

Abstract
Objective
Cerebral edema is common in severe hepatic encephalopathy and may be life-threatening. Bolus 23.4% hypertonic saline (HTS) improves surveillance neuromonitoring scores, although its mechanism of action is not clearly established. We investigated the hypothesis that bolus HTS decreases cerebral edema in severe hepatic encephalopathy utilizing a quantitative technique to measure brain and CSF volume changes.
Design
Retrospective analysis of serial computed tomography (CT) scans and clinical data for a case-control series was performed.
Setting
Intensive care units of a tertiary care hospital.
Patients
Patients with severe hepatic encephalopathy treated with 23.4% HTS and control patients who did not receive 23.4% HTS.
Methods
We used clinically obtained CT scans to measure volumes of the ventricles, intracranial CSF, and brain using a previously validated semi-automated technique (Analyze Direct; Overland Park, KS). Volumes before and after 23.4% HTS were compared with Wilcoxon signed-rank test. Associations between total CSF volume, ventricular volume, serum sodium, and Glasgow Coma Scale Scores were assessed using Spearman correlation.
Results
Eleven patients with 18 administrations of 23.4% HTS met inclusion criteria. Total CSF (median 47.6 [35.1–69.4] to 61.9 [47.7–87.0] mL, p<0.001) and ventricular volumes (median 8.0 [6.9–9.5] to 9.2 [7.8–11.9] mL, p=0.002) increased and Glasgow Coma Scale Scores improved (median 4 [3–6] to 7 [6–9], p=0.008) after 23.4% HTS. In contrast, total CSF and ventricular volumes decreased in untreated control patients. Serum sodium increase was associated with increase in total CSF volume (r=0.83, p<0.001) and change in total CSF volume was associated with ventricular volume change (r=0.86, p<0.001).
Conclusions
Total CSF and ventricular volumes increased after 23.4% HTS, consistent with a reduction in brain tissue volume. Total CSF and ventricular volume change may be useful quantitative measures to assess cerebral edema in severe hepatic encephalopathy.
Intracranial hypertension (IH) and diffuse cerebral edema occur in 86–95% of comatose patients with hepatic encephalopathy and contribute to mortality (1, 2). Hyperosmolar agents (mannitol or hypertonic saline [HTS]) are mainstay treatments of cerebral edema and IH both in severe hepatic encephalopathy (grade 3 [somnolent but responsive] or 4 [coma] on the four-grade West Haven Criteria [3]) and in other acute brain diseases. The principal proposed mechanism of action of hyperosmolar agents is that a functional blood brain barrier allows an osmolar gradient to develop between blood and brain parenchyma resulting in efflux of water from brain tissue with reduction in brain volume (4, 5). The reduction of brain volume within the rigid cranium allows displaced cerebrospinal fluid (CSF) to return from the spinal subarachnoid space such that the total sum of the volumes of the individual components of the intracranial space remains constant (Monro-Kellie doctrine) and intracranial compliance improves (4, 6, 7). Since displacement of CSF occurs before blood or brain displacement during pathologic processes (4) one may be able to measure changes in intracranial CSF volume to evaluate the severity of cerebral edema and quantify changes in intracranial tissue volume.
Studies of hyperosmolar therapy in humans have most frequently investigated mannitol and have concentrated on focal brain diseases such as trauma and stroke, with fewer data for diseases associated with diffuse cerebral edema such as severe hepatic encephalopathy. In the case of hyperosmolar therapy and severe hepatic encephalopathy, studies have generally reported either intracranial pressure (ICP) measurements or qualitative radiographic signs of cerebral edema (8, 9). However, ICP change is an indirect measure of volume change and because CSF displacement to the spinal subarachnoid space has a buffering effect it is possible for cerebral edema to progress without clearly pathologic ICP elevation (4). Furthermore, invasive ICP monitoring is controversial in hepatic encephalopathy management because of the potential hemorrhagic complications of monitor placement, failure of several studies to identify a survival benefit, and recent evidence that invasive monitoring may be associated with worse outcome for some patients (3, 10). Additionally, the subdural and intraparenchymal ICP monitors most commonly used in liver failure may not accurately reflect compression of brain structures, such as the thalami and brainstem, distant from the device’s pressure transducer (11). A quantitative radiographic maker of brain tissue volume applied to serial neuroimaging studies could potentially provide insight in to the evolution of cerebral edema and the effect of hyperosmolar therapy on brain volume. We sought to use a validated neuroimaging analysis technique and a quantitative marker of brain tissue volume to investigate the hypothesis that bolus 23.4% HTS decreases brain volume in severe hepatic encephalopathy patients.
Methods
Cohort
We retrospectively identified a treatment group of all patients ≥18 years of age admitted to our institution between January 1st, 2012 and November 1st, 2014 who received at least one intravenous bolus of 23.4% HTS for treatment of life-threatening cerebral edema and had a diagnosis of acute or acute-on-chronic liver failure. Bolus 23.4% HTS administration was identified using electronic query of medication administration records as previously described (12). The resulting patient list was manually reviewed to confirm time of 23.4% HTS administration and to identify those diagnosed with acute or acute-on-chronic liver failure as documented by an attending intensivist or hepatologist. Liver failure was confirmed by identifying uncompensated liver dysfunction, specifically coagulopathy and encephalopathy (3). Predetermined exclusion criteria were: pregnancy, lack of head computed tomography (CT) within 24 hours prior to (pre-HTS) or 24 hours following (post-HTS) administration of 23.4% HTS, hemicraniectomy, acute focal lesion (e.g. ischemic or hemorrhagic stroke), and prior administration of mannitol.
