

Abstract
Del(13)Svea36H (Del36H) is a deletion of ∼20% of mouse chromosome 13 showing conserved synteny with human chromosome 6p22.1-6p22.3/6p25. The human region is lost in some deletion syndromes and is the site of several disease loci. Heterozygous Del36H mice show numerous phenotypes and may model aspects of human genetic disease. We describe 12.7 Mb of finished, annotated sequence from Del36H. Del36H has a higher gene density than the draft mouse genome, reflecting high local densities of three gene families (vomeronasal receptors, serpins, and prolactins) which are greatly expanded relative to human. Transposable elements are concentrated near these gene families. We therefore suggest that their neighborhoods are gene factories, regions of frequent recombination in which gene duplication is more frequent. The gene families show different proportions of pseudogenes, likely reflecting different strengths of purifying selection and/or gene conversion. They are also associated with relatively low simple sequence concentrations, which vary across the region with a periodicity of ∼5 Mb. Del36H contains numerous evolutionarily conserved regions (ECRs). Many lie in noncoding regions, are detectable in species as distant as Ciona intestinalis, and therefore are candidate regulatory sequences. This analysis will facilitate functional genomic analysis of Del36H and provides insights into mouse genome evolution.
The Del(13)Svea36H mutation (referred to hereafter as Del36H) is a microscopically visible deletion of ∼20% of mouse chromosome 13 (Arkell et al. 2001). Mice that are heterozygous for Del36H display a phenotype that varies with genetic background and that can involve reduced size, craniofacial malformation, eyes open at birth, and a mild tail kink. These mice may model some aspects of human genetic disease, because the Del36H region shows conserved synteny with regions of human chromosome 6p22.1-6p22.3 and 6p25 that are lost in some deletion syndromes (Davies et al. 1999). Furthermore, several disease loci map to this region in humans: two eye defects (iridogoniodysgenesis and Axenfeld-Rieger anomaly; Mears et al. 1998; Nishimura et al. 1998), haemochromatosis (Feder et al. 1996), dyslexia (Grigorenko et al. 2003), and schizophrenia susceptibility (Straub et al. 2002).
Mice with interstitial chromosome deletions like Del36H are potent experimental tools for functional genomics. In particular, they can be used to reveal recessive phenotypes due to mutations that map to a specific chromosomal region. However, the positional candidate approach to identifying mutations in genes underlying mutant phenotypes remains nontrivial, especially for point mutations such as those induced by ENU (Brown and Hardisty 2003). A prerequisite for effective mutation detection using this approach is a comprehensive gene list, with exhaustive annotation of exons and regulatory elements. A limited catalog of the genes deleted in Del36H can be found in genetic and radiation hybrid maps (Arkell et al. 2001; Avner et al. 2001; Hudson et al. 2001), and an automatically annotated genomic sequence is available (Waterston et al. 2002), but the current public mouse genome assembly is a mixture of draft and finished sequence and, by definition, draft genomic sequence contains gaps and regions of lower sequence quality. These artefacts can influence gene annotation and, therefore, the subsequent design of mutation detection assays. Manual annotation, in contrast, should provide a gold standard reference set.
As well as being an invaluable resource for functional genomics, a large genomic region of this kind provides an opportunity to investigate the organization and evolution of a significant piece of the mouse genome. Such studies also rely on high-quality sequence and manual gene annotation to avoid errors in sequence alignment, identification of coding and pseudogenes, classification of repetitive elements, and so on. The accumulating information on genome sequences from a number of species raises many questions about genome evolution. Important among these are the relative roles of whole-genome, segmental, and individual gene duplication, and the mechanisms underlying these processes (Lynch and Conery 2000; Dehal et al. 2001; Eichler and Sankoff 2003; Friedman and Hughes 2004); the usefulness of inter-genome comparisons for identifying selectively conserved regions in genomes, including not only genes, but regulatory regions and functional RNA genes (Mallon et al. 2000; Dehal et al. 2001; Dermitzakis et al. 2002; Kondrashov and Shabalina 2002; Margulies et al. 2003; Frazer et al. 2004); the roles of repeated (transposable element-like) and repetitive (satellites, microsatellites, and minisatellites) sequences in genome evolution (Toth et al. 2000; Hancock 2002; Babcock et al. 2003; Alba and Guigo 2004; Han et al. 2004; Kazazian Jr. 2004); and the characteristics of sites of evolutionary chromosome breakpoints (Puttagunta et al. 2000; Dehal et al. 2001; Pevzner and Tesler 2003).
Here, we describe the genomic architecture of Del36H based on 12.66 Mb of finished DNA sequence, annotated using a combination of manual annotation with synteny and comparative sequence analysis. We find that the region is gene rich, primarily as the result of high gene densities in regions containing gene families that are smaller or absent in the orthologous human regions, and which appear to contribute to the special requirements of the lifestyle of the mouse. We consider forces and processes that may have contributed to the expansion of these gene families during evolution. We also identify a segment of Del36H containing two nearby evolutionary breakpoints, and show that these lie in a gene desert, a potentially optimal site for chromosome breakage. Finally, we consider the evolutionary dynamics of Evolutionarily Conserved Regions (ECRs; Mallon et al. 2000) within Del36H and their potential application to the identification of regulatory, and potentially other functional sequences within noncoding regions of the mouse genome.
RESULTS
Mapping, Sequencing, and Annotation
Physical mapping of the region was completed as described in the Methods, resulting in the production of a sequence-ready map. This map comprises a minimal tiling path (MTP) of 95 clones from the C57BL/6 BAC library. Sequence assembly of the finished sequence generated from the MTP produced a single contig of 12,660,359 bp that extends across chromosome bands A3.1-A4, encompassing both light and dark staining Giemsa bands. Waterston et al. (2002) concluded that the mouse genome, as a whole, is 14% smaller than the human genome. We were able to identify human sequences apparently homologous to 12,176,528 bp of Del36H. The equivalent human region is 10,421,640 bp long, 14.4% shorter than in mouse.
Sequencing was carried out to 10-fold coverage, with an estimated error rate of <1 per 105 bases. On this basis, we expect fewer than 127 sequencing errors in our finished sequence. Exons make up 2.2% of the region. Assuming that about two-thirds of errors in protein-coding regions result in errors in amino acid assignment, this suggests no more than one to two protein sequence errors in coding regions. In addition, other errors may affect regulatory regions. Individual clone sequences have been submitted to the public databases (see Methods for accession nos.).
Each finished clone was subjected to high-quality manual annotation as described in the Methods, resulting in the identification of 201 genes falling into one of the two categories, known genes or novel CDS genes, encoding transcripts containing an open reading frame (we describe this set of genes collectively as ORF genes), and a further 35 novel transcript genes with significant support, but no transcript ORF (see Ashurst and Wilming 2002 for definitions of the different annotation classes of genes used in this study). In addition, a further 95 pseudogenes were identified. The features, including gene annotation of the region, are summarized in Figure 1. The annotated sequence and features can be accessed online via the VEGA database and browser (http://vega.sanger.ac.uk/Mus_musculus/).
Figure 1.

Pictorial representation of the annotation of the Del36H sequence contig. (Top to bottom) Yellow blocks represent the finished sequence of individual genomic clones forming the tiling path across this region; a black line or block at the start and/or end of a clone represents redundant overlapping sequence. Five tracks show in green the distribution of various types of repeats as defined by RepeatMasker. (Red) The distribution of the fraction of G and C nucleotides, between 0.3 (30%) and 0.7 (70%); (purple) the Relative Simplicity Factor scores; (blue) the cumulative signs plot of RSF scores. Alternating gray and black blocks indicate the segments of interest; odd-numbered segments contain gene clusters and evolutionary breakpoints, even-numbered segments lie between and around these. Arrows show the position of annotated genes, their relative transcriptional orientation, and their type; only genes of type “known” have their gene symbol shown. Dark blue: known genes; light blue: novel CDS genes; orange: novel transcript genes; green: pseudogenes; gray: putative genes.
A number of gene family clusters were identified through annotation (see Gene Clusters section). Of these, three [the vomeronasal receptor (VnRs), serpin, and prolactin gene families] contain more members in mouse than in human. Two other clusters, a split histone cluster and a cluster of three forkhead box (Fox) genes, are roughly the same size in human and mouse, whereas the butyrophilin family is smaller in mouse. On the basis of the presence of these gene clusters, plus the region containing the evolutionary breakpoints between mouse and human, the region was subdivided into 10 subregions (segments 0-9; Fig. 1), which we call segments here for clarity. Segment 1 is defined as containing the histone and vomeronasal receptor clusters, which are interspersed; segment 3 contains the prolactin gene cluster; segment 5 contains the breakpoints; segment 7 contains the Fox gene cluster; and segment 9 the serpin gene cluster. Other segments correspond to subregions lying outside of these regions of interest.
Manually annotated genes were compared with automatic predictions in Ensembl (version 16.30.1, based on the NCBI build 30 composite assembly http://www.ensembl.org/Mus_musculus/whatsnew/v16_30_1.html) (Hubbard et al. 2002; Table 1). Comparisons are represented as sensitivities and specificities based on the ability of Ensembl to predict our annotated genes. The comparisons produced reasonably high sensitivities for protein-coding regions, but Ensembl showed less agreement with manual annotations for nontranslated exons and at the transcript level. Specificities were uniformly lower than sensitivities. Almost all of the overpredicted Ensembl genes overlapped pseudogenes.
Table 1.
Comparison of Ensembl and Manual Gene Annotations of the Region
| Match type | Sensitivitya | Specificitya |
|---|---|---|
| Gene level | 0.94 | 0.83 |
| Transcript level | 0.75 | 0.72 |
| Annotated exons covered completely: all exons | 0.76 | 0.74 |
| Annotated exons covered completely: coding exons | 0.90 | 0.75 |
| Annotated exons with exact matches at boundaries: all exons | 0.63 | 0.57 |
| Annotated exons with exact matches at boundaries: coding exons | 0.85 | 0.71 |
Calculations of sensitivity and specificity represent the success of Ensembl in predicting genes identified by our manual annotation process. Thus, a specificity of 0.83 indicates that 83% of positives in the Ensembl process are true positives by our criteria.
Del36H Landscape
Del36H contains a noticeably higher density of annotated genes (one ORF gene per 63.0 kb, or one gene per 53.6 kb if novel transcript genes are included) than the draft mouse genome (Waterston et al. 2002), for which one gene per 113.6 kb has been reported. One gene per 119 kb was reported in mouse chromosome 16 (Mural et al. 2002). Despite Del36H being longer than the identifiably homologous regions of the human genome, the overall annotated gene density in the human regions is lower than in the mouse region (one per 93.0 kb compared with one per 68.0 kb for the corresponding mouse region), consistent with gain of genes in the mouse sequence. ORF genes averaged 4.1 exons per gene (4.2 per gene for known genes and 3.8 per gene for novel CDS genes), whereas novel transcript genes averaged 2.7, putative genes 2.5, and pseudogenes 1.4 exons per gene. These averages are lower than the 8.3-9 suggested for the mouse genome as a whole (Waterston et al. 2002), reflecting the high frequency of single exon genes in Del36H.
