

Abstract
The retinoblastoma tumor suppressor gene plays important roles in cell cycle control, differentiation and survival during development and is functionally inactivated in most human cancers. Early studies using gene targeting in mice suggested a critical role for pRb in erythropoiesis, while more recent experiments have suggested that many of the abnormal embryonic phenotypes in the Rb null mouse result from a defective placenta. To address this controversy and determine whether Rb has cell intrinsic functions in erythropoiesis, we examined the effects of Rb loss on red cell production following acute deletion of pRb in vitro and under different stress conditions in vivo. Under stress conditions, pRb was required to regulate erythroblast expansion and promote red cell enucleation. Acute deletion of Rb in vitro induced erythroid cell cycle and differentiation defects similar to those observed in vivo. These results demonstrate a cell intrinsic role for pRb in stress erythropoiesis and hematopoietic homeostasis that has relevance for human diseases.
Keywords: cell cycle, enucleation, erythropoiesis, homeostasis, pRb
Introduction
The retinoblastoma tumor suppressor gene (Rb) is essential for normal mouse development (Clarke et al, 1992; Jacks et al, 1992; Lee et al, 1992) and Rb null mice die around E14.5 of gestation exhibiting defects in lens, placental, muscle, hematopoietic and nervous system development (Morgenbesser et al, 1994; Macleod et al, 1996; Zacksenhaus et al, 1996; Wu et al, 2003). The hematopoietic defect in Rb null mice is the least well characterized of these developmental phenotypes and is marked by a failure to produce enucleated red blood cells (Clarke et al, 1992; Jacks et al, 1992; Lee et al, 1992).
Abnormalities in Rb null erythropoiesis have been attributed to non-cell autonomous effects and to defects in paracrine signaling (Whyatt and Grosveld, 2002). In particular, Rb-deficient cells were able to contribute normally to the peripheral blood of adult Rb null chimeric mice (composed of wild-type and Rb−/− cells) (Robanus-Maandag et al, 1994; Williams et al, 1994). Although these studies did not determine the relative contribution of Rb−/− cells to the bone marrow, or to specific lineages and stages of differentiation, they suggested that the red cell differentiation defects observed in fetal liver (FL) (low red cell count and a failure to enucleate) could be overcome by the presence of wild-type cells (Robanus-Maandag et al, 1994; Williams et al, 1994). Recent data have suggested that reduced nutrient exchange between maternal and fetal circulations, due to abnormal placental development, may explain non-cell autonomous defects in the Rb null mouse (Wu et al, 2003). Certainly, apoptosis in the developing nervous system of Rb null embryos appears to be the consequence of hypoxia, as shown recently when Rb loss was conditionally targeted to the nervous system (Ferguson et al, 2002; Macpherson et al, 2003). However, it is still not clear to what extent erythroid defects in Rb null mice contribute to non-cell autonomous defects in other tissues or whether aberrant placental function plays a causative role in red cell defects.
To determine the exact nature of the red cell defect and the extent to which such defects were cell intrinsic, we examined the effects of Rb loss on erythropoiesis in vitro and in vivo. Our results showed that loss of Rb led to a block to erythroid differentiation, which is attributable to a unique cell intrinsic defect in erythroblast expansion and red cell enucleation under stress conditions.
Results
Erythroid defects in the Rb null fetal liver
Erythropoiesis occurs in the FL between E11.5 and E16.5 of mouse embryonic development (Godin and Cumano, 2002). Erythropoiesis can be assessed in primary tissues by quantitating expression of c-Kit, CD71 and TER119 during the differentiation of erythroblasts to erythrocytes (Kina et al, 2000; Socolovsky et al, 2001). When we examined the profile of cKit, CD71 and TER119 expression in E12.5 FL, we observed increased relative numbers of cKit-expressing cells and fewer TER119hi-expressing cells in Rb null FL (Figure 1B, D and F) compared to wild-type littermate FL (Figure 1A, C and E). Specifically, there was a marked increase in the percentage of cKit+CD71+ cells (21.0%, Figure 1B), CD71+TER119− cells (43.5%, Figure 1D) and cKit+TER119− cells (34.5%, Figure 1F) compared to wild type (9.4%, Figure 1A; 14.5%, Figure 1C; 13.3%, Figure 1E), and a decreased percentage of cKit−CD71+ (68.2%, Figure 1B), CD71+TER119+ (34.7%, Figure 1D) and cKit−TER119hi (30.6%, Figure 1F) relative to wild type (83.8%, Figure 1A; 74.2%, Figure 1C; 69.4%, Figure 1E). These results demonstrated that loss of Rb resulted in a block to fetal erythropoiesis as cells became cKit− and TER119hi.
Figure 1.
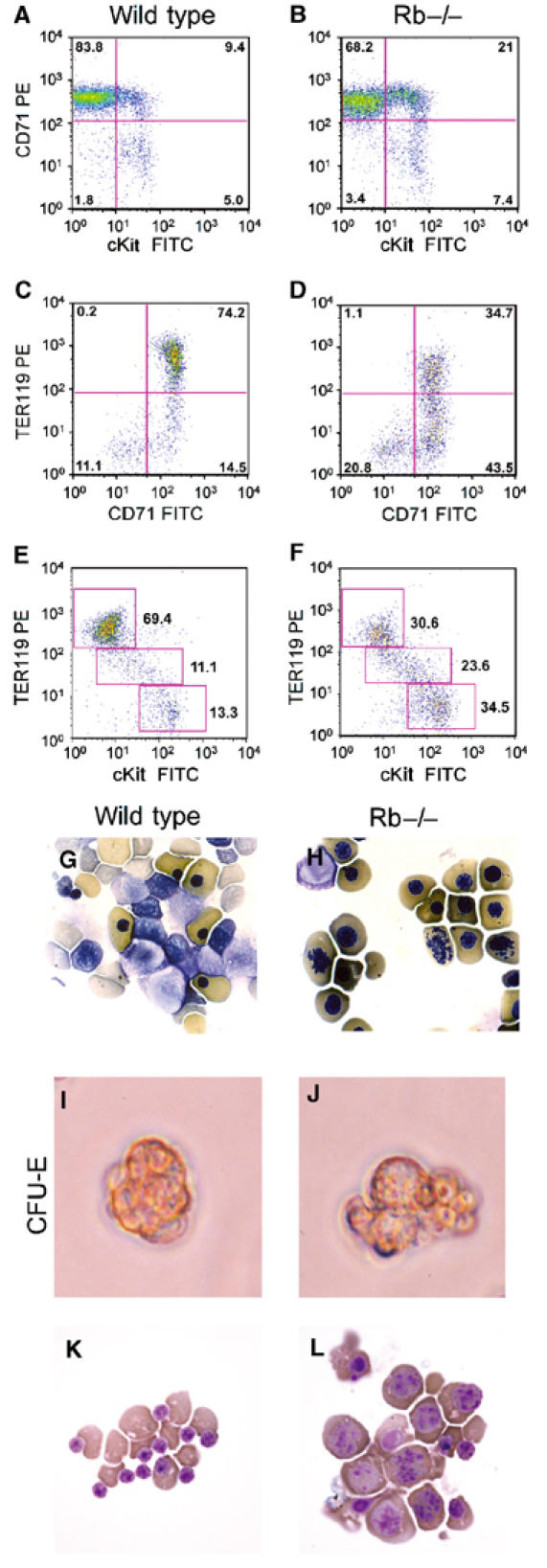
Defective erythroid differentiation in Rb null FL and in vitro. FACS analysis (10 000 events) of E12.5 wild-type (A, C, E) and Rb−/− (B, D, F) FL labeled with fluorescent antibodies to CD71 and cKit (A, B), CD71 and TER119 (C, D) and cKit and TER119 (E, F). Less compact nuclei, disorganized chromatin structure and decreased enucleation in red cells of Rb null E13.5 embryos (H) compared to wild type (G). Day 2 CFU-Es are shown from wild-type (I) and Rb null (J) E12.5 FL. Cytospin of cells from day 2 CFU-Es, wild-type (K) or Rb null (L) E12.5 FL.
