

Abstract
Control of ligand-gated ion channel (LGIC) expression is essential for the formation, maintenance and plasticity of synapses. Treatment of mouse myotubes with proteasome inhibitors increased the number of surface nicotinic acetylcholine receptors (AChRs), indicating LGIC expression is regulated by the ubiquitin–proteasome system (UPS). Elevated surface expression resulted from increased AChR delivery to the plasma membrane and not from decreased turnover from the surface. The rise in AChR trafficking was the direct result of increased assembly of subunits in the endoplasmic reticulum (ER). Because proteasome inhibitors also blocked ER-associated degradation (ERAD) of unassembled AChR subunits, the data indicate that the additional AChRs were assembled from subunits normally targeted for ERAD. Our data show that AChR surface expression is regulated by the UPS through ERAD, whose activity determines oligomeric receptor assembly efficiency.
Keywords: acetylcholine receptor, endoplasmic reticulum-associated degradation, ubiquitin–proteasome system
Introduction
Cells respond to extracellular stimuli by regulating the number and/or activity of surface receptors they express. This regulation is particularly important in neurons and skeletal muscle, where the trafficking and targeting of postsynaptic ligand-gated ion channels (LGICs) are critical determinants of synaptic efficacy at the neuromuscular junction (NMJ) (reviewed in Sanes and Lichtman, 2001) and at central excitatory synapses (Beattie et al, 2000; Man et al, 2000; Wang and Linden, 2000; Malinow and Malenka, 2002). The molecular controls of LGIC surface expression remain poorly defined but a number of recent studies have implicated ubiquitylation and the ubiquitin–proteasome system (UPS) in the modulation of synaptic strength by regulating the molecular composition at both pre- (Speese et al, 2003; Zhao et al, 2003) and postsynaptic (Burbea et al, 2002; Ehlers, 2003; Zhao et al, 2003) elements.
Ubiquitin is a 76 aa peptide conjugated to proteins by a multistep enzymatic cascade (reviewed in Glickman and Ciechanover, 2002) with a known role in regulating surface protein expression. Monoubiquitylation, the attachment of a single ubiquitin, is a signal for plasma membrane receptor endocytosis (reviewed in Bonifacino and Weissman, 1998; Hicke, 2001). The synthesis of ubiquitin polymers on proteins (polyubiquitylation) directs substrates to 26S proteasomes where they are degraded (UPS; reviewed in Ciechanover and Schwartz, 1998). Proteasome inhibitors increase expression of several different proteins at the plasma membrane (Staub et al, 1997; Musil et al, 2000; van Kerkhof et al, 2000; Malik et al, 2001; Shenoy et al, 2001; Melman et al, 2002), but as the UPS influences a wide array of cellular functions, precisely which aspect is critical for surface receptor expression remains unclear.
One well-defined role for the UPS is as part of endoplasmic reticulum-associated degradation (ERAD), which ensures fidelity in the secretory pathway (Brodsky and McCracken, 1999). As part of this mechanism, the UPS degrades membrane proteins that misfold in the endoplasmic reticulum (ER) like the mutant cystic fibrosis transmembrane regulator (CFTRΔF508) (Ward et al, 1995), as well as unassembled subunits of oligomeric receptors (Yu et al, 1997; Tiwari, 2001). Proteasome inhibitors prevent CFTRΔF508 degradation but cannot rescue its maturation (Ward et al, 1995), owing to its aggregation in the cytosol (Johnston et al, 1998). Whether UPS inhibition can rescue unassembled subunit maturation (i.e. oligomeric receptor formation) is not known since those studies of unassembled subunit ERAD have only been performed under conditions where assembly cannot occur. Subunit assembly/maturation efficiency in the ER only ranges between 20 and 40% for a variety of oligomeric receptors/ion channels (Merlie and Lindstrom, 1983; Schmidt and Catterall, 1986; Ward and Kopito, 1994), suggestive of a potentially significant but uncharacterized role for ERAD in oligomeric receptor assembly.
In this study, we describe a novel point of regulation for oligomeric receptor expression by the UPS through ERAD of unassembled subunits. Using the muscle-type nicotinic acetylcholine receptor (AChR), a hetero-oligomeric ion channel assembled in the ER from individual subunits (α, β, γ/ɛ, δ) and whose biogenesis is well characterized (reviewed in Green and Millar, 1995; Green, 1999; Keller and Taylor, 1999), we show that ERAD occurs concurrently with receptor assembly, degrading both assembly-competent and misfolded subunits. Consequently, UPS-mediated degradation modulates AChR assembly efficiency and in doing so regulates the number of mature receptors reaching the plasma membrane.
Results
Proteasome inhibitors increase surface AChR expression
C2C12 myotubes constitutively expressing AChRs were used to determine the role of the UPS in AChR assembly and expression. Surface AChR expression assayed in myotubes by 125I-α-bungarotoxin (125I-Bgt) binding (Figure 1A) exhibited a marked increase (30–45%) over untreated myotubes following treatment with various proteasome inhibitors. An increase was also observed for AChRs stably expressed in NIH3T3 cells (B23ɛ; Green and Claudio, 1993) treated with lactacystin (LACT) (Figure 1A). To test whether treatment with proteasome inhibitors affects AChR internalization as with other surface proteins (van Kerkhof et al, 2000; Shenoy et al, 2001), the rate of AChR turnover from the plasma membrane was measured (Figure 1B), and was unchanged in myotubes treated with LACT (half-life (t1/2)=18.1 h) compared to untreated myotubes (t1/2=17.9 h). These half-life values are similar to values reported previously for AChRs expressed in BC3H-1 cells (Hyman and Froehner, 1983). In contrast, disrupting lysosomal proteases by ammonium chloride (NH4Cl) markedly slowed the rate of AChR turnover (t1/2=47.7 h), consistent with previous studies (Devreotes and Fambrough, 1975; Libby et al, 1980). These data show that surface AChRs are not degraded directly by proteasomes and that the increase in expression is not the result of decreased receptor turnover.
Figure 1.
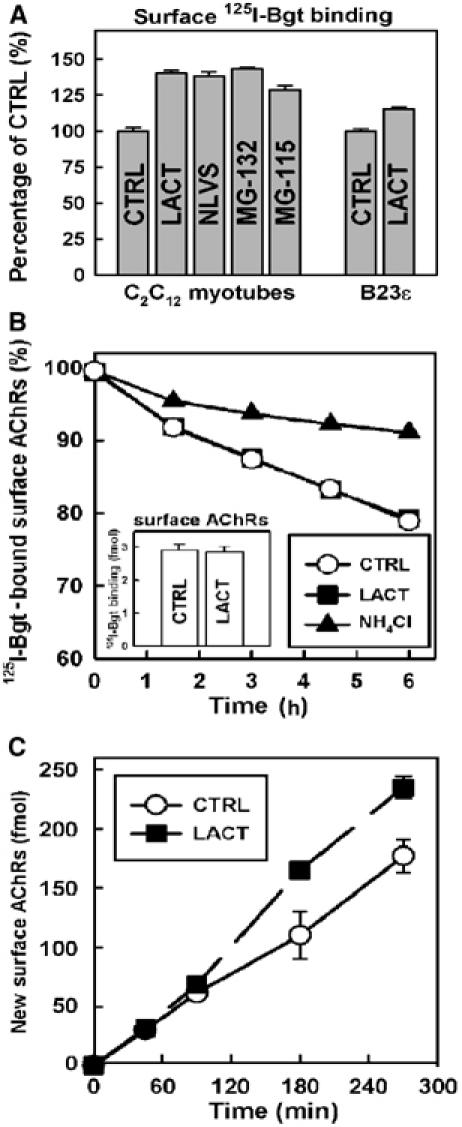
Proteasome inhibitors increase surface AChR expression. (A) C2C12 myotubes and α2βɛδ AChR-expressing B23ɛ cells incubated with proteasome inhibitors (LACT, NLVS, MG-115 and MG-132) for 4.5 h were bound with 125I-Bgt to assess surface AChR expression. Values were normalized to 125I-Bgt binding to untreated cells (P<0.005). (B) Surface AChR turnover rates inferred from 125I-Bgt degradation and release into growth media containing vehicle (CTRL ), LACT (•) or NH4Cl (▴). t1/2 values were estimated from single exponential curve fits (CTRL: 17.9 h; LACT: 18.1 h; NH4Cl: 47.7 h). The inset graph shows total 125I-Bgt binding for CTRL and LACT-treated cells at 0 h. (C) Delivery of newly assembled AChRs to the cell surface as measured by 125I-Bgt binding at designated time points during treatment with LACT. Data are shown for CTRL (○) and LACT (•) in fmol of receptor. All graphs represent mean and s.e.m. values (n=3).
