

Abstract
Objective(s):
To understand the role of mitochondrial respiration in cisplatin sensitivity, we have employed wild-type and mitochondrial DNA depleted Rho0 yeast cells.
Materials and Methods:
Wild type and Rho0 yeast cultured in fermentable and non-fermentable sugar containing media, were studied for their sensitivity against cisplatin by monitoring growth curves, oxygen consumption, pH changes in cytosol/mitochondrial compartments, reactive oxygen species production and respiratory control ratio.
Results:
Wild-type yeast grown on glycerol exhibited heightened sensitivity to cisplatin than yeast grown on glucose. Cisplatin (100 μM), although significantly reduced the growth of wild- type cells, only slightly altered the growth rate of Rho0 cells. Cisplatin treatment decreased both pHcyt and pHmit to a similar extent without affecting the pH difference. Cisplatin dose-dependently increased the oxidative stress in wild-type, but not in respiration-deficient Rho0 strain. Cisplatin decreased the respiratory control ratio.
Conclusion:
These results suggest that cisplatin toxicity is influenced by the respiratory capacity of the cells and the intracellular oxidative burden. Although cisplatin per se slightly decreased the respiration of yeast cells grown in glucose, it did not disturb the mitochondrial chemiosmotic gradient.
Keywords: Cisplatin, Mitochondria, Oxidative phosphorylation, Respiration, Rho0
Introduction
Cisplatin (CDDP, cis-diamine dichloro platinum) is one of the most widely utilized anticancer drugs against malignancies of ovaries, testicles as well as head and neck (1, 2). The therapeutic potential of CDDP is constrained by the extremely undesirable effects and drug resistance (3).
Cisplatin loses chloride moieties and assumes positive charge inside the cell, tending to accumulate in the negatively charged mitochondrial compartment and reacts with nucleophilic sites to form DNA crosslinks (4). The formation of DNA adducts induces nucleotide excision repair mechanism, but few Pt-DNA adducts cannot be repaired and accumulation of these adducts correlates with CDDP cytotoxicity (5). The DNA lesions induced by CDDP impair cell cycle progression as well as cell division, in addition to inhibiting mRNA synthesis and promoting apoptosis (6).
Mitochondria are subcellular organelles involved not only in energy metabolism, but are critical effectors of major apoptotic pathways through generation of free radical species (7, 8). CDDP binds mitochondrial DNA more efficiently than nuclear DNA (9). It was proposed that less efficient DNA repair mechanisms in mitochondria may be responsible for inducing cytotoxicity and cell death (10-13) and altered mitochondrial function is implicated in tumor drug resistant tumors (14). Further, adverse effects of CDDP are intricately associated with mitochondrial DNA damage and dysfunction (15). Cisplatin strongly suppressed mitochon -drial DNA replication and transcription. It was also demonstrated that CDDP impairs mitochondrial respiration and oxidative phosphorylation (16). Although cisplatin impaired mitochondrial respiration and inhibited OXPHOS system, their causal role in production of ROS in inducing cell death or resistance remains unknown.
Production of several free radicals including peroxide, superoxide and hydroxyl radicals is catalyzed by the redox-active enzymes such as CYP450 isoforms, nitric oxide synthase (NOS), NADPH oxidase, NOX (17-20). These oxidants are highly reactive towards various cellular biomolecules. Cytochrome bc1 electron transfer complex of the electron transport chain is capable of producing ROS (17). The levels of these free radical species are generally controlled and minimized by antioxidant defense such as superoxide dismutase, metallothionein, GSH (21). Cells exhibiting impaired electron transfer due to loss of cytochrome c (copper deficiency), or mutations in complex III and IV produce higher levels of hydrogen peroxide (22). In this study, we have attempted to understand the importance of mitochondria in cisplatin sensitivity/resistance, using WT and Rho0 (mitochondrial DNA depleted and respiration deficient) cells.
Materials and Methods
Chemicals
CDDP was dissolved in DMSO to prepare a 500 mM stock solution. A 4 mM stock solution of 2’, 7’-Dichloro -fluorescein diacetate (Molecular Probes) was prepared in ethanol. These reagents were stored in a refrigerator to ensure no direct exposure to light. Other fine chemicals were procured from Sigma Aldrich.