FL erythropoiesis in Rb null mice is marked by reduced red blood cell count, an increased proportion of which are nucleated (Clarke et al, 1992; Jacks et al, 1992). Closer inspection revealed a failure of Rb null red cell nuclei to compact and significant numbers of erythroblasts with condensed chromosomes in their benzidine-positive cytoplasm (Figure 1H) compared to wild type (Figure 1G). Similar defects in nuclear condensation and enucleation were observed in Rb null erythroblasts derived from in vitro colony assays (Figure 1L). When cKit+CD71+ erythroid progenitors from either wild-type or Rb−/− E12.5 FL were plated in semisolid media, they gave rise to similar numbers of CFU-E erythroid colonies by 2 days in culture. However, Rb null CFU-Es (Figure 1J) were qualitatively different from wild-type CFU-Es (Figure 1I). Rb−/− CFU-Es were more varied in size, were physically disorganized and displayed irregular shape (Figure 1J), in contrast to wild-type CFU-Es (Figure 1I). When erythroblasts from day 2 CFU-Es were examined cytologically, we observed high levels of red cell enucleation in wild-type CFU-E cultures (Figure 1K). In contrast, Rb null CFU-E-derived erythroblasts were deficient for enucleation (Figure 1L). Furthermore, Rb null erythroblasts were increased in size, showed increased nuclear to cytoplasmic ratio, and condensed chromosomes were visible in many cells (Figure 1L) reminiscent of observations in vivo (Figure 1H). These observations suggest that in vitro erythroid differentiation recapitulates the defects observed in vivo.
Rb null erythroblasts exhibit defects in cell cycle exit during terminal differentiation
To determine whether Rb null erythroblasts undergo normal cell cycle exit during terminal differentiation, disaggregated wild-type and Rb null E12.5 FL cells were labeled with antibodies to cKit, CD71 and TER119 and sorted by FACS. Purified populations were fixed, stained with propidium iodide (PI) and analyzed for DNA content as a measure of cell cycle phase distribution. As shown in Figure 2A, we observed three distinct populations when we examined the TER119 expression profile of cKit+CD71+ E12.5 wild-type FL erythroblasts (red profile): TER119neg (39.0%), TER119lo (36.0%) and TER119hi (24.7%). In Rb null FL (blue profile), TER119hi cells made up only 10.7% of cKit+CD71+ erythroblasts and Rb null cKit+CD71+ erythroblasts accumulated as TER119lo cells (66%, Figure 2A, blue profile). These data suggest that Rb null cells are defective for the end stages of erythroid differentiation including upregulation of TER119 from TER119lo to TER119hi that normally accompanies cell cycle exit (Kina et al, 2000).
Figure 2.
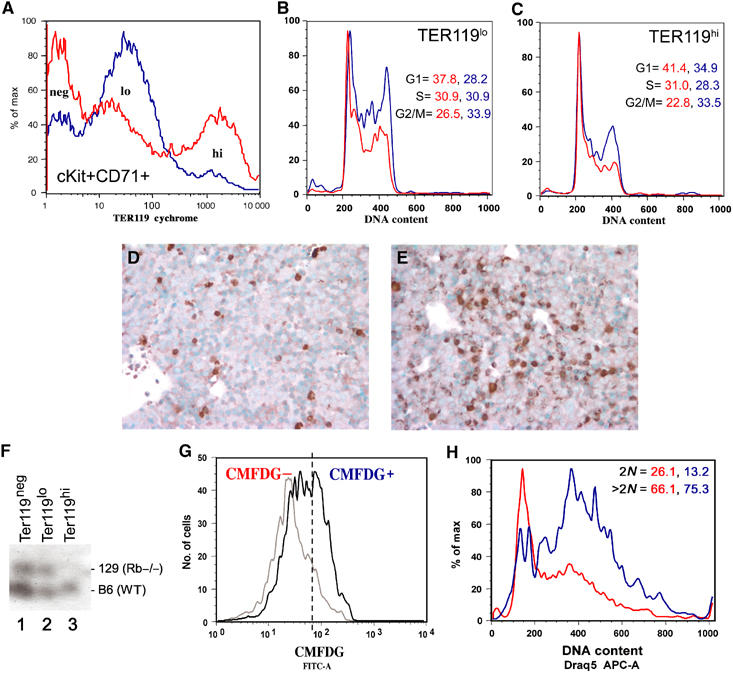
Rb null erythroblasts show deregulated cell cycle. Wild-type (red) and Rb−/− (blue) E12.5 cKit+CD71+ erythroblasts can be subdivided into TER119neg, TER119lo and TER119hi populations (A, 10 000 events). PI staining of TER119lo- (B) and TER119hi- (C) expressing cells from E12.5 wild-type (red profile) or Rb null FL (blue profile) revealing % cells in each population with G0/G1 (2N), S (2N–4N) or G2/M (4N) DNA content. Immunohistochemical staining of wild-type (D) and Rb−/− (E) FL for PH3 expression. GPI analysis of contribution by Rb null cells to the TER119neg, TER119lo and TER119hi populations in an E12.5 chimeric mouse embryo (F). FACS profile of TER119lo E12.5 chimeric (bold profile) and E12.5 wild-type control FL (gray profile) stained with CMFDG to distinguish β-galactosidase-expressing Rb−/− TER119lo erythroblasts (CMFDG+) from nonexpressing wild-type TER119lo erythroblasts (CMFDG−) (G). FACS analysis of CMFDG− (red) and CMFDG+ (blue) cells labeled with Draq5 to assess DNA content as a measure of cell cycle profile (H).
When we examined the cell cycle of E12.5 cKit+CD71+ erythroblasts, we observed that wild-type cells accumulated in G0/G1 with a 2N DNA content as they passed from the TER119lo state of differentiation (Figure 2B, red profile, 37.8%) to TER119hi (Figure 2C, red profile, 41.4%), whereas Rb null erythroblasts retained an abnormally high 4N DNA content (33.9% as TER119lo; 33.5% as TER119hi), consistent with accumulation in the G2/M phases of cell cycle (Figure 2C, blue profile). As shown in Figure 2A, very few Rb null erythroblasts had made the transition from TER119lo to TER119hi, consistent with an arrest of Rb null erythroblasts in G2/M at the TER119lo stage of erythroid maturation. Consistent with arrest in M phase, we detected substantially increased numbers of cells expressing high levels of phosphohistone H3 (PH3) in the Rb null FL (Figure 2E) compared to wild-type FL (Figure 2D). These results suggested that pRb was required to ensure proper cell cycle arrest of FL erythroblasts prior to, or coincident with, upregulation of TER119 expression. Loss of Rb resulted in mitotic arrest, a failure to upregulate TER119 and to enucleate.