To determine whether increased AChR surface expression in the presence of LACT was due to increased receptor trafficking, we monitored the arrival of AChRs at the cell surface by 125I-Bgt binding. Prebinding unlabeled Bgt to the surface ensured that 125I-Bgt only bound to newly inserted AChRs and not those pre-existing at the plasma membrane. As in Figure 1A, the number of AChRs at the cell surface increased after treatment with LACT (32.6±3.3%; Figure 1C). Note that increased AChR appearance at the cell surface was not observed until ∼90 min post-treatment. Since AChR assembly in the ER requires ∼90–120 min (Merlie and Lindstrom, 1983; Ross et al, 1991; Green and Claudio, 1993), these data indicate that proteasome-dependent degradation affects AChRs prior to their transport to the plasma membrane.
Proteasome inhibitors increase AChR assembly
The early events in AChR biogenesis were investigated by pulse–chase analysis of myotubes (Figure 2A–C). To make certain that proteasome inhibitors did not alter AChR subunit synthesis rates, LACT was added immediately after the pulse-labeling of subunits, ensuring that the measurements were only post-translational events. The high-affinity interaction between Bgt and AChRs permitted efficient separation of surface receptors from intracellular receptors. Surface AChR–Bgt complexes were immunoprecipitated by anti-Bgt antibodies and analyzed by SDS–PAGE to assess the arrival of pulse-labeled AChRs to the cell surface (Figure 2A and B). LACT treatment increased the number of cell surface AChRs, as illustrated by a comparison of the α subunit band intensities for the LACT-treated and untreated myotubes (Figure 2B). As in Figure 1C, increased AChR surface expression was not apparent until ∼90 min and was ∼40% greater than untreated myotubes by 4 h of treatment with LACT. In repeated trials of this experiment with a 4.5 h LACT treatment, α and β subunit band intensities increased by 37.7±2.3 and 41.5±11.5% (Figure 2C), respectively, confirming that LACT increased the number of assembled AChRs delivered to the plasma membrane. Interestingly, the ratio of α:β subunit band intensities was unaffected by LACT (untreated: 2.2±0.2; LACT: 2.1±0.2). The 2:1 ratio indicates that the additional surface AChRs containing labeled subunits were assembled with the proper stoichiometry in the presence of LACT (Figure 2C).
Figure 2.
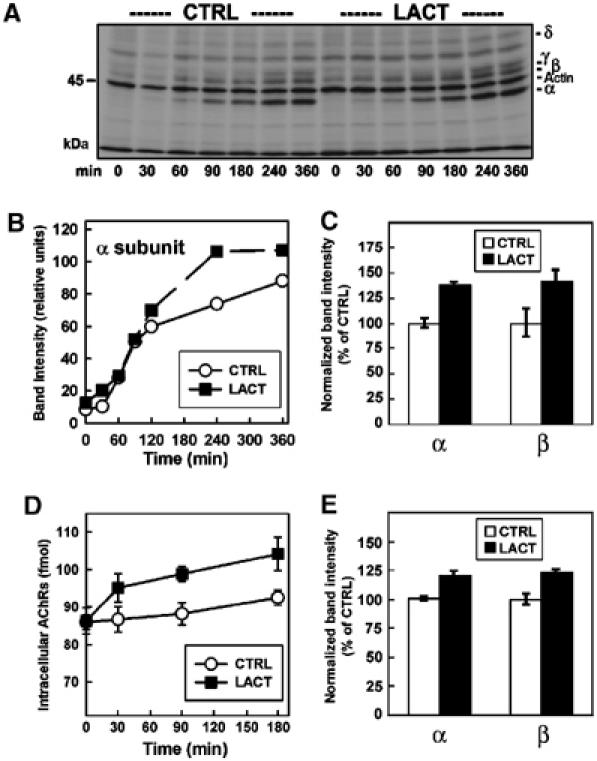
Proteasome inhibitors increase assembly and delivery of AChRs to the cell surface. (A) Pulse–chase assay in myotubes with LACT and unlabeled Bgt. Radiolabeled AChRs inserted into the plasma membrane were immunoprecipitated as part of receptor–toxin complexes by anti-Bgt. Individual subunits were resolved by SDS–PAGE. (B) α subunit band intensities from (A) are quantified and shown for CTRL (○) and LACT (•). (C) The experiment described in (A, B) was repeated in triplicate for 0 and 4.5 h time points. The α and β subunit band intensities were quantified and are shown as the percentage of CTRL (n=3). (D) Time course of 125I-Bgt binding to intracellular myotube AChRs in the presence of LACT. TX-100-soluble fractions were bound overnight with 125I-Bgt and immunoprecipitated by mAb35. Data are presented as the amount of AChR (fmol, n=3). (E) Affinity purification of newly assembled AChRs by Bgt-Seph from myotubes pulse labeled and chased in the presence of LACT for 90 min (n=3). Band intensities for α and β were quantified and are shown as in (C).
By masking surface Bgt-binding sites with unlabeled Bgt prior to solubilization, intracellular complexes could be specifically bound with 125I-Bgt and immunoprecipitated by an α subunit-specific monoclonal antibody (mAb35) to measure intracellular Bgt-binding site formation. In contrast to the increase in surface AChRs detectable only after ∼90 min (Figures 1C, 2A and B), the number of intracellular Bgt-binding sites increased within 30 min of exposure to LACT, reaching ∼23% over untreated myotubes by 180 min (Figure 2D). The overall increase in intracellular Bgt-binding sites was less than that observed for the surface sites. However, this measurement significantly underestimates the actual percent increase with LACT treatment because it includes Bgt-binding sites formed prior to the start of our assay. Because Bgt-binding sites form on the AChR during assembly in the ER (Merlie and Lindstrom, 1983; Green and Wanamaker, 1998), the relatively rapid effect of LACT suggests that a UPS-mediated degradation event occurs early during AChR biogenesis. This result was confirmed by metabolic labeling of myotubes using the pulse–chase protocol described in Figure 2A. After anti-Bgt immunoprecipitations to isolate surface receptors, intracellular AChRs were affinity-purified using Bgt-Sepharose (Bgt-Seph). Quantification of intracellular α and β subunits bound to Bgt determined 90 min after the labeling revealed increases of 20.5±3.5 and 22.8±4.4%, respectively (Figure 2E). Again, the ratio of α and β subunit band intensities was ∼2:1, indicating that labeled subunits were assembled into α2βγδ complexes by 90 min but remained intracellular. The rapid increases in intracellular Bgt-binding sites as well as in newly assembled AChRs resulting from LACT treatment demonstrate that proteasome activity affects AChR assembly in the ER.