Analysis of the ORF gene distribution between the 10 segments (Table 2) showed a statistically highly significant nonrandom distribution of the numbers of genes (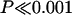 ; χ2 test, 9 df [degrees of freedom]). This reflected high gene numbers in segments 0 and 1 (approximately one every 20 kb) and very low numbers (of the order of one every 300 kb) in segments 4 and 8. Densities of gene counts do not take into account the sizes of the genes—a low numerical gene density could reflect an overrepresentation of long genes and vice versa. We therefore also estimated the proportion of the regions included within ORF genes. This was 28.87% for Del36H as a whole—percentage coverages for individual segments are shown in Table 2. Segments containing gene families (segments 1, 3, 7, and 9) had the lowest percentage coverages in the region, below 20% in all cases. In contrast, segments 4 and 8 had percentage coverages around 50%. Thus, segments rich in gene families contained numerous short genes, in agreement with the observation in Caenorhabditis elegans that duplicated genes tend to be shorter than average (Katju and Lynch 2003).
; χ2 test, 9 df [degrees of freedom]). This reflected high gene numbers in segments 0 and 1 (approximately one every 20 kb) and very low numbers (of the order of one every 300 kb) in segments 4 and 8. Densities of gene counts do not take into account the sizes of the genes—a low numerical gene density could reflect an overrepresentation of long genes and vice versa. We therefore also estimated the proportion of the regions included within ORF genes. This was 28.87% for Del36H as a whole—percentage coverages for individual segments are shown in Table 2. Segments containing gene families (segments 1, 3, 7, and 9) had the lowest percentage coverages in the region, below 20% in all cases. In contrast, segments 4 and 8 had percentage coverages around 50%. Thus, segments rich in gene families contained numerous short genes, in agreement with the observation in Caenorhabditis elegans that duplicated genes tend to be shorter than average (Katju and Lynch 2003).
Table 2.
Gene Distributions of the Del36H Region and Its Segments
| Gene distribution
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Known
|
Novel
|
Novel transcript
|
Putative
|
Pseudogene
|
Total ORF genes
|
Total
|
||||||||
| Region | Number | % of Contig | Number | % of Contig | Number | % of Contig | Number | % of Contig | Number | % of Contig | Number | % of Contig | Number | % of Contig |
| 0 | 16 | 20.29 | 6 | 4.38 | 0 | 0.00 | 0 | 0.00 | 1 | 0.10 | 22 | 24.67 | 23 | 24.77 |
| 1 | 70 | 12.55 | 32 | 3.41 | 11 | 1.48 | 0 | 0.00 | 54 | 2.10 | 102 | 15.96 | 167 | 19.54 |
| 2 | 11 | 17.38 | 6 | 12.38 | 9 | 1.47 | 0 | 0.00 | 12 | 0.22 | 17 | 29.76 | 38 | 31.45 |
| 3 | 18 | 13.12 | 6 | 3.42 | 1 | 1.23 | 0 | 0.00 | 4 | 1.10 | 24 | 16.54 | 29 | 18.87 |
| 4 | 6 | 49.76 | 1 | 0.03 | 3 | 1.55 | 2 | 0.13 | 4 | 0.09 | 7 | 49.79 | 16 | 51.56 |
| 5 | 3 | 20.68 | 0 | 0.00 | 1 | 1.86 | 0 | 0.00 | 2 | 0.12 | 3 | 20.68 | 6 | 22.66 |
| 6 | 2 | 19.04 | 1 | 0.15 | 5 | 8.34 | 1 | 11.98 | 2 | 0.67 | 3 | 19.19 | 11 | 40.18 |
| 7 | 3 | 4.83 | 0 | 0.00 | 3 | 11.44 | 1 | 0.48 | 0 | 0.00 | 3 | 4.83 | 7 | 16.75 |
| 8 | 2 | 52.32 | 1 | 1.95 | 2 | 2.06 | 0 | 0.00 | 1 | 0.10 | 3 | 54.27 | 6 | 56.43 |
| 9 | 11 | 13.65 | 6 | 5.21 | 0 | 0.00 | 0 | 0.00 | 15 | 4.25 | 17 | 18.86 | 32 | 23.11 |
| Total | 142 | 24.15 | 59 | 4.72 | 35 | 1.75 | 4 | 0.80 | 95 | 0.96 | 201 | 28.87 | 335 | 32.38 |
The mean GC content of the sequence (expressed as percentage G+C) is 41.1%, which is very similar to the 42% quoted for the mouse genome as a whole (Waterston et al. 2002) and identical to the 41.1% quoted for mouse chromosome 16 (Mural et al. 2002). %GC for individual BACs ranged from 25.5% (for the clone RP23-58N9) to 53.4% (for RP23-287K23), with a standard deviation of 3.1%. Genes have frequently been found to be associated with regions of high GC-content in genomes, for example, in analyses of isochores, which are regions of homogeneous base composition typically of the order of 100-300 kb long (Bernardi 1995; Nekrutenko and Li 2000). We therefore investigated whether there was a detectable correlation between the GC content of BAC sequences and their gene density. Taking all clones into account, a weakly significant correlation was observed (P < 0.05). However, if two outliers (RP23-287K23 and RP23-58N9, clones with extreme high and low GC content) were excluded, no significant correlation was observed.
The predominant features of eukaryotic genome sequences are repeated sequences. Repeated element densities in Del36H are close to average for the mouse genome as represented by the draft sequence (Waterston et al. 2002). The only exception is the density of SINEs, which is 36% lower than in the genome as a whole (5.8% of the sequence compared with 8.2% in the draft; see Table 3). We investigated the possibility of association between gene duplications and transposable element (TE) density by carrying out Wilcoxon 2-sample tests on the ranks of groups of BACs (corresponding to particular segments) compared with ranks of BACs in the remainder of the sequence. Using this approach, we observed elevated representation of particular classes of repetitive element in the three segments associated with gene families expanded in mouse relative to human, and in segment 0, but not in others. Segments 0, 1 (histones, vomeronasal receptors), and 9 (serpins) were associated with significantly high LTR element densities, whereas segments 3 (prolactins) and 9 showed a high density of LINEs (P < 0.05 after Bonferroni correction). In addition, it is noteworthy that segment 1 is rich in IAP (intracisternal A-particle) elements. These have been found previously to be associated with genome rearrangements, for example, in leukemia (Ishihara et al. 2004), and are rare in the mouse genome, which is thought to contain only around 1000.
Table 3.
Repeat Distribution and G+C Content Across the Region and in Each Segment
| Repeat Coverage (% of Sequence)
|
|||||||
|---|---|---|---|---|---|---|---|
| Segment | Length | SINEs | LINEs | LTR | DNA elements | Total | % GC |
| 0 | 436,413 | 8.33 | 13.41 | 13.22 | 0.63 | 35.60 | 41.05 |
| 1 | 2,218,902 | 5.19 | 19.65 | 15.53 | 0.22 | 40.60 | 41.18 |
| 2 | 3,122,470 | 7.94 | 17.59 | 9.73 | 0.52 | 35.78 | 40.86 |
| 3 | 1,094,034 | 1.51 | 35.84 | 11.49 | 0.26 | 49.10 | 37.30 |
| 4 | 2,100,944 | 9.85 | 14.31 | 6.91 | 1.08 | 32.15 | 42.29 |
| 5 | 496,671 | 5.33 | 15.71 | 11.65 | 0.82 | 33.51 | 42.35 |
| 6 | 809,098 | 3.58 | 9.02 | 8.20 | 1.32 | 22.12 | 42.56 |
| 7 | 252,320 | 3.81 | 7.44 | 9.71 | 2.35 | 23.31 | 45.31 |
| 8 | 1,031,448 | 5.09 | 8.60 | 6.60 | 1.17 | 21.46 | 40.77 |
| 9 | 1,098,059 | 3.39 | 33.54 | 18.15 | 0.23 | 55.31 | 40.67 |
| Total | 12,660,359 | 6.14 | 18.67 | 11.00 | 0.67 | 36.48 | 41.08 |
The other major class of repeated sequences in genomes is simple sequences, which comprise microsatellites, minisatellites, and related sequences. To produce a summary analysis of the distribution of this class of sequence within Del36H, we used the program SIMPLE (Tautz et al. 1986; Hancock and Armstrong 1994). SIMPLE measures the simple sequence content of sequences relative to random sequences of the same dinucleotide composition. It does this by measuring the reoccurrence of motifs within 64-bp sliding windows by awarding points to a window, depending on the number of times the sequence motif at its center reoccurs in the window. The simplicity factor (SF) for the sequence is then the average score for a window within the sequence, whereas the relative simplicity factor (RSF) is the ratio of the SF to SFs obtained for random sequences with the same dinucleotide composition as the tested sequence. A random sequence has an RSF of 1 and a repetitive sequence an RSF > 1. SIMPLE was applied to individual BAC sequences within Del36H. The mean RSF for BACs was 1.607 ± 0.164 (SD), which lies between previous estimates for the mouse genome based on multiple smaller genomic fragments (Hancock 1995, 2002). Relationships between RSF and gene density or base composition of individual BACs were investigated by regression analysis. Gene density showed a significant negative correlation with RSF (P < 0.01). Base composition did not show a significant correlation with RSF, but did show a highly significant positive rank correlation ( ), despite the fact that the SIMPLE algorithm compensates for the base composition of the tested sequence (Tautz et al. 1986; Hancock and Armstrong 1994).
), despite the fact that the SIMPLE algorithm compensates for the base composition of the tested sequence (Tautz et al. 1986; Hancock and Armstrong 1994).
The distribution of BAC RSF scores along the sequence is represented in Figure 1. The plot shows regions of relatively high and low repetitiveness. This is shown most clearly by the cumulative signs plot in Figure 1. This was produced by assigning each clone a score of +1 or -1, depending on whether its RSF was higher or lower than the mean for Del36H. The value plotted is a cumulative sum of these values, which shows changes of direction when groups of BACs have RSFs of different signs to their preceding neighbors. The plot indicates that Del36H contains domains of relatively high and low RSF ∼2.5 Mb long. This order of substructure is at a larger scale than that conventionally attributed to isochores, which are usually defined as being of the order of 100-300 kb long (Bernardi et al. 1985; Nekrutenko and Li 2000). Recent attempts to define isochores computationally have produced evidence that some may be of the order of megabases long (Oliver et al. 2001; Li 2002) but IsoFinder (Oliver et al. 2001; http://bioinfo2.ugr.es/isochores/) predictions of isochores in Del36H do not exceed 200 kb in length. The simple sequence domains are of comparable size to chromosome bands. Unfortunately, boundaries of chromosome bands are currently not well defined at the sequence level, so it is not possible to test for a relationship between these simplicity domains and chromosome bands at this time.
Sites of Evolutionary Rearrangement
Homology of Del36H to regions of the human genome was established initially by BLAST analysis of individual genes to identify the chromosomal location of human orthologs (identified as the human sequence with the lowest BLAST E score). This confirmed conserved synteny to human chromosomes 6p22.1-6p22.3 and 6p25.2-6p25.3. The orientations of the two parts of Del36H homologous to human chromosome 6 are opposed—the 5′ part of Del36H is in the same orientation as in human, with its 5′ end nearer the centromere, whereas the 3′ end of Del36H, which is closer to the telomere in mouse, corresponds to a region closer to the centromere in human (see Fig. 2A). Loss of conserved synteny, and therefore the locus of chromosome reorganization between the two species, lies within four BACs (RP23-372H19, RP23-380L6, RP23-279E14, RP23-217J3), a region 49.7 kb long. Further analysis (Fig. 2B) identified an additional short region with homology to human chromosome 3q24 within this region. We are therefore able to define two regions not showing a detectable homologous relationship to any human chromosome and a novel syntenic relationship for the region.