Differentiation defects in Rb null red cells are cell intrinsic
To determine whether defects in cell cycle control during end-stage Rb null erythroid differentiation were cell intrinsic or not, we examined Rb null erythroblasts from E12.5 FL derived from chimeric mouse embryos generated by injection of wild-type blastocysts with Rb null-LacZ+(ROSA 26) transgenic ES cells (Lipinski et al, 2001). TER119neg-, TER119lo- and TER119hi-expressing erythroblasts were sorted from chimeric FLs at E12.5 and glucose phosphate isomerase (GPI) assays were conducted to determine the relative contribution of Rb null cells to each distinct erythroid population. As shown in Figure 2G in a representative GPI assay, the contribution of Rb null cells to the TER119lo population (lane 2, 51.6%) was greater than the contribution to the TER119neg population (lane 1, 18.9%), while the contribution to the TER119hi end-stage erythroid population (lane 3, 1.1%) was negligible. These percentages reflect the data shown in Figure 2A indicating that even in chimeric FL with comparatively low overall chimerism, Rb null erythroblasts show defects in end-stage erythroid maturation to TER119hi status and accumulate as TER119lo-expressing cells.
We compared the cell cycle status of Rb null TER119lo erythroblasts (5-chloromethylfluoroscein di-β-galactosipyranoside (CMFDG)-positive, Figure 2H) with wild-type (CMFDG-negative, Figure 2H) TER119lo erythroblasts from the same E12.5 chimeric FL. As shown in Figure 2I, increased numbers of Rb null (75.3%, CMFDG+, blue profile) erythroblasts possessed a greater than 2N DNA content compared to wild type (66.1%, CMFDG−, red profile), suggesting that Rb null TER119lo erythroblasts in the context of a low chimeric FL demonstrated similar cell cycle defects to that seen in the homozygous Rb mutant FL at E12.5 (Figure 2B and C), namely a G2/M arrest. These observations suggested that the erythroid cell cycle defect observed in the Rb null FL was cell intrinsic and independent of the placental defect since extraembryonic tissues in chimeric mice are blastocyst derived. Furthermore, the presence of wild-type cells was unable to confer the ability on Rb null erythroblasts to exit cell cycle or to upregulate TER119, suggesting that the erythroid defect in Rb null chimeric embryos was cell intrinsic.
To further explore the cell intrinsic nature of red cell defects in Rb null erythropoiesis, we examined the effects of acute deletion of Rb from erythroblasts in vitro. E12.5 FL erythroblasts were recovered from Rbflox/flox embryos (Marino et al, 2000), infected with either control retrovirus (pMSCV-IRES-GFP) or with Cre recombinase-expressing retrovirus (pMSCV-Cre-IRES-GFP) and then cultured for 2 days in the presence of dexamethasone, Kit ligand and erythropoietin to promote proliferation of erythroid progenitors (Bauer et al, 1999). GFP-positive TER119lo erythroblasts were FACS sorted for control cells (Figure 1A) and Cre-expressing cells (Figure 1B). Sorted cells were analyzed by PCR for the floxed allele to confirm that Cre-mediated deletion had taken place successfully in the pMSCV-Cre-IRES-GFP-infected cells (Figure 3C). Efficient (100%) deletion of the floxed allele was measured by reduction of the 700 bp PCR product (lanes 2 and 3) to a 260 bp PCR product (lanes 4 and 5) in the Cre-expressing cells (lanes 4 and 5) but not in the control virus-infected cells (lanes 2 and 3).
Figure 3.
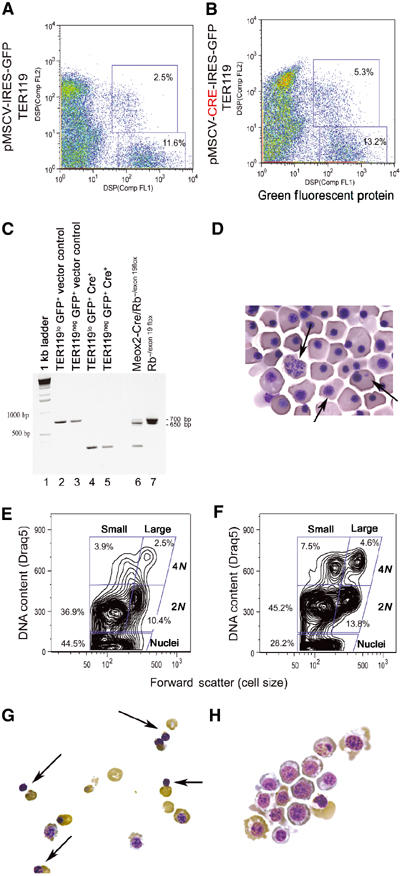
Acute inactivation of pRb in cultured erythroblasts. FACS sort profile of E12.5 FL erythroblasts harvested from Rbflox/flox mice, infected in vitro with control pMSCV-IRES-GFP retrovirus (A) or with Cre recombinase-expressing pMSCV-Cre-IRES-GFP (B) and sorted as GFP-positive/TER119neg- or GFP-positive/TER119lo-expressing cells. PCR analysis (C) of DNA harvested from sorted GFP-positive/TER119neg- (lanes 3 and 5) or GFP-positive/TER119lo- (lanes 2 and 4) expressing cells from either control virus-infected (lanes 2 and 3) or Cre-expressing virus-infected cells (lanes 4 and 5). Excision of floxed exon 19 reduces the size of the Rb PCR product in Rbflox/flox erythroblasts from 700 to 260 bp in erythroblasts infected with Cre-expressing retrovirus (lanes 4 and 5) but not with control retrovirus (lanes 2 and 3). PCR analysis of DNA from E13.5 Rb−/flox FL carrying an Rb null allele (650 bp) and a floxed exon 19 allele (700 bp) (lane 7) and following in vivo excision of the floxed exon 19 on the Meox2-Cre transgenic background generates a 260 bp fragment (lane 6). Peripheral red blood cells from Meox2-Cre/Rb−/exon 19flox E13.5 embryos show abnormal nuclear structure and reduced enucleation (D, arrows). FACS profile of cultured TER119lo/GFP+ erythroblasts following Cre-mediated excision of exon 19 (F) or control cultures (E) labeled with Draq5. Cytospin analysis of TER119lo/GFP+ erythroblasts following 2 days in culture showing extensive red cell enucleation (G, arrows) in the control cultures but not in the Cre-expressing cultures (H).
After 2 days in differentiation medium, Rbflox/flox erythroblasts in which exon 19 had been acutely deleted (RbΔexon19) showed a marked decrease in enucleation rate (28.2%, Figure 3F) compared to control Rbflox/flox erythroblasts (44.5%, Figure 3E). This is supported by cytological analysis showing extensive enucleation in control cultures (Figure 3G, arrows) but not in RbΔexon19 erythroblasts (Figure 3H). RbΔexon19 erythroblasts also showed increased cell size and nuclear:cytoplasmic ratio (Figure 3H) compared to control erythroblasts (Figure 3G), which is reminiscent of the defects observed in vivo (Figure 1H) and in vitro (Figure 1L) for constitutively Rb null erythroblasts. In addition to the enucleation defect, RbΔexon19 erythroblasts showed increased numbers of cells with a 4N DNA content (12.1%, Figure 3F) compared to control Rbflox/flox erythroblasts (6.4%, Figure 3E). These observations are consistent with those obtained from both homozygous Rb mutant FLs (Figure 2B and C) and Rb null chimeric FLs (Figure 2I) and suggest that the accumulation of Rb null erythroblasts with G2/M DNA content is determined by cell intrinsic defects.