Using an electronic record of neurology consultations during the study period we identified a separate control group with acute or acute-on-chronic liver failure and severe hepatic encephalopathy who did not receive 23.4% HTS. This group met the same study criteria as the treatment group except the imaging requirement was the availability of at least two head CTs separated by no more than 24 hours and the first performed within 48 hours of admission.
Surveillance neurologic exams and neuroimaging
Consistent with the practice of several members of the US Acute Liver Failure Study Group, we infrequently use invasive ICP monitors to manage severe hepatic encephalopathy (13). In these patients we implement a strategy of hourly surveillance neurologic examinations and serial neuroimaging similar to the approach previously described (14). The frequency of neuroimaging is guided by degree of neurologic impairment, prior imaging results, the clinical course of the patient’s liver failure, and the development of factors that may confound the neurologic exam (e.g. severe sepsis, seizures, medication toxicity). Our practice is to repeat neuroimaging within 6 to 12 hours after 23.4% HTS such that post-treatment imaging is available to guide future therapy.
Use of hypertonic saline
Intravenous 23.4% HTS is institutionally restricted to the urgent treatment of life-threatening cerebral edema or IH and is administered at the discretion of an attending intensivist. Our neurointensivists institute therapy for cerebral edema in liver failure if a patient manifests a cerebral herniation syndrome or other acute neurologic deterioration in the context of existing severe hepatic encephalopathy (15, 16) and the absence of a more likely explanation for neurologic decline (e.g. severe sepsis, seizure, medication toxicity). We do not consider life-threatening edema to be present if patients are easily arousable or capable of localization on neurologic exam. Patients may receive repeat doses of 23.4% HTS if they demonstrate additional events of neurologic decline that meet the above criteria, and the decision to re-administer 23.4% HTS may be modified by knowledge of prior clinical response and intervening serial neurologic exam and neuroimaging data.
Others have used 23.4% HTS as the initial form of HTS therapy or have added it to ongoing infusion of 3% HTS (15), and we follow a similar practice. We administer 23.4% HTS as a 30 mL bolus over 10 minutes via central venous catheter to target an acute serum sodium increase of 5 mEq/L (15). Following 23.4% HTS we check serum sodium 1 hour later and then monitor serum sodium at least every 6 hours. Based on these sodium levels a continuous infusion of 3% HTS may be initiated and titrated to maintain target serum sodium. Infusion of 3% HTS to maintain steady serum sodium levels may be necessary to avoid the theoretical risk of rebound cerebral edema with acute serum sodium decline (4).
Data collection
Demographic and clinical data
We used retrospective review of the electronic medical record to collect key demographic and clinical data including serum sodium and Glasgow Coma Scale (GCS) scores; a standardized exam scale ranging from unresponsiveness [3] to alert and oriented [15] and consisting of motor, verbal, and eye subscores; at each neuroimaging and HTS administration time point. GCS scores are assessed and documented hourly in severe hepatic encephalopathy patients. We collected bilirubin, international normalized ratios (INR), and creatinine values nearest the time of 23.4% HTS (initial CT scan for controls) in order to calculate the model for end-stage liver disease (MELD) score (a standard model of liver failure severity and mortality) (17). We collected Acute Physiology and Chronic Health Evaluation IV Acute Physiology Score (APS) scores as a measure of overall illness severity. APS is a widely used measure in which increasing scores suggest greater illness severity (18). We also reviewed electronic medication administration records and calculated the total milliequivalents of sodium delivered in any form (23.4% HTS bolus, 3% HTS or 0.9% saline infusions, medications, etc) between CT scans.
Determination of Intracranial Volumes
We analyzed digital imaging and communications in medicine (DICOM) format CT scans acquired as 5-mm-thick contiguous slices using Analyze Direct 11.0 (Overland Park, KS). Scans were analyzed blinded to order of acquisition, demographics, and clinical data by trained imaging analysts (EML and ALR). Analyze Direct is a semi-automated computer program that uses a pixel thresholding technique based on signal intensity thresholds between structures to define regions of interest and compute volumes. Regions can be manually edited if thresholding includes incorrect pixels. For each CT scan we used threshold signal intensity between brain, cranium, and CSF to define regions that incorporated all the intracranial CSF and then determined volumes of interest using the software algorithm. The pixel threshold values corresponding to CSF were established for each CT scan by adjusting the software’s threshold setting such that a region of interest encompassing the CSF-filled ventricle but excluding brain tissue was defined on a scan slice passing through the middle of the lateral ventricles. The resulting threshold values corresponding to CSF signal were then used to include the intracranial CSF of the sulci, cisterns, and ventricles on each scan slice. We used a similar approach to determine the volume of the ventricular system for each scan. We determined brain tissue volume by measuring the volume of the entire intracranial space and subtracting the total intracranial CSF volume as previously been described by others (19, 20). Pixel thresholding has previously been used to measure intracranial CSF volumes (19–21) and accurately measures volumes with excellent inter-rater agreement (κ=0.97) (22).
Statistical analysis and protocol approvals
Data are presented as number or median (interquartile range). To assess changes in total CSF, ventricular, and brain tissue volumes and clinical and laboratory variables between paired CT scans we used the nonparametric related-samples Wilcoxon signed-rank test since change in total CSF volume was not normally distributed. Control and treatment group variables were compared using Mann-Whitney U test. To identify associations between variables we determined Spearman’s correlation coefficients (r). We considered correlation coefficients of absolute value ≤0.35 weak, 0.36 to 0.67 moderate, and 0.68 to 1.0 strong correlations (23). We considered two-tail p≤0.05 significant. To account for disproportionate influence from outliers, we performed all statistical analysis for both the entire cohort and the cohort after removing outliers, defined as scan pairs with total CSF volume change exceeding 1.5 times the interquartile range outside the hinges of the interquartile range. Standard statistical software was used (IBM SPSS v.21; Armonk, NY).