Figure 2.

Location of region on mouse and human chromosomes and dotplot of the breakpoint region. (A) The location of the contig on mouse chromosome 13 and the equivalent regions on human chromosome 6. The segments of interest (0-9) are indicated. Note that on the human chromosome, the regions are split into two blocks, breaking in segment 5. Positions and relative sizes of regions are only approximate; human and mouse chromosomes are shown to different scales. (B) Del36H segment 5 has homology with human chromosome 6p25 and 6p22 at its edges, as well as 3q24 in the center. Between these stretches of homology are two stretches of sequence, BP-1 and BP-2, that do not have any detectable homology with human sequence.
Studies by Puttagunta et al. (2000) and Pletcher et al. (2000) on evolutionary breakpoints in mouse chromosome 10 showed an association with a region of very high (60%-70%) repeated sequence content and Dehal et al. (2001) found a concentration of LINEs and LTR elements at evolutionary breakpoints. Consideration of the gross distribution of repeated elements, including simple repeats, and base composition in segment 5 (the breakpoint region) as a whole compared with the rest of the Del36H sequence, showed no apparent associations. BP1, one of the two sequences apparently without a human homolog (see Fig. 2B), contains a relatively high concentration of LTR elements, 19.91%, which is atypical for segment 5 as a whole, although a number of individual clones show higher LINE densities. BP2, the other unique region, does not show an unusual TE composition.
Gene Clusters
Gene clusters were identified in three of the segments, as mentioned previously.
Segment 1: Cluster of Vomeronasal Receptor, Butyrophilin, and Histone Genes
The two large histone clusters on human chromosome 6 (Albig and Doenecke 1997) are also present on mouse chromosome 13 (Wang et al. 1996). As in human, the mouse butyrophilin cluster (Rhodes et al. 2001) is located between the two histone clusters, although in mouse, the large vomeronasal receptor cluster next to it further separates the two histone clusters. The histone clusters contain genes from all five histone families (H1, H2A, H2B, H3, H4). In mouse, we identify 57 genes, including three pseudogenes, whereas the human clusters contain a total of 66 genes, of which 11 are pseudogenes. Discounting the pseudogenes, both species have virtually the same number of histone genes (54 in mouse, 55 in human). However, aligning the mouse and human clusters shows that these are not all equivalent; positionally, not all 54 mouse genes can be matched up to a corresponding ortholog in human. There are four genes unique to mouse and two that have a human pseudogene as an ortholog. Conversely, there are six human unique genes and one human gene that has a mouse ortholog degenerated into a pseudogene. Of the pseudogenes, one, an H2a family pseudogene, is conserved between mouse and man. Despite these differences, the overall organization (order, orientation) is very similar (see Fig. 3). The genomic organization of the human histone cluster is in agreement with that published earlier (Marzluff et al. 2002). However, the organization of the mouse cluster is different from that described by Marzluff et al. (2002). We find two more H2b genes, and one each of H2a and H4 family genes, all in the centromeric cluster. Marzluff et al.'s (2002) assignment of mouse or human unique genes is different from that presented here, owing to incorrect mouse-human alignment of part of the centromeric cluster (Marzluff et al. 2002, their Fig. 2B); the human cluster is incorrectly reversed in orientation, and four mouse histone genes are missing. In addition, one of Marzluff et al.'s (2002) unique mouse genes has an orthologous pseudogene in man.
Figure 3.

Gene clusters. Panels showing the relative organization of genes in several of the gene clusters present in the region, comparing mouse to human. Human sequences shown at the top, mouse at the bottom, with the relevant chromosomes indicated. Where necessary, human sequence has been reoriented to align with mouse sequence (which is shown in centromere-telomere orientation as in Fig. 1). Figures are not to scale and only show selected genes, not noncluster genes or clusters interspersed within some of these clusters. However, conserved genes and clusters within or immediately neighboring clusters are shown with their current mouse gene symbols below the clusters, with an arrow indicating transcriptional orientation. Phylogenetic trees for three of the clusters (A, B, C) are also shown. (A) The vomeronasal 1 receptor (V1r) cluster. Orange ovals indicate where it could not conclusively be determined whether the pseudogene derived from family h or i. Black dots in the phylogenetic tree mark previously unknown family members. The human sequence has only five V1r genes, all degenerated into pseudogenes, whereas in mouse, the equivalent region houses 67 copies, just over half of which (34) are pseudogenes. An asterisk marks a human pseudogene that is displaced from the V1R cluster and actually located in the histone cluster (asterisk in D). (B) The prolactin cluster. Circled in the phylogenetic tree are the solitary family members in mouse and human, the Gh (growth hormone) gene (yellow) on mouse chromosome 11 and the PRL (prolactin) gene (green) on human chromosome 6. In mouse, the prolactin cluster has greatly expanded (to 26 members, with three pseudogenes), whereas in human, the growth hormone cluster has modestly expanded to an estimated five members. (C) The serpin cluster. In human, only one copy (encircled in the phylogenetic tree) of a member of three subfamilies [SERPINB1 (yellow), SERPINB6 (blue), and SERPINB9 (green)] is present (plus a SERPINB9 pseudogene), whereas in mouse, all three subfamilies have expanded, approximately maintaining the order, although not orientation, of the subfamily members. In mouse, a complete b1-b9-b6 block may have inserted itself in reverse orientation within the b9 cluster after individual genes were locally duplicated and inserted, giving rise to the current configuration. The b9 gene shown with a white oval is a partial gene (only the first two exons, including the first coding exon), and probably qualifies as a pseudogene. The human pseudogene is actually located between the two conserved genes shown at bottom, RIPK1 and NQO2. (D) The histone and butyrophilin clusters. Histone family 1 (Hist1) comprises members of all five histones H1, H2a, H2b, H3, and H4. The clusters are of comparable size in mouse and human, part of the slightly larger size in human mostly accounted for by a higher number of pseudogenes. Genes appear to have been duplicated in several groups of genes, containing successively smaller numbers of genes with conserved relative orientations and order as follows: a group of a single copy of each of the five genes in order and orientation H2b--H2a+-H3+-H1--H4+ or its reverse complement (white lollipops), this group minus H1 (yellow), then minus H3 (green), and finally a two-member group also missing H4 (black). A gray background area splitting the histone gene cluster in two contains the butyrophilin (Btn) cluster, represented in human by seven genes and in mouse by only two. The V1r cluster shown in A is actually located immediately left of the Btn cluster, further dividing the two parts of the Hist1 cluster. An olfactory receptor gene cluster is located to the left of the histone gene cluster and an Slc17a cluster within the telomeric end of the telomeric cluster. In both mouse and man, this cluster consists of four genes with conserved orientation. The mouse equivalent of the human TRIM38 gene is positionally conserved, but has the opposite transcriptional orientation. At top, the human HMGN4 gene located between ABT1 and the butyrophilin gene cluster is not present in mouse, and an asterisk shows the position within the centromeric histone gene cluster (i.e., outside of the V1R cluster) of a human V1R pseudogene marked by an asterisk in A.
On human chromosome 6, there are five pseudogenes derived from vomeronasal receptors, which are members of a large G-protein-coupled receptor (GPCR) superfamily (for review, see Harmar 2001). These are expressed in sensory neurons of the vomeronasal organ, a part of the accessory olfactory system, which is responsible for the detection of chemical cues such as pheromones (for review, see Keverne 1999). In contrast, the orthologous region on mouse chromosome 13 contains 67 vomeronasal receptor genes, belonging to two families, V1rh and V1ri. Just over half (34) of these genes are pseudogenes by our criteria (Ashurst and Wilming 2002; see Methods). Of the 33 putatively expressed genes, four have not previously been described. There are 12 known Vnr gene families, totalling 137 members (Rodriguez et al. 2002). V1rh has 21 known members and V1ri has 10. We can account for all of these known members, but we find V1rh1 and V1ri8 to be pseudogenes. We add one new member to the V1rh family and three new members to the V1ri family. Alignment of mouse V1rh and V1ri proteins against the human genome in Ensembl (release 20) shows that there are no other human genes orthologous to these families. We did, however, detect a small cluster of expressed vomeronasal receptor genes on human chromosome 19 with genes similar to mouse V1re, V1rf, and V1rg.
Human chromosome 6 contains eight butyrophilin family genes, seven located at the centromeric end of the extended MHC region, the other at the telomeric end (Rhodes et al. 2001). The function of butyrophilin proteins remains uncertain, although they are known to form part of the human milk-fat globule membrane, and may be receptors (Peterson et al. 2001). The solitary BTNL2 gene on human chromosome 6 is represented by an estimated five genes in the equivalent region on mouse chromosome 17. In contrast, we find that the cluster of seven butyrophilin genes (BTN1A1, BTN2A1, BTN3A3, BTN2A3, BTN3A1, BTN2A2, BTN3A2) in the human region with conserved synteny to Del36H is represented in mouse by only two genes (Btn1a1 and a gene similar to BTN2A2). The transcriptional orientation and gene order are conserved in mouse compared with members 1A1 and 2A2, as is most of the immediate genomic context. Both regions are flanked by ABT1 and an H4 gene in conserved orientation, although the human high-mobility group protein gene HMGN4 (HMG17L3) does not have a mouse equivalent at the corresponding location, and various putative novel genes and a pseudogene are not conserved (see Fig. 3D).
Segment 3: Prolactin Gene Cluster
The prolactin family consists of a variety of related proteins currently known as Gh (growth hormone), Prl (prolactin), Csh (chorionic somatomammotropin), Prlp (prolactin-like protein), and Pl (placental lactogen; Forsyth and Wallis 2002). As their names suggest, they are hormones involved in various aspects of pregnancy, lactation, and growth. The genes are located in two groups on mouse chromosomes 11 and 13 (Jackson-Grusby et al. 1988) and the homologous human chromosomes 17 and 6 (George et al. 1981; Owerbach et al. 1981). In mouse, the chromosome 13 group consists of a cluster of 26 related genes (including three pseudogenes), whereas on human chromosome 6, there is only one gene (PRL). The reverse is true for the five-member group in human chromosome 17, which on mouse chromosome 11 is represented by only one gene (Gh). For both clusters, the genomic context (flanking genes) and orientation are conserved between human and mouse.
Wiemers et al. (2003) also describe 26 prolactin genes in this region, but their gene set differs from ours. We find 23 genes and three pseudogenes, whereas Wiemers et al. (2003) describe 26 true genes. Four of the genes described by Wiemers et al. (2003) are closely related copies of a gene they call PLF (our bM20A22.4), but they were only able to map one copy and speculated the remaining copies (which they inferred on the basis of cDNA and EST data) to lie in a gap between two clones, RP23-231P12 and RP23-20A22. This gap is closed in our sequence, and no such copies are found. None of the cDNAs or ESTs they attribute to these genes exactly match our sequence; however, the best matches are to our gene bM20A22.4. It is possible that the additional genes reported by Wiemers et al. (2003) represent sequence or copy-number polymorphism in the region, as the sequences used in their study were from more than one mouse strain.