Cre-mediated deletion of exon 19 in vitro was markedly more efficient than the deletion effected in vivo in the FL by crossing Rb−/flox mice to Meox2-Cre mice (lane 6), suggesting that these mice are chimeric for Rb loss. Nevertheless, we consistently observed red cell enucleation defects (Figure 3D) at E13.5 in Meox2-Cre/Rb−/exon 19flox embryos that mirrored those seen in the E13.5 homozygous null embryos (Figure 1H).
Mice rescued with Rb null FL die exhibiting anemia
Further evidence to support a cell intrinsic aspect to the erythroid defect came from hematopoietic reconstitution experiments carried out using wild-type, Rb heterozygous, Rb homozygous mutant or Rb null chimeric FL as donor tissue (Figure 4). All host mice rescued with wild-type FL survived beyond 12 months postirradiation (Figure 4A, red profile). In contrast, none of the mice rescued with Rb null FL survived more than 5 months after irradiation (Figure 4A, blue profile). Interestingly, heterozygosity for pRb reduced the reconstituting potential of FL (Figure 4A, green profile), suggesting that Rb may be haploinsufficient for aspects of hematopoietic homoeostasis.
Figure 4.
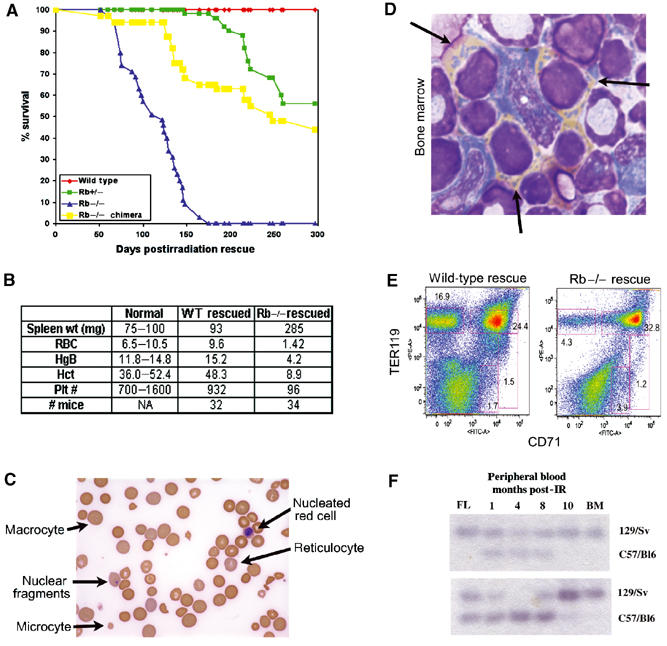
Decreased survival and aplastic anemia in mice reconstituted with Rb null FL. Survival curve of mice reconstituted with wild-type (32, red), Rb null (34, blue), Rb heterozygous (34, green) or Rb null chimeric (24, yellow) FL (A). Rb null rescued hosts at experimental end point (loss of >20% of their body weight) and matching wild-type rescued hosts were assessed for blood cell parameters including red blood cell number (RBC, × 106/ml), hemoglobin levels (HgB, g/l), hematocrit (Hct, %) and platelet numbers (Plt, × 103/ml). Spleen weights in mg are also indicated. Normal value ranges for each parameter are also shown (B). Peripheral blood of mice reconstituted with Rb null FL showing the appearance of abnormal erythrocytes (C). Cytospin of bone marrow from mice transplanted with Rb null FL shows the increased presence of benzidine-positive (yellow stain) nucleated erythroblasts with condensed nucleic acid in the cytoplasm (D, arrows). FACS analysis of bone marrow from representative wild-type and Rb null rescued mice at experimental end point (5 months post-IR) (E). Donor FL, peripheral blood (1, 4, 8 and 10 months post-IR rescue) and bone marrow (end point) of recipient mice were assessed by GPI analysis, for contribution by wild-type and Rb null cells (F).
Toward the end of their lives, mice rescued with Rb null FL showed a hunched appearance, overall weight loss and a general failure to thrive. Blood analysis of these animals revealed a dramatically reduced hematocrit, reduced hemoglobin (Figure 4B) and abnormal erythrocyte morphology evident in all blood smears (Figure 4C). These defects in adult erythroid differentiation were very similar to those observed in Rb null fetal erythropoiesis.
Most mice rescued with chimeric FL (Figure 4A, yellow profile) lived longer than mice reconstituted with Rb null FL (blue profile), but not as long as wild-type FL reconstituted mice (red profile). We observed that there was an initial decrease in contribution by Rb null cells to peripheral blood in all chimeric rescued mice, as measured by GPI contribution (Figure 4F). However, by 4–8 months postirradiation, Rb null cells reappeared in the peripheral blood and by experiment end point, showed high contribution to blood and bone marrow, irrespective of initial FL chimerism (Figure 4F). The dynamic contribution to peripheral blood by Rb null cells over time may reflect a growth advantage conferred on erythroid progenitors by Rb loss under conditions of stress. However, these mice ultimately suffered the same erythroid defects as Rb null rescued mice (low hematocrit, low hemoglobin levels, abnormally shaped RBCs) and also died of anemia.
Progressive hematopoietic depletion in mice reconstituted with Rb null FL
Bone marrow from mice reconstituted with Rb null FL or with Rb null chimeric FL contained increased numbers of benzidine-positive, nucleated red cells indicative of a maturation defect similar to that observed in the Rb null FL (Figure 4D). In addition to an enucleation defect, FACS analysis revealed that erythroblasts from bone marrow reconstituted with Rb null FL failed to downregulate the CD71 transferrin receptor (Figure 4E) in contrast to wild-type erythroblasts (Socolovsky et al, 2001). Loss of Rb resulted in markedly reduced numbers of bone marrow CD71−TER119hi erythroid cells (4.3%, Figure 4E) compared to wild type (16.9%, Figure 5A) and increased numbers of CD71+TER119hi erythroblasts (32.8%, Figure 4E) compared to wild type (24.4%, Figure 4E). The failure to downregulate CD71 in bone marrow erythroblasts suggested a critical role for Rb in the maturation of bone marrow red cells.
Figure 5.
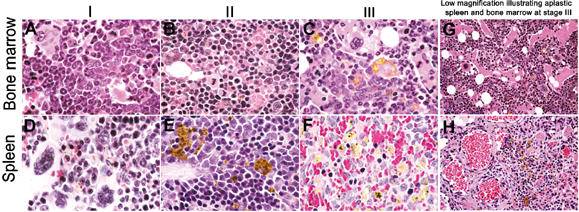
Bone marrow and spleen aplasia in mice rescued with Rb null FL. H&E-stained sections of bone marrow from mice rescued with Rb null FL showed various stages following adoptive transplant. Stage I: We initially observe reduced erythroid contribution and increased relative numbers of myeloid elements in the bone marrow (A), which is accompanied by extramedullary hematopoiesis to the spleen (D). Stage II: Loss of myelopoiesis from the bone marrow as defective erythropoiesis takes over (B). If this is also accompanied by displacement from the spleen (E), the mouse succumbs to anemia, prior to Stage III: the complete exhaustion of all elements from the bone marrow (C, G) and spleen (F, H).
More detailed histological examination of bone marrows and spleens from mice transplanted with Rb null FL (Figure 5) or Rb null chimeric FL revealed discrete changes in lineage contribution to these organs over time. Bone marrow pathologies could be subdivided into three categories: (I) reduced erythroblast numbers (and progenitors) but abundant production of granulocytes and megakaryocytes (Figure 5A); (II) erythroblast hyperplasia (Figure 5B) and (III) bone marrow depletion marked by extensive spaces and bone deposits (Figure 5C and G).
