

Abstract
The prediction of subcellular localization of proteins from their primary sequence is a challenging problem in bioinformatics. We have created a Bayesian network localization predictor called PSLT that is based on the combinatorial presence of InterPro motifs and specific membrane domains in human proteins. This probabilistic framework generates a likelihood of localization to all organelles and allows to predict multicompartmental proteins. When used to predict on nine compartments, PSLT achieves an accuracy of 78% as estimated by using a 10-fold cross-validation test and a coverage of 74%. When used to predict the localization of proteins from other closely related species, it achieves a prediction accuracy and a coverage >80%. We compared the localization predictions of PSLT to those determined through GFP-tagging and microscopy for a group of human proteins. We found two general classes of proteins that are mislocalized by the GFP-tagging strategy but are correctly localized by PSLT. This suggests that PSLT can be used in combination with experimental approaches for localization to identify proteins for which additional experimental validation is required. We used our predictor to annotate all 9793 human proteins from SWISS-PROT release 41.25, 16% of which are predicted by PSLT to be present in more than one compartment.
Eukaryotic proteins are organized into organelles and suborganelles that generate appropriate environments for their specialized functions. Thus, subcellular localization often offers important clues toward determining the function of an uncharacterized protein. The mechanisms of targeting of proteins to various subcellular localizations have been widely studied, and the predominant mechanisms uncovered so far involve specific amino acid sequence motifs. The consequences of mislocalization and mistargeting are manifested in a number of human genetic diseases, including cystic fibrosis (Skach 2000), Wilson's disease (Payne et al. 1998), and juvenile pulmonary emphysema (Parfrey et al. 2003).
There are numerous experimental approaches that attempt to determine both the subcellular localization of a protein and the amino acid motifs responsible for this targeting. Although these methods are capable of determining the linear amino acid motifs that are necessary for targeting, they are generally not able to help determine structural requirements and are generally not suited for use in a high-throughput fashion. The latter point is important because high-throughput proteomic efforts are now able to identify the most abundant proteins of an organelle (Bell et al. 2001; Michaud and Snyder 2002; Huh et al. 2003; Taylor et al. 2003). However, the localizations of proteins identified by these approaches are prone to error (i.e., they may have high rates of false-positive and/or false-negative entries). Furthermore, the sensitivity of these approaches is not sufficient to detect the full protein complement of organelles due to, for example, the low abundance of some proteins. Because the cDNA sequences of most human proteins are now available, bioinformatic predictors of subcellular localization offer a complementary and comprehensive approach that can help resolve such noisy proteomic data sets. This will provide a clearer, more complete picture of basic cellular organization and may also shed more light on the mechanisms of subcellular targeting.
The existing bioinformatics localization predictors in the literature can be broadly grouped into three categories.
Predictors based on amino acid composition. Several machine learning-based classification approaches have been used to predict subcellular localization based uniquely on amino acid composition, including neural networks (Reinhardt and Hubbard 1998) and support vector machines (Hua and Sun 2001). Several subsequent localization methods also incorporate additional information such as so-called quasi-sequence order effects (Chou 2001; Cai et al. 2002). These methods have the advantage of achieving a very high coverage but generally do not address the problem of multicompartmental proteins. This category also includes predictors such as SignalP (Nielsen et al. 1997), MitoProt (Claros and Vincens 1996), TargetP (Emanuelsson et al. 2000), and Predotar (www.inra.fr/predotar/), which aim at identifying specific signal sequences for the ER, mitochondria, and/or chloroplast and can in some cases predict proteins to be in several compartments simultaneously.
Predictors that determine protein localization by integrating various protein characteristics, including targeting motifs of different organelles. Such predictors include PSORT (Nakai and Kanehisa 1992) and a Bayesian framework (Drawid and Gerstein 2000). PSORT is a publicly available integrated expert system based on the sequential application of if/then rules relating to amino acid composition and the presence of targeting signals to various organelles. PSORT was further refined into a more probabilistic framework based on the k--nearest-neighbors method (Horton and Nakai 1996). The integrated Bayesian system created by Drawid and Gerstein for the prediction of yeast protein localization is based on prior knowledge of the proportion of proteins in the different compartments considered. This framework can address the problem of multicompartmental proteins, since a probability of localization can be assigned to all proteins in all compartments. However, because it requires the knowledge of several types of protein characteristics (thus requiring extensive experimental data such as expression data of cells under different conditions as well as knockout mutation viability information), it is not well suited for the annotation of unknown proteins generated by the numerous high-throughput genomics and proteomics projects.
Homology-based predictors. Such methods include phylogenetic profiling of proteins (Marcotte et al. 2000), which can be applied to the prediction of proteins in organelles of endo-symbiotic origins, and a protein domain projection method, which uses a measure of the co-occurrence of SMART motifs to predict localization (Mott et al. 2002). Support vector machines and a new hybridization approach have also been used to classify proteins into subcellular compartments (Chou and Cai 2002, 2003). The most recent addition to this category is the Proteome Analyst, which uses a naïve Bayesian network to predict localization based mostly on the SWISS-PROT keywords and annotations that can be extracted from the closest homologs of the query protein (Lu et al. 2004). The Proteome Analyst achieves high prediction accuracies by using the annotation of very closely related sequences. This is in contrast to most previous predictors, which are trained and tested on data sets consisting of proteins of limited pairwise sequence identity (when training and predicting on proteins from more than one organism).
Existing predictors have several shortcomings. Most localization prediction methods achieve high accuracy for the most populated compartments, such as the nucleus and cytosol, but are generally less accurate on the numerous compartments containing fewer individual proteins. Many existing predictors use only three or four different subcellular localizations. Moreover, the sets of proteins used to train these methods often do not contain transmembrane proteins because the localization of these proteins is believed to be already elucidated (Huang and Li 2004). However, although there are accurate predictors of transmembrane domains in proteins (Krogh et al. 2001), these do not predict the organellar location of transmembrane proteins. Last, very few predictors deal with the issue of multicompartmental proteins (proteins that may be localized to different organelles). Currently, there is no precise estimate of how many proteins are multicompartmental.
This article presents PSLT (Protein Subcellular Localization Tool, pronounced “silt”), a system that addresses the aforementioned issues and problems. PSLT uses the combinatorial presence of InterPro motifs, as well as signal peptides and the number of transmembrane domains in human proteins, to predict the subcellular localization of proteins within a Bayesian framework. InterPro is a database of protein domains, families, functional sites, and posttranslational modifications (Mulder et al. 2003). Collectively, we refer to these objects as InterPro motifs, all of which can be used alone or in combination to predict subcellular localization. Amino-terminal signal peptides are frequently responsible for targeting of nascent polypeptides to the ER, allowing for subsequent transport through the secretory pathway (von Heijne 1990; Rapoport 1992). When considered in combination with the presence of transmembrane domains, signal peptides can also facilitate the prediction of subcellular localization.