Segment 9: Serpin Gene Cluster
On human chromosome 6, there is a cluster of three proteinase inhibitor genes (serpin-b), belonging to three different families, and one pseudogene. These genes are members of the ovalbumin (ov)-like serpins, which are implicated in the regulation of tumor progression, inflammation, and cell death. Kaiserman et al. (2002) have shown that the mouse serpins within Del36H show restricted patterns of expression, with many found in reproductive tissue. On mouse chromosome 13, this cluster is represented by multiple genes for each family, totalling 17 genes and 10 pseudogenes (see Kaiserman et al. 2002). The disparity in gene numbers means that there is no meaningful conservation of orientation, but the flanking genes are conserved (see Fig. 3C). Analysis of the draft rat genome also reveals more serpin genes in this region than in human, but there are differences in the copy numbers of the different serpin-b subfamilies between rat and mouse (Puente and López-Otín 2004).
A second serpin cluster (serpin-a) is present on mouse chromosome 12 and human chromosome 14. The Ensembl annotation of this cluster indicates mild expansion in mouse (∼18 genes vs. 10 human genes). The two expanded family members (serpina1, serpin-a3) seem to have expanded in situ, maintaining the general order between mouse and man.
Mechanisms of Expansion
Local gene clusters, to be distinguished from larger scale duplications arising from segmental duplication (Bailey et al. 2002), could arise by transposition, including retrotransposition, or by the action of unequal crossing-over. Clustering appears more likely to arise as a result of unequal crossing-over, but could also arise if there were strong target sites for transposition. Unequal crossing-over is likely to leave traces of local clustering of genes due to tandem amplification. To test for this statistically, we used the runs test (see Sokal and Rohlf 1995). This test identifies whether a sequence of binary characters (e.g., members of one gene family or another, or genes on one strand or the other) contains fewer (or more) runs of a single character than would be expected by chance. Tandem arrangements of genes produced by unequal crossing-over would be expected to show fewer such runs than expected by chance using this test.
The two subfamilies of VnR genes in Del36H (V1rh and V1ri) appear to be randomly dispersed with respect to each other (i.e., show no significant deviation from the expected number of runs of genes of the same type by the runs test) if their strand orientation is ignored. However, the two families combined show significant clustering by strand (P < 0.001; runs test), consistent with an origin by unequal crossing-over. The prolactin genes also show significant clustering by strand (P < 0.001, runs test). The lack of pseudogenes or gene remnants near the solitary Prl gene on human chromosome 6 and the Gh gene on mouse chromosome 9 suggests that the common ancestor had one gene each of Prl and Gh, which in human and mouse, expanded differentially, with humans expanding the Gh line and mice the Prl line. The serpins do not show significant clustering by the runs test. However, although there is some mixing of the three families (identified in Fig. 3) in the mouse serpin cluster, the order of the three families is broadly maintained between mouse and man, suggesting a localized mechanism of gene duplication, such as UCO.
The proportion of pseudogenes within a given gene family can reflect at least two evolutionary processes. Firstly, it reflects the strength of purifying selection acting on the family. If purifying selection has acted strongly, few, if any, of a familiy's members should accumulate deleterious mutations, whereas weak or no purifying selection would allow the accumulation of pseudogenes (see Zhang and Webb 2003). Secondly, gene conversion has been shown to result in the homogenization of sequences within some gene families (including the VnR gene family—see Lane et al. 2004), increasing sequence similarity and potentially eliminating pseudogenes. Both of these processes should give rise to a positive correlation between the accumulation of synonymous mutations within coding regions and the accumulation of pseudogenes. The three expanded gene families in Del36H do not show such a correlation. By our criteria (Ashurst and Wilming 2002), three of the 26 prolactin genes are pseudogenes (12%), whereas 37% (10/27) of the serpins and 51% (34/67) of the V1rs are. However, Table 4 shows that the vomeronasal receptor genes have, on average, accumulated fewest synonymous mutations, as judged both by the mean difference between sequence pairs and the maximum value of Ks observed within the family. The mean Ka/Ks for this family is also not exceptionally high. On the other hand, the prolactins, which have the lowest proportion of pseudogenes, are more divergent, as judged from pairwise Ks values, but have a higher mean pairwise Ka/Ks. The proportion of pseudogenes in these gene families therefore appears to reflect different levels of purifying selection and/or homogenization acting on them. It is noteworthy in this context that Wallis (1993, 1996) has suggested that positive selection has acted on prolactins. This is consistent with high (greater than one) Ka/Ks ratios seen in two pairwise comparisons of Del36H prolactin genes. The highest pairwise Ka/Ks ratio seen in these analyses was seen for a pair of serpin genes, raising the possibility that positive selection may also have acted on this gene family.
Table 4.
Synonymous and Nonsynonymous Divergence Within Gene Families in Del36H
| Ks
|
Ka
|
Ka/Ks
|
|||||
|---|---|---|---|---|---|---|---|
| Family | No. | Mean | Max | Mean | Max | Mean | Max |
| Histone H1 | 6 | 1.231 | 2.582 | 0.211 | 0.408 | 0.170 | 0.263 |
| Histone H2a | 13 | 0.135 | 0.638 | 0.012 | 0.071 | 0.060 | 0.223 |
| Histone H2b | 15 | 0.119 | 0.365 | 0.015 | 0.086 | 0.095 | 0.324 |
| Histone H3 | 9 | 0.082 | 0.122 | 0.002 | 0.003 | 0.021 | 0.068 |
| Histone H4 | 11 | 0.206 | 0.362 | 0.001 | 0.004 | 0.007 | 0.027 |
| Prolactins | 23 | 1.196 | 3.262 | 0.698 | 0.988 | 0.616 | 1.110 |
| Serpins | 16 | 0.965 | 3.572 | 0.297 | 0.576 | 0.382 | 1.374 |
| Vomeronasal Receptors | 33 | 0.772 | 1.645 | 0.285 | 0.504 | 0.392 | 0.731 |
Evolutionarily Conserved Regions
We have previously demonstrated the utility of using short (50 bp) regions of interspecies conservation (Evolutionarily Conserved Regions; ECRs) to identify candidate transcriptional units in the Bpa/Str region of the mouse X chromosome (Mallon et al. 2000). Dermitzakis et al. (2002) described an analysis of human-mouse ECRs from human chromosome 21, which suggested that a significant proportion were noncoding and potentially regulatory elements. We noted previously that ECRs corresponding to different classes of sequence (for example coding exons, 5′ and 3′ UTRs) typically showed different mean conservation and variance (see also Makalowski et al. 1996). Dermitzakis et al. (2003) have suggested that ECRs corresponding to noncoding regions (which we call NC-ECRs and they call CNGs) can be highly conserved between multiple mammalian species and show patterns of evolution that might be expected in transcription factor binding sites. A multispecies comparative analysis by Margulies et al. (2003) showed that 70% of the bases in multispecies conserved sequences (MCS), which are comparable to ECRs, were within noncoding regions. Initial characterization of MCSs revealed sequences that corresponded to clusters of transcription factor binding sites, noncoding RNA transcripts, and other candidate functional elements (Margulies et al. 2003).
For the Del36H region, we have extended ECR analysis to consider not only mouse-human but also mouse-Rat, mouse-Takifugu, mouse-Tetraodon, mouse-Zebrafish, and mouse-Ciona intestinalis ECRs. Our aim in doing this was to gain an insight into the degree to which the conservation corresponding to a particular ECR has decayed over relatively long periods of evolutionary time, potentially providing useful additional information on the function of the ECR. Other species (chimpanzee, cattle, chicken, and sea urchin) did not show significant matches to Del36H, most likely due to lack of data at the time of analysis in most cases.
Numbers of coding (i.e., exonic) and noncoding (nonexonic) ECRs (C- and NC-ECRs; see Methods for details) found for each species pair and in each segment of Del36H are given in Table 5. Overall numbers of ECRs decrease with evolutionary distance, but decrease much more sharply from the mouse/rat comparison to the mouse/human comparison than from mouse/human to the more distant comparisons. This suggests that many of the mouse/rat ECRs reflect neutral (rather than selective) sequence conservation, despite the higher stringency used in the comparison. Mouse/fish and mouse/Ciona comparisons produced similar numbers of ECRs, suggesting that ECRs observed in one species over this relatively long evolutionary time-scale (700-1100 Myr) are likely to be broadly conserved.
Table 5.
Evolutionary Conserved Regions in Each Segment
| Rat
|
Human
|
Zebrafish
|
Takifugu
|
Tetraodon
|
Ciona intestinalis
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ECRs | Ca | NCb | ECRs | C | NC | ECRs | C | NC | ECRs | C | NC | ECRs | C | NC | ECRs | C | NC | |
| 0 | 342 | 118 | 224 | 26 | 23 | 3 | 39 | 21 | 18 | 6 | 5 | 1 | 8 | 7 | 1 | 14 | 11 | 3 |
| 1 | 1286 | 631 | 645 | 143 | 103 | 40 | 261 | 114 | 147 | 138 | 83 | 55 | 131 | 78 | 53 | 33 | 20 | 13 |
| 2 | 3409 | 1185 | 2224 | 255 | 85 | 170 | 122 | 88 | 34 | 131 | 118 | 13 | 87 | 75 | 12 | 8 | 7 | 1 |
| 3 | 363 | 134 | 229 | 1 | 0 | 1 | 10 | 0 | 10 | 0 | 0 | 0 | 8 | 0 | 8 | 0 | 0 | 0 |
| 4 | 3147 | 1222 | 1925 | 360 | 205 | 155 | 31 | 17 | 14 | 45 | 29 | 16 | 36 | 35 | 1 | 12 | 9 | 3 |
| 5 | 591 | 169 | 422 | 25 | 19 | 6 | 29 | 20 | 9 | 7 | 3 | 4 | 8 | 7 | 1 | 0 | 0 | 0 |
| 6 | 944 | 362 | 582 | 55 | 21 | 34 | 26 | 26 | 0 | 32 | 28 | 4 | 27 | 26 | 1 | 0 | 0 | 0 |
| 7 | 764 | 73 | 691 | 73 | 27 | 46 | 33 | 21 | 12 | 27 | 12 | 15 | 18 | 16 | 2 | 11 | 7 | 4 |
| 8 | 1707 | 484 | 1223 | 171 | 49 | 122 | 38 | 17 | 21 | 45 | 37 | 8 | 11 | 8 | 3 | 5 | 4 | 1 |
| 9 | 286 | 124 | 162 | 7 | 6 | 1 | 40 | 35 | 5 | 29 | 26 | 3 | 13 | 0 | 13 | 22 | 0 | 22 |
| Total | 12,839 | 4502 | 8327 | 1116 | 539 | 578 | 629 | 359 | 270 | 460 | 341 | 119 | 347 | 252 | 95 | 105 | 58 | 47 |
| %NC | 64.9 | 51.8 | 42.9 | 25.9 | 27.4 | 44.8 | ||||||||||||
Number of ECRs in coding (exonic) regions.
Number of ECRs in noncoding (nonexonic) regions.
The proportion of noncoding ECRs also decreased with increasing evolutionary distance, reflecting higher conservation of exons than noncoding regions. In the mouse/rat comparison, 65% of ECRs corresponded to noncoding regions. This dropped to 52% in mouse/human, 42%-45% in mouse/zebrafish and mouse/Ciona, and 27% in the mouse/pufferfish comparisons.
To further characterize the evolutionary dynamics of ECRs, the ECRs identified in the mouse/human comparison were tested to determine how many of them appeared in other pairwise comparisons. Results of this analysis are presented in Table 6. A total of 91.6% of the mouse/human ECRs were also seen in rat, but these made up only 8% of mouse/rat ECRs, suggesting a high false-positive rate (with respect to their ability to detect selectively conserved sequences) in the latter comparison. Around 10%-15% of mouse/human ECRs were also observable in fish—the lower proportion for Tetraodon may reflect incompleteness of data. However, these only made up about 25%-35% of the ECRs observed in these species, again suggesting a high false-positive rate.