Strains
The haploid BY4741 (MATα; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0) wild-type strain was obtained from IMTECH, Chandigarh, India. Wild-type strain was depleted of mtDNA by continuous culturing for 3 days in YPD medium containing ethidium bromide (10 mg/ml). Cells were streaked in parallel on YPD and YPEG [1% (w/v) yeast extract, 2% (w/v) bactopeptone, 3% (v/v) ethanol, 3% (v/v) glycerol] plates to obtain individual colonies. Rho0 strain, as expected, grew only on YPD plates as small colonies (23). Absence of mtDNA was verified by DAPI staining.
Growth conditions and cisplatin treatment
WT and Rho0 cells were diluted in minimal medium and grown at 30 °C (OD600 ~0.2). Cells were treated with varying concentrations of CDDP or equal volume of vehicle, DMSO. Optical density of cell cultures was monitored at 600 nm.
Measurement of cytosolic pH (pHcyt and pHmit)
pHcyt and pHmit were accurately determined by using pHluorin gene construct as mentioned elsewhere (24). Cells made to express pHluorin either in cytosol or mitochondrial compartments using specific targeting signals were grown in black microplates with clear bottom. The minimal medium was buffered to the desired pH of 5.0, using 25 mM sodium citrate. Fluorescence intensity was measured at λem=512 nm after excitation at dual wavelengths, 390 and 470 nm. pH values corresponding to the pHluorin emission intensity were derived from a calibrated standard curve.
Determination of the rate of oxygen uptake
Effect of cisplatin on oxygen consumption was measured at 30 °C using a Clark-type oxygen electrode. Reaction was initiated using 0.5% (w/v) glucose solution to the reaction mixture consisting of 100 mM potassium phosphate buffer (pH 3.0 or 5.0 or 7.0), 10 mM MgSO4 and ~6 x106 cells. Increasing concentrations of cisplatin were added to the reaction mixture after 2 min of incubation. Oxygen consumption was determined using the standard dissolved oxygen concentration of 236 μM in air-saturated water at 30 °C.
Colony viability curve
Exponential phase yeast cells grown to reach an optical density of 0.2 at 600 nm were treated with varying concentrations of cisplatin. After 6 hr of incubation, approximately 300 cells equivalent to 1x10-5 OD units were streaked on to YPD plates. The number of colonies was determined after the cells were allowed to multiply at 30 °C for 3 days.
Oxidative stress
Actively growing yeast cells of wild-type and Rho0 strains were allowed to reach optical density values of 0.2 and were treated with 0–100 μM cisplatin. To detect the levels of ROS, 10 μM of the ROS-sensitive fluorescent dye, 2’,7-dichlorofluorescein diacetate was added. After 3 hr of incubation at 30 °C with DCF, the cultures were centrifuged (3 min at 3000 r.p.m.), and cell pellets were washed and resuspended in 1 ml water. Fluorescence (λex=485 nm, λem=535 nm) intensity was measured and adjusted for the cell density.
Measurement of ROS using EPR spectroscopy
The spin trap, 5, 5-dimethyl-1-pyrroline-N-oxide (DMPO) was procured from Aldrich. 1 mM Diethylene traimine penta acetic acid (DTPA) was from Sigma. The reaction mixture consisted of DMPO (80 mM) in air saturated DMSO solution (containing 10% buffered aqueous solution of 0.05 M sodium borate-carbonate buffer, pH 10.0) incubated with either WT or Rho0 yeast cultures for 30 min. Individual yeast cell pellets were housed in the EPR TM cavity using an EPR aqueous flat cell (60 x 17 x 0.25 mm, inner diameter) for the measurement of free radical generation. The EPR spectra were recorded with a Bruker D-200 ER spectrometer. The EPR parameters were set at 100 K Hz, X band microwave frequency, 9.78 GHz; microwave power 20 mW; modulation amplitude 0.63G; time constant 0.32S, scan time 500 s; and receiver gain 5 x 105.
Results
Generation of Rho0 (ρ0) cells
Wild-type yeast cells that were treated with ethidium bromide to deplete mitochondrial DNA were found to grow as small colonies on non-fermentable carbon source glycerol. Loss of mitochondrial DNA in Rho0 cells was confirmed as the absence of DAPI staining in the cytosol (Figure 1).
Figure 1.