The study was approved by our institutional review board with waiver of consent for retrospective review.
Results
Over the study period there were 65 administrations of 23.4% HTS in 30 patients. Seventeen of these patients had acute or acute-on-chronic liver failure. Of these, two (12%) were excluded because they were admitted and received 23.4% HTS for neurologic exam findings before imaging could be performed. Four (24%) patients were excluded because they were too unstable to transport for post-HTS imaging within 24 hours. The remaining 11 (65%) patients met inclusion criteria for the treatment group and contributed 18 pairs of pre- and post-HTS CT scans. Pre-HTS scans were performed 2.6 (1.3–5.0) hours before and post-HTS scans were performed 6.1 (4.7–8.0) hours after 23.4% HTS. During the same period, eight additional patients with 12 CT scan pairs had acute or acute-on-chronic liver failure with severe hepatic encephalopathy but did not receive 23.4% HTS and formed the control group.
Demographic and clinical characteristics of the groups are summarized in (Table 1). All patients demonstrated coagulopathy (INR>1.5) and severe hepatic encephalopathy (GCS scores ranging from 3 to 13, consistent with West Haven grade 3 or 4) at the time of initial CT. All treatment group patients required initiation of renal replacement therapy as determined by a board-certified nephrologist within 24 hours of admission. Ten treatment group patients received continuous renal replacement therapy and one received a single treatment of intermittent hemodialysis before transitioning to continuous renal replacement therapy. Two treatment group patients received 3% HTS infusion before 23.4% HTS (one patient at 50 mL per hour for 2 hours and one at 40 mL per hour for 3 hours). Ten treatment group patients received 3% HTS infusion between CT scans.
Table 1.
Baseline demographic and clinical data.
| Patient | Age (years) |
Sex | Diagnosis | Peak Ammonia (mcg/dL) |
Pre- HTS GCS Score |
Pre- HTS MELD |
Pre- HTS INR |
Pre- HTS Sodium (mEq/L) |
Admit Cr (mg/dL) |
LVEF (%), Diastolic Function |
|---|---|---|---|---|---|---|---|---|---|---|
| Treatment Group | ||||||||||
| 1 | 27 | F | Acute-on-chronic, autoimmune hepatitis | 437 | 4 | 32 | 2.1 | 140 | 1.88 | 65%, normal |
| 2 | 24 | M | Acute-on-chronic, transplant rejection | 274 | 6 | 42 | 3.1 | 139 | 1.37 | 70%, normal |
| 3 | 69 | F | Acute-on-chronic, NASH cirrhosis | 317 | 3 | 31 | 1.6* | 137 | 4.28 | 65%, Grade 1 dysfunction |
| 4 | 48 | F | Acute, acetaminophen | 257 | 3 | 30 | 2.4 | 135 | 4.70 | 40%, Grade 1 dysfunction |
| 5 | 20 | M | Acute, MDMA intoxication | 203 | 5 | 29 | 2.2 | 140 | 4.50 | 55%, normal |
| 6 | 30 | F | Acute, acetaminophen | 586 | 9 | 37 | 2.8 | 145 | 2.95 | 65%, normal |
| 7 | 44 | F | Acute, autoimmune hepatitis | 264 | 8 | 46 | 4.7 | 140 | 0.90 | 70%, normal |
| 8 | 55 | F | Acute-on-chronic, alcoholic | 228 | 3 | 41 | 2.6 | 136 | 1.21 | 70%, Grade 1 dysfunction |
| 9 | 39 | M | Acute, likely viral | 250 | 6 | 53 | 12.0 | 131 | 1.93 | 65%, normal |
| 10 | 24 | M | Acute, unknown intoxication | 441 | 4 | 30 | 2.0 | 143 | 3.32 | 40%, Grade 1 dysfunction |
| 11 | 26 | M | Acute, acetaminophen | 445 | 7 | 39 | 7.4 | 141 | 2.14 | 60%, normal |
| Control Group | ||||||||||
| 1 | 47 | F | Acute, acetaminophen | 305 | 3 | 44 | 8.6 | 143 | 2.79 | 60%, normal |
| 2 | 58 | F | Acute-on-chronic, alcoholic | 493 | 5 | 36 | 2.8 | 134 | 2.49 | 70%, normal |
| 3 | 22 | F | Acute, Wilson’s disease | 105 | 10 | 37 | 2.3 | 141 | 2.84 | 65%, normal |
| 4 | 41 | M | Acute, acetaminophen | 360 | 13 | 31 | 6.4 | 138 | 0.93 | 70%, normal |
| 5 | 54 | M | Acute-on-chronic, hepatitis C virus | 235 | 7 | 42 | 2.5 | 140 | 4.75 | 65%, normal |
| 6 | 73 | F | Acute-on-chronic, hepatitis B virus | 127 | 13 | 48 | 12 | 134 | 1.44 | 70%, normal |
| 7 | 52 | F | Acute, alcohol and acetaminophen | 550 | 9 | 31 | 6.7 | 144 | 1.00 | 60%, normal |
| 8 | 20 | F | Acute, acetaminophen | 1012 | 13 | 45 | 11.3 | 136 | 2.09 | 75%, indeterminate |
Abbreviations: HTS=23.4% hypertonic saline, GCS=Glasgow Coma Scale, MELD=model for end-stage liver disease, INR=international normalized ratio, Cr=serum creatinine, LVEF=left ventricular ejection fraction by transthoracic echocardiogram, F=female, M=Male, NASH=nonalcoholic steatohepatitis, MDMA=3,4-methylenedioxy-N-methylamphetamine “ecstasy”
Fresh frozen plasma had been administered prior to this value.