Unassembled AChR subunits are targets for ERAD
How inhibiting proteasome-mediated degradation leads to increased AChR assembly is unclear. It has long been observed that assembly of AChRs is an inefficient process, incorporating only 20–30% of synthesized subunits (Merlie and Lindstrom, 1983; Ross et al, 1991; Green and Claudio, 1993). Unassembled subunits do not progress beyond the ER (Blount and Merlie, 1988; Claudio et al, 1989; Sumikawa and Miledi, 1989; Gu et al, 1991) and are degraded rapidly (Claudio et al, 1989; Blount and Merlie, 1990). From the pulse–chase assays in Figure 2, we estimate that AChRs assemble in C2C12 myotubes with ∼30% efficiency. The residual AChR subunits (∼70%) are presumed to be unassembled or misfolded, two forms that are known to be rapidly degraded from the ER. The similarities shared with unassembled T-cell receptor subunits (e.g. TCRα) suggest that the majority of AChR subunits in myotubes may be targeted for ERAD. If so, then the increase in AChR assembly could be the result of a decrease in subunit degradation.
To explore this possibility, we tested whether the UPS, through ERAD, degrades unassembled AChR subunits. In C2C12 myotubes and B23ɛ cells, AChR subunit assembly and degradation occur concurrently. To study subunit degradation independent of assembly, we generated cell lines stably expressing individual wild-type or HA-tagged subunits. The effect of LACT treatment on subunit degradation was assayed by pulse–chase analysis (Figure 3). Individual subunits were degraded rapidly and with similar but distinct kinetics. Utilizing half-life values (t1/2) as a quantitative measure of degradation (Figure 3A, CTRL), unassembled α subunits had the slowest rate of degradation, followed by δ, ɛ-HA, β-HA and γ-HA. Note that these rates are comparable to those observed for Torpedo (Claudio et al, 1989) and mouse α subunits (Blount and Merlie, 1988). The presence of an HA epitope tag did not alter subunit degradation kinetics as the rates obtained for α-HA and untagged α subunits did not differ significantly (data not shown). Treatment with LACT slowed degradation of all singly expressed AChR subunits (Figure 3A, LACT), increasing mean t1/2 values by as much as four-fold.
Figure 3.
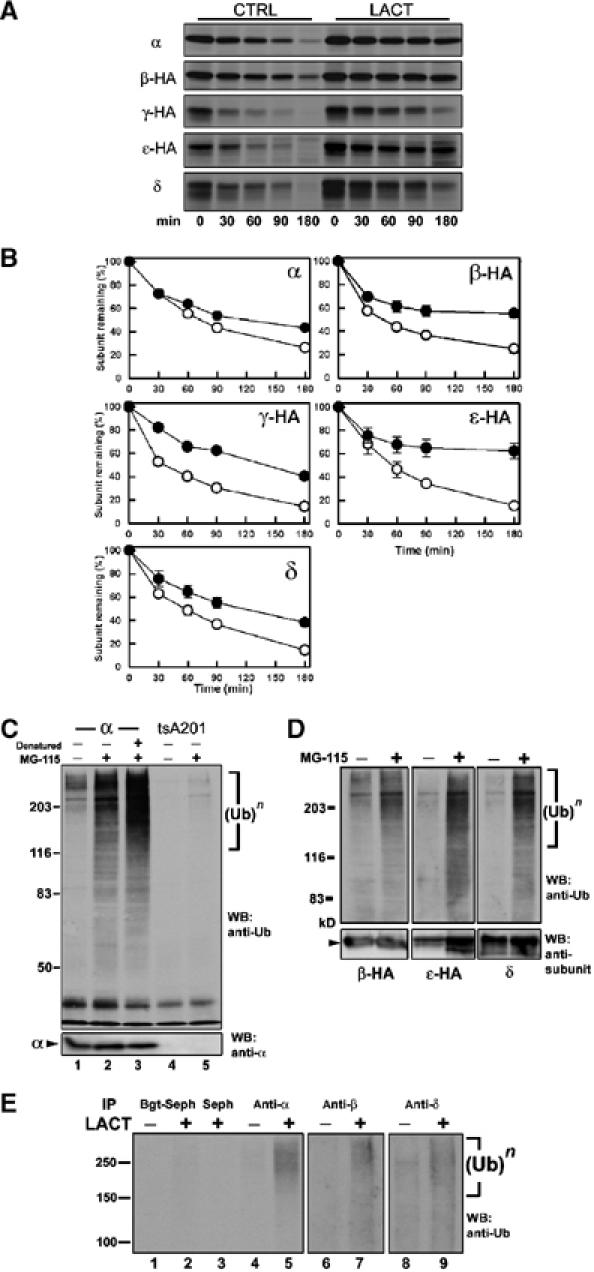
Unassembled AChR subunits are degraded by ERAD. (A) Representative autoradiographs from pulse–chase assays of AChR subunit-expressing cell lines show degradation of each subunit in the absence (left) and presence (right) of LACT. (B) Composite degradation rates for unassembled AChR subunits. Subunit band intensities were quantified for CTRL (○) and LACT (•) and plotted as a percentage of the starting amount (time=0). Data are shown as the mean±s.e.m. of at least three experiments. t1/2 values for CTRL (α: 77 min; β-HA: 42 min; γ-HA: 30 min; ɛ-HA: 57 min; δ: 57 min) and LACT (α:123 min; β-HA: >180 min; γ-HA: 120 min; ɛ-HA: 180 min; δ: 111 min) were determined from the composite data. (C) Western blot of α subunit-expressing cells treated for 4.5 h with vehicle (CTRL, lane 1), MG-115 (lane 2), or treated with MG-115 and denatured by SDS prior to immunoprecipitation (lane 3). tsA201 cells treated with vehicle (CTRL, lane 4) or MG-115 (lane 5). Samples were immunoprecipitated by mAb P22 and probed with anti-Ub (top) and anti-α (bottom). (D) Western blot of β-HA, ɛ-HA or δ subunits from stably expressing cells treated with MG-115 probed with anti-Ub (top) and subunit-specific antibodies (bottom), as above. (E) Anti-Ub Western blot of assembled and unassembled AChR subunits in myotubes treated for 4.5 h with LACT. Assembled AChRs and complexes were affinity purified from cell lysates by Bgt-Seph (lanes 1 and 2) while residual non-Bgt binding/unassembled subunits were collected from SDS-denatured secondary supernatants by immunoprecipitation with mAbP22 (α, lanes 4 and 5), mAb148 (β, lanes 6 and 7) or mAb88b (δ, lanes 8 and 9).
Integral membrane ERAD substrates can be retrotranslocated from the ER, deglycosylated, and accumulate in the cytoplasm with proteasome inhibition (Wiertz et al, 1996; Yu et al, 1997; Petaja-Repo et al, 2001). In the presence of LACT, we were unable to detect deglycosylated α subunit forms that would have indicated exposure of the N-terminal glycosylation site (N143) to cytosolic N-glycanase (Figure 3A). The increased t1/2 values measured during LACT treatment represent an extended presence of subunits with appropriate molecular weights and glycosylation profiles. Furthermore, all subunits remained associated with membrane fractions upon cellular fractionation (data not shown). These results indicate that proteasomes degrade unassembled AChR subunits from the ER and imply that subunit retrotranslocation is a process coupled tightly to proteasome activity.