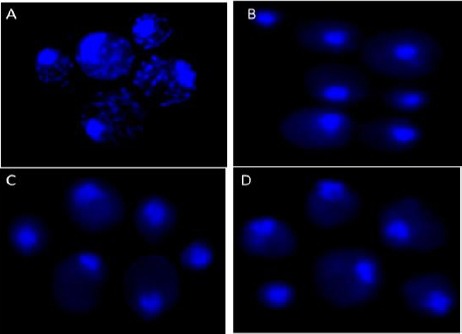
Depletion of mtDNA in Rho0 mutants. Haploid BY4741 cells treated for 3 days with EtBr and parental cells were stained with DAPI to visualize DNA by fluorescence microscopy. Panel A, parental BY4741 cells; panel B-D, Different isolates of EtBr treated cells showing loss of mitochondrial DNA
Wild-type yeast cells are more sensitive to cisplatin
Mitochondria are critical mediators of cisplatin toxicity (9, 25, 26). To investigate whether mitochondrial respiration influences the toxicity of cisplatin, yeast growth on medium containing fermentable or non-fermentable substrates and treated with increasing concentrations of CDDP were monitored. In parallel experiments, cells lacking mitochondrial DNA (Rho0) were also treated with cisplatin.
When glucose is available, the wild-type cells utilize glycolytic pathway to obtain energy for cellular requirements. When glycerol serves as the substrate, cells tend to depend more on respiration than glycolytic pathway (27). Growth rates of cells grown in glucose and exposed to 50 μM cisplatin declined significantly compared to untreated cells (Figure 2A). However, on glycerol, growth rates further declined with 50 μM CDDP (Figure 2B). Growth of wild-type yeast on the non-fermentable substrate glycerol was more CDDP-sensitive than on the fermentable substrate glucose, indicating that mitochondrial respiration influences cisplatin cytotoxicity.
Figure 2.
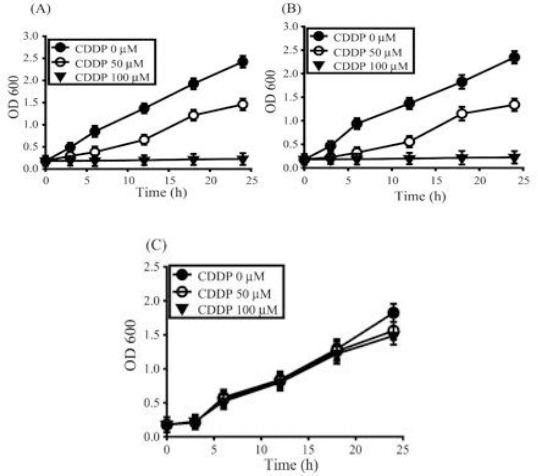
Respiration affects cisplatin sensitivity. The wild-type yeast strain grown on (A) fermentable substrate or (B) non-fermentable substrate and (C) Rho0 strain were incubated with 0 μM (●), 50 μM (○), or 100 μM (▼) CDDP. Data represents the mean±SD of n=3 independent cell isolates whose growth rate was determined by OD600 values
Cells forced to respire exhibited less growth rates and increased toxicity to cisplatin at low concentration. At high concentrations of cisplatin (100 μM), cell growth was totally suppressed both on glucose and glycerol.
To further probe if respiration efficiency influences cisplatin sensitivity, we studied the susceptibility of Rho0 strain to cisplatin. Mutant Rho0 cells owing to loss of mitochondrial DNA and devoid of respiratory enzymes can grow on fermentable substrate such as glucose, but display slow growth rates compared to wild-type. Whereas wild type cells barely grew when challenged with 100 μM cisplatin, the growth of mutant Rho0 cells was minimally affected (Figure 2C), indicating that respiration indeed regulates cisplatin toxicity.
Cisplatin suppresses mitochondrial respiration, but does not dissipate the proton gradient
It is proposed that cisplatin cytotoxicity is dependent on its ability to uncouple oxygen uptake from ATP synthesis by dissipation of H+ gradient. By utilizing pHluorin expression in cytosolic and mitochondrial compartments (24), we measured pHcyt and pHmit in buffered medium. Cisplatin (100 μM) exposure in buffered medium exhibited similar cytotoxicity as 50 μM cisplatin in non-buffered medium. Cisplatin, being a weak acid, exhibits significantly greater uptake from acidic milieu, resulting in increased intracellular accumulation and heightened cellular toxicity.
Actively growing wild-type cells in glucose-containing medium displayed a pHcyt of 6.9 and pHmit of 7.2. After addition of 100 μM CDDP, pHcyt and pHmit values decreased quickly within 30 min (Figure 3A), but remained stable at 6.4 and 6.6 until 6 hr (data not shown). Earlier study indicated that the uncoupler carbonylcyanide m-chlorophenylhydrazone (CCCP) diminishes the difference between pHcyt and pHmit (24). As cisplatin influenced the pH change in both compartments to the same extent and maintained the pH difference, we reasoned that cisplatin does not uncouple respiration.