The control group did not differ from the treatment group in terms of age, peak serum ammonia, serum sodium at initial CT, admission serum creatinine, MELD score, or central venous pressure at initial CT (all p >0.35) but did have higher GCS scores (10 [5–13] vs. 5 [3–7]; p=0.05) and GCS motor subscores (5[3–6] vs. 2[1–4]; p=0.004) at initial CT. APS scores did not differ between control and treatment patients (112 [70–128] vs. 104 [84–113]; p=0.71). Six control patients required continuous renal replacement therapy at the recommendation of a nephrologist and 3 received an infusion of 3% HTS which began after initial CT scan in each case. No patient in either treatment or control group received mannitol, neurosurgical intervention, or invasive ICP monitoring during their hospitalization.
(Figure 1 A–D) demonstrates the CSF and ventricular volume changes and corresponding GCS score changes for a patient treated with 23.4% HTS and for a control group patient. Key quantitative neuroimaging, laboratory, and clinical data for the treatment and control groups corresponding to paired CT scans are summarized in (Table 2). Total intracranial CSF volume, ventricular system volume, and serum sodium increased significantly after treatment with 23.4% HTS, while calculated median brain volume remained unchanged. GCS scores (4 [3–6] to 7 [6–9]; p=0.008) and GCS motor subscores (2 [1–4] to 4 [3–5]; p=0.011) improved significantly after 23.4% HTS. In contrast, the control group total intracranial CSF volume and ventricular volume decreased significantly between CT scans with no change in serum sodium or GCS scores. The change in total intracranial CSF volume (−10.4 [−19.4– −3.0] vs. 11.8 [6.2–17.2] mL, p<0.001) and ventricular volume (−2.6 [−4.9– −0.6] vs. 1.6 [0.6–2.9] mL, p=0.001) between serial CT scans differed significantly between the control and treatment groups.
Figure 1. A–D. Serial CT scans demonstrating change in CSF and brain tissue volumes and corresponding Glasgow Coma Scale Scores in a patient treated with 23.4% hypertonic saline and in a control patient.
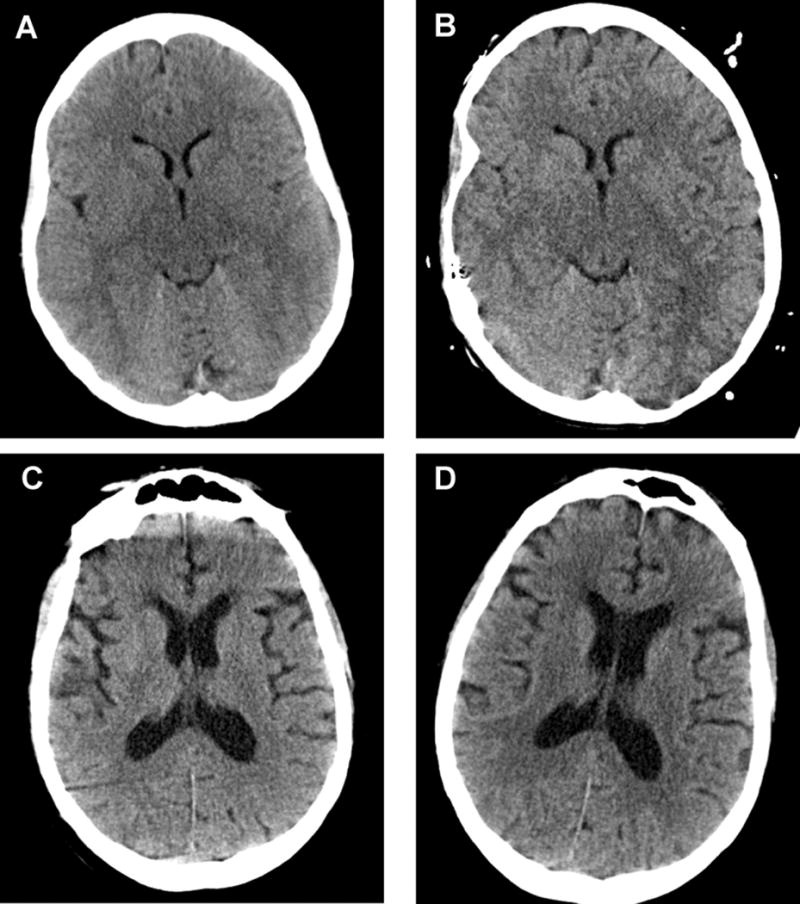
A. Treatment group patient before 23.4% hypertonic saline (Total CSF volume: 33.0 mL, Ventricular volume: 6.4 mL, Glasgow Coma Scale: 4)
B. Treatment group patient after 23.4% hypertonic saline (Total CSF volume: 48.3 mL, Ventricular volume: 8.0 mL, Glasgow Coma Scale: 6)
C. Control group patient initial CT scan (Total CSF volume: 205.1 mL, Ventricular volume: 56.7 mL, Glasgow Coma Scale: 7)
D. Control group patient repeat CT scan (Total CSF volume: 182.7 mL, Ventricular volume: 47.2 mL, Glasgow Coma Scale: 6)
Table 2.