Table 6.
Numbers of Coincident ECRs Across Species
| Region | Humana | Rat | Zebrafish | Fugu | Tetraodon | Ciona |
|---|---|---|---|---|---|---|
| 0 | 26 | 20 | 7 | 1 | 5 | 1 |
| 1 | 143 | 132 | 69 | 64 | 34 | 10 |
| 2 | 255 | 233 | 20 | 35 | 28 | 0 |
| 3 | 1 | 1 | 0 | 0 | 0 | 0 |
| 4 | 360 | 351 | 20 | 29 | 22 | 11 |
| 5 | 25 | 10 | 6 | 1 | 3 | 0 |
| 6 | 55 | 56 | 9 | 9 | 5 | 0 |
| 7 | 73 | 72 | 12 | 15 | 11 | 3 |
| 8 | 171 | 144 | 8 | 8 | 1 | 0 |
| 9 | 7 | 5 | 0 | 2 | 0 | 0 |
| Total | 1116 | 1024 | 151 | 164 | 109 | 25 |
| % Conservedb | 91.7 | 13.5 | 14.7 | 9.8 | 2.2 | |
| % of Totalc | 8.0 | 24.0 | 35.7 | 31.4 | 23.8 |
ECRs found in the mouse-human comparison, against which other species pairs were compared to identify coincident ECRs.
Percentage of human/mouse ECRs also seen in the target species.
Percentage of all ECRs seen in the target species that are also seen in the human/mouse comparison.
Distributions of both coding and noncoding ECRs across the sequence were nonrandom with respect to expected numbers on the basis of the lengths of the 10 segments in mouse (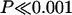 , χ2, 9 df). Highly significant nonrandomness was also seen for the other species comparisons. Segments 3 and 9 showed ECR contents of <10% of expected values (in the case of segment 3, the observed value was close to 1% of expectation). Segment 7 showed a threefold elevation of ECR density, whereas segments 4 and 8 showed approximately twofold elevations. Observed/expected ratios for C- and NC-ECRs correlated significantly (r = 0.73; P < 0.02).
, χ2, 9 df). Highly significant nonrandomness was also seen for the other species comparisons. Segments 3 and 9 showed ECR contents of <10% of expected values (in the case of segment 3, the observed value was close to 1% of expectation). Segment 7 showed a threefold elevation of ECR density, whereas segments 4 and 8 showed approximately twofold elevations. Observed/expected ratios for C- and NC-ECRs correlated significantly (r = 0.73; P < 0.02).
Similar results were obtained if expected ECR frequencies were calculated on the basis of ORF gene content, except that in this case, segments 0 and 1 were also deficient in ECRs, and segments 4 and 8 showed nine- to 10-fold excesses.
ECRs and Gene Identification
A subsequent analysis of mouse-human ECRs allowed us to identify additional and variant exons. We also identified a novel SCAN domain containing C2H2 type zinc-finger protein (933017L02Rik) that was missed in the original manual annotation. ECRs, therefore, proved valuable for detecting some exons that may have been missed during clone-based manual annotation, particularly when genes crossed clone boundaries.
DISCUSSION
Manual annotation of the finished Del36H genomic sequence allowed us to carry out a comprehensive analysis of the region, as well as producing an invaluable resource for mutation detection. Comparisons of the manual annotation with Ensembl annotation (Table 1) confirmed that the manual annotation will be important for mutation detection, as automated annotation predicts the precise coordinates of genes, and particularly exons, less well.
Evolutionary Rearrangement
Parts of Del36H are orthologous to two large and one smaller block of the human genome, 6p22.1-6p22.3, 3q24, and 6p25.3. The two regions of Del36H orthologous to human 6p lie in opposite orientations in mouse. On the basis of comparisons of syntenic associations that are widespread in mammalian orders, Murphy et al. (2001) proposed a hypothetical ancestral placental mammalian karyotype. In this, the segment corresponding to human chromosome 6 is present as a single block, as is that corresponding to chromosome 3. Thus, the organization of Del36H appears to be the result of at least two recombination events between two ancestral blocks. This is consistent with the conclusion that there has been more chromosome breakage in the rodent lineage than in the primates (Bourque et al. 2004). However, it is not clear from this whether the Del36H arrangement represents a deletion in mouse or whether human chromosome 6 contains an insertion between 6p22.3 and 6p25.3. Repetitive elements have been implicated in chromosome breakage in human disease and during evolution (Murphy et al. 2001), with some evidence that the two types of breakage site may coincide and be associated with pseudogene insertions (Kost-Alimova et al. 2003). However, we found no strong association between evolutionary breakage in this region and repetitive elements. Segment 5 of Del36H, which contains the two evolutionary breakpoints, has a very low gene density, probably containing no more than three genes in 496 kb. A region such as this may be a prime candidate for genome rearrangement, because breaks are unlikely to impact coding or regulatory regions of genes.
Gene Family Evolution
Data on the gene content of the Del36H sequence provide us with insight into the origins of its high gene density. The region contains representatives of six gene families, the vomeronasal receptors, butyrophilins, histones, prolactins, Fox transcription factors, and serpins. The segments within which these gene families lie have high gene densities; the remainder of Del36H has a gene density half the average value estimated by Waterston et al. (2002). The three gene families expanded in Del36H relative to human contain 67 more genes than are present in the human regions. Thus, there is an association between accumulation of these genes and the high gene density of the region as a whole.
Examination of the patterns of organization of these gene families shows variation, but also some underlying similarities. Segment 1 is of particular interest in this respect, because it contains three interspersed gene families. This region is dominated by two histone gene arrays interrupted by the VnR and butyrophilin genes. On a gross scale, the histone arrays comprise all five classical histone types (Fig. 3D). The arrays show evidence of expansion, most likely by some recombinational process such as unequal crossing-over. In the distal mouse cluster, the arrangement {H2b-reverse + H2a-forward + H3-forward + H1-reverse + H4-forward} is repeated several times, although because of the mouse-human divergence, some members of the block have degenerated into pseudogenes, genes have been lost, or other blocks have inserted in one or the other organism. Subsets of the basic block missing cumulatively H1, H3, and H4 also appear to have been duplicated. This appears to indicate expansion of groups of ancestral histone genes before the divergence of primates and rodents. The expanded gene families in Del36H also show patterns of organization consistent with amplification by recombination, as the VnR and prolactin genes show clustering by DNA strand. This was not detectable for the serpin genes, although in this case, the order of the three families is broadly maintained between mouse and man, suggesting a series of individual gene duplication events and not block duplication of larger units.
Gene Factories
A tempting conceptual model of the forces underlying the expansion/contraction of gene families in localized regions of genomes is that gene family expansion during evolution is driven by natural selection, favoring genes that make a specific contribution to the lifestyle of the species concerned (Dehal et al. 2001; Waterston et al. 2002; Emes et al. 2003). In Del36H, three such gene families are represented as follows: vomeronasal receptors, relating to mate choice using pheromones; prolactins, relating to the requirement in mice to suckle numerous offspring (in comparison to humans); and serpins, belonging to a class expressed in reproductive tissue (Kaiserman et al. 2002), which is again highly active and rapidly developing in mice compared with humans. Expansion of these gene families could have two advantageous consequences. Firstly, it could allow the rapid production of large amounts of protein by mobilizing multiple genes simultaneously. Secondly, it could allow evolutionary diversification of the gene family, allowing an individual to express or potentially express multiple variants of a gene type with subtly different functional characteristics. Expansion of these gene families could form part of a wider process of adaptation by gene duplication (Dehal et al. 2001; Emes et al. 2003).
An implicit assumption of such a model is that the duplication of genes is more or less equally likely for any gene (potentially a founder of a gene family). This might be because the genomic processes giving rise to gene families act more or less uniformly across the genome, or it might reflect the potential for any gene to find itself in a region predisposed to gene duplication, giving rise to evolutionarily rapid expansion. We call this latter model the “gene factory” hypothesis. Segment 1 of the Del36H region appears to be a candidate gene factory, because it contains a number of gene families. Locally accelerated gene duplication could be driven by high concentrations of TEs, which are recombinogenic (e.g., Burwinkel and Kilimann 1998; Deininger and Batzer 1999; Hill et al. 2000), and expanded gene families in Del36H are associated with high TE concentrations. Concentrations of particular classes of element would be expected to have more effect than a general increase in density of all elements, as this would increase the chance of out-of-phase recombination during meiosis. However, it is not clear whether some elements would have greater effects than others—a possible class of candidate elements are IAP elements, which are relatively common in segment 1 of Del36H and are associated with c. 10% of spontaneous mouse mutants (Ostertag and Kazazian Jr. 2001), whereas L1 elements have recently been implicated in the diversification of the rodent V1R gene family (Lane et al. 2004), although we did not find a significant association of LINEs with segment 1 compared with other parts of Del36H.
An alternative explanation of the observed association between gene clusters and TE density is that TE insertion is favored by processes such as recombination, which also produce gene clusters. Frequent strand breakage in a locality might favor invasion by TEs. If this hypothesis were true, we would expect to find the same proportions of TE classes associated with each gene cluster, but we find the different gene cluster segments to be enriched in different classes of TE. However, differences in the sequence characteristics of the different segments might be invoked to explain this. Further sequence and experimental analysis will be required fully to resolve these possibilities.
The three expanded (relative to human) gene families in Del36H contain considerably different proportions of pseudogenes. If gene family expansion in a lineage is adaptive (Dehal et al. 2001; Waterston et al. 2002; Emes et al, 2003), we might expect selection against pseudogenization within families, but the vomeronasal receptor (V1R) gene family in particular contains numerous pseudogenes. Our preliminary analysis suggests no relationship between the amount of neutral change, as measured by synonymous mutations, that has accumulated in these gene families and the proportion of pseudogenes. Ka/Ks ratios for all three gene families were also not low, suggesting that negative selection on them is not strong, although there is a possibility that positive selection has played a role in the evolution of the prolactin and serpin families. Thus, gene family expansion, at least in Del36H, appears to affect genes that are not subject to very strong negative selection. Gene duplication may then be followed by pseudogenization or evolution of new functions, as predicted by gene duplication theory (Ohno 1970; Lynch and Conery 2000). This process is akin to the birth and death evolution model of Nei and coworkers (Nei and Hughes 1992; Nei et al. 1997). The vomeronasal receptor family, which has a high proportion of pseudogenes, may represent a special case, as individual genes appear to be expressed in individual neurons of the vomeronasal organ (Keverne 1999). Gene duplication, combined with rapid evolution, could provide an increased repertoire of receptors, which could provide a selective advantage, but copies that are not useful to the organism would not be subject to purifying selection.
Our definition of a pseudogene implies only that the gene region concerned is not capable of making a complete protein. Recent evidence has shown that in some cases, RNA transcribed from genes of this kind may have regulatory roles (Hirotsune et al. 2003). It may also encode truncated proteins. We are unable to distinguish any such cases from wholly nonfunctional gene copies in this analysis, particularly in the absence of published cDNA or EST sequences. This is the case in particular for the V1r genes, which were particularly problematic, as they are also single exon genes. The current evidence for these genes is based mostly on similarity to genomic translations derived from other members of the same cluster, running the risk of an escalating cascade of self-confirmation.