PSLT uses a Bayesian framework to integrate the presence or absence of combinations of motifs in a statistically coherent manner. The accuracy of PSLT is estimated to be 78% using a 10-fold cross-validation test and >85% using an independent data set test. When used to predict the localization of the independent set of human proteins from the LIFEdb project (Simpson et al. 2000), PSLT was capable of detecting several examples of possible GFP-localization bias, indicating the proteins for which additional experimental validation of localization is required. PSLT was used to annotate all 9793 human proteins contained in SWISS-PROT release 41.25. The results suggest that at least 16% of human proteins are multicompartmental. These annotations are available at www.mcb.mcgill.ca/~hera/PSLT.
RESULTS
Statistical Tests of Accuracy
The prediction accuracy of PSLT is assessed by three distinct approaches: a self-consistency test, a 10-fold cross-validation test, and independent data set tests. The different data sets used to train and test PSLT are shown in Table 1. With respect to the self-consistency test (also known as the resubstitution test), the accuracy of the predictor is evaluated by using the same data set used for training. As shown in Table 2, the overall prediction accuracy of PSLT using the self-consistency test on the Hera (Human Endoplasmic Reticulum Aperçu) data set (see Methods) is 90% and the coverage is 88%.
Table 1.
Number of Proteins per Compartment in the Data Sets
| Hera human data set | Yeast data set | LIFEdb GFP data set | Mouse data set | |
|---|---|---|---|---|
| ER | 333 | 184 | 49 | 77 |
| Golgi | 90 | 75 | 20 | 61 |
| Cytosol | 349 | 213 | 90 | 333 |
| Nucleus | 581 | 462 | 81 | 598 |
| Peroxisome | 37 | 46 | 1 | 18 |
| Plasma membrane | 205 | 141 | 14 | 36 |
| Lysosome | 91 | 20 | 1 | 46 |
| Mitochondria | 218 | 345 | 31 | 179 |
| Secreted | 294 | 14 | 0 | 454 |
| Multicompartmental | 18 | 112 | 105 | 293 |
| Total | 2216 | 1612 | 392 | 2095 |
Table 2.
Prediction Accuray of PSLT on Human Proteins
| Self-consistency test (second-best test)
|
10-fold cross validation test (second-best test)
|
|||||
|---|---|---|---|---|---|---|
| Sensitivitya | PPVb | Sensitivitya | PPVb | |||
| ER | 82 (95)% | 93 (98)% | 69 (82)% | 83 (85)% | ||
| Golgi | 84 (91)% | 87 (98)% | 60 (64)% | 74 (81)% | ||
| Cytosol | 90 (98)% | 85 (96)% | 65 (83)% | 68 (78)% | ||
| Nucleus | 96 (99)% | 93 (97)% | 93 (97)% | 84 (93)% | ||
| Peroxisome | 73 (94)% | 89 (94)% | 43 (64)% | 50 (67)% | ||
| Plasma membrane | 94 (98)% | 86 (96)% | 89 (91)% | 77 (89)% | ||
| Lysosome | 81 (96)% | 90 (99)% | 60 (71)% | 71 (76)% | ||
| Mitochondria | 88 (98)% | 85 (97)% | 67 (76)% | 61 (72)% | ||
| Extracellular | 95 (100)% | 91 (99)% | 89 (93)% | 87 (91)% | ||
| Overall accuracy | 90 (97)% | 78 (86)% | ||||
| Coverage | 88 (88)% | 74 (74)% | ||||
Sensitivity calculated as TP/(TP + FN).
PPV indicates positive predictive value (calculated as TP/[TP + FP]).
In the 10-fold cross-validation test, the data set is randomly partitioned into 10 distinct nonoverlapping sets of proteins. Nine of these sets are used to train the predictor. The prediction accuracy of the predictor is evaluated on the remaining excluded group. This procedure is repeated 10 times. The cross-validation prediction accuracy shown in Table 2 is the average of the 10 experiments. The overall prediction accuracy using the 10-fold cross-validation test on the Hera human data set is 78% and the coverage is 74%.
The third approach used to assess the prediction accuracy of PSLT is an independent data set test. In this test, PSLT is trained by using the entire Hera human data set and tested independently by using the LIFEdb GFP human data set. As shown in Table 3, the overall prediction accuracy of PSLT using this independent test is 55%. The coverage of the GFP data set is 50%. We note that many of the proteins in the LIFEdb GFP data set are hypothetical proteins derived from cDNA sequence data and have not been previously studied. Their subcellular localization was experimentally determined by tagging GFP to their N and C termini (in two separate experiments) and by visualizing the resulting protein localization by microscopy (Simpson et al. 2000). This experimental method will produce many localization errors because the terminal GFP tags can mask known localization signals in the proteins, as well as modify expression levels. For example, a lumenal endoplasmic reticulum protein possessing both an N-terminal signal peptide and a C-terminal KDEL retrieval sequence will probably be mislocalized in both the C- and N-terminal GFP tagging constructs.
Table 3.
Prediction Accuracy of PSLT on an Independent Human Data Set
| Independent test of PSLT on all proteins of LIFEdb data set (second-best test)
|
Independent test of PSLT on subset of LIFEdb proteins verified by literature (second-best test)
|
Concordance between LIFEdb GFP-based annotation and the literature
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Sensitivitya | PPVb | Sensitivitya | PPVb | Sensitivitya | PPVb | ||||
| ER | 32 (57)% | 47 (60)% | 100 (100)% | 100 (100)% | 100% | 38% | |||
| Golgi | 25 (42)% | 50 (71)% | 0 (100)% | 0 (0)% | 75% | 75% | |||
| Cytosol | 44 (77)% | 63 (77)% | 82 (82)% | 82 (82)% | 64% | 74% | |||
| Nucleus | 85 (88)% | 66 (89)% | 100 (100)% | 89 (94)% | 84% | 76% | |||
| Peroxisome | — | 0 (0)% | 100 (100)% | 67 (67)% | 67% | 100% | |||
| Plasma memb | 54 (57)% | 39 (53)% | 73 (82)% | 100 (100)% | 20% | 75% | |||
| Lysosome | — | 0 (0)% | 100 (100)% | 100 (100)% | 0% | 0% | |||
| Mitochondria | 25 (33)% | 43 (57)% | 100 (100)% | 100 (100)% | 50% | 40% | |||
| Extracellular | — | — | 100 (100)% | 33 (50)% | 0% | 0% | |||
| Overall accuracy | 55 (72)% | 87 (88)% | 59% | ||||||
| Coverage | 50 (50)% | 67 (67)% | — | ||||||
Sensitivity calculated as TP/(TP + FN).
PPV indicates positive predictive value (calculated as TP/[TP + FP]).