Treatment and control group neuroimaging, laboratory, and clinical data for paired serial CT scans.
| 23.4% HTS Treatment Group | |||
|---|---|---|---|
| Pre-23.4% HTS Bolus | Post-23.4% HTS Bolus | P value | |
| Measured Total Intracranial CSF Volume (mL) | 47.6 (35.1–69.4) | 61.9 (47.7–87.0) | <0.001 |
| Measured Ventricular Volume (mL) | 8.0 (6.9–9.5) | 9.2 (7.8–11.9) | 0.002 |
| Calculated Brain Volume (mL) | 1400.2 (1150.6–1444.4) | 1408.7 (1126.0–1458.7) | 0.65 |
| Measured Intracranial Skull Volume (mL) | 1442.1 (1238.9–1504.3) | 1478.0 (1206.5–1526.3) | 0.37 |
| Serum Sodium (mEq/L) | 141 (139–145) | 148 (143–149) | <0.001 |
| Serum Ammonia (mcg/dL) | 220 (157–260) | 182 (105–247) | 0.018 |
| Arterial Partial Pressure Carbon Dioxide (mmHg) | 33 (28–41) | 32 (30–40) | 0.65 |
| Central Venous Pressure (mmHg)* | 8 (5–14) | 9 (8–12) | 0.44 |
| GCS total score | 4 (3–6) | 7 (6–9) | 0.008 |
| GCS motor subscore | 2 (1–4) | 4 (3–5) | 0.011 |
| Control Group | |||
| Initial CT Scan | Repeat CT Scan | P value | |
| Measured Total Intracranial CSF Volume (mL) | 97.0 (47.9–133.5) | 91.0 (41.7–106.1) | 0.022 |
| Measured Ventricular Volume (mL) | 16.5 (10.7–31.0) | 15.3 (11.2–28.6) | 0.028 |
| Calculated Brain Volume (mL) | 1324.8 (1101.8–1421.0) | 1341.4 (1093.0–1411.5) | 0.58 |
| Measured Intracranial Skull Volume (mL) | 1420.0 (1171.9–1460.2) | 1398.6 (1162.9–1459.7) | 0.70 |
| Serum Sodium (mEq/L) | 139 (134–141) | 137 (134–140) | 0.35 |
| Serum Ammonia (mcg/dL) | 181 (92–313) | 109 (75–217) | 0.18 |
| Arterial Partial Pressure Carbon Dioxide (mmHg) | 30 (27–37) | 30 (26–32) | 0.80 |
| Central Venous Pressure (mmHg)** | 9 (9–12) | 9 (8–15) | 0.72 |
| GCS total score | 10 (5–13) | 10 (6–13) | 0.83 |
| GCS motor subscore | 5 (3–6) | 4 (3–6) | 0.79 |
CSF=cerebrospinal fluid, HTS=hypertonic saline, GCS=Glasgow coma scale
Complete central venous pressure data available for 15 pairs of CT scans.
Complete central venous pressure data available for 11 pairs of CT scans.
There was a positive correlation between the degree of change in serum sodium and the degree of change in total intracranial CSF volume (r=0.83, p<0.001). Similarly an association was found between change in serum sodium and change in ventricular volume (r=0.64, p<0.001). There was no correlation between the change in total CSF volume and the milliequivalents of sodium administered between CT scans (r= −0.11, p=0.66). The changes in total intracranial CSF volume and the corresponding changes in ventricular volume were positively correlated (r=0.86, p<0.001). In addition, there was a moderate positive correlation between the degree of change in serial GCS scores and the corresponding degree of change in total intracranial CSF volumes (r=0.44, p=0.018) and ventricular volumes (r=0.39, p=0.034). The degree of change between serial GCS motor subscores was likewise positively correlated with the degree of change in total intracranial CSF volumes (r=0.38, p=0.048) and ventricular volumes (r=0.36, p=0.048).
Two pairs of CT scans in the treatment group (total CSF volume increase of 67.6 mL and 46.8 mL) met criteria as outliers. Exclusion of these two CT pairs from statistical analysis yielded similar statistically significant results and associations between variables (Supplemental Tables e1–e2).
Discussion
In this retrospective study of patients with severe hepatic encephalopathy, bolus 23.4% HTS was associated with a significant increase in total intracranial CSF volume, ventricular volume, and GCS scores, while those who did not receive 23.4% HTS demonstrated a significant decrease in total and ventricular CSF volumes. These data suggest that total intracranial CSF volume change and ventricular volume change (which strongly correlates with total intracranial CSF volume change) can both serve as quantitative markers of brain tissue volume change and may non-invasively gauge the effect of therapies directed at cerebral edema. Our results are a reasonable extension of the Monro-Kellie doctrine in that a therapy intended to reduce brain volume led to increases in CSF volume and support the hypothesis that 23.4% HTS therapy leads to brain tissue volume reduction in severe hepatic encephalopathy.
Our findings are in agreement with the theorized osmotic gradient mechanism of action for HTS leading to reduction in brain volume (4). Studies in brain-injured rabbits and non-injured rats suggest that hypertonic saline results in reduction in cerebral water content (5, 24), which one would expect to result in reduced brain tissue volume. In human traumatic brain injury 20% HTS boluses appear to decrease the volume of non-contused tissue as assessed on serial CT scans (20), and in patients comatose from focal brain injury invasive measurements of brain tissue thermal conductivity suggest a reduction in brain water content after bolus hyperosmolar therapy (mannitol or 23.4% HTS) (25).
We found total intracranial CSF volume change after 23.4% HTS is associated with the magnitude of serum sodium change effected but not the total milliequivalents of sodium delivered. This observation is consistent with animal evidence in which hypertonic saline results in brain water reduction proportionate to the increase in serum sodium (5) and human evidence in which the magnitude of serum sodium change after 23.4% HTS predicts clinical response in those treated for transtentorial herniation (15).
Our treatment group experienced a 3 point improvement in median GCS scores after 23.4% HTS and we found a moderate association between the magnitude of CSF volume change and the corresponding change in GCS score on paired CT scans. These findings support a pathophysiologic mechanism relating brain tissue edema to neurologic deterioration and the potential benefit of targeting that mechanism with edema reducing therapies. The complex and multifaceted pathophysiology of hepatic encephalopathy is a potential reason for our finding of moderate rather than strong association between CSF volume change and GCS change. Growing evidence suggests that processes such as systemic inflammation, cytokine derangements, multi-system organ failure, and neurotransmitter abnormalities affect brain function during liver failure by mechanisms independent of cerebral edema formation (26, 27) and these mechanisms would be expected to impact neurologic function in addition to the effects of cerebral edema. Treatment of neurologic deterioration during liver failure may benefit from therapies directed both at cerebral edema and at other pathologic mechanisms. The relationships between CSF and brain tissue volume, HTS therapy, serum sodium change, and processes such as systemic inflammation and the clinical impact of those relationships are areas for further investigation.