Although TEs show associations with gene clusters within Del36H, simple sequences show the opposite relationship, that is, they correlate negatively and significantly with gene density over the region. They also tend to lie in blocks around 2.5 Mb long interspersed with similar length regions containing relatively fewer simple sequences. These correspond to the low gene density regions lying between the gene-rich segments of Del36H. This pattern presumably reflects the effective exclusion of micro- and minisatellite-like sequences by sequences that are mostly not internally repetitive (coding regions and TEs). Whether the periodicity of gene clusters seen within Del36H also occurs in the rest of the mouse genome, remains to be investigated.
ECRs
Sequence conservation across species may be a valuable indicator of both coding and regulatory regions (Mallon et al. 2000). Here, we have evaluated the level of conservation of ECRs in comparisons between mouse and a number of other complete or partially complete chordate genomes. We have found what appears to be a massive overrepresentation of ECRs in the mouse/rat comparison, despite raising the stringency of our search to 85% match over 100 bp. Fine tuning of this criterion may reduce the number of these matches to a number more comparable to those found for other species, but it may be that rat is too closely related to mouse to be of use in such studies. Numbers of ECRs observed for the human/mouse comparison were similar to those observed for fish and Ciona, suggesting that there is a core of ECRs that shows strong conservation over long periods of evolutionary time. Many of these are noncoding, raising the possibility that they represent strongly conserved regulatory elements. Some of them may also be examples of a newly discovered class of ultraconserved elements (Bejerano et al. 2004).
The distribution of ECRs across Del36H is not homogeneous. This was observed whether expected values were calculated on sequence length or ORF gene content. When gene content is taken into consideration, the three segments containing expanded gene families (particularly segment 3, which contains the prolactin cluster) contain a very low ECR concentration. In contrast, other segments, notably 4 and 8, showed relatively high ECR contents. This could result from differences in ECR conservation in different parts of the genome, due to differences in selective pressure or other processes. However, it might also reflect the specialized mode of evolution of regions containing multiple copies of gene family members. Gene duplication may not have duplicated regulatory elements along with coding sequences. This would result in fewer ECRs per gene and could be one of the causes of pseudogenization, which itself would reduce the number of significant PIPMAKER hits. The inverse of this is that positively selected genes may also not be detected in PIP analysis if they have diverged considerably more than genes under purifying selection. Finally, the pattern could result from differences in ECR number per gene. However, observed/expected ratios of C- and NC-ECR content in the different regions correlated significantly with one another, indicating that genes tend to have similar numbers of NC- and C-ECRs and providing further evidence that NC-ECRs may be useful for identifying regulatory regions.
Conclusions
We have sequenced and annotated to a high standard a 12.66-Mb segment of the mouse genome corresponding to Del36H. We have identified a number of interesting sequence features in the region, including a number of gene families that are more extensive in mouse than in human, and association of the loci of these gene families with high concentrations of TEs. We suggest that the locations of these expanded gene families may represent gene factories that may be driven by high densities of recombinogenic TEs. Simple sequences within the region are arranged in ∼2.5-Mb blocks, probably reflecting the exclusion of this class of sequence from regions with high gene and TE densities. We also identified two points of breakage/rearrangement between the mouse and human genomes, although we have been unable to identify unambiguous sequence features that might have been involved in these rearrangements. Finally, we have investigated the potential of ECRs as tools to detect gene regulatory regions, and conclude that they appear to have potential for identifying conserved regulatory regions in genes, especially if present in a range of chordate species.
This analysis will form an invaluable starting point both for further biological analysis of genes located in this region of the mouse genome and for further analysis of the mouse genome sequence itself.
METHODS
Physical Mapping
The process of map construction evolved during the project, as large-scale public data resources became available. Initially, the RPCI23 BAC library (Osoegawa et al. 2000) was replicated, archived, and arrayed onto high-density filters (Dunham et al. 1999) at the MRC Rosalind Franklin Centre for Genomics Research (formerly the MRC Human Genome Mapping Project Resource Centre). Markers for genes, expressed-sequence tag (EST), sequence-tagged-site (STS), and microsatellites known to map in Del36H (Arkell et al. 2001) were selected for use in physical mapping. PCR amplicons or overgos were designed for the markers, purified, and radioactively labeled, and then pools of up to 12 probes used as hybridization probes (Ross et al. 1999) to screen the BAC library. High-density filters were obtained either from Childrens Hospital Oakland Research Institute (CHORI) or from the MRC Rosalind Franklin Centre for Genomics Research. Bacteria representing positive coordinates were grown and arrayed at low density on nylon filters (Dunham et al. 1999) using a Biomek 2000 (Beckman). These were then screened by hybridization with individual probes as a secondary screen and to identify potential clone overlaps. All BACs were restriction-digest fingerprinted (Marra et al. 1997) and contigs constructed at high stringency using a combination of fingerprint and marker content data in FPC (Soderlund et al. 2000).
As the project progressed, we exploited two further mapping approaches, using human-mouse conserved synteny and the availability of nearly complete fingerprint data for the RPCI23 and RPCI24 BAC libraries. Human chromosome 6p21.3-6ptel shows conserved synteny with proximal mouse chromosome 13 (Stephenson and Lueders 1999). Repetitive elements in selected tile-path clones from the human genome sequence were masked using RepeatMasker (A.F.A. Smit and P. Green, unpubl.) and tested for alignment with mouse BAC insert end, gene, and EST sequences, using the BLAST server at the MRC Rosalind Franklin Centre for Genomics Research (http://www.hgmp.mrc.ac.uk/Registered/Webapp/blast/). Mouse sequences with alignments with BLAST significance values of <10-4 were used to design overgos or PCR amplicons and then used in hybridization screening as described above. Selected markers were mapped (Arkell et al. 2001) using the T31 radiation hybrid panel (McCarthy et al. 1997) to ensure localization to the Del36H interval. Finally, FPC (Soderlund et al. 2000) was used to “walk” across gaps between clone contigs by high-stringency comparisons with the public fingerprint database (http://www.bcgsc.ca/lab/mapping/mouse). As an independent test of localization, selected BACs were labeled and used in fluorescent in situ hybridization (FISH) with metaphase chromosomes (Buckle and Rack 1993). Sequencing was performed as described (Lander et al. 2001; Waterston et al. 2002).
Annotation and Analysis
The finished genomic sequence in the form of BACs was analyzed using an automatic Ensembl pipeline with modifications to aid the manual curation process. The G+C content of each clone sequence was determined and putative CpG islands marked. Interspersed repeats were detected using RepeatMasker (A.F.A. Smit and P. Green, unpubl.) and simple repeats using Tandem Repeats Finder (Benson 1999). The combination of the two repeat types was used to mask the sequence. This masked sequence was searched against vertebrate cDNAs and expressed sequence tags (ESTs) using WUBLASTn (http://blast.wustl.edu) and matches were cleaned up and aligned using est2genome (http://www.rfcgr.mrc.ac.uk/Software/EMBOSS/Apps/est2genome.html). A protein database combining nonredundant data from SWISS-PROT and TrEMBL was searched using WUBLASTx (http://blast.wustl.edu). Ab initio gene structures were predicted using FGENESH (Salamov and Solovyev 2000) and GENSCAN (Burge and Karlin 1997). By use of modified Acedb software (Durbin and Thierry Mieg 1991), the predicted gene structures were manually annotated according to the human annotation workshop (HAWK) guidelines (Ashurst and Wilming, 2002) and gene categories used were as described therein as follows: Known genes are identical to known mouse cDNA or protein sequences and should have an entry in MGD, Locuslink, or GDB. Novel CDS genes have an ORF, are identical to spliced ESTs or have some similarity to other genes or proteins. Novel transcripts are similar to novel genes, but no ORF can be determined. Putative genes are identical to spliced mouse ESTs, but do not contain an ORF. Pseudogenes (processed or unprocessed) are nonfunctional copies of genes with in-frame stop codons and/or frameshifts disrupting the ORF. Multiple alignments and bootstrapped trees were generated by ClustalX (Thompson et al. 1997) using default parameters (1000 iterations for the trees).
The individual clone sequences were submitted to the GenBank/EMBL/DDBJ databases under the following accession numbers: BX001068, BX296547, AL589742, AL589651, AL589879, AL606968, AL590614, AL645683, AL645546, AL645667, AL645686, AL645923, AL606472, AL606513, AL627387, AL669819, AL714025, AL732361, AL592149, AL590388, AL606464, AL683873, AL590864, AL589744, AL591851, AL513014, AL589699, AL645533, AL589735, AL589766, AL670757, AL645748. AL606965, BX119969, AL611951, AL731562, AL590870, AL837512, AL589722, AL645663, AL662807, AL731657, AL844606, AL591843, AL589679, AL662813, AL592443, AL590522, AL732408, AL627326, AL606783, AL590616, AL607105, AL590503, AL590626, AL606488, AL606528, AL606511, AL646015, AL645746, AL589701, AL645587, AL512647, AL513025, AL450321, AL645749, AL607131, AL731649, AL611944, AL731659, AL645745, AL606764, AL606496, AL606782, AL606525, AL645662, AL589738, AL590625, AL645783, AL645697, AL645643, AL645763, AL645664, AL645825, AL589737, AL645799, AL645808, AL645693, AL606533, AL589871, AL645704, BX545914, AL450331, AL513022, and AL450406.
For ECR analysis, individual finished BAC sequences were masked using RepeatMasker (A.F.A. Smit and P. Green, unpubl.). The masked sequences were then BLASTed against the most current versions of the genome sequence databases for nine species as follows: human, chimpanzee, cattle, rat, Takifugu, Tetraodon, Chicken, Ciona, and Sea Urchin using BLASTN (Altschul et al. 1990). High scoring pairs (HSPs) with an E-value of <1 × 10-25 (1 × 10-100 for rat) were then extracted and compared with the set of genome sequences using PIPMAKER or MULTIPIPMAKER (Schwartz et al. 2000). Concise output files were then processed using a perl script to extract ECRs with predefined minimum lengths and percentage matches to the target genome as follows: Mouse-Human (50 bp in length and 85% identity), Mouse-Rat (100 bp, 85%), Mouse-Takifugu rubripes (50 bp, 50%), Mouse-Tetraodon nigroviridis (50 bp, 50%), Mouse-Zebrafish (Danio rerio) (50 bp, 50%), and Mouse-Ciona Intestinalis (50 bp, 50%). Coding (C-) ECRs are defined as ECRs that significantly overlap an mRNA or, in the absence of mRNA sequence, a protein-coding region. All other ECRs are defined as noncoding (NC-) ECRs.
Sequence repetition across the region was analyzed using the program SIMPLE (v 3.0; Alba et al. 2002). Briefly, the algorithm passes a 65-bp window along a sequence, awarding a score to each window, depending on the frequency at which the motif at the center of the window occurs within the window. The mean score for the sequence is compared with a mean score generated for 10 randomized sequences of the same base and dinucleotide composition, rendering a measure of sequence repetition (Relative Simplicity Factor, RSF) for the region. The method compensates for base composition and sequence length by simulating sequences of the same length and base dinucleotide composition as the original (Hancock and Armstrong 1994).
Synonymous and nonsynonymous sequence differences were derived from ClustalX multiple alignments using the program DnaSP (Rozas et al. 2003).