Polyubiquitylation targets ERAD substrates to 26S proteasomes for degradation. To assess subunit polyubiquitylation, individual subunits were immunoprecipitated from stably expressing cells treated with proteasome inhibitors, and immunoblotted with anti-ubiquitin (anti-Ub) (Figure 3C). Polyubiquitylated proteins appear as high-molecular-weight smears of ubiquitin immunoreactivity. Only after treating with proteasome inhibitors were polyubiquitylated α subunits readily apparent (compare lanes 1 and 2, data not shown). Comparable results were observed for all other AChR subunits (Figure 3D). The lysomotropic agents, NH4Cl and chloroquine, which blocked surface turnover (Figure 1B), had no effect on subunit polyubiquitylation (data not shown), and denaturation with 1% SDS did not disrupt α subunit polyubiquitylation, confirming a covalent modification (Figure 3C, lane 3).
AChR subunit ubiquitylation was also assayed in C2C12 myotubes (Figure 3E). After solubilization, assembled subunits were removed by affinity purification with Bgt-Seph (Figure 3E, lanes 1 and 2). Residual and presumably unassembled subunits were denatured prior to immunoprecipitation (Figure 3E, α: lanes 4 and 5, β: lanes 6 and 7, δ: lanes 8 and 9). As with individually expressed subunits (Figure 3C and D), polyubiquitylated forms of unassembled α, β and δ were detected in myotubes following LACT treatment, indicating that subunit degradation through the UPS is occurring during receptor assembly.
Assembly-competent subunits are targeted for ERAD
To test whether proteasome inhibition permitted the maturation of misfolded subunits, we evaluated biochemical and pharmacological properties of AChRs assembled in the presence of LACT. We began by measuring the dissociation rate of 125I-Bgt bound to surface AChRs. The toxin dissociation kinetics (Figure 4A) for receptors assembled while myotubes were treated with LACT did not differ significantly from untreated myotubes, with estimated t1/2 values of 8.4 days (CTRL) and 9.2 days (LACT). We next tested whether AChRs expressed during treatment with LACT or NLVS sediment similarly on sucrose gradients (Figure 4B). Conformationally mature AChRs are released from the ER as α2βγδ pentamers and sediment at a 9S peak on sucrose velocity gradients after solubilization in Triton X-100 (Merlie and Lindstrom, 1983). AChR pentamers not comprised of the full subunit complement dissociate in Triton X-100, as do partially assembled AChR complexes (Green and Claudio, 1993). Surface AChRs from untreated and LACT (or NLVS)-treated myotubes migrated in a single peak at 9S and their sedimentation was indistinguishable (Figure 4B). These results together with our finding of a 2:1 ratio of α:β subunits for surface and intracellular AChRs (Figure 2C and E) indicate that AChRs assembled in the presence of proteasome inhibitors have the expected size and subunit stoichiometry.
Figure 4.
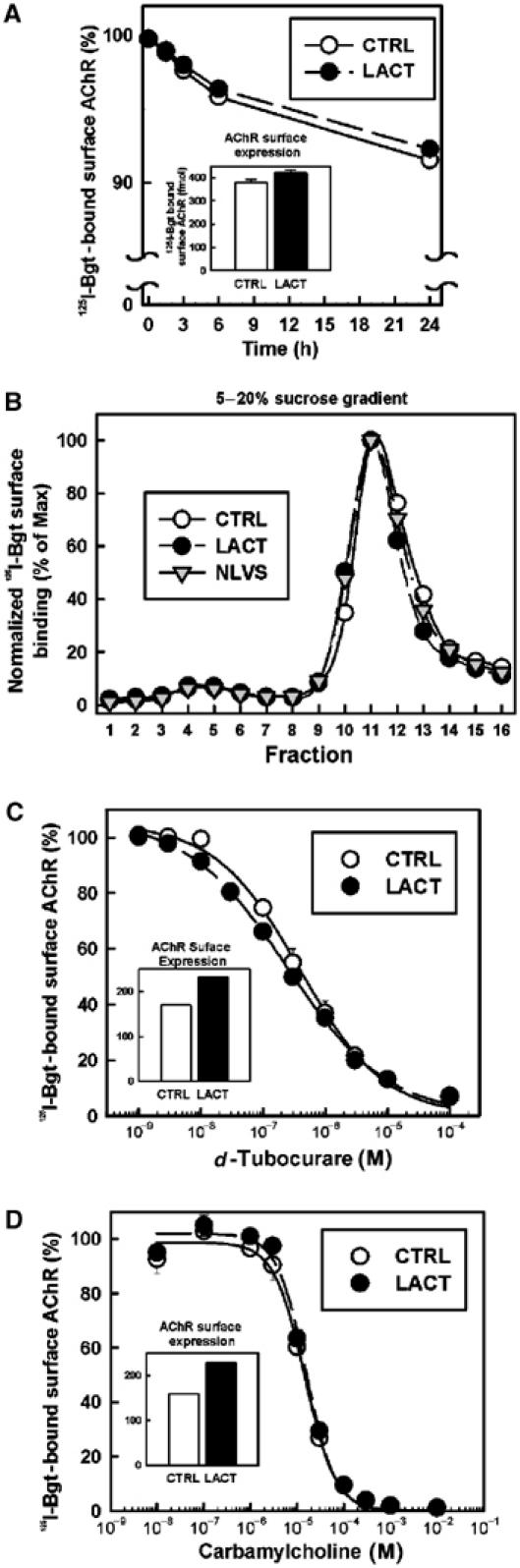
Structure and pharmacology of AChRs assembled in the presence of LACT. Proteasome inhibitor treatment increased surface AChR expression in C2C12 myotubes. The inset graphs in panels A, C and D display increased surface AChR expression (in fmol) with LACT for each experiment (∼25–40% above CTRL). (A) Bgt affinity for surface AChRs measured by the rate of 125I-Bgt release at 4°C, following 4.5 h LACT treatment of myotubes to surface AChR expression (with 125I-Bgt included during the final 2 h). Expressed as the percentage of cell surface-bound 125I-Bgt remaining (n=3), t1/2 values were estimated to be 8.4 days (CTRL, ○) and 9.2 days (LACT, •) by single exponential curve fits. (B) Sucrose gradient sedimentation of surface AChRs expressed during 4.5 h of proteasome inhibitor treatment. Newly inserted surface AChRs were bound with 125I-Bgt using the technique described in Figure 2C and separated on 5–20% sucrose gradients. The quantity of bound 125I-Bgt in each fraction (total 18) was normalized to the peak fraction (11). Surface AChRs expressed in both LACT- (•) and NLVS- (▾) treated myotubes sedimented identically to CTRL (○) at a 9S peak (fractions 10–12) but had 26% (LACT) and 23% (NLVS) more 125I-Bgt binding than CTRL. After incubation with LACT, the ligands (C) dTC and (D) carb were competed against the initial rate of 10 nM 125I-Bgt binding. Graphs are plotted as mean and s.e.m. values (n=3). IC50 values and Hill coefficients were calculated from curve fits using a three-parameter Hill equation.