The complex, multifaceted pathophysiology of hepatic encephalopathy presents a clinical challenge in the management of acute liver failure. Over time patients with acute liver failure may acquire factors such as sepsis, renal failure, and medication and metabolic toxicities that complicate neurologic assessment and present a challenge to identifying the causes of neurologic deterioration. In our clinical practice we have utilized qualitative serial neuroimaging to investigate cerebral edema evolution as a potential contributor in these situations. However, prior studies attempting to radiographically detect brain volume changes in liver failure patients required highly-trained clinicians to detect qualitative changes on CT scans and did not clearly demonstrate how those qualitative assessments might be used to gauge the effect of therapy (9, 28). While additional investigation is needed, our technique of volumetric analysis of the CSF space may represent a means to serially evaluate and compare the state of brain edema and intracranial compliance quantitatively between time points. This quantitative imaging data combined with knowledge of corresponding neurologic function and response to therapy at prior time points may help the physician identify instances of neurologic deterioration with potential to benefit from cerebral edema directed therapies. The contentious role of ICP monitoring in hepatic encephalopathy management (3, 10, 16), failure of studies to show a benefit of invasive ICP monitoring and an association with worse outcome in some patients with liver failure (3, 10), and a large clinical trial that showed equivalence of invasive monitor-guided therapy to therapy guided by surveillance neurologic exams and neuroimaging in traumatic brain injury (29) suggest that there may be a growing role for non-invasive approaches to evaluating disease evolution in select patients.
There are limitations to our study. Despite demonstrating an increase in total CSF volume, calculated median brain volume determined by subtracting measured total CSF volume from measured total intracranial volume did not decrease after 23.4% HTS. The underlying reason is likely to be the large relative volume of the total intracranial space and the small relative changes in volume affected by HTS, as compared to the large relative change in volume in CSF. While pixel thresholding is an accurate method of measuring the volume of intracranial regions of interest it does so with an error of 2–6% of the region’s total volume (22); when an error of 6% is introduced to the measurement of a large volume, such as the entire intracranial volume or the entire brain, the absolute error may be large. Since even a few milliliters of volume change in the patient with severe intracranial pathology may have a clinically meaningful effect (30), radiographic techniques with relatively large absolute volume errors are likely limited as markers of meaningful change in cerebral edema. Measuring the much smaller CSF space uncovers a larger relative change in volume. Since the total intracranial volume is fixed, any change in the total CSF volume between serial CT scans must be equivalent to the change in intracranial tissue volume (Monro-Kellie doctrine) (4, 7). As such, we believe that CSF volume analysis results in a more accurate, robust reflection of brain volume changes in individual patients.
Our technique is limited by the inability of CT scans to distinguish intravascular blood within the confines of the brain from the brain parenchyma itself, so the change in CSF volume is equivalent in magnitude to the sum of the change in intracranial blood and brain parenchyma volumes. Though our technique cannot attribute the proportion of intracranial tissue volume change due to brain parenchyma versus intracranial blood there is evidence that intracranial blood volume does not change significantly in response to 23.4% HTS (25, 31). Additionally, we only assessed the effect of 23.4% HTS on edema in severe hepatic encephalopathy and the effect may differ in other diseases.
Our preference to avoid ICP monitor placement in liver failure patients because of its contentious position in clinical practice and potential for complications is a study limitation. We are unable to determine if neurologic improvement after 23.4% HTS was associated with reducing ICP below a critical pathologic threshold or if relieving tissue compression and distortion by reducing edema even in the absence of marked ICP elevation can yield improvements in neurologic exam. Future work might investigate the relationship between CSF volume changes measured by our technique and both invasive (ICP measurement or jugular venous oxygenation) and noninvasive (transcranial Doppler or near-infrared spectroscopy) technologies in use for neurologic monitoring.
Our method did not allow for rigorous specification of the timing of CT scan acquisition. While future research might more rigorously specify the timing of imaging studies the level of acuity in these patients may make strict timing of neuroimaging difficult to implement.
The retrospective nature of our study limits our ability to account for confounders. We attempted to account for confounding by assessing a control group of patients with severe hepatic encephalopathy treated in the same clinical setting but who did not receive 23.4% HTS. We also attempted to account for the effect of outliers.
Conclusion
In patients with cerebral edema and severe hepatic encephalopathy, 23.4% HTS administration was associated with measurable increases in total intracranial CSF volume and ventricular volume, and by the Monro-Kellie Doctrine a commensurate decrease in brain tissue volume, as well as improvement in GCS scores. In contrast, untreated subjects experienced reductions in ventricular and total CSF volume with no change in GCS scores. Quantitative total CSF and ventricular volumes may provide a non-invasive means of cerebral edema evaluation in severe hepatic encephalopathy.
Supplementary Material
CSF=cerebrospinal fluid, HTS=hypertonic saline, GCS=Glasgow coma scale
CSF=cerebrospinal fluid
Acknowledgments
Funding
Departmentally funded.