Acknowledgments
We thank Tracy Marsland and Anne Southwell for help with physical and RH mapping of the Del36H region, Jacky King for technical assistance, Colin Beechey and Ruby Banerjee for FISH mapping of BACs, Stephen Keenan and Eduardo Eyras for their help with the analysis, and the Sanger Institute mouse mapping and sequencing groups. The RPCI23 BAC library was kindly provided by P. de Jong (CHORI). This study describes research funded as part of the MRC UK Mouse Sequencing Consortium. The consortium comprises: MRC Mammalian Genetics Unit, Harwell, Oxfordshire (A.-M. Mallon, J. Weekes, M. Cadman, R. McKeone, C. Sellick, M.A. Strivens, J.M. Hancock, P. Denny, and S.D.M. Brown); The Sanger Institute, Hinxton Genome Campus (L. Wilming, J. Ashurst, S. Peyrefitte, L. Matthews, S. Gregory, and J. Rogers); MRC Rosalind Franklin Centre for Genomics Research, Hinxton Genome Campus (P. North, N. Leaves, J. Greystrong, M. Coppola, S. Manjunath, M. Campbell, M. Smith, G. Strachan, C. Tofts, E. Boal, V. Cobley, G. Hunter, C. Kimberley, D. Thomas, L. Cave-Berry, P. Weston, M.R.M. Botcherby, and R.D. Campbell); MRC Human Genetics Unit, Western General Hospital, Edinburgh (I. Jackson, S. Cross, S. White, R. Edgar, M. Taylor, and P. Gautier); MRC Prion Unit, St. Mary's Hospital Medical School, London (H. Hummerich and M. Iravani); School of Biochemistry & Molecular Genetics, University of New South Wales, Sydney (P. Little).
Footnotes
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2478604. Article published online before print in September 2004.
References
- Alba, M.M. and Guigo, R. 2004. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 14: 549-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alba, M.M., Laskowski, R.A., and Hancock, J.M. 2002. Detecting cryptically simple protein sequences using the SIMPLE algorithm. Bioinformatics 18: 672-678. [DOI] [PubMed] [Google Scholar]
- Albig, W. and Doenecke, D. 1997. The human histone gene cluster at the D6S105 locus Hum. Genet. 101: 284-294. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Arkell, R.M., Cadman, M., Marsland, T., Southwell, A., Thaung, C., Davies, J.R., Clay, T., Beechey, C.V., Evans, E.P., Strivens, M.A., et al. 2001. Genetic, physical, and phenotypic characterization of the Del(13)Svea36H mouse. Mamm. Genome 12: 687-694. [DOI] [PubMed] [Google Scholar]
- Ashurst, J. and Wilming, L. 2002. Genomic sequence annotation guidelines. http://www.sanger.ac.uk/HGP/havana/docs/guidelines.pdf
- Avner, P., Bruls, T., Poras, I., Eley, L., Gas, S., Ruiz, P., Wiles, M.V., Sousa-Nunes, R., Kettleborough, R., Rana, A., et al. 2001. A radiation hybrid transcript map of the mouse genome. Nat. Genet. 29: 194-200. [DOI] [PubMed] [Google Scholar]
- Babcock, M., Pavlicek, A., Spiteri, E., Kashork, C.D., Ioshikhes, I., Shaffer, L.G., Jurka, J, and Morrow, B.E. 2003. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 13: 2519-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, J.A., Gu, Z., Clark, R.A., Reinert, K., Samonte, R.V., Schwartz, S., Adams, M.D., Myers, E.W., Li, P.W., and Eichler, E.E. 2002, Recent segmental duplications in the human genome. Science 297: 1003-1007. [DOI] [PubMed] [Google Scholar]
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W.J., Mattick, J.S., and Haussler, D. 2004. Ultraconserved Elements in the Human Genome. Science 304: 1321-1325. [DOI] [PubMed] [Google Scholar]
- Benson, G. 1999. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27: 573-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi, G. 1995. The human genome: Organization and evolutionary history. Annu. Rev. Genet. 29: 445-476. [DOI] [PubMed] [Google Scholar]
- Bernardi, G., Olofsson, B., Filipski, J., Zerial, M., Salinas, J., Cuny, G., Meunier-Rotival, M., and Rodier, F. 1985. The mosaic genome of warm-blooded vertebrates. Science 228: 953-958. [DOI] [PubMed] [Google Scholar]
- Bourque, G., Pevzner, P., and Tesler, G. 2004. Reconstructing the genomic architecture of ancestral mammals: Lessons from human, mouse and rat genomes. Genome Res. 14: 507-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S.D.M. and Hardisty, R.E. 2003. Mutagenesis strategies for identifying novel loci associated with disease phenotypes. Semin. Cell. Dev. Biol. 14: 19-24. [DOI] [PubMed] [Google Scholar]
- Buckle, V.J. and Rack, K.A. 1993. Fluorescent in situ hybridization. In Human Genetic Disease analysis, a practical approach, 2nd edition (ed. K.E. Davies), pp. 59-80. IRL Press, Oxford, UK.
- Burge, C. and Karlin, S. 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78-94. [DOI] [PubMed] [Google Scholar]
- Burwinkel, B. and Kilimann, M.W. 1998. Unequal homologous recombination between LINE-1 elements as a mutational mechanism in human genetic disease. J. Mol. Biol. 277: 513-517. [DOI] [PubMed] [Google Scholar]
- Davies, A.F., Mirza, G., Sekhon, G., Turnpenny, P., Leroy, F., Speleman, F., Law, C., van Regemorter, N., Vamos, E., Flinter, F., et al. 1999. Delineation of two distinct 6p deletion syndromes. Hum. Genet. 104: 64-72. [DOI] [PubMed] [Google Scholar]
- Dehal, P., Predki, P., Olsen, A.S., Kobayashi, A., Folta, P., Lucas, S., Land, M., Terry, A., Ecale Zhou, C.L., Rash, S., et al. 2001. Human chromosome 19 and related regions in mouse: Conservative and lineage-specific evolution. Science 293: 104-111. [DOI] [PubMed] [Google Scholar]
- Deininger, P.L. and Batzer, M.A. 1999. Alu repeats and human disease. Mol. Genet. Metab. 67: 183-193. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Reymond, A., Lyle, R., Scamuffa, N., Ucla, C., Deutsch, S., Stevenson, B.J., Flegel, V., Bucher, P., Jongeneel, C.V., et al. 2002. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420: 578-582. [DOI] [PubMed] [Google Scholar]
- Dermitzakis, E.T., Reymond, A., Scamuffa, N., Ucla, C., Kirkness, E., Rossier, C., and Antonarakis, S.E. 2003. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs). Science 302: 1033-1035. [DOI] [PubMed] [Google Scholar]
- Dunham, I., Dewar, K., Kim, U.J., and Ross, M.T. 1999. Bacterial cloning systems. In Genome analysis: A laboratory manual series, cloning systems, Vol. 3 (ed. J. Roskams) pp. 1-86. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Durbin, R. and Thierry Mieg, J. 1991. A C. elegans Database. Documentation, code and data available from anonymous FTP servers at lirmm.lirmm.fr, cele.mrc-lmb.cam.ac.uk and ncbi.nlm.nih.gov.
- Eichler, E.E. and Sankoff, D. 2003. Structural dynamics of eukaryotic chromosome evolution. Science 301: 793-797. [DOI] [PubMed] [Google Scholar]
- Emes, R.D., Goodstadt, L., Winter, E.E., and Ponting, C.P. 2003. Comparison of genomes of human and mouse lays the foundation of genome zoology. Hum. Mol. Genet. 12: 701-709. [DOI] [PubMed] [Google Scholar]
- Feder, J.N., Gnirke, A., Thomas, W., Tsuchihashi, Z., Ruddy, D.A., Basava, A., Dormishian, F., Domingo Jr., R., Ellis, M.C., Fullan, A., et al. 1996. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13: 399-408. [DOI] [PubMed] [Google Scholar]
- Forsyth, I.A. and Wallis, M. 2002. Growth hormone and prolactin-molecular and functional evolution. J. Mamm. Gland Biol. Neoplasia 7: 291-312. [DOI] [PubMed] [Google Scholar]
- Frazer, K.A., Tao, H., Osoegawa, K., de Jong, P.J., Chen, X., Doherty, M.F., and Cox, D.R. 2004. Noncoding sequences conserved in a limited number of mammals in the SIM2 interval are frequently functional. Genome Res. 14: 367-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, R. and Hughes, A.L. 2004. Two patterns of genome organization in mammals: The chromosomal distribution of duplicate genes in human and mouse. Mol. Biol. Evol. 21: 1008-1013. [DOI] [PubMed] [Google Scholar]
- George, D.L., Phillips III, J.A., Francke, U., and Seeburg, P.H. 1981. The genes for growth hormone and chorionic somatomammotropin are on the long arm of human chromosome 17 in region q21 to qter. Hum. Genet. 57: 138-141. [DOI] [PubMed] [Google Scholar]
- Grigorenko, E.L., Wood, F.B., Golovyan, L., Meyer, M., Romano, C., and Pauls, D. 2003. Continuing the search for dyslexia genes on 6p. Am. J. Med. Genet. 118B: 89-98. [DOI] [PubMed] [Google Scholar]
- Han, J.S., Szak, S.T., and Boeke, J.D. 2004. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429: 268-274. [DOI] [PubMed] [Google Scholar]
- Hancock, J.M. 1995. The contribution of slippage-like processes to genome evolution. J. Mol. Evol. 41: 1038-1047. [DOI] [PubMed] [Google Scholar]
- ____. 2002. Genome size and the accumulation of simple sequence repeats: Implications of new data from genome sequencing projects. Genetica 115: 93-103. [DOI] [PubMed] [Google Scholar]
- Hancock, J.M. and Armstrong, J.S. 1994. SIMPLE34: An improved and enhanced implementation for VAX and Sun computers of the SIMPLE algorithm for analysis of clustered repetitive motifs in nucleotide sequences. Comput. Appl. Biosci. 10: 67-70. [DOI] [PubMed] [Google Scholar]
- Harmar, A.J. 2001. Family-B G-protein-coupled receptors. Genome Biol. 2: REVIEWS3013.1-3013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, A.S., Foot, N.J., Chaplin, T.L., and Young, B.D. 2000. The most frequent constitutional translocation in humans, the t(11;22)(q23;q11) is due to a highly specific Alu-mediated recombination. Hum. Mol. Genet. 9: 1525-1532. [DOI] [PubMed] [Google Scholar]
- Hirotsune, S., Yoshida, N., Chen, A., Garrett, L., Sugiyama, F., Takahashi, S., Yagami, K., Wynshaw-Boris, A., and Yoshiki, A. 2003. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 423: 91-96. [DOI] [PubMed] [Google Scholar]
- Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., Clark, L., Cox, T., Cuff, J., Curwen, V., Down, T., et al. 2002. The Ensembl genome database project. Nucleic Acids Res. 30: 38-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, T.J., Church, D.M., Greenaway, S., Nguyen, H., Cook, A., Steen, R.G., Van Etten, W.J., Castle, A.B., Strivens, M.A., Trickett, P., et al. 2001. A radiation hybrid map of mouse genes. Nat. Genet. 29: 201-205. [DOI] [PubMed] [Google Scholar]
- Ishihara, H., Tanaka, I., Wan, H., Nojima, K., and Yoshida, K. 2004. Retrotransposition of limited deletion type of intracisternal A-particle elements in the myeloid leukemia cells of C3H/He mice. J. Radiat. Res. (Tokyo) 45: 25-32. [DOI] [PubMed] [Google Scholar]
- Jackson-Grusby, L.L., Pravtcheva, D., Ruddle, F.H., and Linzer, D.I. 1988. Chromosomal mapping of the prolactin/growth hormone gene family in the mouse. Endocrinology 122: 2462-2466. [DOI] [PubMed] [Google Scholar]