The spleen was enlarged in most of the Rb null rescued mice, from early to late stages following transplant, weighing approximately three to four times more than spleens from control mice rescued with wild-type FL (Figure 4B). Extramedullary hematopoiesis (EMH) in the spleen is indicative of defects in bone marrow hematopoiesis. We noted that multilineage EMH (Figure 5D) was most frequently observed in mice during the early stages of reconstitution with Rb null FL and not in moribund mice.
As mice became moribund, the spleens of most Rb null rescued mice (26 out of 34) showed loss of follicular structure as erythroid cells (judged by their size and dark staining nuclei) outpopulated other elements, including B cells. However, erythropoiesis in the spleens of these mice was ineffective (Figure 5E shows extensive hemosiderin deposition). Given the timing of the changes in bone marrow and spleen pathology, we propose that initial ineffective erythropoiesis in the bone marrow promotes multilineage EMH in the spleen, which is also ineffective due to defects in end-stage erythroid differentiation. This is followed by a period in which erythroblasts expand in the bone marrow and spleen as homeostatic mechanisms respond to anemia.
A significant proportion of mice (eight mice out of 34) rescued with Rb null FL showed very small spleens when they became moribund. These spleens were found to be aplastic and showed loss of hematopoietic progenitors (Figure 5F and H). Mice that showed fibrotic spleens also had aplastic bone marrow (Figure 5C and G).
These observations suggested that loss of Rb conferred a cell intrinsic growth advantage on CD71+ erythroblasts, which coupled with defective red cell maturation depleted the bone marrow and spleen of hematopoietic progenitors and cell types of other lineages.
Age-dependent erythroid defects in adult Rb null chimeric mice
All of our data presented so far suggested that loss of pRb precipitated cell intrinsic defects in red cell maturation. However, these observations appear to contradict previous results showing high contribution of Rb null cells to the peripheral blood of adult Rb null chimeras with no apparent defects in red cell maturation (Robanus-Maandag et al, 1994; Williams et al, 1994). We re-examined erythropoiesis in adult Rb null chimeras and observed effective contribution to the peripheral blood of young mice by Rb null cells, consistent with published data (Robanus-Maandag et al, 1994; Williams et al, 1994). However, as adult chimeric mice approached 4 months of age and became sick with pituitary tumors (marked by increased coat pigmentation), we noticed substantial increases in peripheral blood contribution by Rb null cells, reduced hematocrits and increasing numbers of morphologically abnormal red cells, similar to that observed in transplanted mice (Figure 4C).
When we examined the contribution of Rb null cells to specific lineages within the bone marrow of 4-month-old adult chimeras by FACS, we observed that in contrast to Mac1+F4/80+ macrophages (Figure 6B) and Mac1+Gr1+ granulocytes (Figure 6C), CD71+TER119+ erythroblasts were almost exclusively Rb null (Figure 6A). Rb null cells consistently contributed more highly to the CD71+TER119+ compartment irrespective of the overall chimerism of the bone marrow. In the example shown, the overall bone marrow chimerism was 20% but the contribution to the CD71+TER119+ population by Rb null cells was 76%. However, Rb−/− CD71+TER119+ erythroblasts (Figure 6F, red box) were unable to progress to the CD71−TER119+ state (Figure 6F, black box) and we observed reduced numbers of enucleated red cells in the bone marrow. These results clearly demonstrate that there are intrinsic requirements for pRb in erythropoiesis, since the presence of wild-type cells in the bone marrow could not rescue red cell maturation.
Figure 6.
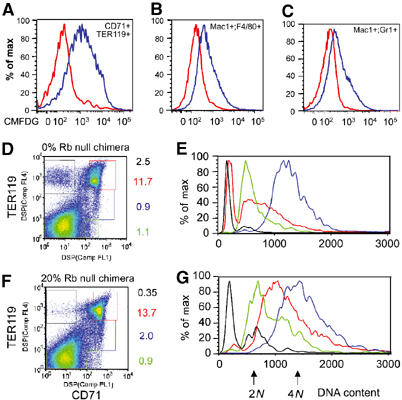
Adult chimeric mice show age-dependent onset of erythroid defects. Expression of β-galactosidase from the ROSA26-LacZ transgene allowed Rb null cells to be distinguished from wild-type cells by labeling with the fluorescent substrate CMFDG (A–C). At least 10 000 events were collected for both wild-type and mutant samples. In the bone marrow of the 20% chimera shown (measured by bone marrow GPI), we observed approximately 27% contribution to Mac1+F4/80+ cells (B, macrophages) and 21% Rb null contribution to Mac1+Gr1+ cells (C, granulocytes) consistent with the overall chimerism of the bone marrow, but 76% contribution to CD71+TER119+ cells (A, erythroblasts). (D–G) Chimeric (150 000 events) and nonchimeric bone marrow (150 000 events) was labeled with fluorescent antibodies to CD71 and TER119 (D, F), analyzed by FACS and the DNA content of specific populations (green=CD71loTER119neg, blue=CD71+TER119lo, red=CD71+TER119hi, black=CD71−TER119hi) was assessed by Hoechst staining (E, G).
Red cell enucleation is Rb dependent
We investigated in more detail whether there was a defect in cell cycle exit of end-stage red cells as we described for FL erythroblasts. During normal bone marrow erythropoiesis illustrated in Figure 6D with FACS data from a 0% chimera, erythroblasts progressed from being CD71loTER119− (green box) to CD71+TER119lo (blue box) to CD71+TER119+ (red box) to CD71−TER119+ (black box), as described previously (Socolovsky et al, 2001). By examining the DNA content of cells in these individual populations, we observed that cells undergo synchronous cell cycling before enucleation (Figure 6E). Cells progressed from being predominantly in G1 as CD71loTER119− erythroblasts (green profile, Figure 6E), accumulated in G2 as CD71+TER119lo erythroblasts (blue profile, Figure 6E) before extruding their nuclei as CD71+TER119hi cells (red profile, Figure 6E) to become enucleated CD71−TER119+ erythroblasts with a Hoescht-negative DNA content (black profile, Figure 6E). These observations suggested that normal bone marrow erythroblasts mature by passing through synchronous cell cycle, accumulate in G2/M and reduce DNA content by passing through a 2N–4N state (red profile). The last stage in maturation is marked by downregulation of CD71 and a further reduction in DNA content that presumably reflects enucleation.
Loss of Rb subverts this process and there is a failure to produce enucleated red cells (black box, Figure 6G). When we examined how Rb null erythroblasts progressed through the last cell cycle, we observed that there was less synchrony within each erythroblast population such that CD71loTER119− erythroblasts showed greater numbers of cells in S and G2/M of the cell cycle (green profile, Figure 6G). As cells upregulated CD71 and TER119 to become CD71+TER119lo, Rb−/− cells progressed into G2/M (Figure 6G, blue profile) but the profile was not as tight as that observed for wild-type CD71+TER119lo erythroblasts (Figure 6E, blue profile). The biggest cell cycle difference between wild-type and Rb null erythroblasts was seen for the CD71+TER119+ population that is over-represented in the Rb null erythroid compartment (Figure 6F, red box). These cells appeared to accumulate with 2N–4N DNA content, did not progress to a Hoescht-negative DNA content and failed to lose their nuclei (Figure 6G). The block to enucleation is associated with a failure to downregulate CD71 and the cells were arrested at this stage of erythroid maturation (Figure 6F).