Each AChR has two agonist-binding sites to which ACh binds cooperatively to activate the receptor. Bgt binds to AChRs at locations that overlap with the agonist-binding sites and both sites form during AChR assembly (Green and Wanamaker, 1998). As a test of ligand-binding site fidelity, we measured the affinities of the competitive antagonist d-tubocurare (dTC; Figure 4C) and the agonist carbamylcholine (carb; Figure 4D) by competition with the competitive antagonist 125I-Bgt. dTC normally binds to the two agonist-binding sites with a 50- to 100-fold difference in affinity (Sine, 1993; Steinbach and Chen, 1995) while the binding of agonists such as carb is cooperative if receptor activation occurs. Both dTC and carb were able to block 125I-Bgt binding to AChRs and the dose dependence of that block was comparable, indicating that the affinities of dTC and carb were the same for untreated and LACT-treated receptors. Importantly, the shape of each dose dependence curve for either antagonist or agonist was indistinguishable between LACT-treated and untreated cells. A Hill slope of 0.5 was obtained from fitting the Hill equation to the dTC data in Figure 4C consistent with a 50-fold difference in dTC affinity between the two agonist-binding sites. In contrast, a Hill slope of 1.5 was obtained from the fit of the Hill equation to carb data in Figure 4D, indicating cooperative agonist binding and receptor activation. From these results, the loss of proteasome activity did not appear to affect adversely the fidelity of AChR assembly and indicate that receptors formed were comprised of correctly folded and post-translationally modified subunits.
Discussion
Based on the results described in this paper, we propose that the UPS regulates AChR expression through its role in ERAD. Proteasome inhibitors can prevent internalization of some surface receptors including β2-AR (Shenoy et al, 2001), GHR (van Kerkhof et al, 2000) and IL-2 (Yu and Malek, 2001), while the turnover of others, such as transferrin receptors (van Kerkhof et al, 2000), is unaffected. Like the AChR, internalization and degradation of a glycine receptor (GlyR1) in the same LGIC family did not require proteasome activity (Bèuttner et al, 2001), suggesting that LGIC expression is not regulated by a UPS-dependent mechanism at the plasma membrane.
Ubiquitylation in the Golgi was suggested as a checkpoint for AChR subunit trafficking (Keller et al, 2001), but our data do not support a role for the UPS at this level. At least 90 min were required until the effects of LACT could be detected on the cell surface (Figures 1C and 2B). AChR assembly requires 90–120 min to complete after subunit synthesis (Merlie and Lindstrom, 1983; Ross et al, 1991). If proteasomal degradation regulated trafficking after ER release to the Golgi, we would have expected increased AChR expression to be detectable much earlier, since mature AChRs are trafficked to the surface after release from the ER much more rapidly than the time it takes for them to be assembled (Ross et al, 1991). LACT increased the number of newly assembled AChR complexes (Figure 2E) and also the formation of intracellular Bgt-binding AChRs, a process that occurs during AChR assembly in the ER (Green and Wanamaker, 1998). Little to no lag time was required for LACT to increase Bgt site formation (Figure 2D), arguing strongly that UPS regulation occurs at the ER by altering an early aspect of AChR assembly.
Previous studies have shown that there is rapid degradation of unassembled AChR subunits from the ER (Claudio et al, 1989; Blount and Merlie, 1990), but the mechanism had not been identified. Because proteasome inhibitors extended the lifetimes of unassembled AChR subunits by as much as four-fold (Figure 3), ERAD is at least partially responsible for subunit clearance. Unassembled AChR subunit lifetimes in the ER are shorter (30–77 min) than the residence time of subunits that are assembled and released from the ER (90–120 min). This finding suggests that unassembled subunits are not significantly retained in the ER, but rather are continuously removed through ERAD. We estimated that ∼70% of synthesized α subunits in myotubes are degraded, suggestive of a robust role for ERAD concurrent with receptor assembly. Many large membrane receptors/oligomers share the features of inefficient assembly in the ER and rapid degradation of their constituent subunits including GABAA receptors (Gorrie et al, 1997), voltage-gated Na+ channels (Schmidt and Catterall, 1986), CFTR (Cheng et al, 1990; Jensen et al, 1995), connexin32 (VanSlyke et al, 2000) and ENaC (Valentijn et al, 1998). Given these common features, it is likely that ERAD is a general mechanism responsible for continuously clearing unassembled subunits from the ER during oligomeric receptor/ion channel assembly.
In Figure 5, we propose a model for regulation of AChR assembly in the ER by the UPS that may serve as a model for other ion channels/oligomeric receptors. Following cotranslational insertion into the ER membrane (Figure 5A), a fraction of unassembled subunits interacts with complementary subunits and chaperones (stabilizing factors) that facilitate folding/oligomerization into mature AChR pentamers competent to exit the ER. Concurrently, subunits that misfold or fail to engage a stabilizing factor are targeted for ERAD. Commitment to ERAD comes as the subunit is polyubiquitylated while still integrated in the ER membrane. Polyubiquitylated subunits are then retrotranslocated (perhaps via lateral gating of Sec61 translocons; Plemper et al, 1998) with the help of cytoplasmic proteasomes, which ultimately degrade them. As implied by the model in Figure 5A, subunit assembly and degradation are processes occurring simultaneously in the ER. It is their opposing activities that ultimately determine the net efficiency at which surface receptors can be expressed.
Figure 5.
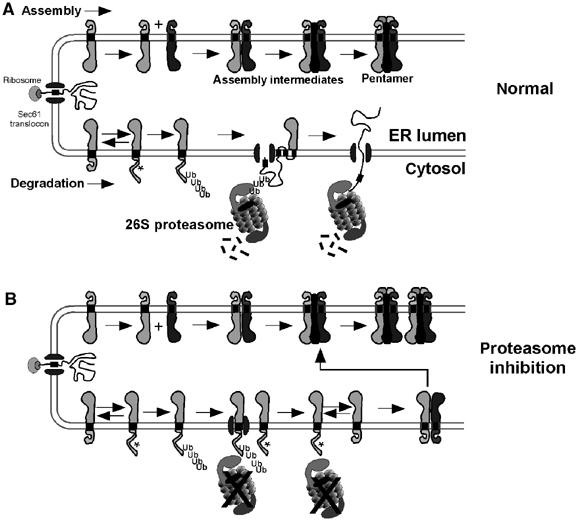
Model for UPS regulation of AChR assembly. (A) Model of the ER illustrating the concurrent processes of assembly (top) and ERAD (bottom) of unassembled AChR subunits. Normally, only between 20 and 40% of subunits assemble into AChRs, while those remaining are retrotranslocated and degraded rapidly by the UPS. Masking or presentation of a putative ‘degron' (indicated by *) on each subunit during folding/oligomerization in the ER determines an assembly or degradation fate, respectively. (B) Loss of proteasome activity results in a decrease of unassembled subunit ERAD and an increase in AChR formation. As subunit retrotranslocation requires proteasome activity, subunits remain integrated in the ER with some retaining the potential for oligomerization into AChRs.