Figure 3.
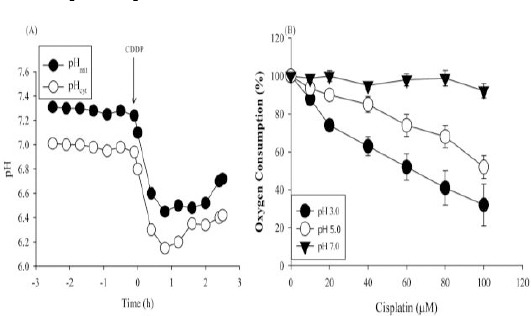
CDDP suppresses oxygen consumption but does not affect the proton gradient. (A) Intracellular pH of cytosolic pHcyt (○) and mitochondrial pHmit (●) compartments of wild-type cells were determined by monitoring the pH-sensitive protein, pHluorin fluorescence, adjusted for cell density. At t = 0 (arrow), 100 μM CDDP was added. Data are expressed as mean±SD (n=3). (B) Oxygen consumption was determined in wild-type cells buffered at pH 3.0, 5.0 and 7.0 with 0–100 μM CDDP. Data are expressed as percentage of oxygen consumption compared with cells incubated without CDDP (100%) (mean ± SD, n=3)
Due to increased lactic acid production, tumor core tends to be more acidic owing to enhanced aerobic and anaerobic glycolysis. Homeostatic mechanisms regulate the extracellular pH below 7.0, while maintaining the intracellular pH in the neutral range above 7.0. Lowered extracelluar pH and maintenance of the pH gradient across the cell membrane significantly influence the sensitivity of tumors to chemotherapeutic drugs. For instance, the acidic milieu of the extracellular medium promotes the uptake of weakly acidic drugs, such as cyclophosphamide and cisplatin and thus increases their cytotoxicity. As CDDP shows enhanced cytotoxicity to yeast in glycerol (respiring) compared to glucose (fermentation) medium, we evaluated if cisplatin affects respiration in yeast. Wild-type cells suspended in potassium phosphate buffer at pH 7.0, 5.0 or 3.0 and treated with increasing concentrations of cisplatin were monitored for oxygen uptake upon addition of glucose (Figure 3B). Basal oxygen consumption did not differ with pH (~7.4 fmol/min/cell) and was considered 100%. CDDP suppressed respiration in a dose-dependent manner, at acidic pH compared to neutral pH. A striking decrease in respiration was evident at pH 3.0. CDDP (100 μM) suppressed respiration by ~13% at pH 5.0, on par with the inhibition by 50 μM CDDP at pH 3.0. Due to comparable growth retardation at pH 3.0 and pH 5.0, it can be argued that growth inhibition correlates with reduced oxygen consumption in this pH range. Collectively, the data suggest that CDDP does not uncouple, but suppresses respiration, with severe inhibition at the acidic pH.
Cisplatin induces the formation of ROS in wild-type but not in Rho0 cells
Because CDDP was found to suppress respiration and as the mitochondrial respiratory chain is known to produce free radicals, we studied the influence of respiration on CDDP-induced generation of free radical species. Wild-type and Rho0 strains were treated with increasing concentrations of cisplatin and incubated for
3 hr in the presence of ROS-sensitive fluorescent compound 2’,7’-dichlorofluorescin diacetate. Basal levels of free radicals produced in untreated cells were set at 100%. Cisplatin treatment resulted in a dose-dependent increase in DCF fluorescence in wild-type cells (Figure 4). DCF fluorescence increased by 2 and 3.5-fold with 60 and 100 μM cisplatin, respectively when compared to untreated wild-type cells. These cisplatin concentrations did not induce DCF fluorescence to the same extent in Rho0 cells, compared to the untreated Rho0 strain (Figure 4). These results were further substantiated by a more sensitive electron paramagnetic resonance (EPR) spectroscopy (Figure 5). No detectable EPR signal was observed with 0 μM CDDP, both for WT and Rho° cells. The EPR spectrum observed with 25 μM and higher concentration of CDDP in both WT and Rho0 cells exhibited a 2x3x3x2 splitting which usually results from the interaction of an uncoupled electron with a primary nitrogen atom along with the secondary beta and gamma-protons. Based on hyper fine splitting and coupling constants, we observed the free radicals as superoxide free radicals. The hyperfine splitting constants of the signal (aN = 13.1 G, βaH=10.4G, and γaH =1.3 G) are consistent with previously reported values for DMPO.O2- in DMSO (28-31). Thus, it follows that CDDP generates superoxide both in WT and Rho0 cells and DMPO is capable of reacting with this primary species to form the spin adduct with the characteristic EPR signal as shown in Figure 5. It is therefore clear that CDDP induces the superoxide radical in wild type cells, but not in Rho0 cells. These results demonstrate a relationship between respiration, oxidative stress and CDDP toxicity.