Because of the possibility of false-positive and false-negative localization annotations of the GFP high-throughput localization data set, we also measured the prediction accuracy of PSLT on the subset of proteins of the LIFEdb data set that have been independently studied by other research groups in a non-high-throughput manner and that have localization that is available in the literature. This subset consists of 82 proteins. As shown in Table 3 in the middle pair of columns, the prediction accuracy of PSLT on this subset of the LIFEdb data set is 87% with a coverage of 67%. This improved performance is much closer to the values obtained by the 10-fold cross-validation test on human proteins from the Hera data set and by the independent test on the mouse data set (see “Generalization to Other Organisms”). In contrast, the concordance of the high-throughput experimental GFP-tagging method with the literature was evaluated to be 59% (as shown in the rightmost pair of columns in Table 3). We decided to further investigate the discrepancy between the prediction accuracy results of PSLT on the full LIFEdb data set and on the subset verified by the literature. Of the 197 proteins from the full LIFEdb GFP data set for which PSLT could predict localization, the PSLT prediction disagrees with the GFP localization results for 89 proteins. Although many proteins in the LIFEdb are annotated “unknown” or “hypothetical,” we found reports of experimental evidence for the localization of 25 of the 89 proteins in the literature. Table 4 shows the comparisons between the LIFEdb localization annotation and the PSLT localization prediction, as well as the information currently available in the literature for these 25 proteins. The available scientific literature confirms the LIFEdb localization annotation of five of the 25 proteins and the PSLT prediction of 20 of the 25 proteins (including two proteins that have been confirmed to be in both the LIFEdb annotated compartment and the compartment predicted by PSLT). Although experimental evidence in the literature could be erroneous or incomplete, these results suggest that the prediction accuracy of PSLT may exceed the prediction accuracy of the LIFEdb GFP data set.
Table 4.
Available Experimental Confirmation Concerning 25 Proteins With Localization That Is Not Agreed Upon by the PSLT Prediction and the LIFEdb GFP Annotation
| Correct LIFEdb GFP annotationa | Correct PSLT predictionb | Bothc | Neitherd | |
|---|---|---|---|---|
| Protein count (%) | 3 (12%) | 18 (72%) | 2 (8%) | 2 (8%) |
Proteins with annotation in the LIFEdb data set that agrees with the information available in the literature.
Proteins with localization predicted by PSLT that agrees with the information in the literature.
Proteins with annotation in the LIFEdb data set and PSLT predicted localization that are confirmed in the literature.
Proteins with annotation in the LIFEdb data set and PSLT predicted localization that both disagree with information in the literature.
We further studied the cases of proteins with LIFEdb localization annotation that disagrees with the PSLT prediction. We note two general recurring cases: (1) proteins that have been shown in the literature to be plasma membrane or secreted proteins but that are annotated as being localized elsewhere in LIFEdb, and (2) proteins predicted by PSLT as peroxisomal but annotated in LIFEdb as localized to another compartment (usually the mitochondria).
With respect to case 1, many proteins predicted to be localized in the plasma membrane or secreted by PSLT are annotated by LIFEdb as being localized elsewhere in the cell, mostly in the ER but also in the cytosol and the nucleus. It is possible that these proteins spend a longer than expected amount of time in the ER (potentially due to an increase in the duration of folding caused by the added GFP tag) or never even succeed in entering the ER. This may explain why such proteins are visualized via microscopy to be localized to the ER or the cytosol when ultimately they are destined for the plasma membrane. This hypothesis may also explain the low proportion of plasma membrane proteins (6% to 7%) in the LIFEdb GFP data set compared with other public localization databases.
With respect to case 2, all proteins predicted by PSLT to be peroxisomal are annotated as being localized elsewhere in the cell by the LIFEdb GFP data set. However, we note that several of these proteins have been confirmed to be peroxisomal in the literature. Perhaps the GFP tag systematically targets the peroxisomal proteins to the mitochondria or elsewhere in the cell. It should be noted, however, that there exist several proteins that are actually annotated to be localized in both the peroxisome and the mitochondria in SWISS-PROT. It could be the case that some of the proteins predicted by PSLT to be peroxisomal and by the GFP localization to be mitochondrial are actually multicompartmental proteins.
In general, disagreements between the prediction of PSLT and one specific experimental approach might warrant further investigation using different experimental techniques to verify the localization (e.g., by immunofluorescence microscopy of normal cells using an antibody specific to the protein of interest). Conversely, when the prediction of PSLT agrees with experimental evidence, our belief that the protein is indeed localized to this compartment should be strengthened.
Comparison Between PSLT and the SMART Domain Projection Method
PSLT uses a Bayesian approach to predict protein localization based on the co-occurrence of protein motifs/domains, making it methodologically similar to the domain projection method described previously (Mott et al. 2002). This method uses the co-occurrence of 300 SMART domains that, when projected onto a two-dimensional space, cluster into three groups corresponding to secreted, cytoplasmic, and nuclear compartments. This domain projection method was evaluated by using a set of diverse eukaryotic proteins containing at least one of the 300 SMART motifs considered and previously annotated with localization information by Meta-A (Eisenhaber and Bork 1998, 1999), revealing a prediction accuracy of 92% and a coverage of 23%. Although methodologically similar to this domain projection method, PSLT can achieve a wider coverage because it uses a Bayesian approach that considers the co-occurrence InterPro and protein membrane domains, thus greatly increasing the feature space. To determine whether PSLT represents a genuine methodological advance over the domain projection method, we tested it using the same data set and the same scoring scheme used to assess the prediction accuracy of the domain projection method. Because this data set annotates proteins as being localized to one or several of only three compartments (cytoplasmic, nuclear, and secreted), PSLT predicted proteins to be in one of these three compartments (the nine compartments PSLT usually predicts on were collapsed into three). By using this test set, PSLT obtains a prediction accuracy of 98% and a coverage of 99% (the coverage is extremely high because this test set is composed only of proteins that contain SMART domains). These results provide a second independent data set test to evaluate the prediction accuracy of PSLT.
Generalization to Other Organisms and Multicompartmental Prediction
PSLT is a predictor of subcellular localization constructed on human sequences. To determine its predictive accuracy when applied to other organisms, we tested it on 2095 mouse proteins and 1612 proteins from yeast described in the Methods section. As shown in Table 5, the overall prediction accuracy of PSLT on this mouse data set is 84% and the coverage is 83%. These high accuracy and coverage values approach those of PSLT in the self-consistency test (Table 2). This is not surprising due to the generally high sequence identity between mouse and human. When PSLT trained on human sequences is used to predict yeast protein localizations, the overall prediction accuracy is 56% and the coverage is 53%. These results indicate the positive relationship between coverage/prediction accuracy and sequence similarity. This relationship is further corroborated by counting the total number of motifs in all proteins of each data set that are also present in proteins in the Hera human data set used to train the predictor. It can be shown that 87% of motifs found in the proteins of the mouse data set are also present in proteins in the Hera human data set as opposed to 70% for the motifs found in the yeast data set and 80% for the LIFEdb data set. Such a predictor achieves a higher prediction accuracy when considering proteins from species that are evolutionarily close to the sequences used to train the predictor. However, as more sequences become available for training, more motifs will be considered and the prediction accuracy and coverage will increase.