Copyright form disclosures: Dr. Liotta is employed by Northwestern University. His institution received grant support from PARTNER II Trial (Edwards Lifesciences), SALUS trial (Direct Flow Medical Inc), and SAGE-547 Clinical Trial (SAGE therapeutics). Dr. Lizza’s institution received grant support from ASHP Education Foundation (New Investigator Research Grant for the study of pharmacist involvement of sedation assessments in the ICU). Dr. Carroll has a patent (unrelated to current work). His institution received grant support from the National Institutes of Health (NIH) and AHA. Dr. Ganger lectured for Gilead. His institution received grant support from ALFSG NIDDK. Dr. Ladner is employed by Northwestern and lectured for Gilead. Her institution received grant support from NIDDK. Dr. Maas consulted for Syregelas Law, is employed by Northwestern University, and received grant support from the NIH. His institution received grant support from the Northwestern Memorial Foundation and PARTNER II Trial, Edwards Lifesciences.
Footnotes
Statistical analysis performed by Dr. Eric M. Liotta and Dr. Andrew M. Naidech. Dr. Naidech possesses formal statistical training and experience and reviewed all quantitative data, statistical analysis, and inferences.
Search terms: Cerebral Edema, Hepatic encephalopathy, [18] Coma, [295] critical care, [149] Gastrointestinal
Author contributions
Eric M. Liotta MD: study conception and design; acquisition, analysis, and interpretation of data; statistical analysis; drafting manuscript; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Bryan D. Lizza PharmD: acquisition of data; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Anna L. Romanova BS: acquisition of data; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
James C. Guth MD: study conception; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Michael D. Berman BS: acquisition of data; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Timothy Carroll PhD: study conception; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Brandon Francis MD MPH: study conception; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Daniel Ganger MD: study conception; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Daniela P. Ladner MD MPH: study conception; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Matthew B. Maas MD: study conception; interpretation of data; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work
Andrew M. Naidech MD MSPH: study conception and design; analysis and interpretation of data; statistical analysis; revision of manuscript for critically important intellectual content; final approval of the submitted version and accountability for all aspects of the work. Dr. Naidech possesses formal statistical training and experience and reviewed the quantitative data and statistical inferences made in this manuscript.
Financial Disclosures:
The authors have no financial disclosures related to this manuscript.
Related to this manuscript:
Eric M. Liotta MD reports no disclosures.
Bryan D. Lizza PharmD reports no disclosures.
Anna L. Romanova BS reports no disclosures.
James C. Guth MD reports no disclosures.
Michael D. Berman BS reports no disclosures.
Timothy Carroll PhD reports no disclosures.
Brandon Francis MD MPH reports no disclosures.
Daniel Ganger MD reports no disclosures.
Daniela P. Ladner MD MPH reports no disclosures.
Matthew B. Maas MD reports no disclosures.
Andrew M. Naidech MD MSPH reports no disclosures.
The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Ede RJ, Gimson AE, Bihari D, Williams R. Controlled hyperventilation in the prevention of cerebral oedema in fulminant hepatic failure. J Hepatol. 1986;2:43–51. doi: 10.1016/s0168-8278(86)80007-1. [DOI] [PubMed] [Google Scholar]
- 2.Raschke RA, Curry SC, Rempe S, et al. Results of a protocol for the management of patients with fulminant liver failure. Crit Care Med. 2008;36:2244–2248. doi: 10.1097/CCM.0b013e31818029a3. [DOI] [PubMed] [Google Scholar]
- 3.Stravitz RT, Kramer AH, Davern T, et al. Intensive care of patients with acute liver failure: Recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;35:2498–2508. doi: 10.1097/01.CCM.0000287592.94554.5F. [DOI] [PubMed] [Google Scholar]
- 4.Ropper AH. Hyperosmolar Therapy for Raised Intracranial Pressure. N Engl J Med. 2012;367:746–752. doi: 10.1056/NEJMct1206321. [DOI] [PubMed] [Google Scholar]
- 5.Toung TJ, Nyquist P, Mirski MA. Effect of hypertonic saline concentration on cerebral and visceral organ water in an uninjured rodent model. Crit Care Med. 2008;36:256–261. doi: 10.1097/01.CCM.0000295306.52783.1E. [DOI] [PubMed] [Google Scholar]
- 6.Poca MA, Sahuquillo J, Topczewski T, Lastra R, Font ML, Corral E. Posture-induced Changes in Intracranial Pressure: A Comparative Study in Patients with and without a Cerebrospinal Fluid Block at the Craniovertebral Junction. Neurosurgery. 2006;58:899–906. doi: 10.1227/01.NEU.0000209915.16235.6D. [DOI] [PubMed] [Google Scholar]
- 7.Mokri B. The Monro-Kellie hypothesis: Applications in CSF volume depletion. Neurology. 2001;56:1746–1748. doi: 10.1212/wnl.56.12.1746. [DOI] [PubMed] [Google Scholar]
- 8.Murphy N, Auzinger G, Bernel W, Wendon J. The Effect of Hypertonic Sodium Chloride on Intracranial Pressure in Patients With Acute Liver Failure. Hepatology. 2004;39:464–470. doi: 10.1002/hep.20056. [DOI] [PubMed] [Google Scholar]