- Kaiserman, D., Knaggs, S., Scarff, K.L., Gillard, A., Mirza, G., Cadman, M., McKeone, R., Denny, P., Cooley, J., Benarafa, C., et al. 2002. Comparison of human chromosome 6p25 with mouse chromosome 13 reveals a greatly expanded ov-serpin gene repertoire in the mouse. Genomics 79: 349-362. [DOI] [PubMed] [Google Scholar]
- Katju, V. and Lynch, M. 2003. The structure and early evolution of recently arisen gene duplicates in the Caenorhabditis elegans genome. Genetics 165: 1793-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian Jr., H.H. 2004. Mobile elements: Drivers of genome evolution. Science 303: 1626-1632. [DOI] [PubMed] [Google Scholar]
- Keverne, E.B. 1999. The vomeronasal organ. Science 286: 716-720. [DOI] [PubMed] [Google Scholar]
- Kondrashov, A.S. and Shabalina, S.A. 2002. Classification of common conserved sequences in mammalian intergenic regions. Hum. Mol. Genet. 11: 669-674. [DOI] [PubMed] [Google Scholar]
- Kost-Alimova, M., Kiss, H., Fedorova, L., Yang, Y., Dumanski, J.P., Klein, G., and Imreh, S. 2003. Coincidence of synteny breakpoints with malignancy-related deletions on human chromosome 3. Proc. Natl. Acad. Sci. 100: 6622-6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E.S., Linton, L.M., Birren, B., Nusbaum, C., Zody, M.C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- Lane, R.P., Young, J., Newman, T., and Trask, B.J. 2004. Species specificity in rodent pheromone receptor repertoires. Genome Res. 14: 603-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. 2002. Are isochore sequences homogeneous? Gene 300: 129-139. [DOI] [PubMed] [Google Scholar]
- Lynch, M.M. and Conery, J.S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151-1155. [DOI] [PubMed] [Google Scholar]
- Makalowski, W., Zhang, J., and Boguski, M.S. 1996. Comparative analysis of 1196 orthologous mouse and human full-length mRNA and protein sequences. Genome Res. 6: 846-857. [DOI] [PubMed] [Google Scholar]
- Mallon, A.M., Platzer, M., Bate, R., Gloeckner, G., Botcherby, M.R., Nordsiek, G., Strivens, M.A., Kioschis, P., Dangel, A., Cunningham, D., et al. 2000. Comparative genome sequence analysis of the Bpa/Str region in mouse and Man. Genome Res. 10: 758-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies, E.H., Blanchette, M., NISC Comparative Sequencing Program, Haussler, D., and Green, E.D. 2003. Identification and characterization of multi-species conserved sequences. Genome Res. 13: 2507-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra, M.A., Kucaba, T.A., Dietrich, N.L., Green, E.D., Brownstein, B., Wilson, R.K., McDonald, K.M., Hillier, L.W., McPherson, J.D., and Waterston, R.H. 1997. High throughput fingerprint analysis of large-insert clones. Genome Res. 7: 1072-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff, W.F., Gongidi, P., Woods, K.R., Jin, J., and Maltais, L.J. 2002. The human and mouse replication-dependent histone genes. Genomics 80: 487-498. [PubMed] [Google Scholar]
- McCarthy, L.C., Terrett, J., Davis, M.E., Knights, C.J., Smith, A.L., Critcher, R., Schmitt, K., Hudson, J., Spurr, N.K., and Goodfellow, P.N. 1997. A first-generation whole genome-radiation hybrid map spanning the mouse genome. Genome Res. 7: 1153-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears, A.J., Jordan, T., Mirzayans, F., Dubois, S., Kume, T., Parlee, M., Ritch, R., Koop, B., Kuo, W.L., Collins, C., et al. 1998. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am. J. Hum. Genet. 63: 1316-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mural, R.J., Adams, M.D., Myers, E.W., Smith, H.O., Miklos, G.L., Wides, R., Halpern, A., Li, P.W., Sutton, G.G., Nadeau. J., et al. 2002. A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science 296: 1661-1671. [DOI] [PubMed] [Google Scholar]
- Murphy, W.J., Stanyon, R., and O'Brien, S.J. 2001. Evolution of mammalian genome organization inferred from comparative gene mapping. Genome Biol. 2: REVIEWS0005.1-0005.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M. and Hughes, A.L. 1992. Balanced polymorphism and evolution by the birth-and-death process in the MHC loci. In 11th Histocompatibility workshop and conference (eds. K. Tsuji, M. Aizawa, and T. Sasazuki) pp. 27-38. Oxford University Press, Oxford, UK.
- Nei, M., Gu, X., and Sitnikova, T. 1997. Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. 94: 7799-7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko, A. and Li, W.H. 2000. Assessment of compositional heterogeneity within and between eukaryotic genomes. Genome Res. 10: 1986-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura, D.Y., Swiderski, R.E., Alward, W.L., Searby, C.C., Patil, S.R., Bennet, S.R., Kanis, A.B., Gastier, J.M., Stone, E.M., and Sheffield, V.C. 1998. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 19: 140-147. [DOI] [PubMed] [Google Scholar]
- Ohno, S. 1970. Evolution by gene duplication. Springer-Verlag, Berlin, NY.
- Oliver, J.L., Bernaola-Galvan, P., Carpena, P., and Roman-Roldan, R. 2001. Isochore chromosome maps of eukaryotic chromosomes. Gene 276: 47-56. [DOI] [PubMed] [Google Scholar]
- Osoegawa, K., Tateno, M., Woon, P.Y., Frengen, E., Mammoser, A.G., Catanese, J.J., Hayashizaki, Y., and de Jong, P.J. 2000. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 10: 16-28. [PMC free article] [PubMed] [Google Scholar]
- Ostertag, E.M. and Kazazian Jr., H.H. 2001. Biology of mammalian L1 retrotransposons. Annu. Rev. Genet. 35: 501-538. [DOI] [PubMed] [Google Scholar]
- Owerbach, D., Rutter, W.J., Cooke, N.E., Martial, J.A., and Shows, T.B. 1981. The prolactin gene is located on chromosome 6 in humans. Science 212: 815-816. [DOI] [PubMed] [Google Scholar]
- Peterson, J.A., Scallan, C.D., Ceriani, R.L., and Hamosh, M. 2001. Structural and functional aspects of three major glycoproteins of the human milk fat globule membrane. Adv. Exp. Med. Biol. 501: 179-187. [DOI] [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc. Natl. Acad. Sci. 100: 7672-7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher, M.T., Roe, B.A., Chen, F., Do, T., Do, A., Malaj, E., and Reeves, R.H. 2000. Chromosome evolution: The junction of mammalian chromosomes in the formation of mouse chromosome 10. Genome Res. 10: 1463-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente, X.S. and López-Otín, C. 2004. A genomic analysis of rat proteases and protease inhibitors. Genome Res. 14: 609-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttagunta, R., Gordon, L.A., Meyer, G.E., Kapfhamer, D., Lamerdin, J.E., Kantheti, P., Portman, K.M., Chung, W.K., Jenne, D.E., Olsen, A.S., et al. 2000. Comparative maps of human 19p13.3 and mouse chromosome 10 allow identification of sequences at evolutionary breakpoints. Genome Res. 10: 1369-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes, D.A., Stammers, M., Malcherek, G., Beck, S., and Trowsdale, J. 2001. The cluster of BTN genes in the extended major histocompatibility complex. Genomics 71: 351-362. [DOI] [PubMed] [Google Scholar]
- Rodriguez, I., Del Punta, K., Rothman, A., Ishii, T., and Mombaerts, P. 2002. Multiple new and isolated families within the mouse superfamily of V1r vomeronasal receptors Nat. Neurosci. 5: 134-140. [DOI] [PubMed] [Google Scholar]
- Ross, M.T., LaBrie, S., McPherson, J., and Stanton Jr., V.P. 1999. Screening large-insert libraries by hybridization. In Current protocols in human genetics (ed. A. Boyl), pp. 5.6.1-5.6.32. Wiley, New York.
- Rozas, J., Sanchez-DelBarrio, J.C., Messeguer, X., and Rozas, R. 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496-2497. [DOI] [PubMed] [Google Scholar]
- Salamov, A. and Solovyev, V. 2000. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10: 516-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, S., Zhang, Z., Frazer, K.A., Smit, A., Riemer, C., Bouck, J., Gibbs, R., Hardison, R., and Miller, W. 2000. PipMaker—A web server for aligning two genomic DNA sequences. Genome Res. 10: 577-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund, C., Humphray, S., Dunham, A., and French, L. 2000. Contigs built with fingerprints, markers, and FPC V4.7. Genome Res. 10: 1772-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R.R. and Rohlf, F.J. 1995. Biometry W.H. Freeman & Co., New York.
- Stephenson, D.A. and Lueders, K.K. 1999. Mouse chromosome 13. Mamm. Genome 10: 954. [DOI] [PubMed] [Google Scholar]
- Straub, R.E., Jiang, Y., MacLean, C.J., Ma, Y., Webb, B.T., Myakishev, M.V., Harris-Kerr, C., Wormley, B., Sadek, H., Kadambi, B., et al. 2002. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am. J. Hum. Genet. 7: 337-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz, D., Trick, M., and Dover, G.A. 1986. Cryptic simplicity in DNA is a major source of genetic variation. Nature 322: 652-656. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., and Higgins, D.G. 1997. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25: 4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth, G., Gaspari, Z., and Jurka, J. 2000. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 10: 967-981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis, M. 1993. Remarkably high rate of molecular evolution of ruminant placental lactogens. J. Mol. Evol. 37: 86-88. [DOI] [PubMed] [Google Scholar]
- ____. 1996. The molecular evolution of vertebrate growth hormones: A pattern of near-stasis interrupted by sustained bursts of rapid change. J. Mol. Evol. 43: 93-100. [DOI] [PubMed] [Google Scholar]
- Wang, Z.F., Tisovec, R., Debry, R.W., Frey, M.R., Matera, A.G., and Marzluff, W.F. 1996. Characterization of the 55-kb mouse histone gene cluster on chromosome 3. Genome Res. 6: 702-714. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Wiemers, D.O., Shao, L.J., Ain, R., Dai, G., and Soares, M.J. 2003. The mouse prolactin gene family locus. Endocrinology 144: 313-325. [DOI] [PubMed] [Google Scholar]
- Zhang, J. and Webb, D.M. 2003. Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc. Natl. Acad. Sci. 100: 8337-8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://vega.sanger.ac.uk/Mus_musculus/; Vega Mouse Genome Browser.
- http://bioinfo2.ugr.es/isochores/; Online Resource on Isochore Mapping.
- http://www.hgmp.mrc.ac.uk/Registered/Webapp/blast/; MRC Rosalind Franklin Centre BLAST interface.
- http://www.bcgsc.ca/lab/mapping/mouse; Michael Smith Genome Sciences Centre Mouse Mapping.
- http://blast.wustl.edu; Washington University BLAST archives.
- http://www.rfcgr.mrc.ac.uk/Software/EMBOSS/Apps/est2genome.html; EMBOSS est2genome page.
- http://www.ensembl.org/Mus_musculus/whatsnew/v16_30_1.html; Ensembl Mouse What's New v16.30.1 (August 4, 2003).