Table 5.
Prediction Accuracy of PSLT on Yeast and Mouse Proteins
| Independent yeast data set test (second-best test)
|
Independent mouse data set test (second-best test)
|
|||||
|---|---|---|---|---|---|---|
| Sensitivitya | PPVb | Sensitivitya | PPVb | |||
| ER | 53 (64)% | 65 (72)% | 67 (78)% | 53 (66)% | ||
| Golgi | 37 (43)% | 53 (79)% | 70 (85)% | 79 (85)% | ||
| Cytosol | 55 (77)% | 18 (35)% | 75 (88)% | 77 (89)% | ||
| Nucleus | 73 (83)% | 79 (88)% | 91 (96)% | 92 (97)% | ||
| Peroxisome | 32 (50)% | 30 (45)% | 57 (71)% | 80 (83)% | ||
| Plasma memb | 72 (74)% | 78 (84)% | 71 (72)% | 56 (79)% | ||
| Lysosome | 40 (52)% | 62 (88)% | 77 (93)% | 72 (87)% | ||
| Mitochondria | 55 (64)% | 70 (83)% | 79 (89)% | 86 (94)% | ||
| Extracellular | 50 (50)% | 25 (33)% | 93 (97)% | 93 (96)% | ||
| Overall accuracy | 56 (71)% | 84 (92)% | ||||
| Coverage | 53 (54)% | 83 (83)% | ||||
Sensitivity calculated as TP/(TP + FN).
PPV : positive predictive value (calculated as TP/(TP + FP)).
Because PSLT is based on a probabilistic framework, it can output the probability that a protein is localized to each compartment (not only the most likely compartment). Although for most proteins there is a single compartment that has a high likelihood, there do exist some proteins for which there is an (almost) equally high likelihood for several compartments. If PSLT is allowed to predict the two most likely compartments for each protein (referred to as the second-best test in Tables 2, 3, 5) in the yeast data set, the accuracy of our framework for predicting the correct localization increases to 71% as determined by an independent data set test with a coverage of 54%. As shown in Table 2, when PSLT is allowed to predict the two most likely compartments for each protein in the Hera human data set, the accuracy is 86% by 10-fold cross validation test and 97% by the self-consistency test. When the second-best test is used on the mouse data set, with PSLT trained on human proteins, the prediction accuracy is 92% as shown in Table 5. The Hera data set contains relatively few multicompartmental proteins (shown in Table 1). It is probable that some of the proteins predicted by PSLT to be multicompartmental are in fact multicompartmental proteins even though they are annotated as residing in only one compartment in public databases.
Because the mouse data set contains a higher proportion of multicompartmental proteins than does the Hera human data set, we use it to further explore the multicompartment prediction potential of PSLT. Because PSLT outputs the likelihood of localization to all compartments studied, it is possible to define proteins to be predicted as multicompartmental, if the likelihood difference between their two most likely compartments is less than a certain percentage of their most likely compartment. If we study the proteins for which the likelihoods of the two highest scoring compartments are within 50% of each other, we identify 20% of the multicompartmental mouse proteins from Table 1 and only 10% of the mouse proteins annotated as being localized to only one compartment in Table 1. Therefore, the true-positive rate is two times greater than the false-positive rate (and could be much greater if some proteins annotated as unicompartmental in SWISS-PROT actually are multicompartmental as predicted by PSLT). This holds true for all thresholds between 25% and 75%. This probably indicates that certain specific combinations of motifs are more frequently used by multicompartmental proteins than unicompartmental proteins.
Human Proteome Annotation
We used PSLT trained on the entire Hera human data set to annotate the 9793 human proteins contained in SWISS-PROT release 41.25; 7366 of the proteins (75%) were predicted by PSLT to be localized to one or several of the nine compartments considered. We took advantage of the multicompartment prediction capabilities of PSLT to identify proteins for which the likelihood of the two highest scoring compartments is within 50% of each other. Table 6 shows the number of proteins predicted to be in each compartment or pairs of compartments (for the multicompartmental proteins). In total, 16% of human proteins are predicted by PSLT to be localized in more than one compartment; this is probably a very conservative estimate. We verified the multicompartmental predictions with subcellular localization annotations from SWISS-PROT when available. The largest group of multicompartmental proteins are those predicted to be cytosolic and nuclear. Many of these proteins are involved in the binding, processing, and transport of pre-mRNA and/or mRNA molecules. This group of proteins also includes signaling regulators such as phosphatases, which are involved in the inactivation of MAP kinases and SMAD regulators. A second large group of predicted multicompartmental proteins are the mitochondrial and cytosolic proteins. Although some of these proteins are confirmed to be localized in both organelles by SWISS-PROT, many proteins in this group represent metabolic enzymes of which some isozymes are mitochondrial and others are cytosolic. This is the case for aspartate aminotransferases, aldehyde dehydrogenases, hydroxymethylglutaryl-CoA synthases, and creatine kinases. In this case, rather than predicting the localization of the specific protein, PSLT predicts the localizations of the family of proteins. PSLT was successful at predicting proteins that are annotated as both cytosolic and peroxisomal according to SWISS-PROT as well as proteins that are present on or that shuttle between the ER and Golgi. A small group of proteins was also predicted to be localized to the ER and the mitochondria, including Bcl2, Bax, and Bak family members, several of which are known to be found on these two organelles (for review, see Breckenridge et al. 2003). These predictions are available at www.mcb.mcgill.ca/~hera/PSLT.
Table 6.
Number of Human Proteins Predicted in Each Subcellular Compartment or Pair of Compartments Considered
| ER | Golgi | Cytosol | Nuclear | Pero | PM | Lyso | Mito | Secreted | |
|---|---|---|---|---|---|---|---|---|---|
| ER | 413 (5.6) | — | — | — | — | — | — | — | — |
| Golgi | 45 (0.6) | 125 (1.7) | — | — | — | — | — | — | — |
| Cytosol | 12 (0.2) | 9 (0.1) | 1168 (15.9) | — | — | — | — | — | — |
| Nuclear | 13 (0.2) | 15 (0.2) | 550 (7.5) | 2003 (27.2) | — | — | — | — | — |
| Pero | 6 (0.1) | 0 (0) | 22 (0.3) | 6 (0.1) | 53 (0.7) | — | — | — | — |
| PM | 93 (1.3) | 8 (0.1) | 8 (0.1) | 3 (<0.1) | 0 (0) | 1256 (17.1) | — | — | — |
| Lyso | 16 (0.2) | 5 (0.1) | 0 (0) | 0 (0) | 16 (0.2) | 1 (<0.1) | 84 (1.1) | — | — |
| Mito | 24 (0.3) | 0 (0) | 82 (1.1) | 8 (0.1) | 23 (0.3) | 2 (<0.1) | 2 (<0.1) | 247 (3.4) | — |
| Secreted | 9 (0.1) | 1 (<0.1) | 8 (0.1) | 20 (0.3) | 0 (0) | 160 (2.2) | 5 (0.1) | 5 (0.1) | 840 (11.4) |
Percentage of predictable proteins predicted to be in each compartment or compartment pair is shown in parentheses.