- 9.Thayapararajah SW, Gulka I, Al-Amri A, et al. Acute Fulminant Hepatic Failure, Encephalopathy and Early CT Changes. Can J Neurol Sci. 2013;40:553–557. doi: 10.1017/s0317167100014657. [DOI] [PubMed] [Google Scholar]
- 10.Karvellas CJ, Fix OK, Battenhouse H, et al. Outcomes and Complications of Intracranial Pressure Monitoring in Acute Liver Failure: A Retrospective Cohort Study. Crit Care Med. 2014;42:1157–1167. doi: 10.1097/CCM.0000000000000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bratton SL, Chestnut RM, Ghajar J, et al. VII. Intracranial Pressure Monitoring Technology. J Neurotrauma. 2007;24:S45–S54. doi: 10.1089/neu.2007.9989. [DOI] [PubMed] [Google Scholar]
- 12.Naidech AM, Garg RK, Liebling S, Levasseur K, Macken MP, Schuele SU, Batjer HH. Anticonvulsant use and outcomes after intracerebral hemorrhage. Stroke. 2009;40:3810–3815. doi: 10.1161/STROKEAHA.109.559948. [DOI] [PubMed] [Google Scholar]
- 13.Vaquero J, Fontana RJ, Larson AM, et al. Complications and Use of Intracranial Pressure Monitoring in Patients With Acute Liver Failure and Severe Encephalopathy. Liver Transpl. 2005;11:1581–1589. doi: 10.1002/lt.20625. [DOI] [PubMed] [Google Scholar]
- 14.Maas MB, Rosenberg NF, Kosteva AR, et al. Surveillance neuroimaging and neurologic examinations affect care for intracerebral hemorrhage. Neurology. 2013;81:107–112. doi: 10.1212/WNL.0b013e31829a33e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koenig MA, Bryan M, Lewin JL, III, et al. Reversal of transtentorial herniation with hypertonic saline. Neurology. 2008;70:1023–1029. doi: 10.1212/01.wnl.0000304042.05557.60. [DOI] [PubMed] [Google Scholar]
- 16.Lee WM, Larson AM, Stravitz RT. AASLD Position Paper: The Management of Acute Liver Failure: Update 2011. Hepatology. 2011 Sep;:1–22. [Google Scholar]
- 17.Wiesner R, Edwards E, Freeman R, et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology. 2003;12:91–96. doi: 10.1053/gast.2003.50016. [DOI] [PubMed] [Google Scholar]
- 18.Zimmerman JE, Kramer AA, McNair DD, Malila FM. Acute Physiology and Chronic Health Evaluation (APACHE) IV: hospital mortality assessment for today’s critically ill patients. Crit Care Med. 2006;34:1297–1310. doi: 10.1097/01.CCM.0000215112.84523.F0. [DOI] [PubMed] [Google Scholar]
- 19.Qureshi AI, Majidi S, Gilani WI, et al. Increased Brain Volume Among Good Grade Patients with Intracerebral Hemorrhage. Results from the Antihypertensive Treatment of Acute Cerebral Hemorrhage (ATACH) Study. Neurocrit Care. 2014;20:470–475. doi: 10.1007/s12028-013-9842-1. [DOI] [PubMed] [Google Scholar]
- 20.Lescot T, Degos V, Zouaoui A, et al. Opposed effects of hypertonic saline on contusions and noncontused brain tissue in patients with severe traumatic brain injury. Crit Care Med. 2006;34:3029–3033. doi: 10.1097/01.CCM.0000243797.42346.64. [DOI] [PubMed] [Google Scholar]
- 21.Liotta EM, Bauer RM, Berman MD, et al. Acute changes in ventricular volume during treatment for hepatic and renal failure. Neurol Clin Pract. 2014;4:478–481. doi: 10.1212/CPJ.0000000000000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Divani AA, Majidi S, Luo X, et al. The ABCs of accurate volumetric measurement of cerebral hematoma. Stroke. 2011;42:1569–1574. doi: 10.1161/STROKEAHA.110.607861. [DOI] [PubMed] [Google Scholar]
- 23.Taylor R. Interpretation of the Correlation Coefficient: A Basic Review. JDMS. 1990;1:35–39. [Google Scholar]
- 24.Bacher A, Wei J, Grafe MR, et al. Serial determinations of cerebral water content by magnetic resonance imaging after an infusion of hypertonic saline. Crit Care Med. 1998;26:208–114. doi: 10.1097/00003246-199801000-00024. [DOI] [PubMed] [Google Scholar]
- 25.Ko SB, Choi HA, Parikh G, et al. Real Time Estimation of Brain Water Content in Comatose Patients. Ann Neurol. 2012;72:344–350. doi: 10.1002/ana.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butterworth RF. The liver-brain axis in liver failure: neuroinflammation and encephalopathy. Nat Rev Gastroenterol Hepatol. 2013;10:522–528. doi: 10.1038/nrgastro.2013.99. [DOI] [PubMed] [Google Scholar]
- 27.Felipo V. Hepatic encephalopathy: effects of liver failure on brain function. Nat Rev Neurosci. 2013;14:851–858. doi: 10.1038/nrn3587. [DOI] [PubMed] [Google Scholar]
- 28.Wijdicks EFM, Plevak DJ, Rakela J, Wiesner RH. Clinical and Radiologic Features of Cerebreal Edema in Fulminant Hepatic Failure. Mayo Clin Proc. 1995;70:119–124. doi: 10.4065/70.2.119. [DOI] [PubMed] [Google Scholar]
- 29.Chesnut RM, Temkin N, Carney N, et al. A Trial of Intracranial-Pressure Monitoring in Traumatic Brain Injury. N Engl J Med. 2012;367:2471–2481. doi: 10.1056/NEJMoa1207363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kerr ME, Weber BB, Sereika SM, Wilberger J, Marion DW. Dose response to cerebrospinal fluid drainage on cerebral perfusion in traumatic brain-injured adults. Neurosurg Focus. 2001;11:1–7. doi: 10.3171/foc.2001.11.4.2. [DOI] [PubMed] [Google Scholar]
- 31.Diringer MN, Scalfani MT, Zazulia AR, et al. Cerebral Hemodynamic and Metabolic Effects of Equi-Osmolar Doses Mannitol and 23.4% Saline in Patients with Edema Following Large Ischemic Stroke. Neurocrit Care. 2011;14:11–17. doi: 10.1007/s12028-010-9465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CSF=cerebrospinal fluid, HTS=hypertonic saline, GCS=Glasgow coma scale
CSF=cerebrospinal fluid