In summary, these data show that loss of Rb leads to age-dependent onset of anemia in adult Rb null chimeric mice and that Rb null erythroblasts fail to enucleate due to defects in the last synchronous cell cycle before terminal differentiation. The presence of wild-type cells in the bone marrow of these adult chimeric mice did not mitigate the development of anemia suggesting that Rb plays a cell intrinsic function in erythropoiesis.
Rb is required for stress erythropoiesis
We were intrigued by the late onset of the erythroid defect in aging Rb null chimeras and by the apparent normality of Rb null erythropoiesis in young chimeric mice. Given that the onset of the erythroid defect in these mice overlapped with the onset of pituitary tumorigenesis, we explored the possibility that the defects in Rb null erythropoiesis (including that occurring in bone marrow transplants, in the Rb null FL or in culture in the presence of glucocorticoids) were triggered by rapid expansion under stress conditions. To test this hypothesis, we took young Rb null chimeras (3–6 weeks of age, prior to the onset of pituitary tumors) and treated them with phenylhydrazine (PHZ) to induce hemolytic anemia (Broudy et al, 1996). We reasoned that if pRb was specifically required for stress erythropoiesis but not for steady-state erythropoiesis, then Rb null cells would respond to anemia in the same way as in bone marrow transplants or in the embryo, by expanding preferentially but with defects in end-stage maturation.
Erythroid expansion in response to hemolytic anemia occurs in the spleen (Broudy et al, 1996). When control mice (wild type) or very low chimeric mice (2% Rb−/−) were injected with PHZ, we observed increased erythropoiesis in the spleen by 2 days after treatment and massive clusters of enucleated red cells (Figure 7A). When we examined the response of the spleen to hemolytic anemia in young adult chimeras of increasing chimerism, it became clear that the extent of erythroid maturation taking place in the spleen was inversely proportional to the extent of Rb−/− chimerism. For example, a 25% Rb−/− chimeric spleen showed intermediate levels of red cell maturation (Figure 7B), while a 60% chimera showed remarkably little end-stage red cell production (Figure 7C).
Figure 7.
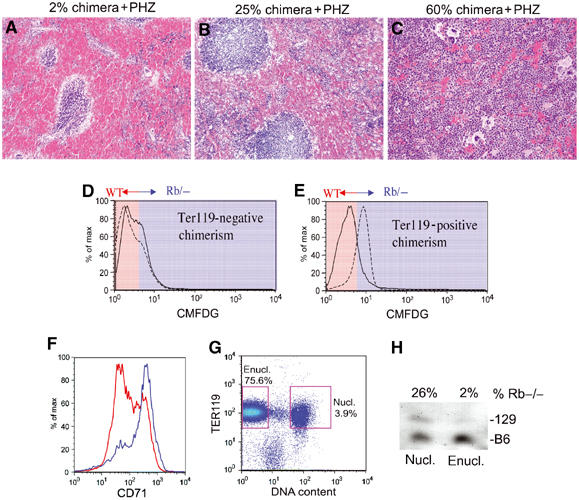
Stress erythropoiesis induces defective red cell maturation in Rb null erythroblasts. Sectioned spleens harvested from young adult chimeras following initial treatment with PHZ to induce hemolytic anemia are shown (Broudy et al, 1996). Spleens shown are from a very low chimera (A, estimated at 2%), low chimera (B, 25%) or medium/high chimera (C, 60%). Nucleated cells from matched low chimeric spleens (5–25% chimeric) were harvested following treatment with either PBS or PHZ, labeled with CMFDG and TER119 and analyzed by FACS (D, E). The dotted line indicates contribution by CMFDG+ Rb null cells and the solid line indicates contribution by CMFDGneg wild-type cells (D, E). Erythroblasts from a medium chimeric spleen (60% chimeric) were harvested following PHZ treatment, labeled with CMFDG, TER119-cychrome and CD71-PE, and gated on TER119+ cells (25 000 events) (F). Enucleated TER119+ (75.6% of total spleen cells) and nucleated TER119+ erythroblasts (3.9% of total spleen cells) were sorted from a low (5% Rb−/− cells) chimeric spleen after PHZ treatment. A total of 250 000 events were collected (G). GPI analysis on these sorted populations revealed that while Rb−/− cells contributed 26% of cells to the nucleated TER119+ fraction, they represented 2% of the enucleated TER119+ fraction of cells (H).
Chimeras (3–6 weeks old) showing identical low contribution of Rb null cells to the peripheral blood were treated with either PBS or PHZ and hematopoietic tissues were harvested after 2 days. In the spleens of all PHZ-treated mice, the overall contribution of Rb null cells to the TER119+ population increased (Figure 7E) in contrast to marginal decreases in contribution to the TER119neg population (Figure 7D). This suggests that loss of Rb conferred a growth advantage on erythroblasts under stress conditions. This effect was consistent for all levels of Rb null chimerism including mice with chimerism as low as 2% contribution (the limit of detection). Importantly, we failed to detect similar changes in contribution by Rb null cells to the TER119+ compartment in the control PBS-treated group of young chimeric mice (Figure 7D and E). These data clearly point to a specific role for Rb in regulating erythroblast expansion in response to anemic stress.
Deregulated expansion of Rb null TER119+ erythroblasts in young PHZ-treated spleens was accompanied by a failure to downregulate CD71 (Figure 7F, blue profile) consistent with the observed defects in red cell maturation in aging chimeras (Figure 6F) and reconstituted hosts (Figure 4E). Furthermore, chimeric mice with medium to high contribution to spleen by Rb null cells failed to recover from hemolytic anemia and were euthanized. These results clearly suggest that pRb is required for end-stage red cell differentiation under stress conditions.
In addition, we sorted nucleated and enucleated TER119+ cells from a 5% Rb−/− chimeric spleen following treatment with PHZ (Figure 7G). GPI analysis on these sorted populations revealed that although the overall chimerism of the spleen was only 5%, Rb null cells contributed approximately 26% to the nucleated erythroid compartment (Figure 7H). This is consistent with Rb loss conferring a growth advantage on erythroblast progenitors under stress conditions. However, Rb null cells failed to contribute to the enucleated fraction under stress conditions (Figure 7H), supporting our hypothesis that Rb is required for end-stage red cell maturation under anemic conditions. These results using chimeras with Rb null cell numbers at the limit of detection suggest that wild-type erythroblasts that make up the vast majority of the red cell compartment in the spleens of these animals were unable to provide a homotypic signal that promotes normal maturation of Rb null red cells.
Discussion
In recent years, the cell autonomous nature of the erythroid defect in Rb null mice has been a matter of scientific intrigue and controversy. Initial observations with Rb knockout mice (Clarke et al, 1992; Jacks et al, 1992; Lee et al, 1992) and more recent in vitro differentiation experiments (Clark et al, 2004) pointed to a critical requirement for Rb during erythroid maturation, while analysis of germline Rb null chimeras showed that Rb null cells could contribute to the peripheral blood and spleen of adult chimeras without any apparent defects in red cell maturation (Robanus-Maandag et al, 1994; Williams et al, 1994). Our current work reconciles these apparent differences by identifying for the first time a key role for pRb in regulating erythroid expansion and maturation specifically under stress conditions, such as occurs in the embryo and in the presence of glucocorticoids (Bauer et al, 1999). Even in adult chimeras with extremely high contribution by Rb null cells to the bone marrow and periphery (as high as 95% Rb−/−), normal red cells are produced at normal levels. This observation argues against the idea of a homotypic signal being produced by wild-type cells that rescues Rb null erythropoiesis, as proposed by Whyatt and Grosveld (2002). Strikingly, we can induce erythroblast expansion and red cell maturation defects in young adult Rb null chimeras by treating with PHZ to provoke hemolytic anemia. Under these stress conditions, Rb null erythroblasts expand preferentially but are defective for end-stage differentiation consistent with defects observed in FL erythropoiesis and in reconstituted mice. Stress-induced defects in Rb null erythroblasts were observed in young mice with chimerism as low as 5% (near the limit of detection), suggesting that paracrine signaling was not capable of imposing normal differentiation on Rb null cells. The presence of at least 95% wild-type cells in the bone marrow and spleens of these animals makes it unlikely that Rb null erythroblasts were defective due to loss of an Rb-dependent homotypic signal. The homotypic signaling model remains an attractive idea to explain defects in GATA-1-deficient erythropoiesis (Whyatt et al, 2000), but our work makes it an unlikely explanation for defects in Rb null erythropoiesis. However, once the identity of the GATA-1-dependent homotypic signal is established, it will be important to revisit Rb null erythropoiesis to examine whether or not its expression or function is disrupted.