Pero indicates peroxisome; PM, plasma membrane; Lyso, lysosome; and Mito, mitochondrion.
Distribution of Motifs in Compartments
The prediction accuracy of PSLT is influenced by how well the different compartments and cellular processes are characterized by InterPro motifs and to what extent the different compartments share motifs. As shown in Table 7, some compartments such as the plasma membrane, nucleus, and extracellular protein group are better covered by InterPro motifs than are other compartments. In fact, >90% of proteins in these organelles contain at least one such motif. Proteins localized to the Golgi apparatus contain the fewest motifs. The average number of motifs per covered protein varies considerably between compartments. The plasma membrane contains by far the most motifs per protein, whereas the lysosome contains the least. The high number of motifs per protein in plasma membrane proteins could reflect the fact that this group is involved in many signaling events and proteins localized here are known to interact with many different proteins. It is also possible that some compartments are not as well characterized by InterPro motifs than others.
Table 7.
Characterization of Motif Distribution in Compartments
| Coveragea | Average number of motifs per covered protein | Proteins predicted on by PSLT using >1 motif | Motif frequencyb | Motif compartment specificity indexc | |
|---|---|---|---|---|---|
| ER | 83.6% | 2.0 | 56% | 2.3 | 0.49 |
| Golgi | 75.0% | 1.7 | 26% | 1.7 | 0.59 |
| Cytosol | 89.4% | 2.2 | 59% | 1.8 | 0.43 |
| Nucleus | 93.2% | 2.3 | 66% | 3.4 | 0.24 |
| Peroxisome | 86.4% | 2.1 | 55% | 1.3 | 0.67 |
| Plasma membrane | 94.1% | 3.4 | 80% | 3.6 | 0.47 |
| Lysosome | 86.8% | 1.4 | 25% | 1.5 | 0.49 |
| Mitochondria | 80.6% | 1.9 | 48% | 1.5 | 0.40 |
| Extracellular | 90.1% | 1.9 | 47% | 2.5 | 0.34 |
The coverage represents the percentage of human proteins in a given compartment that contain at least one InterPro motif.
The motif frequency is calculated as the total number of motifs in all proteins in a given compartment divided by the number of distinct motifs found in proteins in that compartment.
The motif compartment specificity index is calculated as the number of distinct motifs that are unique to a given compartment divided by the total number of distinct motifs in that compartment.
To determine whether PSLT predicts localization based mostly on the co-occurrence of motifs or on the presence of single motifs in proteins, we counted the number of proteins with localization that was predicted by PSLT using more than one motif. As shown in Table 7, in most compartments, >50% of proteins are predicted by the co-occurrence of motifs (as opposed to single motifs). Notable exceptions to this are the Golgi apparatus and the lysosome. In contrast, the localization of 80% of plasma membrane proteins are predicted by the co-occurrence of motifs, which is not surprising given the high average number of distinct motifs per protein in that compartment. It should be noted that PSLT also uses additional motif information (the presence/absence of signal peptides/anchors as well as the number of transmembrane domains) to predict localization, and as such, all proteins are actually predicted based on more than one motif. As proteins in compartments such as the lysosome and the Golgi apparatus become better characterized as a group and as the data sets of these organelles increase in size, the prediction accuracy of PSLT for these organelles should increase.
We also evaluated the extent of motif frequency in compartments. This is defined as the ratio of the total number of occurrences of motifs contained within all proteins in a given compartment to the total number of distinct motifs contained within all proteins in this compartment. Such motif frequency values may give an indication as to the degree of process diversity in the different compartments. As shown in Table 7, the motif frequency of the nucleus and plasma membrane is much higher than is the motif frequency for all other compartments. These two compartments both have large protein families with many members performing similar functions (e.g., the very large receptor families in the plasma membrane or the transcription factor families in the nucleus). Because PSLT predicts localization based on the combinatorial presence of motifs, the performance of the predictor is influenced by the motif frequency of the different compartments. In fact, the motif frequency correlates well with the sensitivity obtained by PSLT for the different compartments.
The extent of motif sharing between the different compartments can also affect the localization prediction accuracy of PSLT. The motif compartment specificity index shown in Table 7 is the ratio of the number of motifs that are unique to a given compartment divided by the total number of motifs in the compartment. The motif compartment specificity index can be an indication of the extent of process sharing as well as protein trafficking and structural motifs shared between the different compartments. The nucleus contains the lowest proportion of compartment-specific motifs. The peroxisome has the highest proportion of compartment-specific motifs; this may indicate that the peroxisome shares few processes with other compartments or that the proteins involved in processes it shares with other compartments are characterized by compartment-specific motifs. The motifs shared by proteins in the largest number of compartments are shown in Table 8. The proline-rich region is present in proteins in all compartments considered. The average number of compartments in which a given motif is present is 1.3.
Table 8.
Most Widely Used InterPro Motifs
| InterPro entry identifier | InterPro entry name | No. of proteins | No. of compartments |
|---|---|---|---|
| IPR000694 | Proline-rich region | 190 | 9 |
| IPR005225 | Small GTP-binding protein domain | 28 | 6 |
| IPR003593 | AAA ATPase domain | 28 | 6 |
| IPR002110 | Ankyrin repeat | 20 | 5 |
| IPR000719 | Protein kinase | 69 | 5 |
| IPR001478 | PDZ/DHR/GLGF domain | 16 | 5 |
| IPR000379 | Esterase/lipase/thioesterase active site | 22 | 5 |
| IPR005834 | Haloacid dehalogenase-like hydrolase | 13 | 5 |
| IPR001687 | ATP/GTP-binding site motif A (P-loop) | 12 | 5 |
Taken together, the ratios in Table 7 are an indication of how InterPro motifs characterize the cell, the various compartments, and processes. The InterPro classification scheme provides a novel way of describing these entities and of evaluating the extent of our knowledge of the different organelles.
DISCUSSION
We present a framework PSLT to predict the subcellular localization of proteins based on InterPro motifs and protein membrane domains. PSLT addresses the problems of low prediction accuracy for underrepresented compartments, the specific organelle prediction of transmembrane proteins, and most importantly, it allows an increased understanding (and prediction of) multicompartmental proteins. PSLT was initially built using only InterPro motifs. The addition of protein membrane domain information, including the presence of signal peptides and the number of transmembrane domains, always improves PSLT under every test we performed and can increase the prediction accuracy by up to 10%. This information is especially important to distinguish between plasma membrane and secreted proteins (data not shown). Because PSLT is built by using a Bayesian framework, it could be easily further improved by incorporating other types of relevant information.