Figure 4.
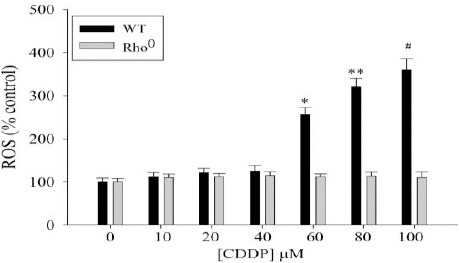
CDDP induces oxidative stress in wild-type but not in Rho0 cells. The wild-type yeast strain (black bars) and Rho0 strain (white bars) were challenged with increasing concentrations of cisplatin for 3 hr. DCF fluorescence was determined and normalized for cell density. ROS formation is expressed as percentage fluorescence compared with untreated wild-type or Rho0 cells (100%) Data are representative of mean ± SD of three different cell isolates
Figure 5.
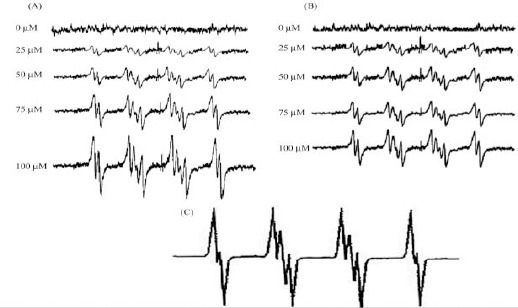
Effect of cisplatin on cellular ROS generation in wild-type and Rho0 yeast. (A) Wild-type or (B) Rho0 yeast cells (1 x 106/ml) were incubated in PBS containing the spin trap DMPO (100 mM) with or without varying concentrations of cisplatin. ESR spectra were then recorded 30 min after the addition of the test agents. (C) The simulated spectrum is shown
Influence of cisplatin on mitochondrial oxidative phosphorylation
Rates of oxygen consumption were determined in isolated yeast mitochondria incubated at 37 °C with increasing cisplatin concentrations for 1, 2 or 3 hr. State 3 and state 4 respiration rates were measured and the respiratory control ratio (RCR) was calculated.
The mitochondrial RCR [respiratory control ratio (state 3/state 4)] is defined as the respiration in state 3ADP divided by that in state 4, and the P/O ratio, mol of ATP synthesized per mol of O (1/2O2) used. Cisplatin challenge led to a dose and time dependent decrease in the RCR (Figure 6). Decrease in RCR implies that the mitochondria have a lowered capacity for substrate oxidation and ATP turnover and increased proton leak.
Figure 6.
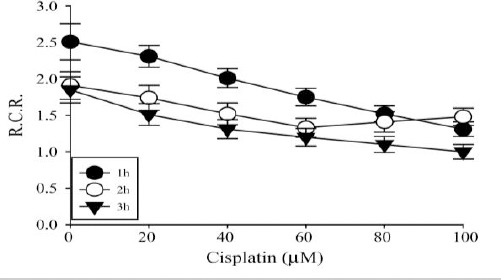
Effect of cisplatin on mitochondrial oxidative phosphorylation. RCR in mitochondria isolated from yeast and incubated at 37 °C for 1, 2 or 3 hr with different cisplatin concentrations: 0, 0.2, 0.4, 0.6, 0.8 and 1.0 mM cisplatin respectively. Data are from three different experiments and expressed as mean ± SD
Discussion
Investigation of the mechanisms of cisplatin-induced cytotoxicity at cellular and subcellular levels in response to cisplatin remains a challenge. Yeast cells have successfully served as tools for evaluation of drug targets (32). Here, we used loss-of-function approach by depletion of mitochondrial DNA in yeast to study the mitochondrial influence in cisplatin cytotoxicity, with a focus on the role of mitochondrial respiration, ROS and oxidative phosphorylation in modulating the cisplatin cytotoxicity.