Proteasome inhibitors caused unassembled subunits to accumulate, resulting in increased AChR assembly efficiency. A model for the causal relationship between these two observations is depicted in Figure 5B. If proteasome activity provided the energy for subunit retrotranslocation as shown in Figure 5A, then inhibiting it could directly affect unassembled subunits levels in the ER membrane. An increase in subunit concentration in the ER favors increased formation of assembly intermediates and consequently AChRs, provided the capacity for oligomerization is retained. Proteasome inhibition causes a number of secreted and single-pass membranes to accumulate cytoplasmically (Wiertz et al, 1996; Yu et al, 1997; de Virgilio et al, 1998; Yang et al, 1998; Shamu et al, 1999), indicating that retrotranslocation occurred even without proteasome activity. Interactions of the substrate polypeptide and polyubiquitin tree with the AAA-ATPase p97/VCP (and its adaptor complex) are thought to be sufficient to cause these substrates' retrotranslocation (Rabinovich, 2001; Ye et al, 2003). In contrast, AChR subunits and other proteins appear to require proteasome activity for retrotranslocation (Plemper et al, 1998; Xiong et al, 1999) and p97/VCP activity alone is not sufficient to compensate for this loss. This stark difference suggests that the general mechanism of ERAD also has substrate-specific functional requirements.
Our data demonstrate that a fraction of subunits accumulating in the ER during proteasome inhibition are capable of assembling into mature, functional AChRs. Akin to these findings, increased gap junction formation in ‘assembly-inefficient' cells was proposed to result from a net increase in connexins available for assembly after proteasome inhibition (Musil et al, 2000). In contrast, proteasome inhibitors fail to rescue surface expression of membrane proteins with mutations that prevent folding in the ER, such as CFTRΔF508 (Jensen et al, 1995; Ward et al, 1995). CFTRΔF508 does not fold properly at 37°C (Denning et al, 1992) and does not remain integrated in the ER membrane, ultimately precluding its maturation. Only because proteasome inhibitors did not adversely affect the integrity of additional AChRs assembled, can we suggest that the constituent subunits possessed the capacity for maturation. Had proteasome inhibitors not been present, subunits would have been targeted to ERAD as if they were terminally misfolded. Our data strongly suggest that the stringency of the ERAD selection process is low, failing to distinguish between subunits capable of assembling and those terminally misfolded. In this broader view, ERAD functions not just in ER quality control but also in the active governing of assembly efficiency and in turn the level of surface receptor expression.
Given their complex structure, it is not clear how AChR subunits are recognized for ERAD. Identification could stem from aberrant trimming of AChR subunit glycans, which would divert subunits to ERAD through interaction with EDEM (Molinari et al, 2003; Oda et al, 2003). Alternatively, subunits may transiently expose a degradation signal/motif or ‘degron' during tertiary rearrangement. A degron is indicated in the model by the ‘*' symbol on the unassembled subunits (Figure 5A). Degrons have been described as either linear (i.e. cl1 (Gilon et al, 2000), PEST (Rechsteiner and Rogers, 1996)) or distributed (Gardner and Hampton, 1999) motifs. For polytopic AChR subunits, a degron could reside in a TM domain (Bonifacino et al, 1991; Wang et al, 2002), or within a partially folded cytoplasmic loop region accessible by ER resident/cytoplasmic ubiquitylation machinery capable of catalyzing subunit polyubiquitylation (as in Figure 5). Polyubiquitylation would preclude subsequent folding/oligomerization steps and divert the subunit to ERAD, perhaps because of engagement with VCP/p97 or 26S proteasomes or by sterically hindering oligomerization. As shown in Figure 5A, oligomerization may obscure each subunit's degron within the assembly intermediate and mature pentameric forms, akin to stabilization of yeast mating factor MATα2 by MATa1 (Johnson et al, 1998). Consistent with this idea, tandem expression of AChR α and δ dramatically increases subunit half-lives compared to expression of each subunit alone (Keller et al, 1998).
The capacity to modulate ion channel/receptor expression at the level of the ER has important consequences for synaptic plasticity and perhaps in the treatment of some pathological conditions. Synaptic plasticity is regulated by an increasing number of pre- and post-translational mechanisms, and modulation of ERAD may be one more to consider. Organelles of the secretory pathway including ER, ER exit sites and Golgi were identified in neuronal dendrites (Horton and Ehlers, 2003). With this apparatus in place at synapses, rapid modulation of LGIC assembly/expression through ERAD represents a potential point for post-translational regulation of synaptic strength. And while rapid modulation may not be as prominent at mature NMJs, where AChRs are relatively stable (t1/2∼10 days), it may play a key role in dynamic synapses such as the developing NMJ or central excitatory/inhibitory synapses. Finally, note that ERAD is also a potential target for therapeutic intervention in diseases characterized by poor LGIC expression (e.g. myasthenia gravis). Current treatments for MG rely on inhibitors of AChesterase, which increase synaptic ACh concentration and enhance activity from residual AChRs left at the NMJ (Lindstrom, 2000). Increasing the efficiency of AChR assembly/expression through small molecule intervention of ERAD, in conjunction with existing treatment, could potentially improve motor function.
Materials and methods
AChR subunit cDNAs
Mouse AChR subunit cDNAs (α and δ) have been reported previously (Eertmoed et al, 1998). Mouse β, γ and ɛ subunit cDNAs were amended with C-terminal hemagglutinin epitope tags (β-HA, γ-HA, ɛ-HA) using standard PCR techniques and cloned into pRBG4.
Cell culture and AChR expression
tsA201 cells were maintained in DME (Gibco)+10% calf serum (CS). Clonal tsA201 lines stably expressing AChR subunits were selected by survival in hygromycin and by immunoreactivity to subunit-specific antibodies. B23ɛ cells are described previously (Green and Claudio, 1993). C2C12 myoblasts (ATCC) were maintained in DME+10% fetal bovine serum (FBS) until they were ∼90% confluent. Cells were then switched to DME+10% heat-inactivated horse serum (HIHS) for 36–48 h followed by starvation for 18 h with DME+2.5% HIHS to induce myotube formation and upregulate AChR expression.
Metabolic labeling and pulse–chase assays
Confluent plates of tsA201 stable cell lines were incubated in Met/Cys-free DME for 10 min. Cells were pulse-labeled by supplementing Met/Cys-free DME with 35S-Met/Cys (NEN EXPRE35S35S: 0.33 mCi/10 cm plate) for 10 min. Rinsing cells twice with ice-cold phosphate-buffered saline (PBS) supplemented with 5 mM Met terminated labeling. Labeled cells were gently harvested and pelleted at 1000 r.p.m. for 10 s. Following resuspension and washing in DME+10% CS supplemented with 5 mM Met, cells were pelleted and resuspended in DME+10% CS+50 mM HEPES+5 mM Met. Suspended cells were divided into five equal volumes and chased in suspension while rotating at 37°C. C2C12 myotubes were labeled similarly but were instead starved for 15 min, labeled for 15 min, and the chase carried out on the plates. For AChR subunit synthesis assays, cells were Met/Cys starved and pulse-labeled for 10 min at each time point.
Inhibitors of protein degradation
The following proteasome inhibitors were used: LACT (10 μM, Kamiya Biochem.), NLVS (10 μM, Calbiochem), MG-115 (10 μM, Calbiochem) and MG-132 (1 μM, Alexis). Inhibitors were added to unassembled subunit pulse–chase assays 5 min into the Met/Cys starvation and also included during the labeling and chase periods. For myotubes pulse–chase assays, inhibitors were included only during the chase, but not the 0 h time point. For 125I-Bgt binding experiments, inhibitors were present continuously. The lysomotropic agents NH4Cl (40 mM) and chloroquine (25 μM, Sigma) were added in similar fashion.