When tested on human proteins, PSLT (based on InterPro motifs and protein membrane domains) achieves an overall prediction accuracy of 78% (with a coverage of 74%) and sensitivity and positive predictive values between 43% and 93% for all compartments considered, including compartments that contain few proteins. When PSLT is allowed to predict the two most likely compartments, its ability to predict at least one compartment well increases to >85% for human proteins. The ability to predict multicompartmental proteins allows us to estimate that at least 16% of human proteins are in fact multicompartmental. When used to predict proteins from closely related species such as the mouse, it achieves high prediction accuracies approaching those of the self-consistency test.
This type of predictor achieves a reasonable prediction accuracy because protein families and functional units are often colocalized in the cell. Even when proteins in different compartments can be characterized by the same InterPro motif and thus share some similar features, it is often the case that they also contain other InterPro motifs capable of differentiating them. Perhaps these additional motifs also modulate their function.
Because PSLT uses InterPro motifs to predict localization, it considers not only known organellar targeting motifs, as several other predictors have done in the past, but also the possible influence on localization of posttranslational modifications and protein-protein interaction domains and their combinations in proteins. Some posttranslational modifications are well known to influence protein localization. The most obvious example of this is probably the addition of lipid anchors to proteins. Phosphorylation has also been shown to cause a change in localization in many proteins, in particular in regulating the nuclear-cytoplasmic shuttling of many proteins (Hood and Silver 1999). In addition, there are several examples of proteins that do not contain any well-studied organellar targeting motifs but are known to be targeted to or retained in specific localizations through protein-protein interaction motifs. This is notably the case for nuclear proteins retained in the cytosol under some circumstances, such as NF-κB by IκB proteins (Karin 1999) or cytosolic signal transducer proteins imported in the nucleus independently of the Nuclear Localization Signal through direct interaction with the nuclear pore complex (Xu and Massague 2004). It is also speculated that some peroxisomal proteins (in particular subunits of oligomers) do not contain known targeting motifs and rely only on protein-protein interaction motifs for import into the peroxisome (Rachubinski and Subramani 1995; Hettema et al. 1999). As well, it is likely that some non-KDEL soluble ER proteins also rely on protein-protein interactions with KDEL-containing proteins to ensure retention in the ER. PSLT not only allows the prediction of these proteins lacking classical organellar targeting sequences but will also provide a method to investigate and better understand the extent of this phenomenon.
METHODS
Data Sets
The Hera database (www.mcb.mcgill.ca/~hera; Scott et al. 2004) was used to generate data sets to train and test PSLT. It currently contains 2216 human proteins annotated with subcellular localization information with a high degree of certainty (classification criteria “c” or “e” in Hera; Scott et al. 2004). These localizations were determined via studies found in the literature and annotations in public databases. Three other data sets were also used as independent testing sets in this study: the LIFEdb GFP human data set (Simpson et al. 2000), a yeast data set, and a mouse data set. All data sets are available at www.mcb.mcgill.ca/~hera/PSLT.
The LIFEdb database contained the experimentally determined subcellular localization information for ∼600 GFP-tagged novel human ORFs at the time of the study. We retrieved all proteins from the LIFEdb Web site (www.dkfz.de/LIFEdb/LIFEdb.aspx) and curated this set by eliminating all proteins (1) that are not annotated as being localized to at least one compartment considered in this study; (2) in which the amino acid sequence does not start with a methionine (i.e., probable partial transcripts); and (3) in which genes have multiple alternative transcripts and in which it was not possible to determine which protein product was actually characterized in the LIFEdb study. This filtering procedure left us with 392 proteins in the LIFEdb data set.
The yeast data set was generated by retrieving all proteins annotated with subcellular localization information from the Saccharomyces Genome Database (SGD; Dwight et al. 2002; www.yeastgenome.org/). Only proteins localized to at least one compartment considered in our present study were kept. The mouse data set contains all mouse proteins from SWISS-PROT release 42.10 that are annotated as being localized in at least one compartment considered in this study and start with a methionine. Table 1 shows the number of proteins in each of the compartments for each of the four data sets. To avoid counting proteins several times, those annotated as being in more than one compartment are identified as multicompartmental in Table 1. Thirteen proteins are present in both the Hera data set and the LIFEdb data set.
Generation of Maximal Motif Sets for Each Compartment
PSLT predicts the subcellular localization of proteins based on the presence of InterPro motifs in their amino acid sequence. Inter-Pro motifs are features present in known proteins but can also be identified in uncharacterized proteins. All proteins considered in this study were thus analyzed for the presence of such motifs by using InterProScan (Zdobnov and Apweiler 2001). We consider that a set of proteins that are all localized to the same compartment are colocalized. A set of motifs that co-occur in a (non-empty) set of colocalized proteins is called a motif set. A maximal motif set is a set of motifs that co-occur in a (nonempty) set of colocalized proteins with the property that none of the remaining motifs co-occur in this set. Note that for each compartment, there may exist more than one maximal motif set. That is, different subsets of colocalized proteins may have different maximal motif sets. Figure 1 illustrates the concept of maximal motif set.
Figure 1.

Visualization of maximal motifs. The hypothetical compartment C illustrated in this figure contains two maximal motif sets: {A,B,C} and {A,D}. Nonempty subsets of these motif sets are themselves considered to be motif sets but are not maximal (such as, e.g., {A,B}). {A} is a nonmaximal motif set that is common to both the {A,B,C} and {A,D} maximal motif sets.
We are interested in finding all maximal motif sets for each compartment. Although this is a computationally intractable problem, we use a dynamic programming approach to find these sets. Intuitively, we begin by identifying all motifs present in at least one protein localized to a given compartment. These are simple motif sets consisting of only one motif. Now, in a dynamic programming fashion, we extend each of the candidate motif sets by exhaustively adding all possible motifs one by one. If any such extended motif set has the property that all members of the motif set occur in a set of proteins that are colocalized, we keep this motif set. Otherwise, we discard this candidate. The algorithm is guaranteed to find the maximal motif sets (although it might require exponential time, the computation was feasible for our human proteins and the InterPro motifs). Throughout this process, each motif set (and all possible subsets of this motif set) are annotated with the proportion of proteins in the compartment under study that contain the motifs.
Likelihood of Localization
Given a protein and a motif set, M, it contains, we estimate its likelihood of being in a specific compartment, C, by using Bayes' rule:
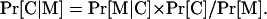 |
Here Pr[M| C] is the probability that the protein contains all motifs in set M given that we know the protein is localized to compartment C. This conditional probability is estimated in a straightforward way from the training set. Also, Pr[M] is the prior probability of a protein-containing motif set M regardless of the localization of the proteins containing M. This prior probability is estimated by determining the presence of InterPro motifs in all 9793 human proteins from SWISS-PROT release 41.25. Lastly, Pr[C] is the prior probability of a protein localizing to compartment C. These compartment priors were initially evaluated by averaging the number of human proteins annotated with localization information in three public databases: SWISS-PROT (Boeckmann et al. 2003), Human Protein Reference Database (www.hprd.org/; Peri et al. 2004), and LIFEdb (Simpson et al. 2000). The compartment priors were subsequently modified; this modification is described in “Compartment Prior Optimization.”