Critical functions of pRb in stress erythropoiesis
Red blood cells are heavily exposed to reactive oxygen species (ROS), such as peroxides and hydroxy radicals, due to the abundance of heme iron and oxygen. This in turn can cause DNA damage if not modulated by the activity of antioxidants such as superoxide dismutase and peroxiredoxins (Finkel and Holbrook, 2000). Oxidative stress dephosphorylates pRb to induce a G1 cell cycle arrest (Esposito et al, 2000). Loss of Rb perturbs DNA damage-induced G1 and intra-S-phase checkpoints resulting in G2/M arrest (Harrington et al, 1998), such as we observe in Rb null erythroblasts. We propose that the unique requirement for pRb in stress erythropoiesis reflects its critical role in regulating DNA damage checkpoints induced by oxidative stress. In the absence of stress, Rb is not critical for erythroid differentiation (thus explaining the absence of a phenotype in young chimeric adult mice), but under stress conditions (hemolytic anemic, tumorigenesis, bone marrow transplant, fetal development, high glucocorticoid levels), pRb is essential. Interestingly, deficiency for DNA damage sensing and repair molecules, such as ATR and FancD2, is associated with anemia and bone marrow failure (D'Andrea and Grompe, 2003; O'Driscoll et al, 2003), such as we observe in Rb null reconstituted mice.
The identification of physiologically relevant E2F target genes in red cell proliferation and maturation will be key to understanding the unique properties of pRb in erythroblast stress response. Our data show that loss of pRb results in a failure of erythroblasts to downregulate the transferrin receptor (CD71) and increased expression of CD71 may contribute to oxidative stress in Rb null red cells by increasing the intracellular levels of ferrous iron. CD71 is upregulated in Rat-1a cells following overexpression of E2f-1 or E2f-3 (Polager et al, 2002), suggesting that it may be a direct E2f target gene. Alternatively, effects of Rb loss on CD71 expression may be indirect and reflect hypoxia or increased oxidative stress (Bianchi et al, 1999). Work is in progress to explore these various possibilities.
A complex developmental phenotype in Rb null mice
The use of conditional targeting has revealed the complexity of developmental defects in Rb null mice (Ferguson et al, 2002; Macpherson et al, 2003; Wu et al, 2003). Defects in the lens and peripheral nervous system appear cell intrinsic as are cell cycle defects in the central nervous system. However, apoptosis in the central nervous system is induced by hypoxia and most likely arises as a consequence of the placental defect, the red cell defect or both (Macpherson et al, 2003). Our data demonstrate that the red cell defect is intrinsic to the erythroid system and occurs in embryos that have a wild-type placenta. The inability of the Rb null erythroid system to cope with hypoxic stress undoubtedly contributes to the timing of the demise of Rb null mice since Rb−/−;E2f1−/− and Rb−/−;E2f2−/− mice live longer in development despite persistent placental defects (A Dirlam and KF Macleod, unpublished). Rb null chimeric and conditional Meox2-Cre/Rbflox/flox embryos show many of the same erythroid defects at E13.5 as the germline homozygous Rb null mutant embryo, including a failure of red cells to enucleate. Given the rapid growth that occurs during embryogenesis, we hypothesize that any diminution of the oxygen supply or transport mechanism is detrimental to the survival of the mouse, whether that be caused by defective placental function or by aberrant red cell development. At later stages of development, such as E16.5, the Rb null embryo is dead making comparisons with chimeric and conditional mutant FLs impossible. Nevertheless, both the chimeric and conditional mouse models show persistent red cell enucleation defects during development and at birth, consistent with a cell intrinsic defect that is independent of the placental defect. We suspect that these defects are less striking at later stages of development due to decreased hypoxic stress, and current work is examining the consequences of specifically rescuing the red cell defect in Rb null mice.
Recent work by Clark et al (2004) also showed that erythroblasts from Rb null FL failed to differentiate properly in vitro but did not exclude the possibility that Rb null erythroblasts had acquired those defects in vivo. We present key experiments here showing that acute deletion of Rb from Rbflox/flox erythroblasts induces deregulated cell cycle and enucleation defects similar to those seen in vivo. This demonstrates for the first time that erythroid defects are cell intrinsic and not the consequence of developmental history in an embryo with a defective placenta.
Intriguingly, we observed reduced reconstitution potential with Rb heterozygous FL compared to wild type. Our unpublished data also identified age-dependent onset of myelodysplasia in adult Rb heterozygous mice that is not associated with loss of the wild-type Rb allele, suggesting that Rb is haploinsufficient for aspects of hematopoietic homeostasis. Work is ongoing to characterize this defect further, but preliminary data show that red cell maturation is not abnormal and that the defect lies at a different point in the hematopoietic hierarchy.
Conclusions
Our data demonstrate a clear cell intrinsic requirement for pRb during stress erythropoiesis to regulate erythroblast expansion and promote end-stage erythroid differentiation, and shed light on how aplastic anemia and myelodysplasia might develop in humans. Furthermore, by examining how pRb regulates the response to oxidative stress in red cells, future work aims to examine the effects of DNA damage on erythroid differentiation.
Materials and methods
Mice and transplantation procedures
All mice were maintained in an SPF barrier facility. Rb heterozygous mice used in these studies were maintained as an inbred 129S/v line of mice. Timed matings of Rb heterozygous mice were set up as described previously (Jacks et al, 1992). Meox2-Cre mice were obtained from Jackson labs and Rbflox/flox mice were obtained from NCI MHCC. Timed matings for conditional targeting of Rb loss to the embryo were carried out by interbreeding Meox2-Cre/Rb+/− mice to Rbflox/flox mice and genotyping embryos for the presence of the null allele and the floxed allele (Marino et al, 2000). PHZ was injected intraperitoneally (60 mg/kg) as described previously (Broudy et al, 1996). Embryos and spleens were fixed in 10% neutral buffered formalin. Chimeric mice were generated by injection of C57B/6 blastocysts with Rb null 129S/v ES cells on a ROSA26 background (Lipinski et al, 2001). FL cells were counted following dissection from each embryo and 2 × 106 cells were injected immediately into lethally irradiated C57B/6 host mice. Host mice were irradiated with 8 Gy followed 3 h later by irradiation with 4 Gy at a dose rate of 52 Gy/h. The genotype of injected FL tissue was determined afterwards by PCR analysis of nonhematopoietic embryonic tissue. Host and donor cell contribution to blood, bone marrow and spleen was determined by GPI analysis or by CMFDG labeling (see below). GPI assays were carried out as described previously (Williams et al, 1994) and plates were quantified by densitometry.