In several cellular systems, including intestinal, renal tissues, CDDP-induced oxidative stress leads to mitochondrial permeability transition, with subsequent release of cytochrome c from the mitochondria, caspase activation and apoptosis (33-37). As the electron transport chain is a significant contributor of cellular oxidative stress (38, 39), we probed the contribution of cellular respiration in CDDP-induced oxidative stress. It was found that when cells are more reliant on respiration, CDDP cytoxicity is enhanced. Rho0 cells devoid of mtDNA and defective in mitochondrial respiration exhibited lowered oxidative stress and decreased cytotoxicity in response to CDDP. Absence of mitochondrial DNA totally suppressed the oxidative burden. These results indicate that defective components of mitochondrial electron transport chain and oxidative phosphorylation may serve to protect cells against cisplatin-induced cell death and suggest a clear association between mitochondrial respiration, oxidative stress and CDDP toxicity. It was previously suggested that cisplatin inhibits mitochondrial function in yeast, which therefore prevents growth on glycerol.
Mitochondrial electron transport chain serves as a major consumer of oxygen by (40). In the present study, cisplatin suppresses oxygen uptake to a certain extent in wild-type yeast, but respiratory inhibition totally suppresses oxygen uptake. Suppression of oxygen consumption by CDDP was also reported in other eukaryotic cells (16). Cisplatin-induced alterations in oxidative metabolism are a result of uncoupling of respiration. Loss of mitochondrial membrane potential in response to CDDP challenge has often been noted in mammalian cells (41, 42), but the mitochondrial permeability transition could also lead to loss of mitochondrial membrane potential. Monitoring of pH changes in cytosol and mitochondria after cisplatin exposure demonstrated an abrupt but equal decrease of pH in both compartments, without disturbing the pH gradient between cytosol and mitochondria. Antimycin A, a complex III inhibitor demonstrated similar pH changes, whereas the uncoupler CCCP indeed results in pH differences and uncoupling (24). Contrary to the uncoupling effect, the present data reveal that cisplatin induces opening of the mitochondrial permeability transition pore.
A strong correlation between oxidative stress and cisplatin sensitivity, implies production of free radical species as the main cause of CDDP toxicity. The yeast experiments described here suggest that an inhibition of electron transport and aerobic respiration per se may not completely describe the consequences of cisplatin interaction with the electron transport. Since yeast cells may survive without aerobic respiration, an inhibition of electron transport does not fully account for the enhanced cytotoxic effect of the drug in normal yeast cells. A proliferation of free radicals as a consequence of cisplatin interaction with the electron transport may explain the cytotoxic effect of the drug in normal yeast cells.
Conclusion
These results clearly suggest that cisplatin toxicity is influenced by the respiratory capacity of the cells and the intracellular oxidative burden after challenging with cisplatin. Although cisplatin per se slightly decreased the respiration of yeast cells grown in glucose, it does not dissipate the proton gradient of the mitochondrial membrane.
Acknowledgment
The study is supported by a startup grant (No.F.30-61/2014) from the University Grants Commission, New Delhi to Dr VLB. Also thanks Ms. SPI Council of Scientific and Industrial Research, India for providing Fellowship.