Subcellular Localization Prediction
PSLT is trained by identifying all maximal motif sets for all nine compartments considered in this study (Table 1). The training step (described above) generates an estimate of Pr[C| M] for each motif set M (and each subset of M). Because Pr[M] is estimated by using all 9793 human proteins from SWISS-PROT release 41.25, Pr[C] is optimized as described in “Compartment Prior Optimization” and Pr[M| C] is estimated by using the training set, the ∑C Pr[C| M] summed over all compartments is not necessarily one. However, we can treat these estimates of Pr[C| M] as likelihoods. These estimates of Pr[C| M] are then used to predict the localization of an unknown protein containing a set of motifs M′ by calculating the likelihood of being in each compartment given M′. If M′ does not match any motif set in a given compartment, then a subset of M′ with the highest localization likelihood is used to predict for that compartment. If no such subset exists, the likelihood of localization to that compartment is zero. The compartment that achieves the highest localization likelihood is predicted to be the subcellular localization of the unknown protein. If several compartments achieve high localization likelihoods, the protein can be predicted to be present in all of these compartments with a specific probability.
The prediction accuracy of PSLT is evaluated in the Results section by using several different tests. The total prediction accuracy is defined as the number of correctly predicted proteins in the test set divided by the total number of proteins in the test set. Because PSLT predicts the localization of proteins based on protein motifs, it will be unable to predict on proteins not containing such motifs or proteins containing motifs not used as motif sets for localization in the training phase of the algorithm. As a consequence, we exclude such proteins from our prediction accuracy statistics. However, for each reported accuracy estimate, the coverage (proportion of predictable proteins in the data sets) is given.
Compartment Prior Optimization
The initial compartment priors were modified by using a genetic algorithm. This algorithm aims to find the compartment priors that optimize the overall prediction accuracy. To assess accuracy during the compartment prior optimization process, we use a threefold cross validation test. Intuitively, the algorithm proceeds by “mutating” a randomly chosen compartment prior. It then readjusts the priors of the remaining compartments and calculates the localization likelihoods as previously explained. If the overall prediction accuracy increases compared to previously created versions of PSLT, this current set of compartment priors is retained by the genetic algorithm. If the overall prediction accuracy does not increase, this current set of compartment priors is retained (i.e., allowed to survive) with probability λ initialized to 0.1 and decreasing with time. The genetic algorithm creates a large population of candidate compartment priors. When the algorithm is allowed to execute for a sufficiently long number of “generations,” most of the candidates tend to converge to similar values. Figure 2 describes the compartment priors that allow to achieve the highest overall prediction accuracy.
Figure 2.

Optimized compartment priors. The compartment priors represent the estimate of the percentage of distinct proteins in each compartment. These compartment priors were optimized for PSLT as explained in the Methods section.
We note that other methods are available, such as structural EM learning, that provide alternative computational approaches in which both the compartment prior optimization and the selection of motif sets could potentially be done simultaneously. However, we chose the above the techniques given the amount of data available and the computational difficulty of these alternative methods.
Additional Protein Characteristics Considered
When available, additional protein characteristics can be used to modify the probability of localization to any given compartment. Such modifications are possible and easy to implement, because PSLT is built as a Bayesian network. If the new information (I) and the motif sets (M) are independent random variables, the new information can be used to modify the likelihood of localization generated by PSLT by simply applying Bayes' rule as follows:
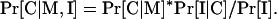 |
Essentially, we are “extending” the Bayesian network to include new information in the initial Bayesian network that uses the presence of motif sets to compute the Pr[C| M] likelihood. However, if the additional information and the motif sets are not independent random variables, the likelihood of localization should be calculated as follows:
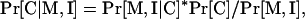 |
which is equivalent to constructing a global Bayesian network that incorporates simultaneously the motif sets and the new additional information.
We investigated the addition of information relating to the presence of signal peptides and the number of transmembrane domains. The presence of a signal peptide and the number of transmembrane domains were respectively evaluated by using SignalP software (Nielsen et al. 1997) and TMHMM software (Krogh et al. 2001). When this additional information is treated as a random variable that is dependent on the motif sets, the coverage of PSLT increases slightly but the prediction accuracy decreases by ∼10% (when compared with the original network using only the motif sets). Furthermore, the time required to train PSLT also increases substantially. However, if this additional information is considered to be independent of the motif sets, the coverage and the time required to train PSLT remain the same and the prediction accuracy increases by several percentage points (depending on the type of evaluation performed and the data set that is being tested). As a consequence, the networks reported in this study are built assuming the independence of the presence of a signal peptide, the number of transmembrane domains in a protein, and the InterPro motif sets. Signal peptides and transmembrane domains will be collectively referred to as protein membrane domains.
Acknowledgments
We are grateful to Dr. Scott Bunnell for critical reading of this manuscript. We wish to thank François Pepin for logistical support, Dr. Ted Perkins for useful discussions, and Dr. Richard Mott for kindly making his testing data set available. This work was supported by grants to D.Y.T. and M.H. from Genome Quebec/Genome Canada as well as to D.Y.T. from the Canadian Institutes of Health Research (CIHR). M.S.S. is a recipient of a Canada Graduate Scholarship (CGS) from CIHR.
Footnotes
[Supplemental material is available online at www.genome.org and www.mcb.mcgill.ca/~hera/PSLT.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2650004.