FACS
Antibodies (BD Pharmingen/Clontech) used for surface labeling of cells are as follows: R-phycoerythrin-conjugated rat anti-mouse CD71 (cat.# 553267), FITC-conjugated rat anti-mouse CD71 (cat.# 553266), R-phycoerythrin-conjugated rat anti-mouse CD117 (c-Kit) (cat.# 5533557), FITC-conjugated rat ant-mouse CD117 (c-Kit) (cat.# 01905B/553354), R-phycoerythrin-conjugated rat anti-mouse TER119 (cat.# 09085B), biotin-conjugated rat anti-mouse TER119 (cat.# 553672), streptavidin-conjugated cychrome (cat.# 13038A), APC-conjugated rat anti-mouse Mac-1 (cat.# 553312), PE-conjugated rat anti-mouse Gr1 (cat.# 553128) and R-phycoerythrin conjugated rat anti-mouse F4-80 (CalTag cat.# RM2904). Single-cell suspensions were antibody labeled and washed in 2% fetal calf serum in PBS. Surface labeled cells were analyzed by FACScan (Becton-Dickinson) and cells were sorted using MoFlo (DAKO Cytomation). Cells were labeled with the fluorescent β-galactosidase substrate CMFDG in the presence of 1 μM chloroquine diphosphate (‘Loading cells using influx reagent' from Molecular Probes, cat.# D-2920). Cells were labeled with 5 μg/ml Hoechst 33342 stain (Sigma cat.# B-2261) or with 5 μM Draq5 (Alexis Corporation). Sorted FL cells were fixed in 70% ethanol at −20°C, RNase treated and labeled with 10 μg/ml PI for 4–16 h before analyzing DNA content by FACS.
In vitro erythroblast culture and conditional targeting
Erythroblasts were seeded at 3 × 104 cells per 3.3 ml of Methocult GF M3334 containing erythropoietin (Stem Cell Technologies) for 2 days to examine CFU-E colony formation. Colonies were picked for cytospin analysis. Erythroblasts were harvested from E12.5 Rbflox/flox embryos (Marino et al, 2000) and infected with Cre recombinase-expressing pMSCV-Cre-IRES-GFP retrovirus or with control virus (pMSCV-IRES-GFP) in expansion culture media including 1 μM dexamethasone, 100 ng/ml Kit ligand and 2 U/ml erythropoietin (Bauer et al, 1999). GFP+ TER119lo cells were FACS sorted away from GFPneg/TER119neg cells after 2 days in expansion culture and cultured in differentiation media containing 1 μM mifepristone and 2 U/ml erythropoietin for a further 2 days.
Histological procedures and immunohistochemistry
Immunohistochemistry for PH3 expression was carried out using a 1:500 dilution of primary antibody (rabbit anti-PH3 from Upstate cat.# 06-570) and developed using Vectastain reagents, as described previously (Macleod et al, 1996). Blood smears and cytospins were benzidine stained for hemoglobin and with Wright–Giemsa to highlight nuclear and cytoplasmic structures. Blood statistics including hematocrit, RBC, RDW, platelet number, and other measurements were carried out using the automated Spectron Corporation SYSMEX K-1000 Hematology Analyzer.
Acknowledgments
We thank Linda Degenstein of the Transgenic Core Facility at the University of Chicago for the generation of chimeric mice. KM is the Raymond Zelko fellow of the Cancer Research Foundation and a V Foundation scholar. TJ is an associate investigator of the Howard Hughes Medical Institute.
References
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schutz G, Beug H (1999) The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev 13: 2996–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Tacchini L, Cairo G (1999) HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res 27: 4223–4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broudy VC, Lin NL, Priestley GV, Nocka K, Wolf NS (1996) Interaction of stem cell factor and its receptor c-kit mediates lodgement and acute expansion of hematopoietic cells in the murine spleen. Blood 88: 75–81 [PubMed] [Google Scholar]
- Clark AJ, Doyle KM, Humbert PO (2004) Cell intrinsic functions of pRb in erythropoiesis. Blood 104: 1324–1326 [DOI] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NMT, van der Valk M, Hooper ML, Berns A, te Riele H (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359: 328–330 [DOI] [PubMed] [Google Scholar]
- D'Andrea AD, Grompe M (2003) The Fanconi anemia/BRCA pathway. Nat Rev Cancer 3: 23–34 [DOI] [PubMed] [Google Scholar]
- Esposito F, Russo L, Russo T, Cimino F (2000) Retinoblastoma protein dephosphorylation is an early event of cellular response to pro-oxidant conditions. FEBS Lett 470: 211–215 [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS (2002) Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21: 3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of aging. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Godin I, Cumano A (2002) The Hare and the tortoise: an embryonic haematopoietic race. Nat Rev Immunol 2: 593–604 [DOI] [PubMed] [Google Scholar]
- Harrington EA, Bruce JL, Harlow E, Dyson N (1998) pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 95: 11945–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmidt E, Bronson R, Goodell M, Weinberg R (1992) Effects of an Rb mutation in the mouse. Nature 359: 295–300 [DOI] [PubMed] [Google Scholar]
- Kina T, Ikuta K, Takayama E, Wada K, Majumdar A, Weissman IL, Katsura Y (2000) The monoclonal antibody TER-119 recognizes a molecule associated with glycophorin A and specifically marks the late stages of murine erythroid lineage. Br J Haematol 109: 280–287 [DOI] [PubMed] [Google Scholar]
- Lee EY-HP, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288–295 [DOI] [PubMed] [Google Scholar]
- Lipinski M, Macleod KF, Crowley D, Mullaney T, Jacks T (2001) Cell-autonomous and non-cell autonomous developmental functions of the retinoblastoma tumor suppressor gene in vivo. EMBO J 20: 3402–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod K, Hu Y, Jacks T (1996) Loss of Rb activates both p53-dependent and -independent cell death pathways in the developing mouse nervous system. EMBO J 15: 6178–6188 [PMC free article] [PubMed] [Google Scholar]
- Macpherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T (2003) Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol 23: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van der Gulden H, Jonkers J, Berns A (2000) Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14: 994–1004 [PMC free article] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA (1994) p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 371: 72–74 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Gen 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Polager S, Kalma Y, Berkovich E, Ginsberg D (2002) E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21: 437–446 [DOI] [PubMed] [Google Scholar]
- Robanus-Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O'Brien J, van Roon M, van der Lugt N, Berns A, te Riele H (1994) Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J 13: 4260–4268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF (2001) Ineffective erythropoiesis in Stat5a−/−5b−/− mice due to decreased survival of early erythroblasts. Blood 98: 3261–3273 [DOI] [PubMed] [Google Scholar]
- Whyatt D, Grosveld F (2002) Cell-autonomous function of the retinoblastoma tumor suppressor protein: new interpretations of old phenotypes. EMBO Reports 3: 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyatt D, Lindeboom F, Karis A, Ferreira R, Milot E, Hendriks R, de Bruijn M, Langeveld A, Gribnau J, Grosveld F, Philipsen S (2000) An intrinsic but cell-nonautonomous defect in GATA-1 over-expressing mouse erythroid cells. Nature 406: 519–523 [DOI] [PubMed] [Google Scholar]
- Williams BO, Schmitt EM, Remington L, Bronson RT, Albert DM, Weinberg RA, Jacks T (1994) Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J 13: 4251–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G (2003) Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947 [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth JD, Philips RA, Gallie BL (1996) pRb controls proliferation, differentiation, and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev 10: 3051–3064 [DOI] [PubMed] [Google Scholar]