References
- 1.Vokes EE, Weichselbaum RR, Mick R, McEvilly JM, Haraf DJ, Panje WR. Favorable long-term survival following induction chemotherapy with cisplatin, fluorouracil, and leucovorin and concomitant chemoradiotherapy for locally advanced head and neck cancer. J Natl Cancer Inst. 1992;84:877–882. doi: 10.1093/jnci/84.11.877. [DOI] [PubMed] [Google Scholar]
- 2.Reedijk J, Lohman PH. Cisplatin: synthesis, antitumour activity and mechanism of action. Pharm Weekbl Sci. 1985;7:173–180. doi: 10.1007/BF02307573. [DOI] [PubMed] [Google Scholar]
- 3.Von Hoff DD, Schilsky R, Reichert CM, Reddick RL, Rozencweig M, Young RC, et al. Toxic effects of cis-dichlorodiammineplatinum(II) in man. Cancer Treat Rep. 1979;63:1527–1531. [PubMed] [Google Scholar]
- 4.Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23–43. doi: 10.1016/s0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 5.Lindauer E, Holler E. Cellular distribution and cellular reactivity of platinum (II) complexes. Biochem Pharmacol. 1996;52:7–14. doi: 10.1016/0006-2952(96)00106-2. [DOI] [PubMed] [Google Scholar]
- 6.Fuertes MA, Castilla J, Alonso C, Perez JM. Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr Med Chem. 2003;10:257–266. doi: 10.2174/0929867033368484. [DOI] [PubMed] [Google Scholar]
- 7.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 8.Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, et al. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- 9.Olivero OA, Semino C, Kassim A, Lopez-Larraza DM, Poirier MC. Preferential binding of cisplatin to mitochondrial DNA of Chinese hamster ovary cells. Mutat Res. 1995;346:221–230. doi: 10.1016/0165-7992(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 10.Preston TJ, Abadi A, Wilson L, Singh G. Mitochondrial contributions to cancer cell physiology: potential for drug development. Adv Drug Deliv Rev. 2001;49:45–61. doi: 10.1016/s0169-409x(01)00127-2. [DOI] [PubMed] [Google Scholar]
- 11.Singh G, Maniccia-Bozzo E. Evidence for lack of mitochondrial DNA repair following cis-dichlorodiammineplatinum treatment. Cancer Chemother Pharmacol. 1990;26:97–100. doi: 10.1007/BF02897252. [DOI] [PubMed] [Google Scholar]
- 12.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 13.Tacka KA, Dabrowiak JC, Goodisman J, Penefsky HS, Souid AK. Effects of cisplatin on mitochondrial function in Jurkat cells. Chem Res Toxicol. 2004;17:1102–1111. doi: 10.1021/tx0499564. [DOI] [PubMed] [Google Scholar]
- 14.Harper ME, Antoniou A, Villalobos-Menuey E, Russo A, Trauger R, Vendemelio M, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002;16:1550–1557. doi: 10.1096/fj.02-0541com. [DOI] [PubMed] [Google Scholar]
- 15.Kruidering M, Van de Water B, de Heer E, Mulder GJ, Nagelkerke JF. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J Pharmacol Exp Ther. 1997;280:638–649. [PubMed] [Google Scholar]
- 16.Garrido N, Perez-Martos A, Faro M, Lou-Bonafonte JM, Fernandez-Silva P, Lopez-Perez MJ, et al. Cisplatin-mediated impairment of mitochondrial DNA, metabolism inversely correlates with glutathione levels. Biochem J. 2008;414:93–102. doi: 10.1042/BJ20071615. [DOI] [PubMed] [Google Scholar]
- 17.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Bridges RJ, Eyer CL. Effect of cytochrome P450 1A induction on oxidative damage in rat brain. Mol Cell Biochem. 2001;223:89–94. doi: 10.1023/a:1017904912654. [DOI] [PubMed] [Google Scholar]
- 19.Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001;22:189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 20.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 21.Guidot DM, McCord JM, Wright RM, Repine JE. Absence of electron transport (Rho 0 state) restores growth of a manganese-superoxide dismutase-deficient Saccharomyces cerevisiae in hyperoxia. Evidence for electron transport as a major source of superoxide generation in vivo. J Biol Chem. 1993;268:26699–26703. [PubMed] [Google Scholar]
- 22.Barros MH, Netto LE, Kowaltowski AJ. H(2)O(2) generation in Saccharomyces cerevisiae respiratory pet mutants: effect of cytochrome c. Free Radic Biol Med. 2003;35:179–188. doi: 10.1016/s0891-5849(03)00307-1. [DOI] [PubMed] [Google Scholar]
- 23.Goldring ES, Grossman LI, Krupnick D, Cryer DR, Marmur J. The petite mutation in yeast. Loss of mitochondrial deoxyribonucleic acid during induction of petites with ethidium bromide. J Mol Biol. 1970;52:323–335. doi: 10.1016/0022-2836(70)90033-1. [DOI] [PubMed] [Google Scholar]
