: 541–542. [DOI] [PubMed] [Google Scholar]
- Londin ER, Adijanto J, Philp N, Novelli A, Vitale E, Perria C et al. Donor splice-site mutation in CUL4B is likely cause of X-linked intellectual disability. Am J Med Genet A 2014; 164A: 2294–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor B, Pichon O, Baron S, David A, Le Caignec C. Deletion of the CUL4B gene in a boy with mental retardation, minor facial anomalies, short stature, hypogonadism, and ataxia. Am J Med Genet A 2010; 152A: 175–180. [DOI] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, O'Meara S, Edkins S, Teague J, Butler A et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet 2007; 80: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet 2007; 80: 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas DA, Slaugh R, Abidi F, Arena JF, Stevenson RE, Schwartz CE et al. A new X linked mental retardation (XLMR) syndrome with short stature, small testes, muscle wasting, and tremor localises to Xq24-q25. J Med Genet 2000; 37: 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, Nakagawa T, Bahi-Buisson N, Haas SA, Hu H, Bienek M et al. Variants in CUL4B are associated with cerebral malformations. Hum Mutat 2015; 36: 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozeva D, Carss K, Spasic-Boskovic O, Tejada MI, Gecz J, Shaw M et al. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat 2015; 36: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Gérard B, Lauer J, Herenger Y, Muller J, Quartier A et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 2014; 51: 724–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badura-Stronka M, Jamsheer A, Materna-Kiryluk A, Sowińska A, Kiryluk K, Budny B et al. A novel nonsense mutation in CUL4B gene in three brothers with X-linked mental retardation syndrome. Clin Genet 2010; 77: 141–144. [DOI] [PubMed] [Google Scholar]
- Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet 2010; 87: 173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravn K, Lindquist SG, Nielsen K, Dahm TL, Tümer Z. Deletion of CUL4B leads to concordant phenotype in a monozygotic twin pair. Clin Genet 2012; 82: 292–294. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Hayabuchi Y, Ono A, Naruto T, Horikawa H, Kohmoto T et al. Detection of 1p36 deletion by clinical exome-first diagnostic approach. Hum Genome Var 2016; 3: 16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Naruto T, Kohmoto T, Komori T, Imoto I. A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Var 2014; 1: 14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Nakagawa R, Naruto T, Kohmoto T, Suga K, Goji A et al. A novel missense mutation of COL5A2 in a patient with Ehlers–Danlos syndrome. Hum Genome Var 2016; 3: 16030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 1998; 23: 198–199. [DOI] [PubMed] [Google Scholar]
- Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C et al. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J 1998; 17: 3484–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry 2016; 21: 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Citations
- Imoto Issei.HGV Database. 2016. 10.6084/m9.figshare.hgv.912. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Imoto Issei.HGV Database. 2016. 10.6084/m9.figshare.hgv.912. [DOI]