Solubilization
Cells were pelleted by brief centrifugation, resuspended, washed once with PBS and solubilized in lysis buffer (150 mM NaCl, 5 mM EDTA pH 7.4, 50 mM Tris pH 7.4, 0.02% NaN3) containing 1% Triton X-100 (TX-100)+NEM (2 mM), PMSF (2 mM), leupeptin (10 μg/ml), TLCK (10 μg/ml), chymotrypsin (10 μg/ml) and pepstatin (10 μg/ml). Following overnight solubilization, samples were centrifuged at 14 000 g for 30 min at 4°C. Subsequent analyses were performed using the TX-100-soluble fraction.
Subunit and AChR immunoprecipitation and purification
Unassembled AChR subunits were immunoprecipitated from precleared lysates using subunit-specific antibodies: α, mAbP21 (V Lennon, Mayo Clinic, Rochester, MN) and δ, mAb88b. HA-tagged subunits were immunoprecipitated by anti-HA mAb12CA5. Subunit–antibody complexes were isolated with Protein G-Sepharose. AChRs were affinity purified from C2C12 lysates by α-bungarotoxin conjugated to Sepharose beads (Bgt-Seph) or immunoprecipitated by mAb35. Following binding of 62.5 nM unlabeled Bgt to intact myotubes, surface AChRs were isolated from cell lysates as toxin–AChR complexes by immunoprecipitating with polyclonal anti-Bgt. Samples were electorphoresed on 7.5% SDS–PAGE gels.
Western blots
Proteins separated by SDS–PAGE were transferred to PVDF membrane and probed with mAbP22 (α), mAb88b (δ), mAb148 (β), anti-HA (Y-11, Santa Cruz Biotech.) and anti-Ub (Stressgen). Membranes to be probed with anti-Ub were boiled in double-distilled H2O for 5 min prior to the blocking step.
125I-α-bungarotoxin binding
Surface AChR expression of C2C12 myotubes was quantified by binding 5 nM 125I-Bgt (NEN) overnight at 4°C, washing three times in PBS and measuring bound radioactivity. Competition assays were performed by preincubating cells with carb or dTC solutions for 15 min, subsequently supplemented with solutions+5 nM 125I-Bgt for 30 min. For surface AChR turnover experiments, myotubes were bound with 10 nM 125I-Bgt in DME (±10 μM LACT) for 1.5 h at 37°C, washed three times in PBS to remove excess toxin and chased at 37°C. At specified times, media were collected and replaced. Collected samples along with harvested cells were counted, summed and turnover expressed as a fraction of 125I-Bgt counts remaining (Devreotes and Fambrough, 1975). Toxin dissociation rates were measured by binding 125I-Bgt in a similar fashion but cells were instead incubated at 4°C in PBS.
Sucrose gradients
C2C12 myotubes were bound with 62.5 nM cold Bgt for 1.5 h at 37°C, washed three times in PBS, then treated with 10 μM LACT (or NLVS) and 10 nM 125I-Bgt for 4.5 h at 37°C. Cells were washed in PBS, harvested and lysed in 1% TX-100 lysis buffer. TX-100-soluble fractions were layered on 5–20% sucrose gradients prepared in 1% TX-100 lysis buffer. Gradients were centrifuged at 40 000 r.p.m. (ω2t=9.0 × 1011) for 14.25 h in a Beckman SW 50.1 rotor. A total of 18 fractions of 300 μl each were collected to determine the amount of 125I-Bgt bound to each fraction.
Quantification
Band intensities of 35S-Met/Cys-labeled AChR subunits were quantified by phosphorimager (Typhoon, Molecular Dynamics). Degradation rates are expressed as a percentage of the value for the 0 h time point. Data are fit by double exponentials and t1/2 values are determined from this approximation. Mean and s.e.m. values of each experiment are shown and t-tests were performed where indicated.
Acknowledgments
We thank N Bence, AJ McClellan, R Kopito, V Berthoud, E Ko and CP Wanamaker for critical reading of the manuscript, and V Lennon for the gift of antibodies. This work was supported in part by an NIH Training grant for JCC and by grants from the National Institutes of Health (NIDA and NINDS) and the Alzheimer's Association (WNG).
References
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291–1300 [DOI] [PubMed] [Google Scholar]
- Bèuttner C, Sadtler S, Leyendecker A, Laube B, Griffon N, Betz H, Schmalzing G (2001) Ubiquitination precedes internalization and proteolytic cleavage of plasma membrane-bound glycine receptors. J Biol Chem 276: 42978–42985 [DOI] [PubMed] [Google Scholar]
- Blount P, Merlie JP (1988) Native folding of an acetylcholine receptor alpha subunit expressed in the absence of other receptor subunits. J Biol Chem 263: 1072–1080 [PubMed] [Google Scholar]
- Blount P, Merlie JP (1990) Mutational analysis of muscle nicotinic acetylcholine receptor subunit assembly. J Cell Biol 111: 2613–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Cosson P, Shah N, Klausner RD (1991) Role of potentially charged transmembrane residues in targeting proteins for retention and degradation within the endoplasmic reticulum. EMBO J 10: 2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Weissman AM (1998) Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol 14: 19–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, McCracken AA (1999) ER protein quality control and proteasome-mediated protein degradation. Semin Cell Dev Biol 10: 507–513 [DOI] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM (2002) Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron 35: 107–120 [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Schwartz AL (1998) The ubiquitin–proteasome pathway: the complexity and myriad functions of proteins death. Proc Natl Acad Sci USA 95: 2727–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio T, Paulson HL, Green WN, Ross AF, Hartman DS, Hayden D (1989) Fibroblasts transfected with Torpedo acetylcholine receptor beta-, gamma-, and delta-subunit cDNAs express functional receptors when infected with a retroviral alpha recombinant. J Cell Biol 108: 2277–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Virgilio M, Weninger H, Ivessa NE (1998) Ubiquitination is required for the retro-translocation of a short-lived luminal endoplasmic reticulum glycoprotein to the cytosol for degradation by the proteasome. J Biol Chem 273: 9734–9743 [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764 [DOI] [PubMed] [Google Scholar]
- Devreotes PN, Fambrough DM (1975) Acetylcholine receptor turnover in membranes of developing muscle fibers. J Cell Biol 65: 335–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eertmoed AL, Vallejo YF, Green WN (1998) Transient expression of heteromeric ion channels. Methods Enzymol 293: 564–585 [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin–proteasome system. Nat Neurosci 6: 231–242 [DOI] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY (1999) A ‘distributed degron' allows regulated entry into the ER degradation pathway. EMBO J 18: 5994–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon T, Chomsky O, Kulka RG (2000) Degradation signals recognized by the Ubc6p–Ubc7p ubiquitin-conjugating enzyme pair. Mol Cell Biol 20: 7214–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Gorrie GH, Vallis Y, Stephenson A, Whitfield J, Browning B, Smart TG, Moss SJ (1997) Assembly of GABAA receptors composed of alpha1 and beta2 subunits in both cultured neurons and fibroblasts. J Neurosci 17: 6587–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WN (1999) Ion channel assembly: creating structures that function. J Gen Physiol 113: 163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green WN, Claudio T (1993) Acetylcholine receptor assembly: subunit folding and oligomerization occur sequentially. Cell 74: 57–69 [DOI] [PubMed] [Google Scholar]
- Green WN, Millar NS (1995) Ion-channel assembly. Trends Neurosci 18: 280–287 [PubMed] [Google Scholar]