- 24.Orij R, Postmus J, Ter Beek A, Brul S, Smits GJ. In vivo measurement of cytosolic and mitochondrial pH using a pH-sensitive GFP derivative in Saccharomyces cerevisiae reveals a relation between intracellular pH and growth. Microbiology. 2009;155:268–278. doi: 10.1099/mic.0.022038-0. [DOI] [PubMed] [Google Scholar]
- 25.Isonishi S, Saitou M, Yasuda M, Tanaka T. Mitochondria in platinum resistant cells. Hum Cell. 2001;14:203–210. [PubMed] [Google Scholar]
- 26.Uslu R, Bonavida B. Involvement of the mitochondrion respiratory chain in the synergy achieved by treatment of human ovarian carcinoma cell lines with both tumor necrosis factor-alpha and cis-diamminedichloroplatinum. Cancer. 1996;77:725–732. [PubMed] [Google Scholar]
- 27.Fendt SM, Sauer U. Transcriptional regulation of respiration in yeast metabolizing differently repressive carbon substrates. BMC Syst Biol. 2010;4:12. doi: 10.1186/1752-0509-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diwu Z, Lown JW. Photosensitization with anticancer agents. 15. Perylenequinonoid pigments as potential photodynamic therapeutic agents: formation of semiquinone radicals and reactive oxygen species on illumination. J Photochem Photobiol B. 1993;18:131–143. doi: 10.1016/1011-1344(93)80055-e. [DOI] [PubMed] [Google Scholar]
- 29.Diwu Z, Lown JW. Photosensitization with anticancer agents. 16. The photo-oxidation of hypocrellin A. A mechanism study using 18O labelling. J Photochem Photobiol B. 1993;18:145–154. doi: 10.1016/1011-1344(93)80056-f. [DOI] [PubMed] [Google Scholar]
- 30.Diwu Z, Zhang C, Lown JW. Photosensitization with anticancer agents. 18. Perylenequinonoid pigments as potential photodynamic therapeutic agents: preparation and photodynamic properties of amino-substituted hypocrellin derivatives. Anticancer Drug Des. 1993;8:129–143. [PubMed] [Google Scholar]
- 31.Diwu Z, Lown JW. Photosensitization with anticancer agents. 17. EPR studies of photodynamic action of hypericin: formation of semiquinone radical and activated oxygen species on illumination. Free Radic Biol Med. 1993;14:209–215. doi: 10.1016/0891-5849(93)90012-j. [DOI] [PubMed] [Google Scholar]
- 32.Bharucha N, Kumar A. Yeast genomics and drug target identification. Comb Chem High Throughput Screen. 2007;10:618–634. doi: 10.2174/138620707782507340. [DOI] [PubMed] [Google Scholar]
- 33.Blanc C, Deveraux QL, Krajewski S, Janicke RU, Porter AG, Reed JC, et al. Caspase-3 is essential for procaspase-9 processing and cisplatin-induced apoptosis of MCF-7 breast cancer cells. Cancer Res. 2000;60:4386–4390. [PubMed] [Google Scholar]
- 34.Kim JS, Lee JH, Jeong WW, Choi DH, Cha HJ, Kim do H, et al. Reactive oxygen species-dependent EndoG release mediates cisplatin-induced caspase-independent apoptosis in human head and neck squamous carcinoma cells. Int J Cancer. 2008;122:672–680. doi: 10.1002/ijc.23158. [DOI] [PubMed] [Google Scholar]
- 35.Park MS, De Leon M, Devarajan P. Cisplatin induces apoptosis in LLC-PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol. 2002;13:858–865. doi: 10.1681/ASN.V134858. [DOI] [PubMed] [Google Scholar]
- 36.Vijayalakshmi B, Sesikeran B, Udaykumar P, Kalyanasundaram S, Raghunath M. Chronic low vitamin intake potentiates cisplatin-induced intestinal epithelial cell apoptosis in WNIN rats. World J Gastroenterol. 2006;12:1078–1085. doi: 10.3748/wjg.v12.i7.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bodiga VL, Bodiga S, Surampudi S, Boindala S, Putcha U, Nagalla B, et al. Effect of vitamin supplementation on cisplatin-induced intestinal epithelial cell apoptosis in Wistar/NIN rats. Nutrition. 2012;28:572–580. doi: 10.1016/j.nut.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20:332–340. doi: 10.1016/j.tem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Rosenfeld E, Beauvoit B. Role of the non-respiratory pathways in the utilization of molecular oxygen by Saccharomyces cerevisiae. Yeast. 2003;20:1115–1144. doi: 10.1002/yea.1026. [DOI] [PubMed] [Google Scholar]
- 41.Pourahmad J, Hosseini MJ, Eskandari MR, Shekarabi SM, Daraei B. Mitochondrial/lysosomal toxic cross-talk plays a key role in cisplatin nephrotoxicity. Xenobiotica. 2010;40:763–771. doi: 10.3109/00498254.2010.512093. [DOI] [PubMed] [Google Scholar]
- 42.Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]