References
- Bell, A.W., Ward, M.A., Blackstock, W.P., Freeman, H.N., Choudhary, J.S., Lewis, A.P., Chotai, D., Fazel, A., Gushue, J.N., Paiement, J., et al. 2001. Proteomics characterization of abundant Golgi membrane proteins. J. Biol. Chem. 276: 5152-5165. [DOI] [PubMed] [Google Scholar]
- Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M.C., Estreicher, A., Gasteiger, E., Martin, M.J., Michoud, K., O'Donovan, O., Phan, I., et al. 2003. The SWISS-PROT protein knowledge base and its supplement TrEMBL in 2003. Nucleic Acids Res. 31: 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge, D.G., Germain, M., Mathai, J.P., Nguyen, M., and Shore, G.C. 2003. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608-8618. [DOI] [PubMed] [Google Scholar]
- Cai, Y.D., Liu, X.J., Xu, X.B., and Chou, K.C. 2002. Support vector machines for prediction of protein subcellular location by incorporating quasi-sequence-order effect. J. Cell. Biochem. 84: 343-348. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. 2001. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43: 246-255. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. and Cai, Y.D. 2002. Using functional domain composition and support vector machines for prediction of protein subcellular location. J. Biol. Chem. 277: 45765-45769. [DOI] [PubMed] [Google Scholar]
- Chou, K.C. and Cai, Y.D. 2003. A new hybrid approach to predict subcellular localization of proteins by incorporating gene ontology. Biochem. Biophys. Res. Commun. 311: 743-747. [DOI] [PubMed] [Google Scholar]
- Claros, M.G. and Vincens, P. 1996. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241: 779-786. [DOI] [PubMed] [Google Scholar]
- Drawid, A. and Gerstein, M. 2000. A Bayesian system integrating expression data with sequence patterns for localizing proteins: Comprehensive application to the yeast genome. J. Mol. Biol. 301: 1059-1075. [DOI] [PubMed] [Google Scholar]
- Dwight, S.S., Harris, M.A., Dolinski, K., Ball, C.A., Binkley, G., Christie, K.R., Fisk, D.G., Issel-Tarver, L., Schroeder, M., Sherlock, G., et al. 2002. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO). Nucleic Acids Res. 30: 69-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber, F. and Bork, P. 1998. Wanted: Subcellular localization of proteins based on sequence. Trends Cell. Biol. 8: 169-170. [DOI] [PubMed] [Google Scholar]
- ____. 1999. Evaluation of human-readable annotation in biomolecular sequence databases with biological rule libraries. Bioinformatics 15: 528-535. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. 2000. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300: 1005-1016. [DOI] [PubMed] [Google Scholar]
- Hettema, E.H., Distel, B., and Tabak, H.F. 1999. Import of proteins into peroxisomes. Biochim. Biophys. Acta 1451: 17-34. [DOI] [PubMed] [Google Scholar]
- Hood, J.K. and Silver, P.A. 1999. In or out? Regulating nuclear transport. Curr. Opin. Cell. Biol. 11: 241-247. [DOI] [PubMed] [Google Scholar]
- Horton, P. and Nakai, K. 1996. A probabilistic classification system for predicting the cellular localization sites of proteins. Proc. Int. Conf. Intell. Syst. Mol. Biol. 4: 109-115. [PubMed] [Google Scholar]
- Hua, S. and Sun, Z. 2001. Support vector machine approach for protein subcellular localization prediction. Bioinformatics 17: 721-728. [DOI] [PubMed] [Google Scholar]
- Huang, Y. and Li, Y. 2004. Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics 20: 21-28. [DOI] [PubMed] [Google Scholar]
- Huh, W.K., Falvo, J.V., Gerke, L.C., Carroll, A.S., Howson, R.W., Weissman, J.S., and O'Shea, E.K. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686-691. [DOI] [PubMed] [Google Scholar]
- Karin, M. 1999. The beginning of the end: IκB kinase (IKK) and NF-κB activation. J. Biol. Chem. 274: 27339-27342. [DOI] [PubMed] [Google Scholar]
- Krogh, A., Larsson, B., von Heijne, G., and Sonnhammer, E.L 2001. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305: 567-580. [DOI] [PubMed] [Google Scholar]
- Lu, Z., Szafron, D., Greiner, R., Lu, P., Wishart, D.S., Poulin, B., Anvik, J., Macdonell, C., and Eisner, R. 2004. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics 20: 547-556. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Xenarios, I., van Der Bliek, A.M., and Eisenberg, D. 2000. Localizing proteins in the cell from their phylogenetic profiles. Proc. Natl. Acad. Sci. 97: 12115-12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud, G.A. and Snyder, M. 2002. Proteomic approaches for the global analysis of proteins. Biotechniques 33: 1308-1316. [DOI] [PubMed] [Google Scholar]
- Mott, R., Schultz, J., Bork, P., and Ponting, C.P. 2002. Predicting protein cellular localization using a domain projection method. Genome Res. 12: 1168-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder, N.J., Apweiler, R., Attwood, T.K., Bairoch, A., Barrell, D., Bateman, A., Binns, D., Biswas, M., Bradley, P., Bork, P., et al. 2003. The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 31: 315-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, K. and Kanehisa, M. 1992. A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14: 897-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, H., Engelbrecht, J., Brunak, S., and von Heijne, G. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10: 1-6. [DOI] [PubMed] [Google Scholar]
- Parfrey, H., Mahadeva, R., and Lomas, D.A. 2003. α1-Antitrypsin deficiency, liver disease and emphysema. Int. J. Biochem. Cell. Biol. 35: 1009-1014. [DOI] [PubMed] [Google Scholar]
- Payne, A.S., Kelly, E.J., and Gitlin, J.D. 1998. Functional expression of the Wilson disease protein reveals mislocalization and impaired copper-dependent trafficking of the common H1069Q mutation. Proc. Natl. Acad. Sci. 95: 10854-10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri, S., Navarro, J.D., Kristiansen, T.Z., Amanchy, R., Surendranath, V., Muthusamy, B., Gandhi, T.K., Chandrika, K.N., Deshpande, N., Suresh, S., et al. 2004. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res 32: D497-D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachubinski, R.A. and Subramani, S. 1995. How proteins penetrate peroxisomes. Cell 83: 525-528. [DOI] [PubMed] [Google Scholar]
- Rapoport, T.A. 1992. Transport of proteins across the endoplasmic reticulum membrane. Science 258: 931-936. [DOI] [PubMed] [Google Scholar]
- Reinhardt, A. and Hubbard, T. 1998. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 26: 2230-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, M., Lu, G., Hallett, M., and Thomas, D.Y. 2004. The Hera database and its use in the characterization of endoplasmic reticulum proteins. Bioinformatics 20: 937-944. [DOI] [PubMed] [Google Scholar]
- Simpson, J.C., Wellenreuther, R., Poustka, A., Pepperkok, R., and Wiemann, S. 2000. Systematic subcellular localization of novel proteins identified by large-scale cDNA sequencing. EMBO Rep. 1: 287-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skach, W.R. 2000. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 57: 825-831. [DOI] [PubMed] [Google Scholar]
- Taylor, S.W., Fahy, E., and Ghosh, S.S. 2003. Global organellar proteomics. Trends Biotechnol. 21: 82-88. [DOI] [PubMed] [Google Scholar]
- von Heijne, G. 1990. The signal peptide. J. Membr. Biol. 115: 195-201. [DOI] [PubMed] [Google Scholar]
- Xu, L. and Massague, J. 2004. Nucleocytoplasmic shuttling of signal transducers. Nat. Rev. Mol. Cell. Biol. 5: 209-219. [DOI] [PubMed] [Google Scholar]
- Zdobnov, E.M. and Apweiler, R. 2001. InterProScan: An integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847-848. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- www.mcb.mcgill.ca/~hera; Human ER Aperçu home page.
- www.dkfz.de/LIFEdb/LIFEdb.aspx; LIFEdb database home page.
- www.yeastgenome.org/; Saccharomyces Genome Database (SGD).
- www.hprd.org/; Human Protein Reference Database home page.
- www.mcb.mcgill.ca/~hera/PSLT; Protein subcellular localization tool.
- www.inra.fr/predotar/; Home page of Predotar, a prediction service for identifying putative mitochondrial and plastid targeting sequences.