- Green WN, Wanamaker CP (1998) Formation of the nicotinic acetylcholine receptor binding sites. J Neurosci 18: 5555–5564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Forsayeth JR, Verrall S, Yu XM, Hall ZW (1991) Assembly of the mammalian muscle acetylcholine receptor in transfected COS cells. J Cell Biol 114: 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2: 195–201 [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD (2003) Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23: 6188–6199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Froehner SC (1983) Degradation of acetylcholine receptors in muscle cells: effect of leupeptin on turnover rate, intracellular pool sizes, and receptor properties. J Cell Biol 96: 1316–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83: 129–135 [DOI] [PubMed] [Google Scholar]
- Johnson PR, Swanson R, Rakhilina L, Hochstrasser M (1998) Degradation signal masking by heterodimerization of MATalpha2 and MATa1 blocks their mutual destruction by the ubiquitin–proteasome pathway. Cell 94: 217–227 [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Ellisman M, Taylor P (2001) Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-subunit. J Biol Chem 276: 18384–18391 [DOI] [PubMed] [Google Scholar]
- Keller SH, Lindstrom J, Taylor P (1998) Inhibition of glucose trimming with castanospermine reduces calnexin association and promotes proteasome degradation of the alpha-subunit of the nicotinic acetylcholine receptor. J Biol Chem 273: 17064–17072 [DOI] [PubMed] [Google Scholar]
- Keller SH, Taylor P (1999) Determinants responsible for assembly of the nicotinic acetylcholine receptor. J Gen Physiol 113: 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Bursztajn S, Goldberg AL (1980) Degradation of the acetylcholine receptor in cultured muscle cells: selective inhibitors and the fate of undegraded receptors. Cell 19: 481–491 [DOI] [PubMed] [Google Scholar]
- Lindstrom JM (2000) Acetylcholine receptors and myasthenia. Muscle Nerve 23: 453–477 [DOI] [PubMed] [Google Scholar]
- Malik B, Schlanger L, Al-Khalili O, Bao HF, Yue G, Price SR, Mitch WE, Eaton DC (2001) ENaC degradation in A6 cells by the ubiquitin–proteasome proteolytic pathway. J Biol Chem 276: 12903–12910 [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka R (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126 [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25: 649–662 [DOI] [PubMed] [Google Scholar]
- Melman L, Geuze HJ, Li Y, McCormick LM, Van Kerkhof P, Strous GJ, Schwartz AL, Bu G (2002) Proteasome regulates the delivery of LDL receptor-related protein into the degradation pathway. Mol Biol Cell 13: 3325–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlie JP, Lindstrom J (1983) Assembly in vivo of mouse muscle acetylcholine receptor: identification of an alpha subunit species that may be an assembly intermediate. Cell 34: 747–757 [DOI] [PubMed] [Google Scholar]
- Molinari M, Calanca V, Galli C, Lucca P, Paganetti P (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Musil LS, Le AC, VanSlyke JK, Roberts LM (2000) Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem 275: 25207–25215 [DOI] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299: 1394–1397 [DOI] [PubMed] [Google Scholar]
- Petaja-Repo UE, Hogue M, Laperriere A, Bhalla S, Walker P, Bouvier M (2001) Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem 276: 4416–4423 [DOI] [PubMed] [Google Scholar]
- Plemper RK, Egner R, Kuchler K, Wolf DH (1998) Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J Biol Chem 273: 32848–32856 [DOI] [PubMed] [Google Scholar]
- Rabinovich E (2001) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Nature 414: 652–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW (1996) PEST sequences and regulation by proteolysis. Trends Biochem Sci 21: 267–271 [PubMed] [Google Scholar]
- Ross AF, Green WN, Hartman DS, Claudio T (1991) Efficiency of acetylcholine receptor subunit assembly and its regulation by cAMP. J Cell Biol 113: 623–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of a post-synaptic apparatus. Nat Rev Neurosci 2: 791–805 [DOI] [PubMed] [Google Scholar]
- Schmidt JW, Catterall WA (1986) Biosynthesis and processing of the alpha subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46: 437–444 [DOI] [PubMed] [Google Scholar]
- Shamu CE, Story CM, Rapoport TA, Ploegh HL (1999) The pathway of US11-dependent degradation of MHC class I heavy chains involves a ubiquitin-conjugated intermediate. J Cell Biol 147: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ (2001) Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294: 1307–1313 [DOI] [PubMed] [Google Scholar]
- Sine SM (1993) Molecular dissection of subunit interfaces in the acetylcholine receptor: identification of residues that determine curare selectivity. Proc Natl Acad Sci USA 90: 9436–9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K (2003) The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13: 899–910 [DOI] [PubMed] [Google Scholar]
- Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D (1997) Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16: 6325–6336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbach JH, Chen Q (1995) Antagonist and partial agonist actions of d-tubocurarine at mammalian muscle acetylcholine receptors. J Neurosci 15 (Part 1): 230–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumikawa K, Miledi R (1989) Assembly and n-glycosylation of all ACh receptor subunits are required for their efficient insertion into plasma membranes. Mol Brain Res 5: 183–192 [DOI] [PubMed] [Google Scholar]
- Tiwari S (2001) Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J Biol Chem 276: 16193–16200 [DOI] [PubMed] [Google Scholar]
- Valentijn JA, Fyfe GK, Canessa CM (1998) Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J Biol Chem 273: 30344–30351 [DOI] [PubMed] [Google Scholar]
- van Kerkhof P, Govers R, Alves dos Santos CM, Strous GJ (2000) Endocytosis and degradation of the growth hormone receptor are proteasome-dependent. J Biol Chem 275: 1575–1580 [DOI] [PubMed] [Google Scholar]
- VanSlyke JK, Deschenes SM, Musil LS (2000) Intracellular transport, assembly, and degradation of wild-type and disease-linked mutant gap junction proteins. Mol Biol Cell 11: 1933–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Zhang L, Yao Y, Viroonchatapan N, Rothe E, Wang ZZ (2002) A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nat Neurosci 5: 963–970 [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ (2000) Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 25: 635–647 [DOI] [PubMed] [Google Scholar]
- Ward CL, Kopito RR (1994) Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269: 25710–25718 [PubMed] [Google Scholar]
- Ward CL, Omura S, Kopito RR (1995) Degradation of CFTR by the ubiquitin–proteasome pathway. Cell 83: 121–127 [DOI] [PubMed] [Google Scholar]
- Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL (1996) Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384: 432–438 [DOI] [PubMed] [Google Scholar]
- Xiong X, Chong E, Skach WR (1999) Evidence that endoplasmic reticulum (ER)-associated degradation of cystic fibrosis transmembrane conductance regulator is linked to retrograde translocation from the ER membrane. J Biol Chem 274: 2616–2624 [DOI] [PubMed] [Google Scholar]
- Yang M, Omura S, Bonifacino JS, Weissman AM (1998) Novel aspects of degradation of T cell receptor subunits from the endoplasmic reticulum (ER) in T cells: importance of oligosaccharide processing, ubiquitination, and proteasome-dependent removal from ER membranes. J Exp Med 187: 835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Malek TR (2001) The proteasome regulates receptor-mediated endocytosis of interleukin-2. J Biol Chem 276: 381–385 [DOI] [PubMed] [Google Scholar]
- Yu H, Kaung G, Kobayashi S, Kopito RR (1997) Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem 272: 20800–20804 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Hegde AN, Martin KC (2003) The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr Biol 13: 887–898 [DOI] [PubMed] [Google Scholar]